

Abstract
This article describes chemoselective and atom-economical methods for the stereoselective assembly of the A- and B-ring subunits of the bryostatins. A Ru-catalyzed tandem alkene-alkyne coupling/Michael addition was developed and applied to the synthesis of the bryostatin B-ring. We explored an acetylide-mediated epoxide-opening/6-exo-dig cyclization route to access the bryostatin A-ring; and the A-ring was eventually furnished via an acid-catalyzed tandem transketalization/ketalization sequence. In addition, a dinuclear zinc-catalyzed methyl vinyl ketone (MVK) aldol strategy was evaluated for the construction of the polyacetate moiety. Utilization of these methods ultimately led to the rapid assembly of a northern bryostatin fragment containing both the A- and B-ring subunits.
Keywords: aldol reaction, asymmetric synthesis, chemoselectivity, diastereoselectivity, enantioselectivity, enynes, Michael addition, natural products
Introduction
As part of our continuing efforts toward the total synthesis of bryostatins,1 we explored new methods and strategies for assembling the A- and B-rings (Figure 1). The structure of the bryostatin B-ring features a 2,6-cis-disubstituted tetrahydropyran with a geometrically defined exocyclic conjugated methyl ester at the C13 position (bryostatin numbering). Controlling the olefin geometry of this α,β-unsaturated ester has been one of the major challenges for syntheses of the B-ring of the bryostatins.2 Use of a chiral Horner-Wadsworth-Emmons (HWE) reagent developed by Evans2a proved to be effective in the syntheses of bryostatin 2 and 3; however, a stoichiometric amount of expensive chiral reagent was required for this non-asymmetric transformation, and the E/Z ratio was only 6:1 to 8:1 (Figure 2). We envisioned that by utilizing our Ru-catalyzed enyne coupling reaction with multi-functionalized substrates, we would be able to generate the required B-ring motif in a highly regio- and chemoselective fashion. A single olefin isomer is expected and this approach would also be catalytic and atom economic.
Figure 1.

Retrosynthetic analysis of the A- and B-ring subunits of the bryostatins.
Figure 2.

Installation of the geometrically defined exo-enoate of the bryostatins.
The bryostatin A-ring region contains multiple 1,3-anti diol units, thus an aldol approach seems to be one of the most straightforward ways to provide this motif. Indeed, in all the previous total syntheses, aldol approaches have been employed for the A-ring synthesis; moreover, these aldol reactions all relied on generation of a stoichiometric amount of metal enolates.3 For example, in Masamune’s synthesis, boron enolates were utilized to form the C9–C10 and C2–C3 bonds;3a–c in Evans’ synthesis, a boron enolate was used to form the C6–C7 bond and a silyl enol ether was used to form the C4–C5 bond;2a–d in Nishiyama/Yamamura’s synthesis of bryostatin 3, a lithium enolate was employed to form the C4-C5 bond.3d–h From an atom-economic viewpoint, use of a catalytic asymmetric direct aldol reaction would allow access to these poly 1,3-diol units in a more efficient manner. Recently, we have developed a highly enantioselective dinuclear Zn-catalyzed direct aldol reaction, in which methyl vinyl ketone (MVK) was used as a bifunctional nucleophile (Figure 2).4 With this method, we envisioned that the C3–C9 fragment along with the C5 and C7 stereocenters would be constructed in a rapid fashion; meanwhile, the C3–C4 olefin could provide a functional handle for further elaboration and union with the other fragments. In this article, we describe our detailed efforts toward developing methods towards atom-economic and stereoselective syntheses of the bryostatin A- and B-rings.
Results and Discussion
First Generation Strategy
Our initial strategy for the synthesis of the northern A,B-rings is shown in Scheme 1. We envisioned that the C9–methyl ketal in compound 2 could arise from the addition of CH3OH to the C9–C10 enol ether in 3 under acidic conditions. Enol ether 3 could be secured through a 6-exo-dig oxypalladation cyclization from homopropargyl alcohol 4. Inspection of intermediate 4 suggested disconnection at the C8–C9 bond, resulting in fragments 5 and 6, with comparable complexity. In the forward direction, when using a nucleophile derived from alkyne 5, attack at the C8 terminus of epoxide 6 was expected to furnish secondary alcohol 4 in the presence of a Lewis acid. Alkyne fragment 5 containing the B-ring subunit would be derived from alkene 7, which can be ultimately prepared from a Ru-catalyzed alkene-alkyne coupling followed by a Pd-catalyzed tandem cyclization. Epoxide fragment 6 could eventually be prepared from a dinuclear zinc prophenol-catalyzed aldol addition between aldehyde 8 and MVK (9).
Scheme 1.

First generation approach for the construction of the A- and B-rings of the bryostatins.
Synthesis of Alkyne Fragment 5
The synthesis of terminal alkyne 5 commenced with (S)-glycidol 12, which was converted to alkynyl alcohol 10 in two straightforward steps: PMB protection followed by epoxide opening with lithium (trimethylsilyl)acetylide (Scheme 2).6 The coupling between alkyne 10 and alkene 11 using the tandem procedure7 of alkene-alkyne coupling followed by Pd AAA went uneventfully to give tetrahydropyran 7. However, excess alkene 11 (5 equiv) was needed for complete consumption of alkyne 10, and tetrahydropyran 7 had the same Rf value as diene 11. Thus, a two-step procedure was used to facilitate product purification. It is noteworthy that the stereochemistry at C11 is under ligand control: when (R,R)-LST standard Trost ligand was employed, the product was obtained as a 97:3 diastereomeric mixture favoring the thermodynamically disfavored 2,6-trans-tetrahydropyran; whereas, the (S,S)-LST gave a 98:2 ratio favoring the cis-tetrahydropyran 7. Interestingly, use of dppe as a ligand gave nearly a 1:1 ratio of a cis:trans isomeric mixture. By this procedure, tetrahydropyran 7 was obtained on gram scale.
Scheme 2.

A tandem Ru-catalyzed ene-yne coupling/Pd-catalyzed allylic alklylation strategy for the formation of the B-ring.
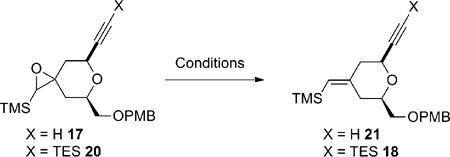
One efficient way to convert alkene 7 to alkyne 5 would be to cleave the terminal alkene oxidatively, followed by alkynylation of the resulting aldehyde, and then to transform the OPMB ether into an olefin (Scheme 3). Unfortunately, the electron-rich disubstituted vinylsilane was also reactive under the dihydroxylation conditions. Under a variety of oxidative cleavage conditions, we were not able to achieve selective oxidation of the terminal olefin with OsO4 (7 → 14).8 Given the electron-richness of the vinylsilane in diene 7 and relative insensitivity of the epoxidation reaction with peracids towards steric factors, it was decided to protect the vinylsilane as an epoxysilane. Treatment of vinylsilane 7 with 1.5 equivalents of m-CPBA at 0 °C gave epoxysilane 16 in 95% yield as a 3:1 diastereomeric mixture (Scheme 4). Oxidative cleavage of the monosubstituted olefin followed by alkyne formation9 went smoothly to deliver alkyne 17 in 83% yield over three steps.
Scheme 3.

Attempted direct functionalization of terminal olefin proved non-selective.
Scheme 4.

Protection of vinyl silane as an epoxide in order to achieve selective functionalization of the terminal olefin.
A deoxygenation reaction at this stage to unmask the exocyclic vinylsilane proved to be non-trivial (Table 1). Martin and Ganem reported a mild Rh-catalyzed deoxygenation of epoxides with dimethyl diazomalonate in refluxing benzene with retention of olefin geometry.10 Under their conditions the deoxygenation of 17 gave the desired vinylsilane 21 as a single isomer (entry 1), albeit in low yield (15%). Switching the solvent to chlorobenzene gave only trace amounts of product (entry 2). Using alumina with a catalytic amount of HgCl2 in refluxing toluene/isopropanol11 led to clean deoxygenation by both TLC and crude 1H NMR analysis (entry 3). Unfortunately, the product was obtained as an inseparable mixture of exocyclic vinylsilane isomers. Deoxygenation employing low valent tungsten, originally reported by Sharpless,12 gave no reaction at −78 °C and decomposition upon warming (entry 4). Treatment with (PhO)3PCH3I13 in the presence of BF3•OEt2 resulted in no reaction at room temperature (entry 5). As the low yield in entry 1 was likely caused by reaction between the terminal alkyne and a Rh-carbenoid, the terminal alkyne was protected with a TES group and the resulting silylalkyne 20 was subjected to the Rh-catalyzed deoxygenation (Rh2(OAc)4, benzene, 80 °C). Gratifyingly, the desired vinylsilane (18) was obtained in 52% yield. With compound 18 in hand, alkyne 5 was obtained smoothly using a four-step sequence of PMB cleavage, IBX oxidation, Wittig olefination, and TES removal.
Table 1.
Deoxygenation Studies for Compounds 17 and 20
 | ||||
|---|---|---|---|---|
| Entry | X | Conditions | Product | Yield |
| 1 | H | 10 mol% Rh2(OAc)4, C6H6, 80 °C | 21 | 15% |
| 2 | H | 10 mol% Rh2(OAc)4, C6H5CI, 80 °C | 21 | trace |
| 3 | H | Al/HgCI2, C6H6/i-PrOH, reflux | 21 + double bond isomer | clean reaction |
| 4 | H | WCI6, 3 equiv. n-BuLi, −78 °C → RT | baseline material | 0% |
| 5 | H | (PhO)3PCH3I, BF3•OEt2, RT | recovered 17 | 0% |
| 6 | TES | 10 mol% Rh2(OAc)4, C6H6, 80 °C | 18 | 52% |
Alternatively, the alkyne B-ring subunit could be synthesized by taking advantage of the internal symmetry present in pyran 7. If the exocyclic double bond geometry is inverted, the terminal alkene can be mapped into the product. In order to obtain alkyne 5 with the desired absolute stereochemistry for the bryostatin synthesis, (R)-glycidol and (R,R)-Trost ligand would be needed.
This route was explored with the previously prepared epoxysilane 16 (Scheme 6). Bromide 24 was obtained from epoxide 16 in a two-step sequence (bromohydrin formation and siloxy elimination) in 81% yield.14 Subsequent oxidation followed by alkyne formation gave alkyne 25, which possessed all the required functionalities for the epoxide coupling strategy (see Scheme 13) but had the opposite stereochemistry.15 Changing the starting glycidol and the Trost ligand would allow access to the desired enantiomer.
Scheme 6.

Inversion of the olefin geometry through manipulation of the epoxide.
Scheme 13.

Functionalization of the vinyl silane.
Efforts Towards the Synthesis of Epoxide Fragment
We envisioned that epoxide fragment 6 could be accessed by an ester aldol or Reformatsky reaction from aldehyde 26 (Scheme 7). Aldehyde 26 could be derived from allyl alcohol intermediate 27 that can be ultimately prepared from diol 28.
Scheme 7.

Retrosynthetic analysis of the 1,3-anti-diol region of the bryostatins.
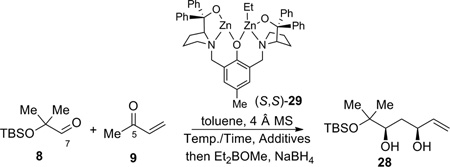
In the forward direction, by utilizing the dinuclear zinc-catalyzed direct aldol addition reaction4 followed by cis-reduction of the crude aldol adduct, diol 28 was rapidly obtained from aldehyde 8 and MVK 9 (Table 2). The yield, dr and enantioselectivity were carefully optimized as shown in Table 2. Initially, the temperature effect was examined (entries 1–3). The reaction showed an inverse relationship between temperature and enantioselectivity, a trait that has been observed previously with this catalyst system.16 The reaction gave better yield at higher temperatures, though at ambient temperature the product was obtained impurely (entry 3). Longer reaction times caused increased decomposition andlower isolated yields (entries 3–5). Increasing the reaction temperature to 50 °C led only to decomposition (entry 6). Surprisingly, using undistilled methyl vinyl ketone at 4 °C led to an improvement in the isolated yield (entry 7). As commercial methyl vinyl ketone contains a small amount of acetic acid and hydroquinone to prevent polymerization, acetic acid is probably responsible for the increase in yield. Indeed, adding an additional 1% acetic acid gave an even better result (entry 8). It is likely that acetic acid could serve as a buffer for the system, thus mitigating the undesired elimination pathway. Lowering the catalyst loading gave a slightly lower yield, with only a modest drop in enantioselectivity (entry 9). Raising the temperature gave poor yield, even at shorter times (entries 10–11). Increasing the added acetic acid to 2% resulted in the observation of only trace product (entry 12). Use of methyl β-hydroxypropionate, an achiral additive that found some success in a similar system gave little conversion.16 From these studies, the most practical set of conditions emerged as shown in entry 8, and the desired reduced adduct 28 was obtained in 52% yield over two steps with 97% ee and 31:1 dr in favor of the desired diastereomer. This method provides a facile route for the installation of two of the stereocenters of epoxide fragment 6.
Table 2.
Dinuclear zinc-catalyzed direct aldol addition reaction optimization.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | mol% cat. | Temp./Time | Additives | Yielda | eeb | drc |
| 1 | 10 | −15 °C/7h | 5 equiv i-PrOH | 20% | 93% | 24:1 |
| 2 | 10 | 4 °C/7h | 5 equiv i-PrOH | 25% | 97% | 22:1 |
| 3 | 10 | 25 °C/7h | 5 equiv i-PrOH | 41% | 97% | 18:1 |
| 4 | 10 | 25 °C/9h | 5 equiv i-PrOH | 25% | 96% | 38:1 |
| 5 | 10 | 25 °C/24h | 5 equiv i-PrOH | 25% | 98% | 26:1 |
| 6 | 10 | 50 °C/9h | 5 equiv i-PrOH | 0% | -- | -- |
| 7 | 10 | 4 °C/7h | 5 equiv i-PrOH, 0.6% AcOH 3% Hydroquinone |
45% | 98% | 35:1 |
| 8 | 10 | 4 °C/7h |
5 equiv i-PrOH, 1.6% AcOH 3% Hydroquinone |
52% | 97% | 31:1 |
| 9 | 5 | 4 °C/7h | 5 equiv i-PrOH, 1.6% AcOH 3% Hydroquinone |
50% | 90% | 25:1 |
| 10 | 10 | 25 °C/7h | 5 equiv i-PrOH, 1.6% AcOH 3% Hydroquinone |
32% | 98% | 17:1 |
| 11 | 10 | 25 °C/4h | 5 equiv i-PrOH, 1.6% AcOH 3% Hydroquinone |
40% | 92% | 50:1 |
| 12 | 10 | 4 °C/20h | 5 equiv i-PrOH, 2.6% AcOH 3% Hydroquinone |
trace | -- | -- |
| 13 | 10 | 4 °C/7h | 16 mol% methyl β-hydroxypropionate |
16% | 84% | 25:1 |
Isolated yield.
ee was determined by chiral HPLC
dr was determined by 1H NMR.
Protection of the less hindered allylic alcohol as a PMB ether was envisioned in order to differentiate the two alcohols, which vary in their steric environment (Scheme 8). After PMB installation, transformation of the more hindered alcohol into a leaving group would allow for epoxide formation. Unfortunately, chemoselective PMB protection of the allylic alcohol proved problematic. For example, treatment of 30 with PMBCl and sodium hydride in DMF, or use of PMB trichloroacetimidate with catalytic triflic acid, gave only recovered starting material. Attempted formation of the tin acetal was unsuccessful, even after reflux for 36 hours. Use of catalytic TBAI to further activate the PMBCl in situ gave no desired product at ambient temperature, but did give the desired product at reflux, though as a mixture of the two isomeric products.
Scheme 8.

Failed elaboration of diol 28.
An alternative approach was undertaken in which the diol was transformed into diastereomeric cyclic sulfites 33, thus activating the C7 alcohol for intramolecular SN2 inversion to form the epoxide, while differentiating the two alcohols by tying them back in a ring and thus preventing attack of the tertiary alcohol at the allylic C5 site (Scheme 9). After generation of the diastereomeric sulfites, a variety of conditions were attempted to desilylate and form the desired epoxide 27. Treatment with an anhydrous fluoride source, tetrabutylammonium triphenyldifluorosilicate (TBAT) or 3HF•NEt3, did not give the desired product or recovered starting material. When TBAF was used as the fluoride source in THF, the desired product was isolated in 18% yield at ambient temperature, and 48% under reflux conditions. The reaction was spot to spot by TLC, but the isolated yield was low. This is likely caused by the volatility of product 27, or the instability of the compound, as it was found to decompose upon storage at −15 °C. In an effort to solve both of these problems, the epoxide was submitted to silylation conditions with TBDPSCl and imidazole in DMF, but this resulted only in decomposition.
Scheme 9.

Epoxide formation and elaboration.
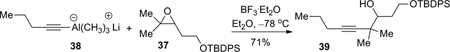
Meanwhile, we conducted a parallel study to investigate the feasibility of the proposed alkyne and epoxide coupling (see Scheme 1). Sterically, although the C8 position is more substituted and therefore more hindered than C7, nucleophilic attack at C7 would also be difficult due to its pseudoneopentyl nature (next to C8). Accordingly, the steric difference between C7 and C8 during nucleophilic attack could be small. On the other hand, when Lewis acids are employed in the reaction, the coordination of Lewis acids to the epoxide will weaken the C8–O bond more than C7–O bond because first, the relief of steric congestion around C8, and second the ability for a tertiary carbon to stabilize positive charge. Therefore, electronically, nucleophilic attack at the C8 terminus of the epoxide would be favored. A recent publication from Zhao and Pagenkopf provided further support for this analysis.17 To this end, model epoxide 37 was synthesized from 3-pentyne-1-ol 34 in three steps (Scheme 10).18 Carbometallation of 3-pentyne-1-ol 34 (TiCl4, Al(CH3)3, CH2Cl2/THF, −78 °C) was clean but incomplete. The product was obtained as a mixture of starting material 34 and olefin 35 in approximately a 1:2 ratio, which was carried over two steps (TBDPS protection and epoxidation) to provide epoxide 37.
Scheme 10.

Preparation of model epoxide 37.
This set the stage for examination of the proposed epoxide opening reaction. Following a procedure from Zhao and Pagenkopf,17 the lithium acetylide trimethylaluminium ate complex 38 (derived from pentyne) reacted with trisubstituted epoxide 37 in the presence of BF3•OEt2, providing the secondary alcohol 39 that indicated the attack at the more hindered position was indeed favored (equation 1). Formation of the other regioisomer could not be completely excluded since the 1H NMR was not very clean; however, the major signal at 3.64 ppm (CDCl3, ddd, J = 1.5, 4.0, 10.5 Hz, assigned to the methine proton next to the hydroxyl group in 39) indicated that the secondary alcohol 39 was the major component (>85%). The use of the trimethylaluminum ate complex is crucial since lithiated pentyne gave no reaction.
 |
(1) |
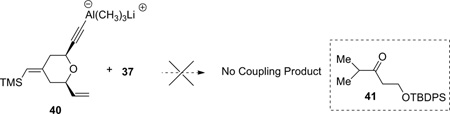
Encouraged by the model study, the coupling between alkyne 5 and epoxide 37 was subsequently attempted. Unfortunately, under the Zhao and Pagenkopf conditions, no reaction occurred. Upon warming, epoxide 37 underwent rearrangement to give ketone 41 (equation 2). Changing the solvent (THF, hexane, and toluene), the Lewis acid ((CH3)3Al, Zn(OTf)2, Al(OTf)3, and B(C6F5)3), or the order of addition did not give promising results.
 |
(2) |
Second Generation Strategy
Although there were still other possible ways to carry out the epoxide opening strategy, the difficulties associated with the ring-opening reaction prompted us to reevaluate our approach toward the northern fragment. At the outset, we decided to form the hindered C8 −C9 bond early in the synthesis. Shown in Scheme 11 is our second-generation strategy. We envisioned that the A-ring moiety along with C9–methyl ketal in 2 could be installed via an acid-catalyzed ketalization with CH3OH from hydroxyketone 42. The presence of the C-O bond at C11, which is β to the C9–ketone, suggested a Michael addition to construct the C11–O bond and the B-ring simultaneously. The requisite α,β-unsaturated ketone 43 contains a 1, 4–diene, which is the characteristic functionality obtained from our Ru-catalyzed alkene–alkyne coupling. Enone fragment 44 could be derived from aldehyde 45 via functional group manipulations, in which the C2–C3 bond could be formed by either an ester aldol or a Reformatsky reaction. As an analogy to our first generation strategy, the aldehyde intermediate (45) would be prepared from diol 46 that could be efficiently synthesized via a dinuclear Zn-catalyzed direct aldol reaction from aldehyde 47 and MVK 9.
Scheme 11.

Retrosynthetic anaylsis featuring an ene-yne coupling/Michael addition.
This new strategy revealed a novel enyne coupling reaction to construct the B-ring of bryostatin, because one of the coupling partners, the β,γ-enone, had never been used in the Ru-catalyzed ene reaction before. One immediate concern was the stability of β,γ-enones in the presence of cationic Ru-catalyst since they are prone to isomerize in the presence of Brönsted or Lewis acids. Thus, we decided to address these issues with a model system (equation 3).15
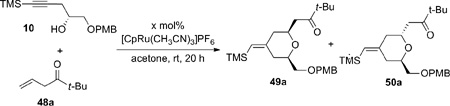 |
(3) |
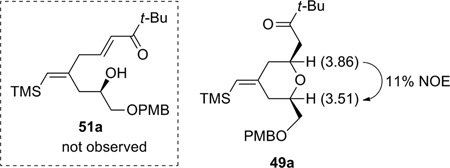
The initial model studies gave surprising but gratifying results (Table 3). When a 1:1 mixture of alkyne 10 and β,γ-enone 48a was treated with 10 mol% [CpRu(CH3CN)3]PF6 in acetone at room temperature, diene 51a was not observed. Instead the cyclized product, 2,6-cis-dihydropyran 49a, was isolated in 31% yield (entry 1). The cis configuration was confirmed by nOe studies. The minor 2,6-trans product 50a was not isolated in this case. To achieve both reasonable conversion and yield, excess enone was employed (entry 2). Higher concentration led to increased conversion, as expected for a typical bimolecular reaction (entry 3). Interestingly, increased catalyst loading did not improve the yield substantially (entry 4). The best yield was achieved using 3 equiv of enone with acetone as the solvent at 0.4 M concentration of alkyne (entry 5) (Recovery of enone 48a was not attempted due to its volatility.)
Table 3.
Ru-catalyzed ene-yne/Michael addition optimization.
| Entry | Concentration (M) | Catalyst loading (x mol%) | 48a:10 | Yield 49a (50a) |
|---|---|---|---|---|
| 1 | 0.1 | 10 | 1 | 31% |
| 2 | 0.1 | 10 | 1.8 | 43% (6%) |
| 3 | 0.4 | 10 | 1.2 | 41% (6%) |
| 4 | 0.1 | 30 | 1 | 36% |
| 5 | 0.4 | 10 | 3 | 68 (15%) |
 | ||||
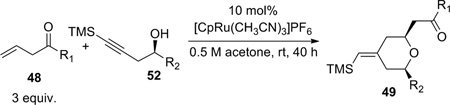
Encouraged by this model study, we decided to probe the scope and limitations of this reaction, with an emphasis on substrates that may prove useful for construction of the A- and B-rings of bryostatin. Accordingly, a variety of homopropargylic alcohols and β,γ-enones were prepared (Scheme 12). In general, the homopropargylic alcohols were synthesized via epoxide opening with lithium (trimethylsilyl)acetylide (A, Scheme 12); β,γ-enones were accessed either through a two-step sequence (allylation then oxidation, B, Scheme 12) or a BiCl3–NaI or BiCl3–ZnI2 catalyzed19 Friedel-Crafts acylation reported by Le Roux and Dubac. (C, Scheme 12).
Scheme 12.

Synthesis of Ru-catalyzed ene-yne coupling substrates.
The propargylic alcohols and β,γ-enones were then subjected to the optimized coupling/cyclization conditions. The results are summarized in Table 4. A variety of functional groups, including p-methoxybenzyl, TBS, and acetyl are tolerated. Since excess enone was necessary to achieve a reasonable conversion and yield, the ability to recover the unreacted enone was crucial. To our delight, enones with tert-alkyl groups could be mostly recovered (entries 3, 5, and 6), which boded well for our bryostatin synthesis. On the other hand, when dec-1-ene-4-one was used, it was recovered together with 15–20% of the α,β-enone isomer as an inseparable mixture (entry 2). Interestingly, complete chemoselectivity was observed for two different types of double bonds (entry 6). No product derived from the coupling of alkyne with the double bond bearing an allylic oxygen was detected. The cis:trans ratios ranged from 5:1–8:1. Preliminary studies suggested the cis- and trans-isomers are not equilibrating under the reaction conditions. Attempts to establish the thermodynamic ratio were thwarted by acid- and base-mediated decomposition.
Table 4.
Tandem alkyne-enone coupling/Michael addition reaction.
 | |||
|---|---|---|---|
| Entry | R1 | R2 | Yield 49 (recovered 48 equiv.) |
| 1 | t-Bu | Et | 80% |
| 2 | n-Hex | Et | 62% |
| 3 |  |
CH2OPMB (10) | 69% (1.8) |
| 4 | 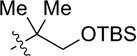 |
CH2OPMB (10) | 58% |
| 5 | 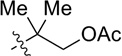 |
CH2OPMB (10) | 77% (1.9) |
| 6 | 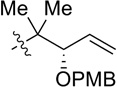 |
CH2OPMB (10) | 39% (1.9) |
The geometrically defined exocyclic vinylsilane resulting from the ruthenium-catalyzed enyne-coupling/Michael addition provides a convenient handle for further functionalization (Scheme 13). Protiodesilylation of 49c went smoothly to give terminal alkene 53 without double bond migration. Vinyl iodide formation also proceeded efficiently and the product was subsequently carbonylated to give α,β-unsaturated methyl ester 54. Furthermore, we were able to invert the double bond geometry through an epoxidation, bromohydrin formation, and siloxy elimination sequence to give inverted bromide 56 (Scheme 14), which was also carbonylated to give methyl enoate 57 with high efficiency. Thus, from one geometrically defined vinylsilane, either geometric isomer of the exocyclic enoate is cleanly available.
Scheme 14.

Inversion of the olefin geometry.
Synthesis of β,γ-Enone Fragment
Efforts were next taken to synthesize the β,γ-enone fragment for the real system. Aldol adduct 58 is available in good yield and excellent enantioselectivity from the dinuclear zinc-catalyzed MVK aldol reaction using aldehyde 47 (containing one more methylene unit than aldehyde 8 used for the epoxide-opening strategy).20 The opposite enantiomer of the catalyst (R,R) was used for this reaction, as the C7 stereocenter would not be inverted using this strategy (Scheme 15).
Scheme 15.

Prophenol zinc-catalyzed asymmetric aldol reaction.
To obtain the desired stereochemistry at C5 position, a trans reduction of β-hydroxyl ketone 58 was required. Evans’ acetoxyborohydride reagent is a commonly employed strategy for the synthesis of anti diols from β-hydroxyketones.21 However, this reaction proceeds slowly at low temperature, but gives poorer selectivities at higher temperatures. In order to balance reactivity and selectivity, a brief survey of reaction conditions was undertaken (Table 5). Using sodium acetoxyborohydride at −10 °C gave incomplete conversion and moderate selectivity (entry 1). Switching to the more reactive tetramethylammonium counterion, the reaction could be lowered to −20 °C, and both the yield and diastereoselectivity were improved (entry 2). Increasing the amount of the reducing agent increased the conversion and yield, though it lowered the diastereoselectivity (entry 3).
Table 5.
Trans reduction of β-hydroxyl ketone 58.
| Entry | Conditions | Yield 46 (Recovered 58)[a] | dr[b] |
|---|---|---|---|
| 1 | NaB(OAc)3H (5 equiv), AcOH/acetone, −10 °C | 46% (11%) | 5:1 |
| 2 | NMe4B(OAc)3H (5 equiv), AcOH/acetone, −20 °C | 54% (26%) | 9:1 |
| 3 | NMe4B(OAc)3H (10 equiv), AcOH/acetone, −20 °C | 69% (0%) | 5:1 |
Isolated yield.
dr was determined by 1H NMR.
Diol 46 was then protected with the cyclopentanone-derived ketal (Scheme 16). Subsequent deprotection of the silyl group provided free alcohol 60, which set the stage for the installation of the β,γ-unsaturated ketone. Unexpectedly, oxidation of the primary alcohol by using IBX or TPAP/NMO caused a rearrangement of the cyclic acetal, and the resultant allylic alcohol was oxidized to give enone 61. Fortunately, oxidation under Moffat-Swern conditions, followed by quenching with allylmagnesium bromide, provided the desired homoallylic alcohol. Subsequent oxidation of the resulting diastereomeric alcohols with Dess-Martin periodinane furnished the desired β,γ-unsaturated ketone 62 in 71% yield over two steps.
Scheme 16.

Prophenol zinc-catalyzed asymmetric aldol reaction.
Proof of Principle: Synthesis of Fragment 132 Containing Both the A- and B-Ring Subunits
At this stage, we investigated the ene-yne coupling/1,4-addition reaction to construct the bryostatin B-ring (Scheme 17). Gratifyingly, submission of β,γ-unsaturated ketone 62 and alkyne 10 to the reaction conditions developed earlier (see Table 3 and 4) afforded the desired tetrahydropyran 63, in spite of the presence of a second terminal olefin. A portion of the starting alkene 62 could be recovered. The low yield of this reaction was attributed to the instability of the ketal moiety to the Lewis acidic ruthenium catalyst, as well as competitive binding of allyl ether double bond with the desired binding of the other. Since this effort was only a proof-of-principle, no effort was made to optimize the yield for this example. Nevertheless, the success of this method in uniting the two components and forming the desired bryostatin B-ring proved the utility of the Ru-catalyzed tandem coupling/cyclization reaction as a viable strategy for the bryostatin synthesis. Subsequently, treatment of tetrahydropyran 63 with a catalytic amount of CSA in MeOH resulted in a tandem transketalization-ketalization sequence, which ultimately provided compound 67 containing both the A- and B-rings of the bryostatins.
Scheme 17.

Completion of the A- and B-rings of the bryostatins.
Conclusion
During our continuing efforts towards the total synthesis of the bryostatin family, we have developed distinct methods for the stereoselective assembly of the A-, and B-ring subunits. In the bryostatin B-ring synthesis, a new method for the stereoselective synthesis of tetrahydropyran rings was disclosed, which features a Ru-catalyzed tandem alkene-alkyne coupling/Michael addition sequence. For the assembly of the bryostatin A-ring subunit, a dinuclear zinc-catalyzed MVK aldol strategy has also been explored for the construction of the polyacetate entities. Furthermore, an acid-catalyzed cascade transketalization/ketalization sequence provided an advanced intermediate containing both the A- and B- rings. All of these highly chemoselective and/or atom-economical methods set the stage for our further advances towards the total synthesis of the bryostatins.22
Experimental Section
Experimental details for the synthesis of the following Compounds and Compounds 16–19, 5, 24, 25, 49b–g, 53, 54–57, 59, 60 and 62 as well as spectral data are available in the Supporting Information.
General Procedure for Carrying out Ru-catalyzed Alkyne-β,γ-enone Coupling Reaction: A point-shaped vial, equipped with a stir bar and capped with a rubber septum, was flame-dried and cooled under argon atmosphere. The septum was temporarily removed to allow the addition of alkyne (0.25 mmol), enone22 (0.75 mmol), and acetone (0.5 mL). After stirring at room temperature for 5 min to ensure the formation of a homogeneous solution, the septum was again removed and [CpRu(CH3CN)3]PF6(11 mg, 0.025 mmol) was added quickly. The vial was sealed and the septum was wrapped with black electric tape. The reaction was stirred at room temperature for 40 h, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel to give the desired product.
Compound 49a: 1H NMR (500 MHz, CDCl3) d 7.25 (d, J = 8.6 Hz, 2H), 6.87 (d, J = 8.8 Hz, 2H), 5.27 (s, 1H), 4.49 (dd, J = 11.8, 15.5 Hz, 2H), 3.88–3.83 (m, 1H), 3.80 (s, 3H), 3.53–3.49 (m, 1H), 3.49–3.42 (m, 2H), 2.91 (dd, J = 5.7, 16.7 Hz, 1H), 2.54 (dd, J = 7.0, 17.0 Hz, 1H), 2.38 (dt, J = 2.0, 13.5 Hz, 1H), 2.24 (dt, J = 2.0, 13.0 Hz, 1H), 1.98 (m, 2H), 1.12 (s, 9H), 0.08 (s, 9H); 13C NMR (125 MHz, CCl3) δ 213.5, 159.1, 152.4, 130.3, 129.2, 123.9, 113.7, 77.4, 75.1, 72.9, 72.8, 55.2, 45.0, 44.2, 43.1, 36.4, 26.1, 0.2; IR (neat film) 1701, 1615, 1585, 1512, 1473, 1460, 1362, 1245, 1099, 1035, 837 cm−1; TLC Rf 0.38 (6% EtOAc in petroleum ether); HRMS calcd for C20H29O4Si [M-C4H9]+: 361.1835. Found: 361.1835.
Compound 50a: 1H NMR (500 MHz, CDCl3) d 7.26 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H), 5.27 (s, 1H), 4.47 (dd, J = 11.6, 13.9 Hz, 2H), 4.36–4.32 (m, 1H), 3.94–3.89 (m, 1H), 3.80 (s, 3H), 3.52 (dd, J = 5.9, 9.8 Hz, 1H), 3.44 (dd, J = 5.5, 9.8 Hz, 1H), 2.72 (d, J = 6.5 Hz, 2H), 2.43 (dd, J = 3.3, 13.1 Hz, 1H), 2.38 (dd, J = 4.4, 13.7 Hz, 1H), 2.21 (dd, J = 6.5, 13.4 Hz, 1H), 2.02 (dd, J = 6.2, 13.2 Hz, 1H), 1.12 (s, 9H), 0.09 (s, 9H); 13C NMR (125 MHz, CCl3) δ 213.4, 159.2, 150.3, 129.4, 125.9, 113.7, 73.1, 72.1, 71.2, 69.6, 55.3, 44.3, 43.8, 40.2, 35.6, 26.2, 26.1, 0.2; IR (film) 1703, 1611, 1582, 1509, 1461, 1364, 1301, 1242, 1170, 1097, 1029, 835 cm−1; TLC Rf 0.28 (6% EtOAc in petroleum ether); HRMS calcd for C20H29O4Si [M-C4H9]+: 361.1835. Found: 361.1833.
Compound 28: Catalyst preparation: To a flame-dried test tube equipped with a stir bar was added S,S-ProPhenol ligand 29 (70.2 mg, 0.11 mmol). The ligand was then azeotroped with benzene 3 times (1 mL each) and then dissolved in toluene (0.40 mL), and diethylzinc was added slowly (1 M in hexanes, 0.20 mL, 0.20 mmol). Isopropanol (0.4 mL, 5.22 mmol) and acetic acid (0.1 mL, 1 M in toluene, 0.01 mmol) were then added and the catalyst was allowed to stir at ambient temperature for 30 min. Aldol reaction: To a flame-dried test tube equipped with 4 Å molecular sieves (203 mg) and a stir bar, was added aldehyde 823 (202.8 mg, 1.00 mmol) and methyl vinyl ketone 9 (undistilled, 0.50 mL, 6.2 mmol). The catalyst was then added and the reaction was stirred at 4 °C for 7 hrs. The reaction was then poured into 1 M pH 7 phosphate buffer, and extracted with petroleum ether/diethyl ether (1:1). The organic layer was washed twice with 1 M pH 7 phosphate buffer and once with brine. The aqueous layers were extracted twice with diethyl ether. The combined organic layers were dried (Na2SO4), filtered, and concentrated in vacuo to give the aldol adduct as a yellow oil, which was used directly in the following reaction. IR (thin film) 3423, 2952, 2929, 2860, 1677, 1590, 1460, 1404, 1363, 1252, 1155, 1122, 1090, 1044, 831, 771 cm−1. 1H NMR (400 MHz, CDCl3); δ 6.38 (dd, J = 10.4, 17.6 Hz, 1H), 6.23 (dd, J = 1.2, 17.6 Hz, 1H), 5.86 (dd, J = 1.2, 10.4 Hz, 1H), 3.82 (dd, J = 2.4, 9.6 Hz, 1H), 2.83 (dd, J = 2.4, 16.4 Hz, 1H), 2.64 (dd, J = 9.6, 16.4 Hz, 1H), 1.23 (s, 3H), 1.21 (s, 3H), 0.85 (s, 9H), 0.10 (s, 3H), 0.09 (s, 3H). The crude aldol adduct from above was dissolved in 20% methanol/THF (10.0 mL) and diethylmethoxyborane (0.8 mL, 6.09 mmol) was added. The reaction vessel was cooled to −78 °C and sodium borohydride (49.2 mg, 1.30 mmol) was added. The reaction was stirred for 2.5 h, quenched with glacial acetic acid (0.1 mL) and warmed to ambient temperature. The reaction mixture was then poured into diethyl ether and washed 4 times with saturated aqueous NaHCO3. The organic layer was dried (MgSO4), filtered, concentrated in vacuo, and then chromatographed gradiently (0–1% v/v diethyl ether in petroleum ether) to give 28 (141.7 mg, 52% yield over two steps, 97% ee, 31:1 dr) as a yellow oil. Rf (10% v/v ethyl acetate in petroleum ether) = 0.8. [α]D25 = −1.2 (CHCl3, c = 1.0). Chiral GC (Cyclosil B): 100 °C for 20 min then ramped up at 5 °C/min to 200 °C): tr (major enantiomer, minor diastereomer) = 35.81 min, tr (minor enantiomer, minor diastereomer) = 35.91 min, tr (minor enantiomer, major diastereomer) = 36.09 min, tr (major enantiomer, major diastereomer) = 36.25 min. IR (thin film) 2958, 2931, 2884, 2858, 1740, 1673, 1648, 1614, 1472, 1463, 1392, 1354, 1326, 1292, 1254, 1216, 1169, 1138, 1091, 1044, 1006, 989, 925, 903, 835, 812, 774, 708, 694, 680, 653 cm−1. 1H NMR (500 MHz, CDCl3); δ 5.85 (ddd, J = 5.4, 10.5, 17.1 Hz, 1H) 5.30 (dt, J = 17.2, 1.5 Hz, 1H), 5.11 (dt, J = 10.5, 1.3 Hz, 1H), 4.39 (m, 1H), 3.67 (dd. J = 2.8, 11.4 Hz, 1H), 2.01 (dt, J = 13.8, 2.8 Hz, 1H), 1.45 (ddd, J = 2.4, 11.4, 13.8 Hz, 1H), 1.23 (s, 3H), 1.16 (s, 3H), 0.83 (s, 9H), 0.07 (s, 6H). 13C NMR (100 MHz, CDCl3); δ 139.33, 114.49, 77.92, 74.70, 71.84, 33.00, 27.24, 25.78, 24.11, 18.11, 7.77, −2.16. Elemental analysis: Calc’d: C, 61.26%, H, 11.02%, found C, 61.47%, H, 11.01%.
Compound 27: Diol 28 (119.3 mg, 0.435 mmol) was dissolved in DCM (3.3 mL, 0.1 M) and triethylamine (0.3 mL, 2.15 mmol) was then added. The reaction was cooled to 0 °C and then thionyl chloride (0.75 M DCM, 1.2 mL, 0.9 mmol) was added. The reaction was stirred for 10 min and then quenched with water, diluted with DCM, washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo to give the crude diastereomeric cyclic sulfites, which were used in the next reaction without purification. The crude cyclic sulfites from above were dissolved in THF (6.5 mL) and then TBAF (1 M THF, 2.2 mL, 2.2 mmol) was added and the solution was heated to reflux for 15 min after which it was cooled to ambient temperature, diluted with diethyl ether, washed with water and brine, dried (Na2SO4), filtered, concentrated in vacuo, and chromatographed gradiently (10–20% v/v ethyl acetate in petroleum ether) to give the title epoxide 27 (29.8 mg, 48% yield over 2 steps) as a dark orange oil. [α]D25 = −3.2 (CHCl3, c = 0.56). IR (thin film) 3426, 3082, 2964, 2926, 1708, 1645, 1459, 1427, 1380, 1325, 1252, 1206, 1123, 1053, 993, 923, 898, 848, 784, 760, 679 cm−1. 1H NMR (500 MHz, CDCl3); δ 5.92 (ddd, J = 5.5, 10.5, 17.5 Hz, 1H), 5.27 (d, J = 17.5 Hz, 1H), 5.12 (d, J = 10.5 Hz, 1H), 4.34 (br q, J = 5.5 Hz, 1H), 2.93 (dd, J = 4.5 Hz, 7.5 Hz, 1H), 2.12 (br s, 1H), 1.81 (ddd, J = 4.5, 7.5, 14.5 Hz, 1H), 1.67 (ddd, J = 4.5, 7.5, 14.5 Hz, 1H), 1.29 (s, 3H), 1.25 (s, 3H). 13C NMR (500 MHz, CDCl3); δ 140.5, 114.7, 70.9, 61.2, 58.3, 35.8, 24.7, 18.9. LRMS Calc’d for C8H15O2 = 143.1, found 143.1.
Compound 58: Catalyst preparation: To a flame-dried test tube equipped with a stir bar was added R,R-ProPhenol ligand 29 (35.9 mg, 0.056 mmol). The ligand was then azeotroped with benzene 3 times (1 mL each) and then dissolved in toluene (0.20 mL), and diethylzinc was added slowly (1 M in hexanes, 0.10 mL, 0.10 mmol). Isopropanol (0.2 mL, 2.6 mmol) was then added and the catalyst was allowed to stir at ambient temperature for 30 min. Aldol reaction: To a flame-dried test tube equipped with 4 Å molecular sieves (110.7 mg) and a stir bar, was added aldehyde 4724 (111.7 mg, 0.52 mmol) and methyl vinyl ketone 9 (freshly distilled, 0.50 mL, 6.2 mmol). The catalyst was then added and the reaction was stirred at 4 °C for 7 hrs. The reaction was then poured into 1 M pH 7 phosphate buffer, and extracted with petroleum ether/diethyl ether (1:1). The organic layer was washed twice with 1 M pH 7 phosphate buffer and once with brine. The aqueous layers were extracted twice with diethyl ether. The combined organic layers were dried (Na2SO4), filtered, and concentrated in vacuo and chromatographed gradiently (2–10% v/v diethyl ether in petroleum ether) to give the title compound 58 (93.1 mg, 63% yield) as a light yellow oil. [α]D24 = −26.0 (CH2Cl2, c = 2.69). HPLC: AD column, 230 nm, 1.0 mL/min., 99.5:0.5 heptane:isopropanol, tr (minor) = 11.124 min, tr (major) = 12.918 min; 94% ee. Rf (20% ethyl acetate/petroleum ether) = 0.65. IR (thin film) 3500, 2926, 2855, 1746, 1681, 1619, 1590, 1464, 1391, 1363, 1252, 1098, 1006, 984, 837, 776, 669 cm−1. 1H NMR (400 MHz, CDCl3); δ 6.37 (dd, J = 10.5, 17.5 Hz, 1H), 6.21 (dd, J = 1.1, 17.5 Hz, 1H), 5.81 (dd, J = 1.1, 10.5 Hz, 1H), 4.03 (dt, J = 9.6, 2.9 Hz, 1H), 3.61 (d, J = 3.2 Hz, 1H), 3.45 (m, 2H), 2.72 (dd, J = 9.6, 16.0 Hz, 1H), 2.63 (dd, J = 2.7, 15.9 Hz, 1H), 0.88 (s, 3H), 0.85 (s, 9H), 0.81 (s, 3H), 0.02 (s, 3H), 0.02 (s, 3H). 13C NMR (100 MHz, CDCl3); δ 201.0, 136.9, 128.5, 74.2, 71.9, 41.9, 38.4, 25.8, 21.8, 19.2, 18.2, −5.6, −5.7. HRMS Calc’d for C15H31O3Si (M+H) = 287.2042, found 287.2014.
Compound 46: Tetramethylammonium triacetoxyborohydride (2.8919 g, 10.99 mmol) was dissolved in acetic acid (5.5 mL) and then cooled to −20 °C. Enone 58 (311.9 mg, 1.089 mmol) was then added as a solution in acetone (5.5 mL) and the reaction was stirred for 17 h. Sodium potassium tartrate was then added as well as DCM and the thick mixture was stirred for 2.5 hrs. The layers were then separated and the aqueous layer was further extracted three times with DCM. The combined organic layers were dried (MgSO4), filtered, concentrated in vacuo, and chromatographed gradiently with 10–20% diethyl ether/petroleum ether to give the title diol 46 (215.6 mg, 69% yield, 5:1 dr, as judged by integration of the olefin peak at 5.11 ppm vs. 5.06 ppm for the diastereomer) as a yellow oil. [α]D24 = −3.62 (CH2Cl2, c = 2.98). Rf (20% v/v ethyl acetate in petroleum ether) = 0.43. IR (thin film) 3452, 2927, 2856, 1744, 1717, 1472, 1404, 1363, 1327, 1293, 1254, 1161, 1094, 1006, 990, 922, 837, 777, 669 cm−1. 1H NMR (400 MHz, CDCl3); δ 5.93 (ddd, J = 5.0, 10.5, 17.2 Hz, 1H), 5.30 (dt, J = 17.1, 1.7 Hz, 1H), 5.11 (dt, J = 10.5, 1.5 Hz, 1H), 4.45 (br s, 1H), 4.09 (d, J = 2.9 Hz, 1H) 3.82 (dt, J = 12.0, 2.7 Hz, 1H), 3.50 (d, J = 9.8 Hz, 1H), 3.46 (d, J = 9.8 Hz, 1H), 3.12 (d, J = 6.4 Hz, 1H), 1.72 (ddd, J = 3.5, 11.0, 14.0 Hz, 1H), 1.56 (ddd, J = 1.4, 6.9, 14.0 Hz, 1H), 0.88 (s, 9H), 0.86 (s, 3H), 0.80 (s, 3H), 0.06 (s, 6H). 13C NMR (100 MHz, CDCl3); δ 141.1, 113.9, 76.3, 73.7, 70.5, 37.8, 37.1, 25.8, 22.3, 18.9, 18.1, −5.7, −5.7. Elemental analysis: Calc’d: C, 62.45; H, 11.18; found C, 62.66; H, 10.97.
Compound 63: Alkene 62 (134.7 mg, 0.484 mmol) and alkyne 106 (49.7 mg, 0.17 mmol) were dissolved in acetone (0.4 mL) in a flame-dried microwave vial. CpRu(MeCN)3PF6 (7.7 mg, 0.018 mmol) was then added and the reaction was stirred at ambient temperature for 22 h. The reaction was then concentrated in vacuo and chromatographed gradiently (5–30% v/v diethyl ether in petroleum ether) to give 63 (17.6 mg, 18% yield, 2.4:1 dr), recovered alkene 62 (39.5 mg, 31% recovery) and recovered alkyne 10 (2.8 mg, 7% recovery). [α]D24 = −3.2 (CH2Cl2, c = 1.24). Rf (10% v/v ethyl acetate in petroleum ether) = 0.55. IR (thin film) 2954, 2895, 2854, 1706, 1618, 1587, 1514, 1466, 1364, 1331, 1302, 1248, 1224, 1173, 1092, 1037, 991, 923, 840, 768, 717, 689 cm−1. 1H NMR (500 MHz, CDCl3); δ 7.23 (d, J = 7.7 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.83 (ddd, J = 17.3, 10.5, 5.9 Hz, 1H), 5.25 (br s, 1H), 5.19 (dt, J = 17.3, 1.6 Hz, 1H), 5.10 (ddd, J = 10.5, 3.3, 1.5 Hz, 1H), 4.49 (d, J = 11.8 Hz, 1H), 4.46 (d, J = 11.8 Hz, 1H), 4.21 (m, 1H), 3.97 (dd, J = 9.5, 6.3 Hz, 1H), 3.83 (m, 1H), 3.78 (s, 3H), 3.48 (m, 1H), 3.45 (dd, J = 10.1, 5.2 Hz, 1H), 3.42 (dd, J = 10.0, 4.3 Hz, 1H), 2.94 (ddd, J = 17.5, 10.0, 5.2 Hz, 1H), 2.58 (dd, J = 17.3, 7.3 Hz, 1H), 2.37 (br d, J = 13.4 Hz, 1H), 2.24 (m, 2H), 1.95 (m, 3H), 1.42–1.86 (m, 8H), 1.30 (s, 3H), 1.30 (s, 3H), 0.06 (s, 9H). 13C NMR (125 MHz, CDCl3); δ 212.5, 159.1, 152.5, 138.4, 130.3, 129.2, 123.8, 115.2, 113.7, 100.5, 77.4, 75.0, 72.9, 72.8, 70.9, 69.8, 68.1, 55.3, 50.6, 45.6, 45.1, 36.5, 32.2, 30.3, 25.3, 24.2, 20.3, 18.9, 0.3. HRMS Calc’d for C33H50O6Si = 570.3377, found 570.3388.
Compound 67: Ketal 63 (16.1 mg, 0.028 mmol) was dissolved in methanol (0.5 mL). Camphor sulfonic acid (0.8 mg, 0.003 mmol) was added at ambient temperature and the reaction was stirred for 3 h. The solution was then poured over aqueous NaHCO3, diluted with diethyl ether, washed with brine, dried (MgSO4), filtered, concentrated in vacuo and chromatographed isocratically (25% v/v diethyl ether in petroleum ether) to give the title compound 67 (10.2 mg, 70% yield) as a yellow oil. [α]D25 = 9.8 (CH2Cl2, c = 0.98). Rf (10% v/v ethyl acetate in petroleum ether) = 0.17. IR (thin film) 3456, 2952, 2925, 2854, 1618, 1514, 1465, 1362, 1302, 1248, 1114, 1038, 843 cm−1. 1H NMR (500 MHz, CDCl3); δ 7.24 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.83 (ddd, J = 17.3, 10.6, 5.1 Hz, 1H), 5.25 (dt, J = 17.2, 1.6 Hz, 1H), 5.21 (br s, 1H), 5.06 (dt, J = 10.6, 1.5 Hz, 1H), 4.53 (d, J = 11.7 Hz, 1H), 4.49 (d, J = 11.7 Hz, 1H), 4.00 (m, 2H), 3.79 (s, 3H), 3.59 (ddt, J = 11.0, 2.2, 5.0 Hz, 1H), 3.4–3.53 (m, 3H), 3.15 (s, 3H), 2.38 (m, 2H), 2.09 (dd, J = 15.9, 5.1 Hz, 1H), 2.05 (m, 2H), 1.94 (m, 2H), 1.78 (dd, J = 16.0, 5.2 Hz, 1H), 1.74 (ddd, J = 12.4, 4.5, 3.1 Hz, 1H), 1.03 (s, 3H), 0.95 (s, 3H), 0.07 (s, 9H). 13C NMR (125 MHz, CDCl3); δ 153.7, 138.4, 130.6, 129.1, 123.0, 114.2, 113.7, 104.1, 77.5, 75.5, 73.0, 72.9, 71.3, 68.9, 55.3, 47.9, 47.0, 43.1, 39.2, 36.6, 35.6, 29.7, 20.6, 15.5, 0.3. HRMS Calc’d for C28H43O6Si (M-CH3) = 503.2829, found 503.2829.
Scheme 5.

Access to the desired enyne through the enantiomer of diene 7.

Acknowledgements
We thank the National Institutes of Health (GM33049) and NSF for their generous support of our programs. We also thank fellowships from Amgen (H.Y.), Bristol-Myers Squibb (H.Y.), NSF (C.S.B.), and ARCS (C.S.B.) for their generous financial support. GD is a Stanford Graduate Fellow. We appreciate the assistance of G. Wuitschik in the model study of the bryostatin B-ring synthesis. Mass spectra were provided by the Mass Spectrometry Regional Center of the University of California-San Francisco, supported by the NIH Division of Research Resources. Palladium and ruthenium salts were generously supplied by Johnson-Matthey.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.Bryostatin 1 isolation and structure determination: Pettit GR, Herald CL, Doubek DL, Herald DL, Arnold E, Clardy J. J. Am. Chem. Soc. 1982;104:6846. For recent reviews on bryostatins, please see: Hale KJ, Manaviazar S. Chemistry: An Asian J. 2010;5:704. doi: 10.1002/asia.200900634. Hale KJ, Hummersone MG, Manaviazar S, Frigerio M. Nat. Prod. Rep. 2002;19:413. doi: 10.1039/b009211h. Wender PA, Baryza JL, Hilinski MK, Horan JC, Kan C, Verma VA. Beyond Natural Products: Synthetic Analogues of Bryostatin 1. In: Huang Z, editor. Drug Discovery Research: New Frontiers in the Post-Genomic Era. Wiley-VCH: Hoboken, NJ; 2007. pp. 127–162. Mutter R, Wills M. Biorg. Med. Chem. 2000;8:1841. doi: 10.1016/s0968-0896(00)00150-4.
- 2. Evans DA, Carter PH, Carreira EM, Charrette AB, Prunet JA, Lautens M. J. Am. Chem. Soc. 1999;121:7540. and earlier references therein. Please see the following references for other examples of stereoselective formation of the exocyclic enoate of the B-ring, including chiral phosphonate reagents: O’Brien M, Taylor NH, Thomas EJ. Tetrahedron Lett. 2002;43:5491. Cuprate additions into enoates: Baron A, Ball M, Bradshaw B, Donnelly S, Germay O, Oller PC, Kumar N, Martin N, O’Brien M, Omori H, Moore C, Thomas EJ. Pure Appl. Chem. 2005;77:103. Intramolecular radical additions to enoates: Maguire RJ, Munt SP, Thomas EJ. J. Chem. Soc., Perkin Trans. 1998;1:2853. Sterically-controlled phosphonate additions: Munt SP, Thomas EJ. J. Chem. Soc., Chem. Commun. 1989;480 Tethered phosphonate reagents: Lampe TFJ, Hoffmann HMR. Tetrahedron Lett. 1996;37:7695.. Use of a symmetrical substrate to ensure one geometrical outcome: Evans DA, Carreira EM. Tetrahedron Lett. 1990;31:4703. Hale KJ, Hummersone MG, Bhatia GS. Org. Lett. 2000;2:2189. doi: 10.1021/ol005850y.
- 3. Blanchette MA, Malamas MS, Nantz MH, Roberts JC, Somfai P, Whritenour DC, Masamune S, Kageyama M, Tamura T. J. Org. Chem. 1989;54:2817. Kageyama M, Tamura T, Nantz MH, Roberts JC, Somfai P, Whritenour DC, Masamune S. J. Am. Chem. Soc. 1990;112:7407. and earlier references cited therein. For a review account of this synthesis by Professor K. Ohmuri and the Keio team, see also: Ohmori K, Ogawa Y, Obitsu T, Ishikawa Y, Nishiyama S, Yamamura S. Angew. Chem., Int. Ed. 2000;39:2290. For a formal synthesis see: Ohmuri K. Bull. Chem. Soc. Jpn. 2004;77:875–885. Manaviazar S, Frigerio M, Bhatia GS, Hummersone MG, Aliev AE, Hale KJ. Org. Lett. 2006;8:4477. doi: 10.1021/ol061626i. and earlier references cited therein.
- 4.Trost BM, Shin S, Sclafani JA. J. Am. Chem. Soc. 2005;127:8602. doi: 10.1021/ja051526s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For other notable synthetic approaches to the A- and B-rings, please see: Lu Y, Krische MJ. Org. Lett. 2009;11:3108. doi: 10.1021/ol901096d. Keck GE, Kraft MB, Truong AP, Li W, Sanchez CC, Kedei N, Lewin NE, Blumberg PM. J. Am. Chem. Soc. 2008;130:6660. doi: 10.1021/ja8022169. Wender PA, DeChristopher BA, Schrier AJ. J. Am. Chem. Soc. 2008;130:6658. doi: 10.1021/ja8015632. Ball M, Bradshaw BJ, Dumeunier R, Gregson TJ, MacCormick S, Omuri H, Thomas EJ. Tetrahedron Lett. 2006;47:2223. Voight EA, Seradj H, Roethles PA, Burke SD. Org. Lett. 2004;6:4045. doi: 10.1021/ol0483044. Vakalopoulos A, Lampe TFJ, Hoffmann HMR. Org. Lett. 2001;3:929. doi: 10.1021/ol015551o. Kiyooka S, Maeda H. Tetrahedron: Asy. 1997;8:3371.
- 6.Trost BM, Machacek MR, Faulk BD. J. Am. Chem. Soc. 2006;128:6745. doi: 10.1021/ja060812g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.A procedure that has combined both the Ru and Pd catalysis in one pot, see: Trost BM, Machacek MR. Angew. Chem., Int. Ed. Engl. 2002;41:4693. doi: 10.1002/anie.200290019.
- 8.This is in striking contrast to what has been accomplished in our mycalamide A synthesis wherein the α,β-unsaturated double bond was selectively dihydroxylated (OsO4, acetone/H2O, 5 °C) in the presence of a similarly disubstituted vinylsilane in high yield. Trost BM, Yang H, Probst GD. J. Am. Chem. Soc. 2004;126:48. doi: 10.1021/ja038787r.
- 9.Roth GJ, Liepold B, Müler SG, Bestmann HJ. Synthesis. 2004;59 [Google Scholar]
- 10.Martin MG, Ganem B. Tetrahedron Lett. 1984;25:251. [Google Scholar]
- 11.Mitchell PWD. Org. Prep. and Proced. Int. 1990;22:534. [Google Scholar]
- 12.Umbreit MA, Sharpless KB. Org. Synth. 1981;60:29. [Google Scholar]
- 13.Yamada K, Goto S, Nagase H, Kyotani Y, Hirata Y. J. Org. Chem. 1978;43:2076. [Google Scholar]
- 14.Chou SSP, Kuo HL, Wang CJ, Tsai CY, Sun CM. J. Org. Chem. 1989;54:868. [Google Scholar]
- 15.For a preliminary report of a portion of our work, see: Trost BM, Yang H, Wuitschik G. Org. Lett. 2005;7:4761. doi: 10.1021/ol0520065.
- 16.Trost BM, Fettes A, Shireman BT. J. Am. Chem. Soc. 2004;126:2660. doi: 10.1021/ja038666r. [DOI] [PubMed] [Google Scholar]
- 17. Zhao H, Pagenkopf BL. Chem. Commun. 2003:2592. doi: 10.1039/b308204k. In the presence of BF3·Et2, the lithium trimethylaluminium ate complex added to the trisubstituted epoxide at the more hindered position in a stereo- and regio-specific manner to give a single stereoisomer.
- 18.Made in analogy to the TBS epoxide of: Fang YF, Oehlschlager AC, Gerogopapadakou NH, Hartman PG, Scheliga P. J. Am. Chem. Soc. 1995;117:670.
- 19.Le Roux C, Dubac J. Organometallics. 1996;15:4646. [Google Scholar]
- 20.Use of the conditions employing acetic acid, which were developed for aldehyde 8, resulted in a lower yield (40%) of aldol adduct 58.
- 21.Evans DA, Chapman KT, Carreira EM. J. Am. Chem. Soc. 1988;110:3560. [Google Scholar]
- 22.Trost BM, Dong G. Nature. 2008;456:485. doi: 10.1038/nature07543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denmark SE, Stavenger RA. J. Am. Chem. Soc. 2000;122:8837. [Google Scholar]
- 24.Sano S, Mori K. Eur. J. Org. Chem. 1999;1679 [Google Scholar]
- 25.Napolitano E, Fiaschi R, Mastrorilli E. Synthesis. 1986;2:122. [Google Scholar]