

Summary
Phosphorylation regulates assembly and disassembly of proteins during endocytosis. In yeast, Prk1 and Ark1 phosphorylate factors after vesicle internalization leading to coat disassembly. Scd5, a protein phosphatase-1 (PP1)-targeting subunit, is proposed to regulate dephosphorylation of Prk1/Ark1 substrates to promote new rounds of endocytosis. In this study we analyzed scd5-PP1Δ2, a mutation causing impaired PP1 binding. scd5-PP1Δ2 caused hyperphosphorylation of several Prk1 endocytic targets. Live-cell imaging of 15 endocytic components in scd5-PP1Δ2 revealed that most factors arriving before the invagination/actin phase of endocytosis had delayed lifetimes. Severely affected were early factors and Sla2 (Hip1R homolog), whose lifetime was extended nearly fourfold. In contrast, the lifetime of Sla1, a Prk1 target, was extended less than twofold, but its cortical recruitment was significantly reduced. Delayed Sla2 dynamics caused by scd5-PP1Δ2 were suppressed by SLA1 overexpression. This was dependent on the LxxQxTG repeats (SR) of Sla1, which are phosphorylated by Prk1 and bind Pan1, another Prk1 target, in the dephosphorylated state. Without the SR, Sla1ΔSR was still recruited to the cell surface, but was less concentrated in cortical patches than Pan1. sla1ΔSR severely impaired endocytic progression, but this was partially suppressed by overexpression of LAS17, suggesting that without the SR region the SH3 region of Sla1 causes constitutive negative regulation of Las17 (WASp). These results demonstrate that Scd5/PP1 is important for recycling Prk1 targets to initiate new rounds of endocytosis and provide new mechanistic information on the role of the Sla1 SR domain in regulating progression to the invagination/actin phase of endocytosis.
Key words: Clathrin, Endocytosis, Protein phosphatase 1, Sla1, Pan1, Las17
Introduction
Clathrin-mediated endocytosis (CME) is a highly dynamic process involving clathrin and a host of other factors, regulatory molecules and actin assembly. Live cell fluorescence microscopy has demonstrated that a highly ordered pathway of assembly and disassembly of these proteins is needed to establish an endocytic site and collect membrane cargo, invaginate the membrane, pinch off the vesicle, uncoat and move the vesicle into the cell. But how this process is coordinated is still not completely understood. Yeast, with its powerful molecular genetic approaches, has become an important model for dissecting the endocytic pathway, since the machinery of CME is conserved throughout eukaryotes and image analysis has demonstrated many parallels in the process from yeast to mammals (Boettner et al., 2012; Conibear, 2010; Perrais and Merrifield, 2005).
In yeast, CME is associated with cortical actin patch structures. First there is an extended immobile phase where coat proteins are recruited in a precise sequence onto a cortical site to collect cargo and prepare the patch for the mobile invagination phase involving actin (see Fig. 2) (for reviews, see Boettner et al., 2012; Weinberg and Drubin, 2012). Clathrin, Ede1 [an Eps15 homology (EH) domain protein] and Syp1 (FCHO1/2 homolog) arrive at the endocytic site up to 1–2 min before the arrival of other coat factors (Boettner et al., 2009; Newpher et al., 2005; Stimpson et al., 2009; Toshima et al., 2006). Next Sla2, a talin-like domain protein related to mammalian Hip1 and Hip1R, is recruited, followed rapidly by late coat factors Pan1, End3 (two other EH domain proteins) and Sla1 (a SH3 domain protein) (Kaksonen et al., 2003; Kaksonen et al., 2005; Newpher and Lemmon, 2006). Other coat module factors include the clathrin adaptors Ent1/2 (epsins) and Yap1801/2, which also bind EH proteins like Pan1 (Aguilar et al., 2003; Wendland and Emr, 1998). Syp1 and Ede1 leave the membrane just prior to the rapid mobile actin phase (Boettner et al., 2009; Stimpson et al., 2009; Toshima et al., 2006), which is associated with recruitment of several actin-remodeling factors (e.g. Arp2/3 complex, capping protein, type 1 myosins, Abp1, etc.). After invagination of about 200 nm and vesicle scission (∼10 sec), clathrin, Sla2 and other coat factors disassemble. This is followed by a fast (∼5 sec) inward long-range movement of the endocytic vesicle with associated Abp1/actin into the cell (Kaksonen et al., 2003; Kaksonen et al., 2005). What regulates the rapid switch from the immobile phase to the actin-dependent mobile phase of endocytosis is still not clear.
Fig. 2.

Endocytic factor patch lifetimes are delayed in scd5-PP1Δ2 cells. (A) Average lifetimes (±s.d.) of endocytic factors were generated from movies of wild-type (green bars) and scd5-PP1Δ2 (orange bars) strains (n≥30 patches for each strain). Most factors were paired with Abp1–RFP marking the actin phase in endocytosis. SCD5 and scd5-PP1Δ2 strains, respectively, were: Ede1–GFP (SL5755, SL 6029), Sla2–GFP (SL5928, SL6023), GFP–Yap1802 (SL6365, SL6366), GFP–Yap1801 (SL5482, SL5441), GFP–Ent2 (SL5481, SL5436), GFP–Ent1 (SL5480, SL5440), Pan1–GFP (SL5425, SL5429), Sla1–GFP (SL5412, SL5411), End3–GFP (SL6039, SL6041), Las17–GFP (SL4819, SL5328), GFP–Scd5 (SL5302), GFP–Scd5-PP1Δ2 (SL5303), Bbc1–GFP (SL5204, SL5265), Myo5–GFP (SL5580, SL6165), and Abp1–RFP (combined data from multiple strains). Clathrin is not shown due to difficulty of estimating lifetimes from wide-field imaging, but estimates are shown for GFP–Clc1 in B for wild type (SL5354) and scd5-PP1Δ2 (SL5356). ***P<0.0001 comparing scd5-PP1Δ2 to wild-type using Student's t-test. (B) Graphical timeline of endocytic factors examined in this study with average lifetimes for wild type strains indicated in parentheses, based on data from A. The three stages of immobile phase recruitment are shown: Early (blue), middle (green), and late stage (purple). The mobile actin phase is shown in red. In the scd5 mutant, timing of most immobile phase factors is extended. The dotted line and times (in seconds) at the left of the wild-type time lines, denote the extended timing seen in most immobile phase factors in scd5-PP1Δ2. See text for further details.
Reversible phosphorylation is a major mechanism for regulating assembly and disassembly of endocytic factors during CME. In resting synapses, several endocytic factors, such as dynamin, amphiphysin, synaptojanin, AP180, Epsin and Eps15, are cytosolic and inactive due to phosphorylation by inhibitory kinases, such as Cdk5 (Cousin et al., 2001; Samuels and Tsai, 2003; Slepnev et al., 1998; Tan et al., 2003; Tomizawa et al., 2003). Upon synaptic transmission an influx of calcium activates the phosphatase calcineurin, which dephosphorylates these components to allow assembly and a burst of compensatory clathrin-mediated endocytosis for recycling of synaptic vesicle membranes (Clayton et al., 2007).
In yeast, endocytosis is regulated by the kinases Ark1 and Prk1, which are homologous with AAK1 and GAK1 kinases involved in CME in animals cells (Smythe and Ayscough, 2003). Prk1 phosphorylates a motif related to Lxx[Q/T]xTG, and known targets include Pan1, Sla1, Ent1/2, Yap1801/2 and Scd5 (Henry et al., 2003; Huang et al., 2003; Watson et al., 2001; Zeng and Cai, 1999; Zeng et al., 2007; Zeng et al., 2001). Also, a number of other immobile phase endocytic factors have Prk1 recognition sites (e.g. Breitkreutz et al., 2010; Mok et al., 2010). Deletion of ARK1 and PRK1 genes together causes an endocytic defect and accumulation of large aggregates containing actin, other endocytic proteins and membranous material in the cytoplasm (Chang et al., 2006; Cope et al., 1999; Sekiya-Kawasaki et al., 2003; Toshima et al., 2005; Watson et al., 2001). A similar phenotype is observed when the Pan1 sites phosphorylated by Prk1 are mutated to Ala (Toshima et al., 2005). Since recruitment of Prk1 and Ark1 during endocytosis is late during the actin phase (Toret et al., 2008; Zeng et al., 2007), these kinases are thought to negatively regulate endocytic factors and/or promote uncoating of endocytic vesicles after scission.
Scd5, in association with protein phosphatase-1 (PP1/yeast Glc7), has emerged as a major candidate to counter Ark1 and Prk1 by dephosphorylating coat factors for new cycles of endocytosis. We identified Scd5 in a screen for multicopy suppressors of clathrin deficiency, and later showed its importance in endocytosis and cortical actin organization (Henry et al., 2002; Nelson et al., 1996). Binding of Glc7 to the canonical [R/K]x0–1[V/I]xF PP1 binding motif of Scd5 (Egloff et al., 1997) is critical for the endocytic function of Scd5 (Chang et al., 2002). Scd5 interacts with a number of endocytic coat factors, and, together with PP1, mediates Pan1 dephosphorylation (Henry et al., 2002; Zeng et al., 2007). scd5 temperature sensitive (ts) mutations are also suppressed by deletion of PRK1 (Henry et al., 2003; Zeng et al., 2007), consistent with Scd5/PP1 recycling Prk1 substrates. Moreover, Scd5 is phosphorylated by Prk1, which is suggested to negatively regulate Scd5 to amplify coat disassembly after scission (Henry et al., 2003; Huang et al., 2003; Zeng et al., 2007).
Scd5 is recruited to cortical endocytic sites late during the immobile stage (Tonikian et al., 2009; Zeng et al., 2007); however, cortical recruitment of Scd5 is not required for efficient endocytosis (Chang et al., 2006). Therefore we set out to examine how impaired PP1/Glc7 targeting by Scd5 affects the dynamics of coat formation and actin-driven vesicle invagination. By analysis of multiple endocytic factors, including several Prk1 substrates, we demonstrate that mutation of the Scd5 PP1 binding site leads to major delays in coat development and progression to the mobile actin phase of internalization. We also provide evidence that the SR-repeat region of Sla1, which is regulated by Scd5/PP1 dephosphorylation, is critical for coordinating this transition.
Results
Prk1 substrates are hyper-phosphorylated in cells mutant for the Scd5 PP1 binding site
Phosphorylation of several immobile phase endocytic factors by Prk1 (and Ark1) promotes coat disassembly at the end of internalization (Chang et al., 2006; Cope et al., 1999; Toret et al., 2008; Toshima et al., 2005; Watson et al., 2001; Zeng et al., 2001). If Scd5 targets PP1/Glc7 to these factors, mutation of the Scd5 PP1 binding site should result in their hyperphosphorylation. We examined several Prk1 targets in scd5-PP1Δ2 cells, where the Scd5 KKVRF PP1 binding motif is mutated to AKAAA. This mutation reduces PP1 binding by about tenfold and causes ts growth and endocytic phenotypes (Chang et al., 2002). We found that all Prk1 substrates tested were hyperphosphorylated in the mutant as compared to wild-type SCD5 cells (Fig. 1A). Each protein showed slower migrating bands on gels in scd5-PP1Δ2 cells, and in the case of the Ent1, Ent2, Yap180 and Yap1802, these collapsed to a single lower band in the presence of calf intestinal phosphatase (CIP; Fig. 1A). For Pan1 and Sla1, a different cell extraction method was used due to their protease sensitivity (Zeng and Cai, 1999), which precluded phosphatase treatment. However, Pan1 and Sla1 are known Prk1 targets, Pan1 has been shown to be dephosphorylated by Scd5/PP1, and another study suggested that Sla1 is a PP1 substrate (Gardiner et al., 2007; Toshima et al., 2005; Zeng and Cai, 1999; Zeng et al., 2007; Zeng et al., 2001). Together, these data support the hypothesis that Scd5 targets PP1 to counter phosphorylation of multiple endocytic targets of Prk1.
Fig. 1.
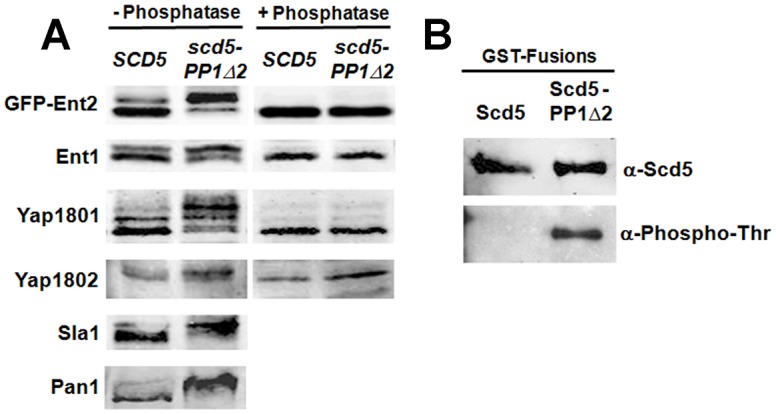
Prk1 substrates are hyper-phosphorylated in scd5-PP1-binding-site mutant cells and Scd5 is regulated by PP1. (A) SCD5 (SL4706) and scd5-PP1Δ2 (SL4823) cells or SCD5 (SL4851) and scd5-PP1Δ2 (SL4852) cells containing pBW56 (CEN, URA3, GFP-ENT2) were grown to log-phase at 25°C. Cell extracts were prepared using glass beads, treated with or without phosphatase, and subjected to immunoblotting for the indicated proteins or GFP (GFP–Ent2). For Pan1 and Sla1, cell extracts were prepared by a TCA precipitation method. (B) pJSC2 (CEN, LEU2, GAL1:GST-SCD5) and pJSC63 (CEN, LEU2, GAL1:GST-scd5-PP1Δ2) were transformed into a wild-type SCD5 strain (SL1462). GST fusions were induced for expression on galactose medium, affinity-purified on glutathione beads, and analyzed by immunoblotting with anti-Scd5 and anti-phosphothreonine antibodies.
Scd5 contains a triple repeat region (3R) with three LxxTxTG motifs that are also subject to Prk1 phosphorylation on the second threonine (Henry et al., 2003; Huang et al., 2003). To determine whether Scd5-targeted PP1 mediates Scd5 dephosphorylation, we expressed GST fusions of Scd5 and Scd5-PP1Δ2 in wild-type SCD5 cells, affinity purified them on glutathione beads, and analyzed their phosphorylation by immunoblotting. While GST–Scd5 was not detected by anti-phosphothreonine antibodies, threonine phosphorylation was clearly seen on GST–Scd5-PP1Δ2 (Fig. 1B). This is consistent with a previous report in scd5 mutant cells (Zeng et al., 2007); however, endogenous wild-type Scd5 was also present in our experiment. Therefore, the hyperphosphorylation of GST–Scd5-PP1Δ2 we saw demonstrates that PP1 dephosphorylates the Scd5 to which it is bound. Thus Scd5 is phosphoregulated in cis by PP1/Glc7.
Endocytic factor patch lifetimes are delayed in scd5-PP1Δ2 cells
Since the Prk1 substrates tested were hyperphosphorylated in scd5-PP1Δ2 cells, we examined how this mutation affects endocytic vesicle progression. Cortical patch dynamics of 15 XFP-tagged endocytic factors involved in each stage of vesicle coat assembly and invagination were analyzed by live cell fluorescence microscopy. In most cases GFP-tagged endocytic factors were paired with Abp1–RFP, an actin phase marker, in both wild-type SCD5 and scd5-PP1Δ2 cells. Nearly all immobile phase endocytic factors displayed delayed lifetimes in scd5-PP1Δ2, with those categorized as early to middle stage coat factors being the most extended (supplementary material Table S3; Fig. 2 and kymograph examples in Fig. 3). The early factor Ede1–GFP was often immobile, with patches persisting the entire length of a 9 min movie in scd5-PP1Δ2, as compared to the lifetimes of Ede1–GFP in wild-type cells (Figs 2, 3; supplementary material Table S3). Similar results were found for GFP–Clc1, marking clathrin, another early endocytic factor (Fig. 3 and data not shown). The lifetime of Sla2–GFP, a middle stage coat factor, was extended approximately fourfold in scd5-PP1Δ2 cells for patches that acquired Abp1–RFP and completed internalization (Fig. 2; supplementary material Table S3; Movie 1).
Fig. 3.

Representative kymographs showing delayed lifetimes of endocytic factors in scd5-PP1Δ2 cells. Kymographs were obtained from the strains indicated in Fig. 2, with most GFP fusions (green) paired with Abp1–RFP (red). However, Sla2–RFP was additionally paired with Myo5–RFP in scd5-PP1Δ2 (SL6175). GFP–Clc1 and End3–GFP were not paired with Abp1–RFP. Time-lapse videos were 9 minutes (1 frame/4 s) for Ede1–GFP and Sla2–GFP. All others were 4 minutes (1 frame/2 s).
In scd5-PP1Δ2 multiple actin events, marked by Abp1–RFP, were observed for Ede1 patches and many elongated Sla2 patches (Fig. 3). Myo5–RFP was also found at the intermediate actin events (Fig. 3), indicating that other mobile phase factors were recruited to these sites. In patches with multiple actin events, the intensity of Sla2 at the cortex often fluctuated, decreasing in coordination with Abp1/actin events without completely disappearing, and then accumulating again (e.g. see Sla2–GFP in Fig. 5A, top panels for scd5-PP1Δ2). Final termination of the Sla2 event usually coincided with an actin event. These results suggest that at least some of these internal actin events were productive internalizations, although abortive internalizations cannot be excluded. In contrast, the intensity of Ede1–GFP, which normally does not internalize with the vesicle, did not fluctuate with the multiple Abp1/actin events. Terminal events where Ede1 disappeared were usually associated with an actin event, although in general these were difficult to capture because of the long Ede1 lifetimes (Fig. 3 and data not shown).
Fig. 5.
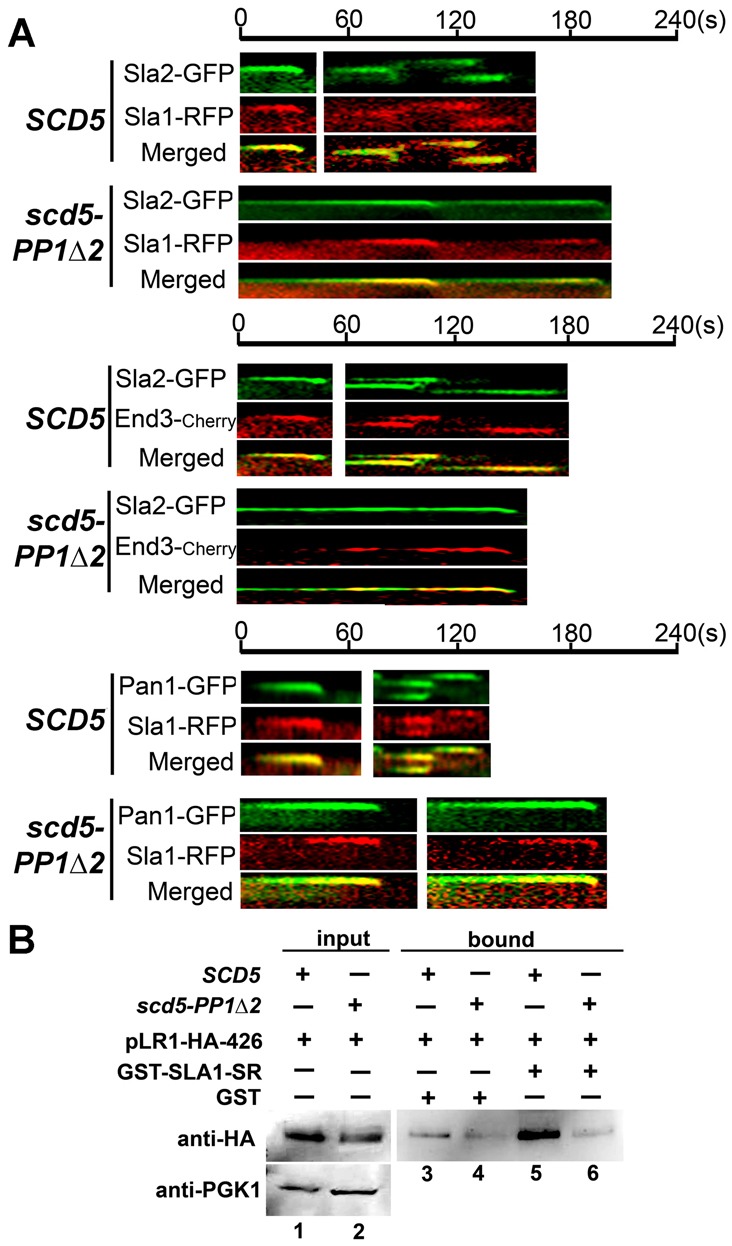
Delayed Sla1 recruitment to endocytic patches is associated with reduced binding to Pan1 in scd5-PP1Δ2. (A) Kymographs of indicated XFP-marked endocytic factors in SCD5 and scd5-PP1Δ2 cells. Top: Sla2–GFP paired with Sla1–RFP in wild-type (SL5839) and scd5-PP1Δ2 (SL5840) cells. Middle: Sla2–GFP paired with End3–Cherry in wild-type (SL6179) and scd5-PP1Δ2 (SL6181) cells. Bottom: Pan1–GFP paired with Sla1–RFP in wild-type (SL6141) and scd5-PP1Δ2 (SL6142) cells. (B) Sla1-SR binding to Pan1 LR1 is impaired in scd5-PP1Δ2. Cell lysates from wild type (SL1463) and scd5-PP1Δ2 (SL4610) expressing pLR1–HA-426 were incubated with bacterially purified GST or GST–Sla1-SR domain immobilized on glutathione beads. Bound fractions (lanes 3–6) and input fractions (lanes 1 and 2, top) were analyzed by immunoblotting with anti-HA antibodies to detect the Pan1-LR1–HA fragment. Anti-PGK1 is a loading control for lysate inputs (lanes 1 and 2, bottom).
We also examined the dynamics of the epsins and AP180s, since they are targets of Prk1 and Scd5/PP1 (Huang et al., 2003; Watson et al., 2001) (Fig. 1). We used N-terminally tagged proteins, so as not to disrupt the C-terminal clathrin-binding motifs. In wild-type cells both GFP–Yap180s had lifetimes similar to Sla2 or late coat factors (Sla1, End3, Pan1) and internalized like these coat module proteins about 200–300 nm before uncoating, presumably just after scission (Figs 2, 3; supplementary material Fig. S1; Table S3). In scd5-PP1Δ2 cells, the Yap1801/2 lifetimes were delayed approximately threefold (Figs 2, 3; supplementary material Fig. S1; Table S3).
GFP–Ent2 behaved similarly to the Yap180s in the scd5 mutant (Figs 2, 3; supplementary material Fig. S2B; Table S3). Surprisingly, GFP–Ent1 demonstrated no delay in scd5-PP1Δ2 cells (Figs 2, 3; supplementary material Fig. S2A; Table S3), even though it has functional overlap with Ent2 and the Yap180s (Aguilar et al., 2006; Aguilar et al., 2003; Maldonado-Báez et al., 2008; Wendland et al., 1999). Also, >80% of GFP–Ent1 patches in wild-type cells internalized >500 nm and disappeared with Abp1/actin, indicating it did not uncoat with the other adaptors and coat factors (supplementary material Fig. S2A,C). This suggests Ent1 may have additional roles in the late stages of CME, and this may be independent of Prk1 and Scd5/PP1 regulation.
Studies using C-terminally tagged epsins and AP180s have given slightly varying dynamics in some cases than we observed using N-terminal tags (Carroll et al., 2012; Toret et al., 2008). It is possible that N-terminal fusions affect the membrane binding ENTH/ANTH domains. But we note that GFP–Ent1 and GFP–Ent2 were previously shown to complement an ent1Δ ent2Δ mutant, so it seems unlikely that their differences are caused solely by the tag (Watson et al., 2001). Moreover, other studies have shown that the two endocytic epsins have functions that are not completely overlapping (Baggett et al., 2003; Maldonado-Báez et al., 2008; Mukherjee et al., 2009; Newpher et al., 2005; Wendland et al., 1999). Further studies will be needed to explain the differences in adaptor behavior during endocytosis.
Late-arriving coat factors Pan1, End3 and Sla1 have direct physical interactions, and Pan1 and End3 form a stoichiometric complex (Tang et al., 2000; Toshima et al., 2007). Also Cai and co-workers showed that the interactions of these three proteins are disrupted by Prk1 phosphorylation (Zeng et al., 2001), and Pan1 interaction with End3 is promoted by Scd5/PP1 dephosphorylation (Zeng et al., 2007). We found Pan1–GFP and End3–GFP had lifetimes approximately twofold longer in scd5-PP1Δ2 compared to in wild-type cells (Figs 2, 3; supplementary material Table S3). The lifetime of Sla1–GFP was also extended in scd5-PP1Δ2 (Figs 2, 3; supplementary material Movie 2; Table S3), but compared to Pan1 and End3, the slowing of Sla1 dynamics was less severe. Representative kymographs show that, though their lifetimes were delayed, Sla1, End3 and Pan1 exhibited single terminal actin-based internalization events, unlike Sla2 and Ede1, which often displayed multiple actin events (Fig. 3). Since the lifetimes of late coat factors, Pan1/End3/Sla1, were much less affected than Sla2, Yap1801/2 and Ent2, we grouped the Yap180s and Ent2 with Sla2 as middle stage coat factors.
Las17, the yeast WASp homolog is a major actin nucleation promoting factor (NPF), but it arrives around the time of the late coat factors in wild-type cells and is subject to inhibition by Sla1 and Syp1 during the immobile phase (Boettner et al., 2009; Kaksonen et al., 2003; Rodal et al., 2003; Sun et al., 2006). In the scd5 mutant Las17–GFP had a delay of about +14 seconds, which was fairly similar to that of Sla1 (Fig. 3; supplementary material Table S3). We also examined dynamics of N-terminally tagged Scd5 (GFP–Scd5) and GFP–Scd5-PP1Δ2, expressed as the sole source of Scd5. GFP–Scd5 arrived at the cortex fairly late in the coat assembly phase and did not invaginate with the vesicle, consistent with a previous study using Scd5–GFP (Tonikian et al., 2009). The lifetime GFP–Scd5-PP1Δ2 was slowed by ∼12 secs, but inhibition of PP1 binding did not prevent Scd5 from being recruited to the cortex (Figs 2, 3; supplementary material Table S3). When components of the actin assembly/fast mobile stage of endocytosis, including Abp1–RFP, Bbc1–GFP, and Myo5–RFP were analyzed in scd5-PP1Δ2 cells, only Abp1–RFP showed a slight delay (Fig. 2; supplementary material Table S3).
Sla1 recruitment to endocytic sites in scd5-PP1Δ2 cells is impaired
The interaction of Sla1, Pan1 and End3 was previously shown to be disrupted by Prk1 phosphorylation (Zeng et al., 2001). Thus we thought their recruitment to the cortex might be diminished in the scd5 PP1 binding site mutant, since they would be hyperphosphorylated and might not assemble efficiently. To analyze this we performed cortical patch to cytosol fluorescence intensity ratio and patch density analyses for a number of coat factors as measures of assembly at endocytic sites. Patch to cytosol fluorescence intensity ratios for Ede1–GFP, Sla2–GFP, Pan1–GFP and End3–GFP in the scd5 mutant were slightly decreased (<45%) compared to wild-type cells, indicating a mild effect on cortical recruitment (Fig. 4A,B; supplementary material Fig. S3A; Table S4). However, intensity ratios were reduced dramatically (threefold) for Sla1 in scd5-PP1Δ2 cells (Fig. 4A; supplementary material Table S4; Movie 2). Images captured at the medial focal plane clearly demonstrated that Sla1–GFP was more cytosolic and less polarized in the mutant (Fig. 4B). Thus, though Sla1, End3 and Pan1 interact in a Prk1 dependent manner, Sla1 recruitment was greatly impaired, while the other coat factors were minimally affected.
Fig. 4.

Cortical localization and patch density of Sla1 are decreased in scd5-PP1Δ2 cells. (A) Average cortical patch to cytosol fluorescence intensity ratios (±s.d.). Strains are: Ede1–GFP (SL6032, SL6029), Sla2–GFP (SL6026, SL6023), Pan1–GFP (SL5425, SL5429), End3–GFP (SL6039, SL6041), and Sla1–GFP (SL5412, SL5411) for wild type (white bars) and scd5-PP1Δ2 (gray bars), respectively; n≥26 cells for each. (B) Example epifluorescence images from the medial focal plane of wild-type and scd5-PP1Δ2 cells indicated in A. Pan1–GFP or Sla1–GFP are shown (see supplementary material Fig. S3A for examples of Ede1, Sla2 and End3). (C) Average patch densities (±s.d.) for Sla2–GFP, Pan1–GFP, End3–GFP and Sla1–GFP in wild type and scd5-PP1Δ2, respectively (strains as in A; n = 30 cells for each). (D) Representative projection images of deconvolved Z-stacks showing patch densities of wild-type and scd5-PP1Δ2 cells expressing Sla2–GFP or Sla1–GFP (strains as in A, see supplementary material Fig. S3B for examples of Pan1 and End3). For graphs, *P = 0.0007, ***P<0.0001, comparing the scd5 mutant to wild type using Student's t-test.
Patch density analysis and projection images of Z-stacks also showed that there were fewer cortical Sla1 patches in scd5-PP1Δ2 than in wild-type cells (Fig. 4C,D; supplementary material Table S5). In contrast, there was a minor to no effect on End3 or Pan1 patch numbers in scd5-PP1Δ2 cells (Fig. 4C; supplementary material Fig. S3B; Table S5), although patches were less polarized typical of most endocytic mutants (e.g. see supplementary material Fig. S3A). There was also a significant increase in Sla2 patch numbers (Fig. 4C,D; supplementary material Table S5), consistent with its severely delayed lifetime and progression defect. Protein levels of Sla2 were similar in the wild-type and scd5 mutant (supplementary material Fig. S3C), so though there were more patches, average Sla2 patch intensity was slightly lower in scd5-PP1Δ2 cells (not shown). Ede1 patch densities were also increased in the scd5 mutant, but patches were too dense for reliable quantification (not shown). Taken together, Sla1 recruitment to cortical sites was most severely affected in the scd5 PP1 targeting mutant, likely due to its hyperphosphorylation and/or that of its binding partner Pan1.
Sla1 spatiotemporal timing is impaired due to inefficient Pan1 binding in scd5-PP1Δ2 cells
Since Sla1–GFP cortical recruitment was the most impaired of the endocytic components in scd5-PP1Δ2 cells, we further dissected its spatiotemporal timing relative to other middle and late stage coat factors (Fig. 5A). When Sla2–GFP was paired with Sla1–RFP in wild-type cells, arrival of Sla1 at the cortex was slightly after, but within 5-10 seconds of that of Sla2 (Fig. 5A) (Carroll et al., 2012; Newpher and Lemmon, 2006). In contrast, Sla1 recruitment was severely delayed in scd5–PP1Δ2 (Fig. 5A, top). However, Sla1 arrival seemed to lead to invagination events, even when Sla2 persisted at the plasma membrane. Similar results were observed for End3–Cherry paired with Sla2–GFP (Fig. 5A, middle). Significantly, Sla1–RFP arrived at the endocytic site after Pan1–GFP in the scd5 mutant, whereas in wild-type cells their recruitment and lifetimes were indistinguishable (Fig. 5A, bottom). This is consistent with our lifetime analysis for Sla1 and Pan1 in the scd5 mutant (Fig. 2; supplementary material Table S3), where Sla1 was recruited ∼18 sec after Pan arrived at endocytic sites.
Since the recruitment of Sla1 to Pan1, and presumably End3, patches was not only diminished, but delayed in the scd5 mutant, we examined whether the interaction of Sla1 with Pan1 was impaired. We expected this might be similar to the effect of Prk1 overexpression, which causes hyperphosphorylation of Sla1 and Pan1 and perturbs their association (Zeng et al., 2001). A cell lysate was generated from wild-type or scd5-PP1Δ2 cells expressing pPan1-LR1–HA, which encodes the first long repeat (LR1) of Pan1 with a C-terminal triple HA tag. Pan1-LR1 mediates the interaction of Pan1 with the C-terminal SR domain of Sla1, and both contain multiple Prk1 phosphorylation sites (Tang et al., 2000; Zeng et al., 2001). Pan1-LR1–HA from the wild-type lysate bound efficiently to GST–Sla1-SR expressed from bacteria; however, binding was at background levels (as compared to GST control) when Pan1-LR1–HA was from the scd5-PP1Δ2 lysate (Fig. 5B, lane 5 versus 6). The reduced interaction of Sla1-SR with Pan1-LR1 is consistent with our in vivo dynamics analysis, and suggests that dephosphorylation by Scd5/PP1 promotes this interaction and is important for progression of endocytic patches to the actin-invagination stage of endocytosis.
Overexpression of SLA1 suppresses defects in Sla2–GFP dynamics in scd5-PP1Δ2
Our data indicate that several Prk1 substrates are hyperphosphorylated in the scd5 PP1 binding site mutant. Therefore we examined whether overexpression of any of these would suppress scd5-PP1Δ2 phenotypes, reasoning that this might provide more dephosphorylated factor for endocytosis. When we overexpressed SLA1, PAN1, ENT1, ENT2 and YAP1802 from 2 µm plasmids, we were unable to detect any suppression of ts growth (data not shown), possibly because Scd5 is essential and is thought to have additional functions in the nucleus (Chang et al., 2006). Though End3 is not a Prk1 target, we tested its gene and obtained similar results. However, when Sla2–GFP and Abp1–RFP dynamics were examined in scd5-PP1Δ2 cells overexpressing these plasmids, we found SLA1 had a dramatic effect on Sla2–GFP in the scd5 mutant. Sla2–GFP lifetimes were reduced from ∼172 s to ∼92 s (P<0.0001; Fig. 6B,E; supplementary material Movie 1), and 81% of patches completed internalization in 3 mins, compared to the mutant alone or transformed with empty vector (≤17% mobile patches; Fig. 6C). There were also significantly fewer patches displaying multiple actin events when SLA1 was overexpressed (24% with SLA1, 2μ versus 52–53% without SLA1 overexpression) (Fig. 6D,E). In contrast, overexpression of other Prk1 targets, PAN1, ENT1/2, and YAP1802 or END3, had no effect on Sla2–GFP/Abp1–RFP dynamics in scd5-PP1Δ2 cells (supplementary material Fig. S4A). All tested factors also had no effect on wild-type cells (Fig. 6; supplementary material Fig. S4B, and data not shown), indicating the lack of suppression was not due to a general negative effect of overexpression. SLA1 had no effect on patch dynamics of later coat factors, Pan1–GFP or End3–GFP, in scd5-PP1Δ2 cells (supplementary material Fig. S4B), but the mobility defect of the early factor Ede1 was also partially suppressed in scd5-PP1Δ2 (supplementary material Fig. S4C). Thus, SLA1 overexpression specifically suppressed the delays from the early/mid coat factor to the late coat factor recruitment stage of internalization and it promoted terminal events when actin arrived.
Fig. 6.

Overexpression of SLA1 or SLA1-SR suppresses Sla2–GFP dynamics in scd5-PP1Δ2. (A) Graphical representation of Sla1 constructs used in B–E. SH3 domains, SHD2, clathrin binding motif (CBM) and C-terminal SR domain repeats are shown. (B–E) Wild-type (SL5928 or SL6026) and scd5-PP1Δ2 (SL6023 or SL6024) strains transformed with no plasmid or 2μ plasmids: pRS426, pJSC66 (YEp-SLA1), pRJC2 (YEp-sla1ΔSR) or pRJC6 (YEp-SR). Similar results were obtained with both strains. (B) Average lifetimes (±s.d.) of Sla2–GFP patches (n≥29; ***P<0.0001 comparing the scd5 transformant to scd5 alone using Student's t-test). (C) ‘% Sla2 mobile patches’ indicates the percentage of Sla2 patches (n = 100) found at the start of the movie that completed internalization in the first 3 min. (D) ‘% multiple actin events’ indicates the percentage of Sla2 patches (n = 100) with multiple actin events in the first 3 min of the movie. (E) Representative kymographs of Sla2–GFP and Abp1–RFP in strains transformed with indicated plasmids.
We next tested whether the Pan1-binding SR domain of SLA1 is important for suppressing the Sla2–GFP/Abp1–RFP dynamics in scd5-PP1Δ2 cells (Fig. 6). Overexpression of sla1ΔSR could not suppress, but overexpression of the SR region alone had a partial, but significant, effect on Sla2–GFP patch lifetime, reducing it to ∼115 s in scd5-PP1Δ2 versus >160 sec in the mutant alone (Fig. 6B,E). The number of Sla2–GFP patches that completed internalization in 3 min was increased to 72% and those that had multiple actin events were decreased from 52–53% to 15% upon overexpression of the Sla1 SR region in the scd5 mutant (Fig. 6C–E). This suggests that the Prk1/Scd5-PP1 regulated SR region of Sla1 is critical for promoting efficient recruitment of Pan1/End3 to Sla2 patches and for driving terminal Sla2 internalization events.
sla1ΔSR causes an endocytic defect, even though Sla1ΔSR–GFP accumulates at the cortex
The failure of overexpression of sla1ΔSR to suppress scd5-PP1Δ2 phenotypes was not surprising, since we deleted the critical binding domain for two important endocytic proteins Pan1 and End3. We thought that Sla1ΔSR might not be recruited to the cortex without its Pan1/End3 interaction region, and that this defect might be more severe than seen in the scd5 mutant, where Sla1 is full length but hyperphosphorylated. To examine this we replaced the genomic copy of SLA1 with sla1ΔSR-GFP. Some nuclear localization of Sla1ΔSR–GFP was observed, likely due to unmasking of a nuclear localization signal (Gardiner et al., 2007). Yet to our surprise, Sla1ΔSR–GFP was recruited to the cell surface as well as or slightly better than Sla1–GFP in scd5-PP1Δ2 (Fig. 7A; supplementary material Fig. S5A). However, the SR truncation was less concentrated in cortical patches and rimmed the cortex, and lifetimes of Sla1ΔSR–GFP, even those for more defined patches, were too extended to quantify (Fig. 7A). Also, few Sla1ΔSR–GFP patches present at the start of movies terminated with Abp1 internalization events, and Sla1-ΔSR–GFP rarely invaginated with the membrane in these few cases (Fig. 7A,B; supplementary material Movie 3).
Fig. 7.
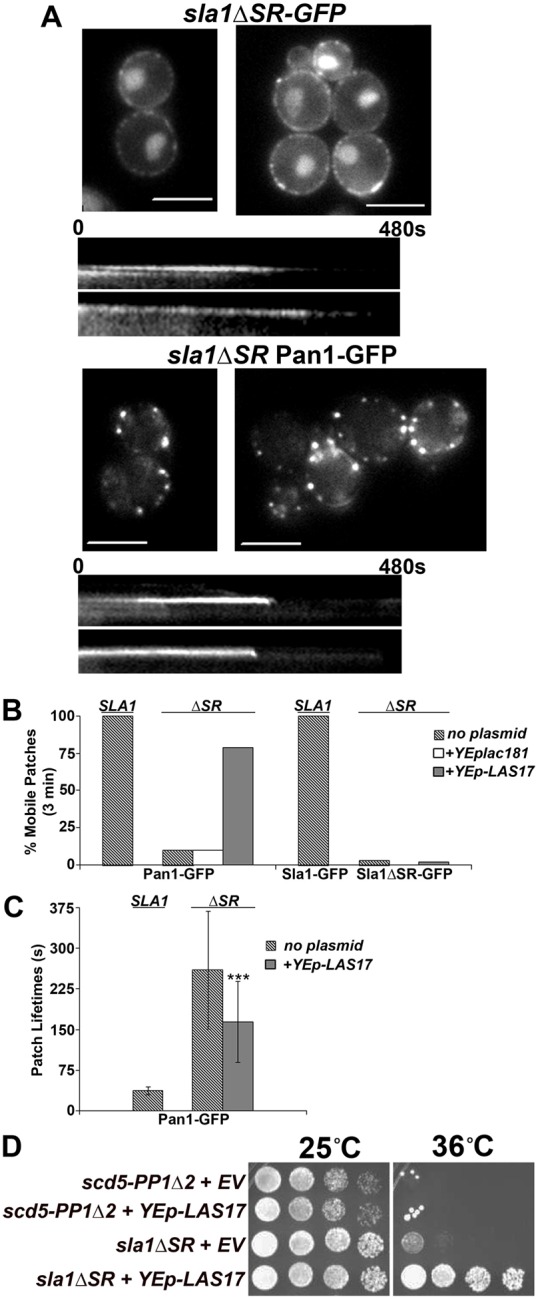
The SR region of Sla1 is crucial for collection of Sla1 in cortical endocytic patches and for endocytic progression, but overexpression of LAS17 suppresses sla1ΔSR. (A) Single frame images and representative kymographs of Sla1ΔSR–GFP (SL6310) and Pan1–GFP in sla1ΔSR (SL6616). Note, some Sla1ΔSR–GFP is in the nucleus due to an exposed nuclear localization signal (Gardiner et al., 2007). Scale bars: 6.6 µm. (B) The percentage of mobile patches (patches completing internalization) within 3 min of the start of the movie for Sla1–GFP (SL5412), Sla1ΔSR–GFP (SL6310), and Pan1–GFP in wild type (SL5425) and sla1ΔSR (SL6616) with and without plasmids, YEplac181(empty vector) or pAM155 (YEp-LAS17). (C) Average patch lifetimes (±s.d.) of Pan1–GFP in wild type (SL5425) and sla1ΔSR (SL6616). sla1ΔSR (SL6616) cells transformed with pAM155 (YEp-LAS17) suppressed Pan1–GFP patch dynamics. n≥30 for each strain; ***P<0.0001 comparing sla1ΔSR transformant to sla1ΔSR alone using Student's t-test. (D) sla1ΔSR causes a temperature-sensitive growth phenotype but is suppressed when overexpressing pAM155 (YEp-LAS17). scd5-PP1Δ2 (SL4610) and sla1ΔSR (SL6216), transformed with YEplac81 (empty vector, EV) or pAM155 (YEp-LAS17) were grown in synthetic drop-out liquid medium to early log phase, concentrated to 1×108 cells/ml, and serial dilutions (1∶6) were pinned onto YEPD plates and grown at 25°C and 36°C for 3.5 days.
In contrast to Sla1ΔSR–GFP, Pan1–GFP patch localization was efficient in sla1ΔSR cells, although often the patches appeared larger than normal (Fig. 7A). Nevertheless, Pan1–GFP internalization was delayed. Lifetimes were generally >4 min and few patches internalized within 3 minutes (Fig. 7A–C; supplementary material Movie 3), although productive invagination and internalization was observed by 8 min in 90% of Pan1–GFP patches (data not shown). In addition, the dynamics of Ede1 and Abp1 were dramatically delayed in the sla1ΔSR strain (supplementary material Fig. S5B,C). Consistent with these endocytic defects, we found that the sla1ΔSR mutation causes ts growth (Fig. 7D).
LAS17 overexpression suppresses sla1ΔSR
Las17 (yeast WASp) arrives at the cortex during the immobile phase of internalization (Kaksonen et al., 2003), but its NPF activity is inhibited by Sla1 and Syp1 during this period (Boettner et al., 2012; Boettner et al., 2009; Rodal et al., 2003). We found that overexpression of LAS17 suppressed the ts growth of sla1-ΔSR at 36°C and partially rescued the Pan1 lifetimes from ∼260 s to ∼165 s, P<0.0001. LAS17 overexpression also increased the number of internalizing patches from 10% to 79%, while Sla1ΔSR–GFP dynamics were unaffected (Fig. 7B,C and data not shown). LAS17 did not suppress scd5-PP1Δ2 cells (Fig. 7D; supplementary material Fig. S5D), so this effect is not general for any endocytic mutant. LAS17 overexpression had the effect of slightly prolonging the lifetime of Abp1 (supplementary material Fig. S5C), but this may represent early or excessive actin assembly due to the additional NPF activity. Overall these findings suggest that the delay in endocytic dynamics of the sla1ΔSR strain is caused in part by constitutive inhibition of Las17.
Discussion
Hyperphosphorylation perturbs endocytic vesicle formation and progression
In budding yeast, the actin regulating serine/threonine kinase family (ARKs), which include Prk1, Ark1 and Akl1, is implicated in actin organization and endocytosis (Cope et al., 1999; Henry et al., 2003; Zeng and Cai, 1999; Zeng et al., 2001). Prk1, the best studied of these kinases, has been shown to phosphorylate a number of downstream targets in the yeast proteome, but the predominant targets are cortical actin patch/endocytic factors, particularly coat module factors that arrive during the immobile phase of internalization (Breitkreutz et al., 2010; Henry et al., 2003; Huang et al., 2003; Mok et al., 2010; Ptacek et al., 2005; Toret et al., 2008; Toshima et al., 2005; Watson et al., 2001; Zeng and Cai, 1999; Zeng et al., 2001). Here we showed that several of these endocytic Prk1 substrates, Pan1, Sla1, Ent1/2, and Yap1801/2, are hyperphosphorylated in scd5-PP1Δ2 cells, thus each of these proteins are likely targets of Scd5/PP1. This is supported by another study on Pan1 and Scd5, which were dephosphorylated by Glc7/PP1 in vitro and hyperphosphorylated in scd5 mutant cells (Zeng et al., 2007). In addition, we found that it is PP1 bound to Scd5 in cis that dephosphorylates Scd5, which has a triple repeat region that is regulated by Prk1 (Henry et al., 2003; Huang et al., 2003). How and when this dephosphorylation regulates Scd5 is still not defined. Nevertheless, these results indicate that PP1 (via its targeting subunit Scd5) is a major opposing phosphatase to Prk1 (and possibly other ARKs) for regulation of actin organization and endocytosis.
Prk1 and Ark1 arrival at cortical patches is late during the mobile actin phase of internalization (Toret et al., 2008; Zeng et al., 2007) and depletion of both proteins leads to accumulation of cortical patch/endocytic factors in large cytoplasmic aggregates containing actin and vesicular material (Chang et al., 2006; Cope et al., 1999; Sekiya-Kawasaki et al., 2003; Watson et al., 2001). Since the major Prk1/Ark1 targets are endocytic coat module proteins, this has led to the proposal that Prk1/Ark1 phosphorylation of these factors promotes their disassembly and/or inactivation after internalization (see model in Fig. 8A). Scd5 targeting of PP1 to counter Prk1/Ark1 would then reverse this phosphorylation and reactivate coat proteins for new rounds of endocytosis. Our live cell imagining results examining 15 endocytic factors showed that the coat assembly/immobile phase of endocytic patch development is dramatically delayed in the scd5 PP1 binding site mutant (Fig. 8B), providing compelling evidence to support this hypothesis. This defect affected the early (clathrin, Ede1) and middle stage coat factors (particularly Sla2) the most, which had lifetimes more than three- to fourfold compared to that of wild-type cells. There was a clear transitional defect from the Sla2 mid-coat phase to the late coat assembly of Sla1/Pan1/End3. We believe this delay was caused, in part, by inefficient recruitment of late stage immobile phase factors to these cortical sites. Sla1 was the most affected of these late factors, in that it arrived after Pan1 and End3 in the scd5 mutant. Also its cortical fluorescence intensities were diminished approximately threefold, while Pan1 and End3 were only slightly affected. A similar difference between Sla1 and Pan1 was seen by Zeng et al. when Prk1 was overexpressed (Zeng et al., 2001). Multiple actin/internalization events were associated with elongated Sla2 and Ede1 (and presumably clathrin patches), but Pan1/End3/Sla1 patches were always found with single internalization/actin events. The partial or complete persistence of Sla2, even after an actin or Pan1/Sla1/End3 event, suggests that the cortical patch was not properly assembled or the delayed arrival of the late coat factors prevented complete consumption of the early/middle stage components.
Fig. 8.
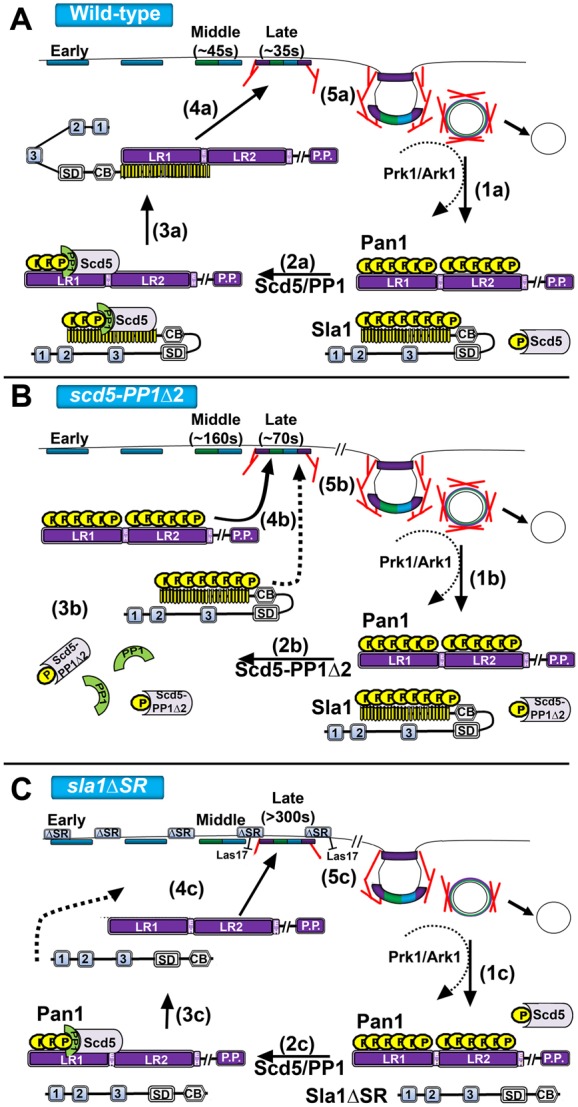
Model of late coat recycling via Scd5/PP1 phosphoregulation. (A) (1a) In wild-type cells, coat factors (Pan1 and Sla1 are shown) are dissembled/inactivated by Prk1/Ark1 phosphorylation after vesicle scission. Sla1 is shown in an auto-inhibited state. (2a) The binding of PP1 activates Scd5/PP1 to target Prk1 substrates for dephosphorylation/activation to promote cortical recruitment and coat assembly. (3a) Dephosphorylation of Sla1 may relieve autoinhibition or may allow binding to Pan1, which could relieve autoinhibition, as well as activation of Pan1. (4a) Dephosphorylated late coat factors (i.e. Pan1 and Sla1) recycle back to the cortex and complete the assembling coat. (5a) Actin-phase factors are recruited and activated (e.g. Arp2/3 complex, Abp1, Myo3/5) thus promoting actin assembly and invagination of the membrane. (B) (2b) In scd5-PP1Δ2 cells, Scd5 can no longer effectively bind PP1/Glc7. (3b) Therefore, late coat factors, which are also Prk1 substrates, remain in their hyperphosphorylated state. (4b) Lifetimes of nearly all endocytic coat factors are delayed. Most notably, Sla1 is not properly recruited to bind and activate the Pan1/End3 complex (4b, dotted line). (5b) This leads to delayed late coat assembly and abnormal internalizing events. (C) (1c–2c) In sla1ΔSR cells, Sla1 is no longer regulated by Prk1 or Scd5/PP1 and is free to diffuse to the cortex, since its inhibitory SR domain is removed. (3c) However, Sla1ΔSR is not collected efficiently in cortical patches because it lacks its Pan1/End3 binding region. (4c) Sla1ΔSR lifetime is slowed considerably and the loss of binding of Sla1 to Pan1 and End3 greatly reduces internalization attempts. (5c) The influx of Sla1ΔSR at the cortex further inhibits Las17 affecting productive progression to the mobile actin phase of endocytosis. (‘//’ denotes the Pan1 coiled-coiled domain and WA domain; proteins not drawn to scale. Note that the plural “positions” of Scd5 on Pan1 and Sla1 are only meant to indicate Scd5/PP1 is removing phosphorylation and not intended to reflect the binding sites on those proteins.)
The types of Sla2 patches seen in scd5-PP1Δ2 cells are similar to events seen in clathrin mutants (Newpher and Lemmon, 2006). In wild-type yeast cells internalization events are extremely efficient and fairly uniform, with almost exclusively terminal events. However, if the immobile coat phase is perturbed (clathrin mutant or scd5), non-terminal events become prevalent. The incomplete turnover of Ede1 and Sla2 at each actin event in scd5-PP1Δ2 also resembles the non-terminal endocytic events seen in animal cells (e.g. see Taylor et al., 2011). Incomplete consumption of coats leaves a coat factor ‘scar’, which can initiate new internalizations. In addition, like in animal cells, even with a delayed immobile phase, the actin/internalization phase, once started, progresses with relatively rapid and normal dynamics. Based on our findings, Sla1is one of the limiting factors required to reach a threshold to progress to this stage.
Positive and negative regulatory roles for Sla1 during endocytosis
Previous studies showed that Prk1 phosphorylation prevents both Pan1-LR1:Sla1-SR and Pan1-LR2:End3 interactions (Zeng et al., 2001). We found that Sla1-SR binding to Pan1-LR1 from scd5-PP1Δ2 cells was also greatly diminished, supporting the notion that Sla1 is not properly incorporated into endocytic sites in scd5-PP1Δ2 cells because of an inability to bind Pan1 (and/or End3) due to hyperphosphorylation of these ARK substrates (see Fig. 8B). Consistent with this, overexpression of SLA1 in scd5-PP1Δ2 cells overcame the progression defects and promoted efficient termination of Sla2 endocytic events. Presumably the increased Sla1 overrode the imbalance between the phosphorylation activity of Prk1 and impaired PP1 targeting, allowing for more dephosphorylated/active Sla1 to incorporate at the cortex with Pan1 and End3. This activity is likely contained in the SR domain of Sla1, since deleting SR (YEp-sla1ΔSR) ablated the suppression, while overexpression of the SR region still partially suppressed Sla2–GFP dynamics in scd5-PP1Δ2 cells. The effect of the SR region may be partial, because overexpression of this domain causes growth impairment in wild-type cells at elevated temperatures (data not shown) and previous studies showed that Sla1-SR is toxic when expressed from a GAL1 promoter (Tang et al., 2000). However, at the temperature we used for imaging, no effect on SCD5 cells was observed.
We postulated that overexpression of sla1ΔSR might not suppress scd5-PP1Δ2 because binding of the SR region to Pan1 and/or End3 is needed for cortical recruitment. However, Sla1ΔSR–GFP was still seen at the cortex, although it rimmed the plasma membrane and was inefficiently collected in patches (see model Fig. 8C). In contrast, Pan1–GFP localized in distinct patches in sla1ΔSR cells. This mutation also severely delayed Ede1, Pan1 and Sla1ΔSR lifetimes and progression of endocytosis. Overall this indicates that the regulatory SR domain is needed to activate targets such as Pan1 and promote internalization.
In contrast to scd5-PP1Δ2, overexpression of the SR region alone could not suppress the sla1ΔSR growth or endocytic defects (not shown). Therefore, one effect of the SR region is to activate Pan1/End3, but the N-terminus of Sla1 appears to have some other negative regulatory function that restrains endocytic progression. The two N-terminal SH3 domains of Sla1 are known to inhibit the WASp Las17 (Rodal et al., 2003), so possibly accumulation of Sla1ΔSR at the cortex constitutively restricts Las17 actin assembly activity (see Fig. 8C). Consistent with this, we found that overexpression of LAS17 could suppress ts growth and Pan1–GFP dynamic defects of the sla1ΔSR mutant.
Curiously, wild-type Sla1–GFP shows diminished recruitment to the cortex in scd5-PP1Δ2, yet retains the N-terminal region that can mediate cell surface localization and inhibit Las17. To reconcile these findings, we hypothesize that phosphorylated Sla1 is not only incompetent for Pan1 binding, but it may be disabled and unable to associate with the cortex through an autoinhibitory mechanism that relies on Prk1 (Fig. 8B). Supporting this, when scd5-PP1Δ2 and sla1ΔSR-GFP were combined, Sla1ΔSR–GFP was still cortical, i.e. it bypassed the effects of the scd5 mutation (data not shown).
Sla1 not only has three SH3 domains and the phospho-regulated SR region, but it contains a SHD1 region that binds and sorts NPFxD cargo (Howard et al., 2002; Mahadev et al., 2007), a variant clathrin TD binding motif and a SHD2/sterile α-motif (SAM) domain that can oligomerize (Di Pietro et al., 2010). Recent work showed that the variant clathrin binding motif is negatively regulated by interaction with SHD2 (Di Pietro et al., 2010). It is proposed that this intramolecular interaction is relieved by binding to clathrin at the endocytic site, which would allow recruitment of Sla1, self-association of the SHD2 region causing concentration of Sla1 at the patch, and binding of Sla1 to its cargo and other endocytic factors. We suggest that the autoinhibition of Sla1 must be relieved to associate with the cortex, inhibit Las17 and activate Pan1, and that both binding to clathrin and dephosphorylation of Sla1 by Scd5/PP1 are critical for this to occur.
A major question that remains is how Sla1 is able to both positively and negatively regulate the endocytic machinery. The spatiotemporal timing of Sla1 is similar to that of Las17, and thus it is positioned to bind and regulate Pan1, also an NPF, while inhibiting Las17. Recruitment to Pan1 itself could open Sla1 to restrain Las17 activity until proper organization of the patch is achieved. Nevertheless, additional regulation via another endocytic protein(s) or mechanism is needed to relieve this Las17 inhibition in order to promote subsequent actin assembly and membrane invagination. Our studies provide new insight into how phosphorylation and Sla1 regulate endocytosis. How an immobile endocytic patch becomes fully mature and the critical transition to the mobile phase is triggered is still one of the least understood aspects of clathrin-mediated endocytosis.
Materials and Methods
Strains, media, growth assays and plasmids
Saccharomyces cerevisiae strains used in this study are listed in supplementary material Table S1. Plasmids are listed in supplementary material Table S2. Yeast growth, mating, sporulation, and tetrad analysis were carried out by standard methods (Guthrie and Fink, 1991). Synthetic medium containing 5-fluoro-orotic acid (5-FOA) was prepared as described in Boeke and colleagues (Boeke et al., 1984). Yeast transformation was performed using the lithium acetate method (Gietz et al., 1995).
Microscopy and image analysis
Cells for live cell imaging of endocytosis were grown to log phase at 25°C in synthetic media, concentrated and mounted on slides in 1.6% agarose, and then imaged at 25°C. All fluorescence lifetimes were calculated from movies acquired on an Olympus BX71 inverted microscope as described in Boettner and colleagues (Boettner et al., 2011) using Slidebook 4.2 for PC platform. For patch dynamics data were combined from at least two experiments, two different spore segregates of the same genotype, or two independent transformants, with at most two patches/cell analyzed. Average patch lifetimes ± s.d. were determined from ≥29 patches for each condition. The Student's t-test was used to calculate P-values. All kymographs, tile-views, projection images and example micrographs were generated in Slidebook and then exported to Adobe Photoshop for figure assembly.
The number of mobile patches was calculated by counting the number of Sla2–GFP or Sla1–GFP patches that were present at the beginning of a time-lapse movie and disappeared within either 3 or 8 min. Patches were scored as having multiple actin events if more than one Abp1–RFP signal appeared during the first three minutes of a single GFP patch lifetime.
Cortical patch to cytosol intensity ratio analysis was carried out on the Olympus fluorescence BX61 upright fluorescence microscope as described before (Newpher and Lemmon, 2006) using Slidebook 4.01 for the Mac for acquisition and analysis. Strains were grown at 25°C to early log phase and single frame images of large budded cells at their medial focal plane were captured. The brightest cortical patch in a cell was identified and the intensity of the brightest pixel was divided by the fluorescence from the mother cell cytosol. A representative background intensity value (outside the cell) was also subtracted from both patch and cytosolic intensities before calculating the patch/cytosol ratio (n≥26 cells for each strain).
Patch densities (patches/surface area µm2) were calculated using optical Z-sections (0.25 µm) of unbudded cells (n = 30 for each strain) as described previously (Newpher and Lemmon, 2006).
Percentage cortical fluorescence was calculated from unbudded and large budded cells grown to log phase at 25°C in synthetic medium. Single frame images (n = 20 cells per strain) were captured at their medial focal plane in Slidebook. ImageJ was used to obtain whole cell fluorescence (WCF) intensity and total internal fluorescence (IF). The latter included all fluorescence below the entire circumference of the cortex. Background fluorescence (using a measurement obtained from outside each cell) was subtracted from the WCF and IF of each cell to generate corrected values. Cortical fluorescence (CF) was obtained by subtracting IF from WCF and then percentage cortical fluorescence was obtained as (CF/WCF)×100. The Student's t-test was used to calculate P-values.
Biochemical methods
Immunoblotting for Prk1 substrates
Cells (1×108) from log phase cultures were harvested, washed with ice-cold water, and resuspended in 0.2 ml of ice-cold RIPA buffer (50 mM Tris-HCl pH 7.5, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 mM sodium pyrophosphate, 150 mM NaCl) plus 1 mM PMSF, a protease inhibitor cocktail and with or without phosphatase inhibitors (Henry et al., 2003). Cell extracts were generated using glass bead lysis, clarified by centrifugation and treated with or without phosphatase (CIP or λ-Ppase; NEB). Samples were analyzed by immunoblotting, with equal loading of gels confirmed by amido black staining. Blots were probed with rabbit anti-Yap180 (1∶5000) (Wendland and Emr, 1998), rabbit anti-Ent1 (1∶20,000) (Aguilar et al., 2003) or goat anti-GFP (for Ent2–GFP; 1∶10,000; Rockland). These were detected with IRDye™800-conjugated goat anti-rabbit IgG (1∶10,000, Rockland) or IRDye™800-conjugated rabbit anti-goat IgG (1∶10,000, Rockland) using the Odyssey Infrared Imaging system (LI-COR).
For Pan1 and Sla1, cells (1×108) were resuspended in 1 ml of ice-cold 10% TCA buffer, incubated on ice for 20 min, and spun for 5 min at 14,000 rpm at 4°C. Pellets were washed twice with cold acetone, dried, and resuspended in 0.2 ml of 50 mM Tris pH 7.5, 1 mM EDTA, 1% SDS for glass bead lysis. Lysates were processed for immunoblotting, probing with rabbit anti-Pan1 (1∶10,000) (Barker et al., 2007) or anti-Sla1 (1∶500) (Warren et al., 2002), and detected as described above.
Purification of GST–Scd5 and GST–scd5-PP1Δ2 from yeast for immunoblotting
pJSC2 (CEN, LEU2, pGAL1:GST-SCD5) and pJSC63 (CEN, LEU2, pGAL1:GST-scd5-PP1Δ2) in SL1462 were grown to log phase in synthetic medium lacking leucine plus 2% raffinose. GST fusion expression was induced by addition of galactose (2% final) for 3 hours at 30°C. Cells (6×108) were harvested, and subjected to glass bead lysis in 1 ml of ice-cold RIPA buffer plus 1 mM PMSF, protease and phosphatase inhibitors. Extracts were cleared by centrifugation at 14,000 rpm for 20 min at 4°C, and diluted to 5 ml with RIPA buffer and incubated with 150 µl of a 75% (vol/vol) slurry of glutathione–Sepharose for 3 h at 4°C. Beads were pelleted, washed 3× with 1 ml RIPA buffer, resuspended in 150 µl 2× SDS-PAGE sample buffer and boiled for 3 min. Samples (22 µl) were subjected to immunoblotting, probing with rabbit anti-Scd5 (1∶6000) or rabbit anti-phosphothreonine (1∶200, Zymed) and detected with IRDye secondaries using the Odyssey.
Purification of bacterially expressed GST fusions and GST pull-downs
GST fusions were expressed in Rosetta E. coli (Agilent Technologies; Santa Clara, CA) and purified as described before (Boettner et al., 2011). To test interaction of the bacterially expressed SR domain with yeast expressed Pan1-LR1–HA, cells (6×108) expressing pLR1–HA-426 (2μ, URA3, HIS3) were subjected to glass bead lysis in 0.5 ml of ice-cold HEKT buffer (20 mM Hepes pH 7.4, 1 mM EDTA acid, 100 mM KCl, 1% Triton X-100) containing protease and phosphatase inhibitors (Henry et al., 2003). Extracts were clarified by centrifugation and 0.2 ml were incubated with 200 nmoles of bacterially purified GST or GST–Sla1-SR domain immobilized on glutathione-Sepharose beads for 1.5 hrs at 4°C. Beads were washed 3× with HEKT buffer and bound proteins were eluted in 160 µL HEKT buffer plus 40 µL of 5× SDS sample buffer and boiled 5 min at 95°C. 30 µl were separated by SDS-PAGE (4–20% gradient gels) and transferred for immunoblotting with rat anti-HA (1∶5000, Roche, 3F10). Detection and quantification were done with the Odyssey. Mouse Anti-PGK1 (1∶1000, Invitrogen, A-6457) was used as a loading control for lysate inputs. Bradford protein assays (Thermofisher) combined with SDS-PAGE densitometry were used to calculate protein concentrations of GST fusions and cell lysates.
Immunoblotting for Sla2-GFP
Cell extracts were prepared as described above, but in the absence of phosphatase inhibitors. Samples were subjected to immunoblotting. Blots were probed with anti-GFP (for Sla2–GFP; 1:5,000; Roche) and anti-Pgk1 antibodies (1:1000, Invitrogen, A-6457). Detection and quantification were done with the Odyssey.
Supplementary Material
Acknowledgments
We thank Clarence Chan, Beverly Wendland, David Drubin, Maribel Geli, Howard Riezman, and Kathryn Ayscough for strains, antibodies, and plasmids. We thank Douglas Boettner for critical reading of this manuscript.
Footnotes
Funding
This work was supported by the National Institutes of Health [grant numbers T32-HL07188 and 5F32-GM087900 to R.J.C, T32-HL007188 to T.L., R01-GM055796 to S.K.L.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.098871/-/DC1
References
- Aguilar R. C., Watson H. A., Wendland B. (2003). The yeast Epsin Ent1 is recruited to membranes through multiple independent interactions. J. Biol. Chem. 278, 10737–10743 10.1074/jbc.M211622200 [DOI] [PubMed] [Google Scholar]
- Aguilar R. C., Longhi S. A., Shaw J. D., Yeh L. Y., Kim S., Schön A., Freire E., Hsu A., McCormick W. K., Watson H. A.et al. (2006). Epsin N-terminal homology domains perform an essential function regulating Cdc42 through binding Cdc42 GTPase-activating proteins. Proc. Natl. Acad. Sci. USA 103, 4116–4121 10.1073/pnas.0510513103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggett J. J., D'Aquino K. E., Wendland B. (2003). The Sla2p talin domain plays a role in endocytosis in Saccharomyces cerevisiae. Genetics 165, 1661–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker S. L., Lee L., Pierce B. D., Maldonado–Báez L., Drubin D. G., Wendland B. (2007). Interaction of the endocytic scaffold protein Pan1 with the type I myosins contributes to the late stages of endocytosis. Mol. Biol. Cell 18, 2893–2903 10.1091/mbc.E07-05-0436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke J. D., LaCroute F., Fink G. R. (1984). A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 197, 345–346 10.1007/BF00330984 [DOI] [PubMed] [Google Scholar]
- Boettner D. R., D'Agostino J. L., Torres O. T., Daugherty–Clarke K., Uygur A., Reider A., Wendland B., Lemmon S. K., Goode B. L. (2009). The F-BAR protein Syp1 negatively regulates WASp-Arp2/3 complex activity during endocytic patch formation. Curr. Biol. 19, 1979–1987 10.1016/j.cub.2009.10.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettner D. R., Friesen H., Andrews B., Lemmon S. K. (2011). Clathrin light chain directs endocytosis by influencing the binding of the yeast Hip1R homologue, Sla2, to F-actin. Mol. Biol. Cell 22, 3699–3714 10.1091/mbc.E11-07-0628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettner D. R., Chi R. J., Lemmon S. K. (2012). Lessons from yeast for clathrin-mediated endocytosis. Nat. Cell Biol. 14, 2–10 10.1038/ncb2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkreutz A., Choi H., Sharom J. R., Boucher L., Neduva V., Larsen B., Lin Z. Y., Breitkreutz B. J., Stark C., Liu G.et al. (2010). A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043–1046 10.1126/science.1176495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll S. Y., Stimpson H. E., Weinberg J., Toret C. P., Sun Y., Drubin D. G. (2012). Analysis of yeast endocytic site formation and maturation through a regulatory transition point. Mol. Biol. Cell 23, 657–668 10.1091/mbc.E11-02-0108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J. S., Henry K., Wolf B. L., Geli M., Lemmon S. K. (2002). Protein phosphatase-1 binding to scd5p is important for regulation of actin organization and endocytosis in yeast. J. Biol. Chem. 277, 48002–48008 10.1074/jbc.M208471200 [DOI] [PubMed] [Google Scholar]
- Chang J. S., Henry K., Geli M. I., Lemmon S. K. (2006). Cortical recruitment and nuclear-cytoplasmic shuttling of Scd5p, a protein phosphatase-1-targeting protein involved in actin organization and endocytosis. Mol. Biol. Cell 17, 251–262 10.1091/mbc.E05-10-0936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton E. L., Evans G. J., Cousin M. A. (2007). Activity-dependent control of bulk endocytosis by protein dephosphorylation in central nerve terminals. J. Physiol. 585, 687–691 10.1113/jphysiol.2007.137539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conibear E. (2010). Converging views of endocytosis in yeast and mammals. Curr. Opin. Cell Biol. 22, 513–518 10.1016/j.ceb.2010.05.009 [DOI] [PubMed] [Google Scholar]
- Cope M. J., Yang S., Shang C., Drubin D. G. (1999). Novel protein kinases Ark1p and Prk1p associate with and regulate the cortical actin cytoskeleton in budding yeast. J. Cell Biol. 144, 1203–1218 10.1083/jcb.144.6.1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin M. A., Tan T. C., Robinson P. J. (2001). Protein phosphorylation is required for endocytosis in nerve terminals: potential role for the dephosphins dynamin I and synaptojanin, but not AP180 or amphiphysin. J. Neurochem. 76, 105–116 10.1046/j.1471-4159.2001.00049.x [DOI] [PubMed] [Google Scholar]
- Di Pietro S. M., Cascio D., Feliciano D., Bowie J. U., Payne G. S. (2010). Regulation of clathrin adaptor function in endocytosis: novel role for the SAM domain. EMBO J. 29, 1033–1044 10.1038/emboj.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff M. P., Johnson D. F., Moorhead G., Cohen P. T., Cohen P., Barford D. (1997). Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J. 16, 1876–1887 10.1093/emboj/16.8.1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner F. C., Costa R., Ayscough K. R. (2007). Nucleocytoplasmic trafficking is required for functioning of the adaptor protein Sla1p in endocytosis. Traffic 8, 347–358 10.1111/j.1600-0854.2007.00534.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R. D., Sugino A. (1988). New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534 10.1016/0378-1119(88)90185-0 [DOI] [PubMed] [Google Scholar]
- Gietz R. D., Schiestl R. H., Willems A. R., Woods R. A. (1995). Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11, 355–360 10.1002/yea.320110408 [DOI] [PubMed] [Google Scholar]
- Grotsch H., Giblin J. P., Idrissi F. Z., Fernandez–Golbano I. M., Collette J. R., Newpher T. M., Robles V., Lemmon S. K., Geli M. I. (2010). Calmodulin dissociation regulates Myo5 recruitment and function at endocytic sites. EMBO J 29, 2899–2914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C., Fink G. R. (1991). Guide to Yeast Genetics and Molecular Biology. San Diego, CA: Academic Press [Google Scholar]
- Henry K. R., D'Hondt K., Chang J., Newpher T., Huang K., Hudson R. T., Riezman H., Lemmon S. K. (2002). Scd5p and clathrin function are important for cortical actin organization, endocytosis, and localization of sla2p in yeast. Mol. Biol. Cell 13, 2607–2625 10.1091/mbc.E02-01-0012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry K. R., D'Hondt K., Chang J. S., Nix D. A., Cope M. J., Chan C. S., Drubin D. G., Lemmon S. K. (2003). The actin-regulating kinase Prk1p negatively regulates Scd5p, a suppressor of clathrin deficiency, in actin organization and endocytosis. Curr. Biol. 13, 1564–1569 10.1016/S0960-9822(03)00579-7 [DOI] [PubMed] [Google Scholar]
- Howard J. P., Hutton J. L., Olson J. M., Payne G. S. (2002). Sla1p serves as the targeting signal recognition factor for NPFX(1,2)D-mediated endocytosis. J. Cell Biol. 157, 315–326 10.1083/jcb.200110027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B., Zeng G., Ng A. Y., Cai M. (2003). Identification of novel recognition motifs and regulatory targets for the yeast actin-regulating kinase Prk1p. Mol. Biol. Cell 14, 4871–4884 10.1091/mbc.E03-06-0362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen M., Sun Y., Drubin D. G. (2003). A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell 115, 475–487 10.1016/S0092-8674(03)00883-3 [DOI] [PubMed] [Google Scholar]
- Kaksonen M., Toret C. P., Drubin D. G. (2005). A modular design for the clathrin- and actin-mediated endocytosis machinery. Cell 123, 305–320 10.1016/j.cell.2005.09.024 [DOI] [PubMed] [Google Scholar]
- Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen and Pringle J. R. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- Mahadev R. K., Di Pietro S. M., Olson J. M., Piao H. L., Payne G. S., Overduin M. (2007). Structure of Sla1p homology domain 1 and interaction with the NPFxD endocytic internalization motif. EMBO J. 26, 1963–1971 10.1038/sj.emboj.7601646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado–Báez L., Dores M. R., Perkins E. M., Drivas T. G., Hicke L., Wendland B. (2008). Interaction between Epsin/Yap180 adaptors and the scaffolds Ede1/Pan1 is required for endocytosis. Mol. Biol. Cell 19, 2936–2948 10.1091/mbc.E07-10-1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok J., Kim P. M., Lam H. Y., Piccirillo S., Zhou X., Jeschke G. R., Sheridan D. L., Parker S. A., Desai V., Jwa M.et al. (2010). Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signal. 3, ra12 10.1126/scisignal.2000482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee D., Coon B. G., Edwards D. F., 3rd, Hanna C. B., Longhi S. A., McCaffery J. M., Wendland B., Retegui L. A., Bi E., Aguilar R. C. (2009). The yeast endocytic protein Epsin 2 functions in a cell-division signaling pathway. J. Cell Sci. 122, 2453–2463 10.1242/jcs.041137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi S. N., Zahn R., Mitchell D. A., Stevenson B. J., Munn A. L. (1998). The WASp homologue Las17p functions with the WIP homologue End5p/verprolin and is essential for endocytosis in yeast. Curr. Biol. 8, 959–962 10.1016/S0960-9822(98)70396-3 [DOI] [PubMed] [Google Scholar]
- Nelson K. K., Holmer M., Lemmon S. K. (1996). SCD5, a suppressor of clathrin deficiency, encodes a novel protein with a late secretory function in yeast. Mol. Biol. Cell 7, 245–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newpher T. M., Lemmon S. K. (2006). Clathrin is important for normal actin dynamics and progression of Sla2p-containing patches during endocytosis in yeast. Traffic 7, 574–588 10.1111/j.1600-0854.2006.00410.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newpher T. M., Smith R. P., Lemmon V., Lemmon S. K. (2005). In vivo dynamics of clathrin and its adaptor-dependent recruitment to the actin-based endocytic machinery in yeast. Dev. Cell 9, 87–98 10.1016/j.devcel.2005.04.014 [DOI] [PubMed] [Google Scholar]
- Perrais D., Merrifield C. J. (2005). Dynamics of endocytic vesicle creation. Dev. Cell 9, 581–592 10.1016/j.devcel.2005.10.002 [DOI] [PubMed] [Google Scholar]
- Ptacek J., Devgan G., Michaud G., Zhu H., Zhu X., Fasolo J., Guo H., Jona G., Breitkreutz A., Sopko R.et al. (2005). Global analysis of protein phosphorylation in yeast. Nature 438, 679–684 10.1038/nature04187 [DOI] [PubMed] [Google Scholar]
- Rodal A. A., Manning A. L., Goode B. L., Drubin D. G. (2003). Negative regulation of yeast WASp by two SH3 domain-containing proteins. Curr. Biol. 13, 1000–1008 10.1016/S0960-9822(03)00383-X [DOI] [PubMed] [Google Scholar]
- Samuels B. A., Tsai L. H. (2003). Cdk5 is a dynamo at the synapse. Nat. Cell Biol. 5, 689–690 10.1038/ncb0803-689 [DOI] [PubMed] [Google Scholar]
- Sekiya–Kawasaki M., Groen A. C., Cope M. J., Kaksonen M., Watson H. A., Zhang C., Shokat K. M., Wendland B., McDonald K. L., McCaffery J. M.et al. (2003). Dynamic phosphoregulation of the cortical actin cytoskeleton and endocytic machinery revealed by real-time chemical genetic analysis. J. Cell Biol. 162, 765–772 10.1083/jcb.200305077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R. S., Hieter P. (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepnev V. I., Ochoa G. C., Butler M. H., Grabs D., De Camilli P. (1998). Role of phosphorylation in regulation of the assembly of endocytic coat complexes. Science 281, 821–824 10.1126/science.281.5378.821 [DOI] [PubMed] [Google Scholar]
- Smythe E., Ayscough K. R. (2003). The Ark1/Prk1 family of protein kinases. Regulators of endocytosis and the actin skeleton. EMBO Rep. 4, 246–251 10.1038/sj.embor.embor776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stimpson H. E., Toret C. P., Cheng A. T., Pauly B. S., Drubin D. G. (2009). Early-arriving Syp1p and Ede1p function in endocytic site placement and formation in budding yeast. Mol. Biol. Cell 20, 4640–4651 10.1091/mbc.E09-05-0429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Martin A. C., Drubin D. G. (2006). Endocytic internalization in budding yeast requires coordinated actin nucleation and myosin motor activity. Dev. Cell 11, 33–46 10.1016/j.devcel.2006.05.008 [DOI] [PubMed] [Google Scholar]
- Tan T. C., Valova V. A., Malladi C. S., Graham M. E., Berven L. A., Jupp O. J., Hansra G., McClure S. J., Sarcevic B., Boadle R. A.et al. (2003). Cdk5 is essential for synaptic vesicle endocytosis. Nat. Cell Biol. 5, 701–710 10.1038/ncb1020 [DOI] [PubMed] [Google Scholar]
- Tang H. Y., Xu J., Cai M. (2000). Pan1p, End3p, and S1a1p, three yeast proteins required for normal cortical actin cytoskeleton organization, associate with each other and play essential roles in cell wall morphogenesis. Mol. Cell. Biol. 20, 12–25 10.1128/MCB.20.1.12-25.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M. J., Perrais D., Merrifield C. J. (2011). A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLoS Biol. 9, e1000604 10.1371/journal.pbio.1000604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizawa K., Sunada S., Lu Y. F., Oda Y., Kinuta M., Ohshima T., Saito T., Wei F. Y., Matsushita M., Li S. T.et al. (2003). Cophosphorylation of amphiphysin I and dynamin I by Cdk5 regulates clathrin-mediated endocytosis of synaptic vesicles. J. Cell Biol. 163, 813–824 10.1083/jcb.200308110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonikian R., Xin X., Toret C. P., Gfeller D., Landgraf C., Panni S., Paoluzi S., Castagnoli L., Currell B., Seshagiri S.et al. (2009). Bayesian modeling of the yeast SH3 domain interactome predicts spatiotemporal dynamics of endocytosis proteins. PLoS Biol. 7, e1000218 10.1371/journal.pbio.1000218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toret C. P., Lee L., Sekiya–Kawasaki M., Drubin D. G. (2008). Multiple pathways regulate endocytic coat disassembly in Saccharomyces cerevisiae for optimal downstream trafficking. Traffic 9, 848–859 10.1111/j.1600-0854.2008.00726.x [DOI] [PubMed] [Google Scholar]
- Toshima J., Toshima J. Y., Martin A. C., Drubin D. G. (2005). Phosphoregulation of Arp2/3-dependent actin assembly during receptor-mediated endocytosis. Nat. Cell Biol. 7, 246–254 10.1038/ncb1229 [DOI] [PubMed] [Google Scholar]
- Toshima J. Y., Toshima J., Kaksonen M., Martin A. C., King D. S., Drubin D. G. (2006). Spatial dynamics of receptor-mediated endocytic trafficking in budding yeast revealed by using fluorescent alpha-factor derivatives. Proc. Natl. Acad. Sci. USA 103, 5793–5798 10.1073/pnas.0601042103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima J., Toshima J. Y., Duncan M. C., Cope M. J., Sun Y., Martin A. C., Anderson S., Yates J. R., 3rd, Mizuno K., Drubin D. G. (2007). Negative regulation of yeast Eps15-like Arp2/3 complex activator, Pan1p, by the Hip1R-related protein, Sla2p, during endocytosis. Mol. Biol. Cell 18, 658–668 10.1091/mbc.E06-09-0788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A., Brachat A., Alberti–Segui C., Rebischung C., Philippsen P. (1997). Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast 13, 1065–1075 [DOI] [PubMed] [Google Scholar]
- Warren D. T., Andrews P. D., Gourlay C. W., Ayscough K. R. (2002). Sla1p couples the yeast endocytic machinery to proteins regulating actin dynamics. J. Cell Sci. 115, 1703–1715 [DOI] [PubMed] [Google Scholar]
- Watson H. A., Cope M. J., Groen A. C., Drubin D. G., Wendland B. (2001). In vivo role for actin-regulating kinases in endocytosis and yeast epsin phosphorylation. Mol. Biol. Cell 12, 3668–3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg J., Drubin D. G. (2012). Clathrin-mediated endocytosis in budding yeast. Trends Cell Biol. 22, 1–13 10.1016/j.tcb.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland B., Emr S. D. (1998). Pan1p, yeast eps15, functions as a multivalent adaptor that coordinates protein-protein interactions essential for endocytosis. J. Cell Biol. 141, 71–84 10.1083/jcb.141.1.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland B., Steece K. E., Emr S. D. (1999). Yeast epsins contain an essential N-terminal ENTH domain, bind clathrin and are required for endocytosis. EMBO J. 18, 4383–4393 10.1093/emboj/18.16.4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng G., Cai M. (1999). Regulation of the actin cytoskeleton organization in yeast by a novel serine/threonine kinase Prk1p. J. Cell Biol. 144, 71–82 10.1083/jcb.144.1.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng G., Yu X., Cai M. (2001). Regulation of yeast actin cytoskeleton-regulatory complex Pan1p/Sla1p/End3p by serine/threonine kinase Prk1p. Mol. Biol. Cell 12, 3759–3772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng G., Huang B., Neo S. P., Wang J., Cai M. (2007). Scd5p mediates phosphoregulation of actin and endocytosis by the type 1 phosphatase Glc7p in yeast. Mol. Biol. Cell 18, 4885–4898 10.1091/mbc.E07-06-0607 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.