

Investigations into the mechanisms by which heme proteins control ligand affinity and reactivity have been studied for decades, with the globins serving as model systems for histidyl-ligated proteins.[1] The recent discovery of a novel family of heme proteins, Heme Nitric oxide/OXygen (H-NOX) binding domains,[2–4] has provided an opportunity to investigate if factors previously found to control ligand affinity and reactivity in the globins can be generalized between protein folds or if there are additional determinants.
The H-NOX family includes the heme domain of soluble guanylate cyclase (sGC), the mammalian NO receptor,[5] as well as heme proteins from bacteria, such as Thermoanaerobacter tengcongensis (Tt).[3] H-NOX domains are a potentially illuminating choice for further investigations into ligand affinity because sGC does not bind O2, even under ambient conditions, whereas other members of the family, such as Tt H-NOX, bind O2 with very high affinity (Kd = 90 nM).[3, 6] A distinctive feature of the wild type (WT) Tt H-NOX crystal structure was a distal pocket hydrogen bonding network, where a key H-bond of Y140 to the bound O2 was observed and the H-bonds of N74 and W9 to Y140.[4] This hydrogen bonding triad is absent in non-O2 binding H-NOX domains, highlighting hydrogen bonding as a major component of O2 binding. A distal H-bond to O2 has been noted as important for the globins. However, introduction of a tyrosine into sGC does not result in O2 binding,[7] suggesting that there are additional determinants of ligand affinity in H-NOX domains.
To further investigate how the H-NOX fold regulates ligand binding, and how this regulation is similar or dissimilar to that of the globins, site-directed mutagenesis was performed on Tt H-NOX. It has been found that the introduction of phenylalanine mutations into the distal pocket of myoglobin (Mb), at positions such as L29 and V68, dramatically alters the oxygen affinity by decreasing off-heme ligand binding sites and reducing ligand "trapping" (Table 1).[8–10] In addition to altering O2 affinity, reaction of ferrous-oxy proteins with NO, or NO dioxygenation (Scheme 1), in Mb was decreased by the addition of distal pocket bulk (Table 1) due to the blocking off-heme binding sites, preventing NO from readily residing in the heme pocket near the bound O2.[8, 11] Therefore, to probe the effects of distal pocket bulk on O2 affinity and NO reactivity of H-NOX proteins, as compared to the globins, isoleucine 75 and leucine 144 were mutated to phenylalanine since both residues are in the back of the distal pocket (Figure 1), analogous to positions L29 and V68 of Mb.
Table 1.
Kinetic parameters for Tt H-NOX and sperm whale myoglobin.
| Protein | Kd (nM) |
koff (s−1) |
kon (µM−1 s−1) |
kox (s−1, ×10−5) |
k'NO,ox (µM−1s−1) |
|---|---|---|---|---|---|
| Tt wt | 88.2 ± 0.67 | 1.20 ± 0.02 | 13.6 ± 1.0 | stable | 0.051± 0.002 |
| Tt I75F | 497 ± 16 | 11.19 ± 0.12 | 22.5 ± 0.7 | stable | 0.19 ± 0.01 |
| Tt L144F | 2360 ± 50 | 16.06 ± 0.21 | 6.8 ± 0.1 | 1.67 ± 0.03 | 0.64 ± 0.02 |
| Tt I75F/L144F | 11150 ± 330 | 45.7 ± 0.9 | 4.1 ± 0.1 | 17.5 ± 1.7 | 2.0 ± 0.1 |
| Mb | 910a | 15a | 17a | 1.53a | 34a |
| Mb V68F | 2100a | 2.5b | 1.2b | 1.94a | 9.4a |
| Mb L29F | 67b | 1.4b | 21b | 0.014b | 8.1a |
| Mb L29F/V68F | 13a | 0.17a | 13a | 2.9a | |
Scheme 1.
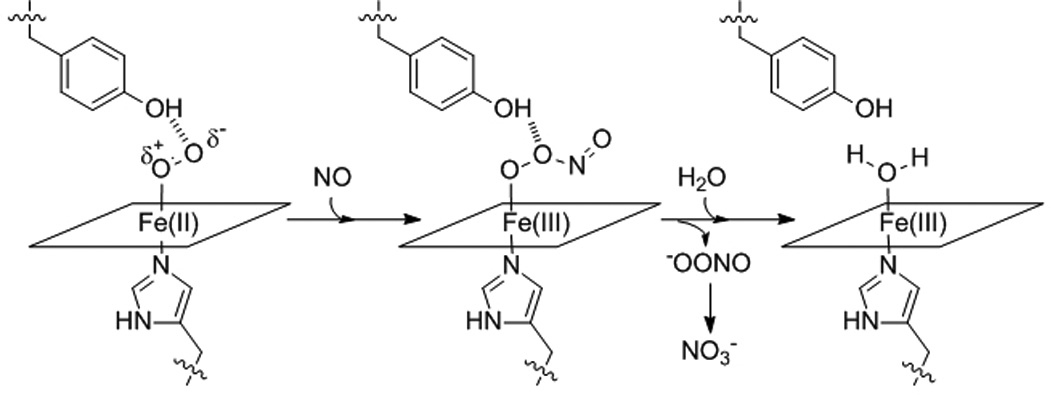
NO dioxygenation reaction.
Figure 1.

Structure of WT Tt H-NOX heme pocket.[4] The heme and key residues are shown in green. L144 and I75 are highlighted in blue.
The I75F, L144F, and I75F/L144F mutants were spectrally similar to WT Tt H-NOX (Figure S1), however, kinetic characterization revealed dramatic changes upon the introduction of distal pocket bulk. The I75F mutation resulted in an approximately 9-fold increase in the O2 off-rate, while the L144F mutation resulted in an approximately 13-fold increase in the O2 off-rate (11.1 9 s−1 and 16.06 s−1, respectively, Table 1). The two mutations had a synergistic effect in the I75F/L144F double mutant, resulting in an O2 off-rate of 45.7 s−1. This represents a 37-fold increase in the O2 off-rate from the introduction of only two mutations, neither of which involved a direct change in the hydrogen bonding network. Typically such large changes in O2 off-rates occur when the primary hydrogen bond donor to the O2 is altered or removed.[9]
There were also significant changes in the O2 on-rate upon introduction of the mutations (Table 1), which, combined with the changes in O2 off-rates, resulted in large decreases in the dissociation constants (Kd) for O2. WT Tt H-NOX has an O2 Kd of ~90 nM, while the I75F mutant was found to have a Kd of 497 nM. The increases in Kd were even more dramatic for the L144F and I75F/L144F mutants (2.36 µM and 11.15 µM, respectively). These represent 6, 27, and 126-fold increases over WT for the I75F, L144F, and I75F/L144F mutations, respectively. To date, these are the largest changes in O2 affinity for Tt H-NOX mutants that stably bind O2. Similar mutations in Mb actually resulted in significantly decreased O2 dissociation rates and very tight binding constants,[8] suggesting that the introduction of distal pocket bulk in Tt results in different changes to the protein structure as compared to Mb.
To understand the cause of the changes in O2 affinity upon introduction of distal pocket bulk, the structure of Tt I75F/L144F was solved (PDB ID 3IQB). In Mb, the mutation of distal pocket residues to phenylalanine resulted in the increased bulk filling cavities in the heme pocket.[11] However, introduction of phenylalanines into Tt H-NOX rearranged the heme pocket, resulting in a more elongated protein structure (~2.5 Å increase in distance from α-helix F to α-helix B, Figure 2A). While an extremely distorted heme was observed in WT Tt H-NOX, the I75F/L144F heme was found to be much flatter (rmsd from planarity of 0.45 vs. 0.20 Å, respectively, Figure 2B) and tilted within the pocket to accommodate the increased bulk (Figure 3A). The addition of the two phenylalanines also altered the conformation of F78, which resulted in a van der Waals contact with α-helix A and a translation of that helix away from the heme (Figure 3B). The combined changes in heme positioning and protein scaffold resulted in a longer Fe(II)-Y140 distance in I75F/L144F, as compared to WT (6.4 Å vs. 5.1 Å, Figure 2B).
Figure 2.

A. Overlay of WT Tt (green) and I75F/L144F (purple) structures. The structures have been aligned along the C-terminal β-sheets since this alignment resulted in the hemes being approximately overlaid. A calculated distance difference matrix (Figure S2) shows that the C-terminal β-sheets change very little between the structures. B. Heme environments of WT and I75F/L144F highlighting changes in inter-atom distances and heme planarity and tilt.
Figure 3.

Heme environment of the I75F/L144F mutant (purple) as compared to WT Tt H-NOX (green). A. Arrangement of F75 and F144 (blue) in the distal pocket. B. Changes in positioning of adjacent residue F78 upon introduction of the phenylalanine mutations.
Besides the changes in distal pocket hydrogen bonding, there were also differences in the proximal pocket of I75F/L144F, as compared to WT Tt H-NOX. The proximal histidine was found to have rotated around the Fe-imidazole bond into a more eclipsed conformation (Figure 2B). An eclipsed histidine conformation has previously been shown to result in decreased O2 binding energy and contribute to lower O2 affinity by decreasing Fe-histidine bond strength.[12] The weaker Fe(II)-histidine bond also likely resulted in the spectrally observed mixture of 5- and 6-coordinate Fe(II)-NO for I75F/L144F. In addition, although within coordinate error of most WT structures,[4, 13] a slight increase in the iron-histidine bond distances was observed that may contribute to the decreased O2 affinity. Calculations previously done on an imidazole-ligated model system found that increasing the Fe-imidazole bond distance by as little as ~0.1 Å, which is difficult to resolve crystallographically, resulted in an approximately 3 kcal/mol decrease in binding energy.[12] The combination of changes to the distal and proximal pockets provides an explanation for the observed 126-fold decrease in O2 affinity for Tt I75F/L144F, as compared to WT. Previous work has found that these factors are important for controlling the energetics of O2 binding in Mb and leghemoglobin,[12] suggesting that these are key determinants of O2 affinity in widely varying protein architectures.
The effects of distal pocket bulk on reactivity of ferrous-oxy Tt H-NOX were further investigated using NO as a probe. The reaction of Fe(II)-O2 protein with NO, (sometimes called NO dioxygenation) (Scheme 1), appears to be an inherent property of the globins and may provide insight as to how the reactivity of the Fe-O2 bond in Tt H-NOX has been altered by the mutations. WT Tt H-NOX was found to have a NO dioxygenation rate of 0.051 µM−1s−1 (Table 1), which is the slowest rate found in the literature.[14, 15] The I75F, L144F, and I75F/L144F mutants were found to have rates of 0.19 µM−1s−1, 0.64 µM−1s−1, and 2.0 µM−1s−1, respectively. All of these rates are among the slowest reported.[14, 15]
It is interesting that the dioxygenation rates for the Tt H-NOX proteins described above are orders of magnitude slower than the globins, even though they both utilize protoporphyrin IX hemes for O2 binding and have similar O2 affinities. Previous work has suggested that the ferric-superoxide-like character of the Fe(II)-O2 complexes and the half life of the O2 on the iron, as the Fe(II)-O2 protein is a required reactant, are important for controlling NO dioxygenation rates.[8, 16]
The O-O stretch, which can be measured by resonance Raman spectroscopy, provides a measurement of the bond order for the bound O2. Tt H-NOX has been found to have an O-O stretch of 1131 ν−1 (vs. O=O stretch of 1556 ν−1), [17] which is very similar to the globins (1122–1155 ν−1) and indicates a metal-superoxide species.[18] Therefore, differences in superoxide character of the bound O2 likely are not fully responsible for the dramatic differences in NO dioxygenation rates between the globins and Tt H-NOX. In addition, long residence time of the O2 at the heme clearly does not dictate NO reactivity in this family of mutants, as WT Tt H-NOX shows the slowest NO dioxygenation rate while having the slowest koff.
Within this family of Tt H-NOX mutants, a possible difference in NO dioxygenation rates is access to the heme-bound O2. The I75F/L144F structure has a small cavity near the bound O2 that could be occupied by NO, while no cavities can be found in the WT structure. Studies with Mb have shown that blocking cavities in the distal pocket can decrease NO reactivity, suggesting that the reverse may be happening upon introduction of distal pocket bulk in Tt H-NOX. In addition, previous work has found that hydrogen bonding of the bound O2 by a tyrosine decreases the NO reactivity of a protein,[15] likely by changing the electronics of the heme iron. WT Tt H-NOX, with a hydrogen bonding tyrosine, has been found to have a reduction potential of 167 mV vs. the standard hydrogen electrode (SHE),[19] as compared to a reduction potential of 59 mV vs. SHE for Mb,[20] which has a hydrogen bonding histidine. The more difficult one electron oxidation of WT Tt H-NOX likely makes formation of the ferric product of NO dioxygenation more unfavorable. Investigations into the heme electronics of the I75F, L144F, and I75F/L144F Tt H-NOX distal pocket mutants are currently underway.
The results reported here show that distal pocket bulk significantly decreases Tt H-NOX O2 affinity. However, in contrast to Mb, the addition of distal pocket bulk results in a more elongated structure and changes to the Y140-Fe bond distance and histidine orientation. The dramatic decrease in O2 affinity in Tt I75F/L144F suggests that these structural features may provide a general method of controlling O2 affinity.
Footnotes
Funding for this work was provided by the National Institutes of Health National Heart Lung and Blood Institute Award F32HL090174 (EEW), NIH grant GM 070671 (MAM), and a grant from the Rogers Family Foundation (MAM). The authors are grateful to Dr. Jay Winkler and the Beckman Instutite Laser Resource Center at the California Institute of Technology for assistance with on-rate measurements and members of the Marletta laboratory for critical reading of this manuscript.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Antonini E, Brunori M. Hemoglobin and myoglobin in their reactions with ligands. Amsterdam: North-Holland Pub. Co.; 1971. [Google Scholar]
- 2.Iyer LM, Anantharaman V, Aravind L. BMC Genomics. 2003;4:5. doi: 10.1186/1471-2164-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karow DS, Pan D, Tran R, Pellicena P, Presley A, Mathies RA, Marletta MA. Biochemistry. 2004;43:10203. doi: 10.1021/bi049374l. [DOI] [PubMed] [Google Scholar]
- 4.Pellicena P, Karow DS, Boon EM, Marletta MA, Kuriyan J. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12854. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Derbyshire ER, Marletta MA. Handb Exp Pharmacol. 2009:17. doi: 10.1007/978-3-540-68964-5_2. [DOI] [PubMed] [Google Scholar]
- 6.Boon EM, Marletta MA. J Inorg Biochem. 2005;99:892. doi: 10.1016/j.jinorgbio.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 7.Rothkegel C, Schmidt PM, Stoll F, Schroder H, Schmidt HH, Stasch JP. FEBS Lett. 2006;580:4205. doi: 10.1016/j.febslet.2006.06.079. [DOI] [PubMed] [Google Scholar]
- 8.Dou Y, Maillett DH, Eich RF, Olson JS. Biophys Chem. 2002;98:127. doi: 10.1016/s0301-4622(02)00090-x. [DOI] [PubMed] [Google Scholar]
- 9.Springer BA, Sligar SG, Olson JS, Phillips GN. Chemical Reviews. 1994;94:699. [Google Scholar]
- 10.Mansy SS, Olson JS, Gonzalez G, Gilles-Gonzalez MA. Biochemistry. 1998;37:12452. doi: 10.1021/bi980516j. [DOI] [PubMed] [Google Scholar]
- 11.Olson JS, Soman J, Phillips GN., Jr IUBMB Life. 2007;59:552. doi: 10.1080/15216540701230495. [DOI] [PubMed] [Google Scholar]
- 12.Capece L, Marti MA, Crespo A, Doctorovich F, Estrin DA. J Am Chem Soc. 2006;128:12455. doi: 10.1021/ja0620033. [DOI] [PubMed] [Google Scholar]
- 13.Nioche P, Berka V, Vipond J, Minton N, Tsai AL, Raman CS. Science. 2004;306:1550. doi: 10.1126/science.1103596. [DOI] [PubMed] [Google Scholar]
- 14.Eich RF, Li T, Lemon DD, Doherty DH, Curry SR, Aitken JF, Mathews AJ, Johnson KA, Smith RD, Phillips GN, Jr, Olson JS. Biochemistry. 1996;35:6976. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]; Gardner PR. J Inorg Biochem. 2005;99:247. doi: 10.1016/j.jinorgbio.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 15.De Jesus-Bonilla W, Jia Y, Alayash AI, Lopez-Garriga J. Biochemistry. 2007;46:10451. doi: 10.1021/bi7003262. [DOI] [PubMed] [Google Scholar]
- 16.Blomberg LM, Blomberg MR, Siegbahn PE. J Biol Inorg Chem. 2004;9:923. doi: 10.1007/s00775-004-0585-5. [DOI] [PubMed] [Google Scholar]
- 17.Tran R, Mathies RA, Marletta MA. [Google Scholar]
- 18.Das TK, Couture M, Ouellet Y, Guertin M, Rousseau DL. Proc Natl Acad Sci U S A. 2001;98:479. doi: 10.1073/pnas.98.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olea C, Boon EM, Pellicena P, Kuriyan J, Marletta MA. ACS Chem Biol. 2008;3:703. doi: 10.1021/cb800185h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varadarajan R, Zewert TE, Gray HB, Boxer SG. Science. 1989;243:69. doi: 10.1126/science.2563171. [DOI] [PubMed] [Google Scholar]