

Abstract
Aminoimidazole carboxamide ribonucleotide transformylase/inosine monophosphate cyclohydrolase (ATIC) is a bifunctional homodimeric enzyme that catalyses the last two steps of de novo purine biosynthesis. Homodimerization of ATIC, a protein-protein interaction with an interface of over 5000 Å2, is required for its aminoimidazole carboxamide ribonucleotide (AICAR) transformylase activity, with the active sites forming at the interface of the interacting proteins. Here, we report the development of a small-molecule inhibitor of AICAR transformylase that functions by preventing the homodimerization of ATIC. The compound is derived from a previously reported cyclic hexa-peptide inhibitor of AICAR transformylase (with a Ki of 17 μM), identified by high-throughput screening. The active motif of the cyclic peptide is identified as an arginine-tyrosine dipeptide, a capped analogue of which inhibits AICAR transformylase with a Ki of 84 μM. A library of non-natural analogues of this dipeptide was designed, synthesized, and assayed. The most potent compound inhibits AICAR transformylase with a Ki of 685 nM, a 25-fold improvement in activity from the parent cyclic peptide. The potential for this AICAR transformylase inhibitor in cancer therapy is assessed by studying its effect on the proliferation of a model breast cancer cell line. Using a non-radioactive proliferation assay and live cell imaging, a dose-dependent reduction in cell numbers and cell division rates was observed in cells treated with our ATIC dimerization inhibitor.
Keywords: AICAR Transformylase, ATIC, Protein-protein interaction Inhibitor, peptide, cancer
Introduction
The activity of many enzymes is controlled by their quaternary structure, via active sites that form at the interface of homodimeric or oligomeric complexes. These enzymes may not only be targeted with substrate analogues, but also by protein-protein interaction inhibitors, enabling an alternative approach to modulating their activity.[1] One such enzyme is aminoimidazole carboxamide ribonucleotide transformylase/inosine monophosphate cyclohydrolase (ATIC), a bifunctional, highly conserved, homodimer that catalyzes the last two steps of de novo purine biosynthesis (Figure 1).[2] Structural studies have shown the ATIC homodimer to have two distinct domains, and a relatively large protein-protein interaction interface of over 5000 Å2.[3] The C-terminal aminoimidazole carboxamide ribonucleotide transformylase (AICAR Tfase) domain catalyzes the transfer of a formyl group from N10-formyl-tetrahydrofolate (10-f-THF) to aminoimidazole carboxamide ribonucleotide (AICAR), and the N-terminal inosine monophosphate cyclohydrolase (IMPCH) domain catalyzes the subsequent dehydrative ring closure to give inosine monophosphate (IMP). The AICAR Tfase active sites are formed at the interface of two interacting ATIC molecules, with each monomer contributing residues to the folate and AICAR binding pockets.[3] Homodimerization is therefore a requisite for the AICAR Tfase activity of ATIC, but not for its IMPCH activity. The significant reliance of rapidly dividing cells on de novo purine biosynthesis for nucleotide production[4] has resulted in much interest and effort towards the development of inhibitors of this enzyme and pathway for use as potential antineoplastic agents.[5]
Figure 1.
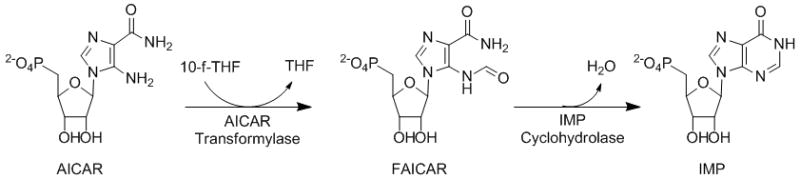
The final two steps of the de novo purine biosythesis pathway catalyzed by ATIC.
ATIC inhibition has also been recently demonstrated to indirectly inhibit cell proliferation and cell cycle progression in human carcinomas by indirectly activating the AMP activated protein kinase (AMPK), highlighting the potential significance of ATIC inhibitors for use in cancer therapy.[6] The development of substrate and cofactor analogues that specifically inhibit ATIC activity has proven challenging however, with folate-based compounds displaying non-specific activity and undesirable side effects.[7] The dimerization requisite for AICAR Tfase activity provides a mechanism by which ATIC may be selectively targeted in the presence of other folate-utilizing enzymes.[8] To this end, we previously reported the screening of a library of 3.2 million cyclic hexa-peptides[9] for inhibitors of ATIC homodimerization.[8a] The most potent inhibitor identified was the cyclic peptide CRYFNV (1), which inhibits AICAR Tfase activity with a Ki of 17 ± 4 μM by preventing ATIC homodimerization.[8a] Although a relatively modest inhibitor for direct use as a therapeutic, a six-membered cyclic peptide that blocks the interaction of the ATIC homodimer holds much potential for further development and evolution into a more potent, small molecule.
Results and Discussion
Identification of the active motif of cyclo-CRYFNV
The development of small molecules from peptides is considered a laborious and time-consuming process.[10] Compared to its linear counterpart, a 6-mer cyclic peptide has limited conformational freedom, with substantially fewer possible structures. This conformational restriction, combined with the sequence homology observed in the nine selected ATIC inhibitors,[8a] indicated that some residues of the cyclic peptide ATIC inhibitor are key to its potency, with others simply functioning as a backbone. To validate this hypothesis and identify the critical motif, we synthesized six analogues of cyclo-CRYFNV, in which each amino acid was sequentially replaced with alanine.
Each alanine-scanning analogue was assayed for inhibition of the AICAR Tfase activity of ATIC. The initially ordered binding of 10-f-THF and AICAR to ATIC[11] result in a dimerization inhibitor effectively acting as a competitive inhibitor with respect to 10-f-THF, and a non-competitive inhibitor with respect to AICAR;[8a] we therefor used Michaelis-Menten kinetics to assess the potency of each analogue.
The data (Table 1) indicated that the neighboring arginine and tyrosine residues are critical for the activity of cyclo-CRYFNV, as replacing either residue resulted in an inactive ATIC inhibitor. In contrast, replacing the other residues (FNVC) with alanine resulted in 2–40 fold loss of activity, suggesting that these amino acids are less critical for inhibitor activity, possibly acting as a backbone to present the RY motif to ATIC. A truncated analogue of cyclo-CRYFNV, in which the FNVC backbone had been removed, was therefore designed (compound 8, Figure 2), synthesized (Scheme 1) and assayed (Table 2) for its ability to inhibit AICAR Tfase.
Table 1.
AICAR Tfase inhibition by CRYFNV alanine-scanning analogues.
| Compound | Sequence | Ki |
|---|---|---|
| 1 | cyclo-CRYFNV | 17 ± 3 μM |
| 2 | cyclo-ARYFNV | 651 ± 25 μM |
| 3 | cyclo-CAYFNV | > 2.5 mM |
| 4 | cyclo-CRAFNV | > 2.5 mM |
| 5 | cyclo-CRYANV | 38 ± 5 μM |
| 6 | cyclo-CRYFAV | 368 ± 29 μM |
| 7 | cyclo-CRYFNA | 252 ± 22 μM |
Figure 2.
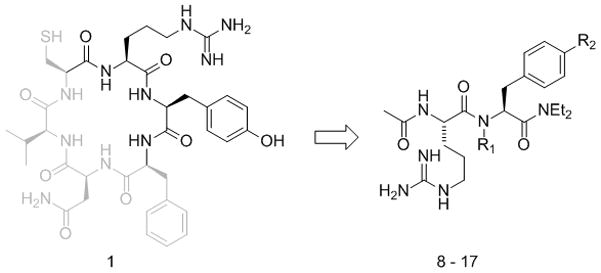
The capped RY dipeptide 8 was designed to mimic the active RY motif (highlighted) of CRYFNV 1. RY analogues 9–17 containing unnatural amino acids were synthesized in an attempt to improve inhibitor activity.
Scheme 1.
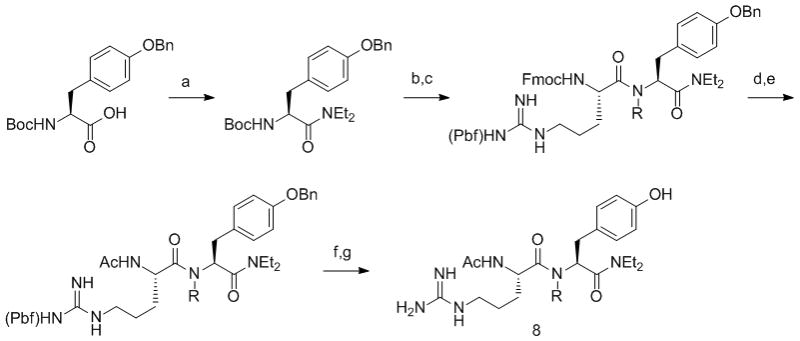
Synthesis of RY dipeptides. Reagents and conditions (R = H, Me). a) EDC, HOBt, Et2NH, CH2Cl2; b) TFA/CH2Cl2; c) Fmoc-Arg(Pbf)-OH, EDC, HOBt, CH2Cl2; d) DBU, EtOAc; e) Ac2O, Et3N, CH2Cl2; f) 10% Pd/C, H2, MeOH; g) TFA/TIS/H2O.
Table 2.
AICAR Tfase inhibition by RY analogues.
| Compound | R1 | R2 | Ki |
|---|---|---|---|
| 8 | H | OH | 84 ± 7 μM |
| 9 | H | H | 339 ± 37 μM |
| 10 | H | F | 135 ± 12 μM |
| 11 | H | OMe | 87 ± 9 μM |
| 12 | H | CN | 56 ± 5 μM |
| 13 | H | B(OH)2 | 59 ± 5 μM |
| 14 | H | NO2 | 685 ± 35 nM |
| 15[a] | H | NO2 | 52 ± 4 μM |
| 16 | Me | OH | 145 ± 14 μM |
| 17 | Me | NO2 | 2.5 ± 0.2 μM |
lysine in place of arginine
The capped RY dimer 8 was found to inhibit the AICAR Tfase activity of ATIC with a Ki of 84 ± 7 μM, suggesting that the RY motif of cyclo-CRYFNV is responsible for its AICAR Tfase activity. The 5 fold loss in activity may be attributed to the increase in conformational freedom of the active motif upon removal of the cyclic backbone; linear-CRYFNV inhibits AICAR Tfase with a Ki of 142 ± 22 μM,[8a] an 8 fold loss compared to the cyclic counterpart, due to the same reason.
Improving the activity of the RY dipeptide with unnatural amino acids
To improve the activity of the capped RY dimer 8, chemical spaces not accessible to our original cyclic peptide library (which is synthesized in vivo using split-inteins[9, 12] and therefore limited to the 20 canonical amino acids) were explored using unnatural amino acids. A series of related compounds containing electron-donating or electron-withdrawing para-substituted tyrosine analogues were synthesized (9–14) to probe the electronic and steric requirements of the tyrosine-binding pocket. These compounds were assayed for AICAR Tfase inhibition (Table 2). Removing the hydroxyl group of tyrosine (compound 9) resulted in a 4 fold loss of activity. Replacing the hydroxyl group with fluorine (compound 10) also resulted in activity loss (1.6 fold), while the p-methoxy derivative (compound 11) had similar levels of activity as the RY dimer. Introducing an electron withdrawing p-nitrile (compound 12), or a p-borono substitution (compound 13) caused a 1.5 fold improvement in activity, while a p-nitro group (compound 14) caused a 120 fold improvement in activity, yielding an AICAR Tfase inhibitor with a Ki of 685 ± 35 nM. Replacing the arginine residue of 14 with lysine (compound 15) resulted in a 75 fold loss of activity.
The effect of N-methylation of the peptide backbone was also explored, as this would be expected to restrict the conformational freedom of the compound, and had been shown to improve the activity of other cyclic peptides[13]. N-methylated analogues of the capped RY dimer 8 and its p-nitro analogue 14 were therefore synthesized and assayed. N-methylation of 8 diminished its activity by 1.7 fold, while N-methylation of 14 resulted in a 3.6 fold loss of activity. The observed loss of activity on methylation may be a result of the steric hindrance associated with the methyl group affecting binding to the active site. The p-nitro-tyrosine analogue of cyclo-CRYFNV was also synthesized and assayed to determine if the increase in potency observed for the RY dipeptide upon introduction of p-nitro group, would also be observed in the parent cyclic peptide. The p-nitro analogue of cyclo-CRYFNV was found to inhibit AICAR Tfase with a Ki of 36 ± 4 μM, a surprising 2 fold reduction in activity compared to the parent compound. Without structural data to identify the binding site of these compounds, the reasons for the observed reduction in activity upon reintroduction of the cyclic peptide backbone remain unclear.
Deciphering the mode of action of compound 14
We previously demonstrated by progress rate analysis that cyclo-CRYFNV is a competitive inhibitor of ATIC with respect to 10-f-THF, and that its inhibition of AICAR Tfase is due to disruption of the ATIC dimer.[8a] To assess if the most potent dipeptide uncovered in this study maintains this mechanism of action, or inhibits AICAR Tfase through a nonspecific aggregate mechanism,[14] 14 was assayed in the presence of 1 mg/mL and 10 mg/mL of bovine serum albumin (BSA). Inhibition by nonspecific aggregators is prevented by the addition of milligram per milliliter concentrations of BSA (prior to the addition of the enzyme and inhibitor);[14] a nonspecific inhibitor would therefore be expected to lose activity under these conditions. The rate of the AICAR Tfase reaction was not affected by the presence of 1 mg/mL of BSA in the assay buffer, but was slowed by around 40% in the presence of 10 mg/mL BSA (Figure 3A). This may be due to the high BSA concentration affecting the ATIC monomer-dimer equilibrium by stabilizing the monomeric form of ATIC, and therefore reducing AICAR Tfase activity. In both cases however, compound 14 at 10 μM and 50 μM continued to inhibit the reaction to a similar extent as observed in the absence of BSA (Figure 3A). This indicated that 14 does not function using a nonspecific aggregate mechanism of inhibition.
Figure 3.

A) The effect of mg/mL quantities of BSA on AICAR Tfase inhibition by 14. The compound continues to inhibit the enzyme regardless of BSA presence, indicating it does not function via nonspecific aggregation. B) and C) Size exclusion chromatography. B) The ratio of monomer to dimer is shown in a 50 μM solution of ATIC C) The same mixture with 10 μM of compound 14 shows the presence of monomeric ATIC at 85 mL.
To further assess if the inhibition of AICAR Tfase by compound 14 is due to its disruption of ATIC homodimerization, size exclusion chromatography was utilized. The large change in molecular mass of ATIC upon homodimerization (from 64 kDa to 129 kDa) enables the ratio of monomer to dimer to be monitored by this technique. ATIC has a Kd of 240 ± 50 nM in the absence of its substrates, with a 50 μM solution containing mainly dimeric species.[15] Size exclusion chromatography of a 50 μM solution of ATIC showed a single peak at 72 mL, corresponding to a mass of ~130 kDa (see Supporting Figure 17 for column calibration), indicating the presence of only dimeric species (Figure 3B).
The experiment was repeated with a 50 μM solution of ATIC treated with 10 μM of compound 14. An additional peak at 85 mL was observed, corresponding to a mass of ~65 kDa, indicating the presence of monomeric ATIC (Figure 3C). As a control, a 50 μM solution of ATIC was treated with 10 μM of the least potent dipeptide inhibitor identified in this study (compound 9, Table 2). As 9 is a significantly weaker ATIC inhibitor than 14, it should not disrupt ATIC dimerization at the concentration used in this experiment. As expected, only a single peak corresponding to dimeric ATIC was observed (supplementary Figure 18). The presence of ATIC monomer in the solution treated with 14, but not in the untreated ATIC solution or that treated with a weak inhibitor, suggests that 14 inhibits AICAR Tfase by perturbing ATIC homodimerization.
Inhibition of ATIC homodimerization in breast cancer cells
Inhibition of purine metabolism has been long proposed as a possible approach to targeting tumor growth.[16] Two pathways operate in cells for purine production; the purine salvage pathway operates in normal cells, whilst de novo purine production is thought to predominate in rapidly dividing cancer cells. Inhibition of de novo purine biosynthesis has therefore been proposed as a possible strategy for the selective inhibition of growth in rapidly dividing cancer cells. The majority of current inhibitors of enzymes in the de novo purine biosynthetic pathway are folate analogues that inhibit multiple enzymes in the cell; the significance of de novo purine production in cancer therapy is therefore difficult to decipher. We therefore used 14 to assess the effect of inhibiting ATIC dimerization on the viability of a model breast cancer cell line; MCF-7 cells were dosed by addition of 100 μM, 250 μM or 500 μM of compound 14 to their growth media. The viability of treated versus untreated cells was determined 48 hours after dosing, using a non-radioactive proliferation assay. A dose-dependent reduction in viability was observed in cells treated with compound 14 (Figure 4A), reaching 40% inhibition at the maximum dose of 500 μM. The effect of ATIC inhibition on cell proliferation was further analyzed in cells treated with 250 μM of 14 using time-lapse microscopy. Images were captured every 45 minutes over 72 hours and analysed. These images showed that cells treated with 250 μM 14 exhibited a 35% reduction in proliferation compared to control-treated cells (Figure 4B and 4C). This data was in agreement with that from the cell viability assays (Figure 4A). Further analysis of the videos indicated that the effect of 14 on cell numbers was through a reduction in cell division rates, rather than an increase in cell death. The MCF-7 breast cancer cell line used retains active cell cycle checkpoints that protect from stress-induced cell death,[17] which may provide a possible explanation for the above observations. Interestingly, indirect inhibition of AICAR Tfase by the antifolate Methotrexate has been proposed as the principal mode of action for its cytotoxicity in MCF-7 cells. Further cell-based studies are necessary to clarify the role of de novo purine biosynthesis in the survival and growth of various cancer cells. Non-folate inhibitors of various enzymes in this pathway, such as compound 14, will be valuable chemical tools for these studies.
Figure 4.

Assessing the proliferation of MCF-7 cells treated with 14. A) MTS assays show a dose-dependent reduction in viable cells 48 hours after treatment. B) Analysis of cell growth by live cell imaging C) Representative images from live cell imaging showing a population of untreated cells over 72 hours, compared to a population of cells treated with 14. The videos shows the reduction in proliferation in cells treated with 14 is due to reduced rate of division rather than increased cell death.
In addition to the above, ATIC inhibition has also recently been proposed to be the primary mode of action for the anticancer drug Pemetrexed;[6a] the rise in unmetabolized AICAR proposed to allosterically activate AMPK by mimicking AMP, causing inhibition of cell proliferation. It is therefore possible that the effect of 14 on cell proliferation is due to AMPK activation, as well as inhibition of de novo purine biosynthesis; this possibility is currently being investigated in our laboratory.
Conclusion
The development of a small molecule inhibitor of ATIC homodimerization with a Ki of 685 nM from a cyclic hexa-pepide is reported. The finding that four of the six amino acids of cyclo-CRYFNV can be eliminated, and that the RY dipeptide is sufficient to disrupt the interaction of a 5000 Å2 interface raises several interesting questions about the possible binding site of the compound, and may be significant for the peptidomimetic evolution of other cyclic peptide inhibitors of protein-protein interactions [18].
We are currently developing non-peptidic analogues of 14 with the aim of further improving the potency of this compound. Structural studies are also underway to determine the binding pocket targeted on ATIC, and to enable further optimization of the inhibitor via traditional medicinal chemistry approaches. Experiments are also underway to decipher the mechanism by which 14 inhibits cell proliferation and to determine the effect of 14 on the proliferation of other cancer cell lines and in vivo tumor models. Whilst the potency of compound 14 may be further improved as a result of these efforts, the compound is sufficiently active in its current form to serve as a tool for investigations into the role of de novo purine biosynthesis in cells.
Experimental Section
General
Amino acids and peptide coupling reagents were obtained from Merck (Novabiochem). All other chemical reagents were obtained from SigmaAldrich or Fisher Scientific and used without further purification. Thin-layer chromatography (TLC) was carried out on Merck silica gel plates with a fluorescence indicator (254 nm). Visualization of TLC plates was carried out by UV light, then by either a cerium sulphate/ammonium molybdate or potassium permanganate stain. Flash chromatography was preformed with 35–70 μm silica gel (Fisher Davisil). Infrared spectra were recorded using a FTIR spectrometer fitted with an ATR accessory. 1H NMR and 13C NMR were recorded using a Bruker DPX400 (400 and 100 MHz). Chemical shifts δ are given in ppm. All enzymatic assays were performed using a Varian Cary 100 Spectrometer. All peptides were purified on a Waters HPLC, using a Waters Alantis Prep T3 OBD 5μm 19 × 100 mm column using a water/acetonitrile at a flow rate of 17 mL min−1. For cell culture experiments, all reagents were obtained from Invitrogen. MCF-7 cells were maintained in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (Autogen Bioclear), penicillin (100 U/ml), streptomycin (100 μg/ml), and L-glutamine (292 μg/ml).
Cyclic Peptide synthesis
The alanine-scanning analogues of CRYFNV were synthesized and purified as previously described for CRYFNV.[8a]
Synthesis of capped dipeptides
The dipeptides in Table 2 were synthesized as outlined for compound 8 in Scheme 1 on a 500 mg scale. Further experimental details and analytical information for compounds 8–13 and 15–17 are provided as supplemental data. The synthesis of compound 14 is detailed below:
Synthesis of Boc-Phe(4-NO2)-diethylamide
Dry CH2Cl2 (8 mL), and Boc-4-nitro-L-phenylalanine (500 mg, 1.61 mmol) were added to a three- neck flask under an inert atmosphere, with vigorous stirring. To this mixture, EDC hydrochloride (309 mg, 1.61 mmol) and HOBt (218 mg, 1.61 mmol) were added, and the solution was stirred for 30min at room temperature. Diethylamine (0.34 mL, 3.38 mmol) was added and the resulting yellow solution was left to stir for 4h. The progress of this reaction was checked by TLC, and was shown to be complete in 4 hours. The reaction was stopped, diluted with CH2Cl2 (30mL) and washed with citric acid (10 mL), NaHCO3 saturated solution (10 mL) and brine (10 mL). The organic phase was dried with anhydrous Na2SO4, filtered and evaporated in vacuo to give a pale yellow oil that solidified upon standing. The crude material was dried on argon stream and re-precipitated using cold hexane to give a white solid that was collected by Buchner funnel (483mg, 82% yield). Rf = 0.82 (EtOAc/Hex 2:1). m.p. 90–90.5 °C. 1H-NMR (300MHz, DMSO-d6): δ = 8.17 (d, 2H, J = 8.8Hz, Ar), 7.57 (d, 2H, J = 8.8Hz, Ar), 7.31 (d, 1H, J = 8.4Hz, NH), 4.57 (q, 1H, J = 7.7Hz, CH2); 2.97–3.41 (m, 6H, CH), 1.30 (s, 9H, tertBut), 1.08 (t, 3H, J = 6.9Hz, CH3), 1.01 (t, 3H, J = 6.9Hz, CH3). 13C-NMR (75MHz, DMSO-d6): 169.7, 154.9, 146.3, 130.8, 123.7, 78.1, 51.1, 41.1, 37.5, 27.9, 14.2, 12.7. HRMS/ES+ (m/z): calculated for C18H27N3O5Na: 388.1843 [M+Na]+ found: 388.1854 [M+Na]+.
Synthesis of fmoc-Arg(Pbf)-CONH-Phe(4-NO2)-L-Diethylamide
Dry CH2Cl2 (6 mL), Boc-4-nitro-L-phenyldiethylamide (350 mg, 1.32 mmol) were added to a flask under an inert atmosphere with vigorous stirring. Trifluoracetic acid (6 mL) was added to this mixture and stirred for 1h. The reaction was monitored by TLC. After this time, the solvent was removed in vacuo and the orange oil co-evaporated with ethylacetate (3 × 10 mL) to give a yellow oil (350 mg, 100% yield) whose identity was confirmed by mass spectrometry. The deprotected amine was added to stirred dry CH2Cl2 (15 mL) in a flask under an inert atmosphere. fmoc-Arg(Pbf)-OH (1.88 mg, 3.0mmol) was added to this solution and the reaction was cooled to −20°C. HOBt (535 mg, 3.96 mmol) and EDC (380 mg, 1.98, mmol) were added, and the reaction was left to stir overnight and allowed equilibrate to room temperature. The reaction was diluted with CH2Cl2 (100 mL), washed with citric acid (20 mL), saturated NaHCO3 solution (20 mL) and brine (20 mL). The yellow solution was evaporated in vacuo to leave a yellow solid, which was purified by column chromatography using CH2Cl2/MeOH 95:5 to give the product (Rf = 0.45) as an off-white solid (520mg, 44% yield). m.p. 110°C decomp. 1H-NMR (300MHz, DMSO-d6): 8.25δ (d, J = 8.4Hz, 1H), 8.03 (d, J = 8.4Hz, 2H), 7.84 (d, J = 7.3Hz, 2H), 7.65 (d, J = 8.8Hz, 2H), 7.46–7.34 (m, 7H), 4.96–4.89 (m, 1H), 4.34–4.22 (m, 3H), 4.04–4.00 (m, 1H), 3.35–2.98 (m, 12H), 2.47 (s, 3H), 2.04 (s, 3H), 1.43 (s br, 10H), 1.07–0.97 (m, 6H). 13C-NMR (75MHz, DMSO-d6): 172.3, 170.1, 158.4, 157.0, 156.7, 147.2, 146.6, 144.8, 144.6, 141.7, 138.2, 132.4, 131.7, 131.6, 128.6, 128.0, 126.1, 125.2, 124.0, 123.9, 121.0, 117.2, 87.2, 66.5, 60.7, 55.1, 50.3, 47.6, 43.4, 42.1, 38.6, 29.2, 19.9, 18.5, 15.2, 13.7, 13.2. LRMS/ES+ (m/z): 896 [M+H]+ 5%, 918 [M+Na]+100%, 1813 [2M+Na]+9.6%. HRMS/ES+ (m/z): calculated for C47H58N7O9S: 896.4011 [M+H]+ found: 896.4010 [M+H]+.
Synthesis of Amino-Arg(Pbf)-CONH-Phe(4-NO2)-L-Diethylamide
Dry ethylacetate(14 mL) was added to fmoc-Arg(Pbf)-CONH-Phe(4-NO2)-diethylamide (520 mg, 0.58 mmol) in a flask under an inert atmosphere and stirred vigorously. To this mixture, DBU (93 mg, 0.1 mL, 0.61 mmoL) was added; the solution was stirred for 1h and the reaction was monitoring by TLC. The solvent was removed in vacuo to give a brown solid, which was purified by column chromatography using CH2Cl2/MeOH 9:1 to give the product (Rf = 0.35) as a white solid (363 mg, 93% yield). m.p. 121°C decomp. 1H-NMR (300MHz, DMSO-d6): 8.31δ (d, J = 8.4Hz, 1H), 8.15 (d, J = 8.4Hz, 2H), 7.51 (d, J = 7.3Hz, 2H), 4.96–4.91 (m, 1H), 3.43–2.94 (m, 13H), 2.54–2.46 (m, 8H overlap DMSO d6), 2.03 (s br, 5H), 1.44 (s br, 10H), 1.07–0.97 (m, 6H). 13C-NMR (75MHz, DMSO d6): 174.3, 169.3, 157.3, 156.0, 146.2, 145.7, 137.2, 134.2, 131.3, 130.7, 124.2, 123.0, 116.2, 86.2, 54.1, 48.7, 42.4, 41.1, 37.9, 32.1, 28.2, 25.3, 18.9, 17.5, 14.3, 12.7, 12.2. LRMS/ES+ (m/z): 674 [M+H]+ 39%, 696 [M+Na]+100%, 1869 [2M+Na]+11%. HRMS/ES+ (m/z): calculated for C32H47N7O7S: 674.3330 [M+H]+ Found: 674.3319 [M+H]+.
Synthesis of Acetyl-Arg(Pbf)-CONH-Phe(4-NO2)-L-Diethylamide
Dry ethylacetate (7 mL) and fmoc-Arg(Pbf)-CONH-Phe(4-NO2)-diethylamide (363 mg, 0.54 mmol) were added to flask under an inert atmosphere and stirred vigorously. Triethylamine (58 mg, 0.08 mL, 0.57 mmoL), and acetic anhydride (58 mg, 0.05 mL, 0.57 mmol) were to theis mixture and stirred overnight. The solvent was removed in vacuo giving a sticky yellow oil that was purified by column chromatography (short path) using CH2Cl2/MeOH 9:1 to give a white solid (Rf = 0.52) in quantitative yield (386mg, 100% yield). m.p. 113°C decomp. 1H-NMR (300MHz, DMSO-d6): 8.36δ (d, J = 8.4Hz, 1H), 8.14 (d, J = 8.8Hz, 2H), 7.89 (d, J = 8.2Hz, 2H), 7.56 (d, 8.8Hz, 2H), 4.92–4.84 (m, 1H), 4.28–4.24 (m, 1H), 3.36–2.99 (m, 16H), 2.54–2.51 (s br, 8H overlap DMSO-d6), 2.45 (s, 3H), 1.94 (s, 3H), 1.84 (s, 3H), 1.55–1.28 (s br, 11H), 1.05–0.96 (m, 6H). 13C-NMR (75MHz, DMSO-d6): 172.1, 170.1, 168.9, 157.3, 155.9, 147.1, 146.7, 138.1, 135.1, 135.1, 133.3, 131.7, 125.2, 124.0, 117.1, 87.2, 50.3, 43.4, 42.0, 38.5, 30.4, 29.1, 26.3, 23.3, 22.0, 18.5, 15.2, 13.6, 13.2. LRMS/ES+ (m/z): 716 [M+H]+ 100%, 1431 [2M+Na]+26%. HRMS/ES+ (m/z): Calcd. for C34H50N7O8S: 716.3436 [M+H]+ Found: 716.3434 [M+H]+.
Synthesis of Acetyl-Arg-CONH-Phe(4-NO2)-L-Diethylamide
To a flask, a cocktail of TFA/TIS/H2O 9.5/0.25/0.25 (22 mL) and acetyl-Arg(Pbf)-CONH-Phe(4-NO2)-diethylamide (386 mg, 0.54 mmol) were added, and stirred at r.t. for 2h. The reaction was monitored by TLC, and the solvent evaporated in vacuo to leave a viscous yellow oil. This was co-evaporated with CH2Cl2 (3 × 5 mL) and MeOH (3 × 5 mL) and the residue dried with an argon stream for ~20min. Diethylether (20 mL) was then added and the white precipitate collected by filtration (263 mg, 100% crude yield). The crude product was purified by reverse phase HPLC (retention time 7.63 minutes) and lyophilized to obtain the product as a white solid (125 mg, 50% yield). m.p. 113°C decomp. 1H-NMR (300MHz, DMSO-d6): 8.31δ (d, J = 8.7Hz, 1H), 8.10 (d, J = 8.6Hz, 2H), 7.91 (d, J =8.1Hz, 2H), 7.65 (t, J = 5.6Hz, 1H), 7.50 (d, 8.6Hz, 2H), 7.17 (s br, 3H), 4.90–4.82 (m, 1H), 4.32–4.23 (m, 1H), 3.37–3.00 (m, 7H), 2.95 (dd, 1H), 1.82 (s, 3H), 1.61–1.50 (m, 3H), 1.00 (t, 7.12Hz, 3H), 0.95 (t, 7.13 Hz, 6H). 13C-NMR (75MHz, DMSO-d6): 171.1,169.1, 169.1, 156.8, 146.3, 145.8, 130.8, 123.1, 51.8, 49.4, 41.4, 40.4, 39.8, 37.6, 29.2, 25.0, 22.4, 14.2, 12.7. LRMS/ES+ (m/z): 464 [M+H]+ 100%. HRMS/ES+ (m/z): Calcd. for C21H33N7O5: 464.2616 [M+H]+ Found: 464.2610 [M+H]+.
Protein expression and enzyme kinetics
Avian and human ATIC fused to an N-terminal 6X histidine tag were expressed from pET28 vectors in BL21(DE3) strains of E. coli, and purified as previously detailed [8a, 19]. AICAR Tfase assays were conducted with avian ATIC and carried out in 1 cm path length quartz cuvettes at 25 °C as previously described [8a, 19]. Each inhibitor was dissolved in DMSO to give a 100 mM stock solution that was further diluted for use in each assay. The data was analyzed using KaleidaGraph (Synergy Software) and Microsoft Excel, by assuming competitive inhibition with 10-f-THF as previously detailed [8a]. Errors in the Ki were calculated by linear regretion. Compounds 1, 8, and 14 were also assayed with human ATIC, with the same Ki (within error) being observed for each compound as for the avian enzyme.
Size exclusion chromatography
Three 50 μM solutions of human ATIC were prepared as above, in 1 mL of buffer (20 mM Tris-Cl, pH 7.5, 150 mM NaCl, 50 mM KCl, 5 mM EDTA, 5 mM dithiothreitol). 10 μM of compound 14 or 9 (negative control) was added to two of these solutions. Each solution was filtered through a Millex GP 0.22 μm filter (Millipore), and loaded onto a HiLoad 16/60 Superdex 200 gel filtration column (GE Healthcare) pre-equilibrated with the above buffer. The flow rate was 1 mL/min and the flow monitored by UV on an AKTAprime FPLC (GE Healthcare). The column was calibrated using a HMW gel filtration calibration kit (GE Healthcare).
Cell proliferation assays
Cells were plated at 2000 cells per well in 96-well plates 24 h prior to dosing with appropriate drug in 100 μl fresh medium, final concentration of solvent DMSO in each well was 0.5%. MTS-based cell proliferation assays were performed 48 h post-dosing according to manufacturer’s instructions (CellTitre Aqueous One Cell Proliferation assay, Promega).
Live-cell imaging
Cells were plated at 10,000 cells per well in 24-well plates. Time-lapse microscopy [17] was initiated on dosing, with images captured every 45 min for 72 hours. Analysis was carried out using Image J software. All conditions were assayed in triplicate, and expressed as means ± S.E.M. Statistical significance was calculated using the one-way ANOVA parametric test and Tukey’s post hoc test using Prism software (GraphPad).
Supplementary Material
Acknowledgments
We thank Dr. Josephine Corsi, Dr. Peter Roach, and Dr. Michael Webb for helpful discussions, and Miss. Abigail Male for the SEC calibration data. This work was funded by National Institute of Health grant GM24129-28, and Cancer Research UK grant 10263.
References
- 1.Cardinale D, Salo-Ahen OM, Ferrari S, Ponterini G, Cruciani G, Carosati E, Tochowicz AM, Mangani S, Wade RC, Costi MP. Curr Med Chem. 2010;17:826–846. doi: 10.2174/092986710790712156. [DOI] [PubMed] [Google Scholar]
- 2.Ni L, Guan K, Zalkin H, Dixon JE. Gene. 1991;106:197–205. doi: 10.1016/0378-1119(91)90199-l. [DOI] [PubMed] [Google Scholar]
- 3.Greasley SE, Horton P, Ramcharan J, Beardsley GP, Benkovic SJ, Wilson IA. Nat Struct Biol. 2001;8:402–406. doi: 10.1038/87555. [DOI] [PubMed] [Google Scholar]
- 4.a Allegra CJ, Hoang K, Yeh GC, Drake JC, Baram J. J Biol Chem. 1987;262:13520–13526. [PubMed] [Google Scholar]; b Bronder JL, Moran RG. Cancer Res. 2002;62:5236–5241. [PubMed] [Google Scholar]
- 5.a Zhang Y, Desharnais J, Marsilje TH, Li C, Hedrick MP, Gooljarsingh LT, Tavassoli A, Benkovic SJ, Olson AJ, Boger DL, Wilson IA. Biochemistry. 2003;42:6043–6056. doi: 10.1021/bi034219c. [DOI] [PubMed] [Google Scholar]; b Marsilje TH, Labroli MA, Hedrick MP, Jin Q, Desharnais J, Baker SJ, Gooljarsingh LT, Ramcharan J, Tavassoli A, Zhang Y, Wilson IA, Beardsley GP, Benkovic SJ, Boger DL. Bioorg Med Chem. 2002;10:2739–2749. doi: 10.1016/s0968-0896(02)00102-5. [DOI] [PubMed] [Google Scholar]; c Desharnais J, Hwang I, Zhang Y, Tavassoli A, Baboval J, Benkovic SJ, Wilson IA, Boger DL. Bioorg Med Chem. 2003;11:4511–4521. doi: 10.1016/s0968-0896(03)00458-9. [DOI] [PubMed] [Google Scholar]; d Marsilje TH, Hedrick MP, Desharnais J, Tavassoli A, Zhang Y, Wilson IA, Benkovic SJ, Boger DL. Bioorg Med Chem. 2003;11:4487–4501. doi: 10.1016/s0968-0896(03)00456-5. [DOI] [PubMed] [Google Scholar]
- 6.a Racanelli AC, Rothbart SB, Heyer CL, Moran RG. Cancer Res. 2009;69:5467–5474. doi: 10.1158/0008-5472.CAN-08-4979. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rothbart SB, Racanelli AC, Moran RG. Cancer Res. 2010;70:10299–10309. doi: 10.1158/0008-5472.CAN-10-1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christopherson RI, Lyons SD, Wilson PK. Acc Chem Res. 2002;35:961–971. doi: 10.1021/ar0000509. [DOI] [PubMed] [Google Scholar]
- 8.a Tavassoli A, Benkovic SJ. Angew Chem Int Ed Engl. 2005;44:2760–2763. doi: 10.1002/anie.200500417. [DOI] [PubMed] [Google Scholar]; b Capps KJ, Humiston J, Dominique R, Hwang I, Boger DL. Bioorg Med Chem Lett. 2005;15:2840–2844. doi: 10.1016/j.bmcl.2005.03.094. [DOI] [PubMed] [Google Scholar]
- 9.Tavassoli A, Benkovic SJ. Nat Protoc. 2007;2:1126–1133. doi: 10.1038/nprot.2007.152. [DOI] [PubMed] [Google Scholar]
- 10.Liskamp RM, Rijkers DT, Kruijtzer JA, Kemmink J. Chembiochem. 2011;12:1626–1653. doi: 10.1002/cbic.201000717. [DOI] [PubMed] [Google Scholar]
- 11.Bulock KG, Beardsley GP, Anderson KS. J Biol Chem. 2002;277:22168–22174. doi: 10.1074/jbc.M111964200. [DOI] [PubMed] [Google Scholar]
- 12.Scott CP, Abel-Santos E, Wall M, Wahnon DC, Benkovic SJ. Proc Natl Acad Sci U S A. 1999;96:13638–13643. doi: 10.1073/pnas.96.24.13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chatterjee J, Gilon C, Hoffman A, Kessler H. Accounts of chemical research. 2008;41:1331–1342. doi: 10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]
- 14.a Coan KE, Shoichet BK. Mol Biosyst. 2007;3:208–213. doi: 10.1039/b616314a. [DOI] [PubMed] [Google Scholar]; b McGovern SL, Caselli E, Grigorieff N, Shoichet BK. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 15.Vergis JM, Bulock KG, Fleming KG, Beardsley GP. J Biol Chem. 2001;276:7727–7733. doi: 10.1074/jbc.M009940200. [DOI] [PubMed] [Google Scholar]
- 16.a Rustum YM, Takita H, Gomez G. Antibiot Chemother. 1980;28:86–93. [PubMed] [Google Scholar]; b Weber G, Nagai M, Natsumeda Y, Ichikawa S, Nakamura H, Eble JN, Jayaram HN, Zhen WN, Paulik E, Hoffman R, et al. Adv Enzyme Regul. 1991;31:45–67. doi: 10.1016/0065-2571(91)90008-a. [DOI] [PubMed] [Google Scholar]; c Natsumeda Y, Prajda N, Donohue JP, Glover JL, Weber G. Cancer Res. 1984;44:2475–2479. [PubMed] [Google Scholar]; d Marijnen YM, de Korte D, Haverkort WA, den Breejen EJ, van Gennip AH, Roos D. Biochim Biophys Acta. 1989;1012:148–155. doi: 10.1016/0167-4889(89)90088-8. [DOI] [PubMed] [Google Scholar]
- 17.Bergman LM, Birts CN, Darley M, Gabrielli B, Blaydes JP. Mol Cell Biol. 2009;29:4539–4551. doi: 10.1128/MCB.00439-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tavassoli A, Lu Q, Gam J, Pan H, Benkovic SJ, Cohen SN. ACS Chem Biol. 2008;3:757–764. doi: 10.1021/cb800193n. [DOI] [PubMed] [Google Scholar]
- 19.Wall M, Shim JH, Benkovic SJ. Biochemistry. 2000;39:11303–11311. doi: 10.1021/bi0007268. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.