

Abstract
Advanced or metastatic prostate cancer is treated by androgen deprivation; however, patients inevitably relapse with castration-resistant prostate cancer (CRPC). CRPC remains dependent on androgen receptor (AR) signaling, which may include constitutive, ligand-independent action of naturally occurring AR splice variants. For example, the AR splice variant AR3 (also termed AR-V7) is expressed in CRPC and is linked to poor prognosis. Vav3, a Rho GTPase guanine nucleotide exchange factor, is an AR coactivator that is up-regulated in human prostate cancer compared with benign tissue and in preclinical models of CRPC. Vav3 confers castration-resistant growth to androgen-dependent human prostate cancer cells. Despite the importance of AR coactivators in promoting CRPC, the potential role of these regulatory proteins in modulating AR splice variant activity is unknown. We examined the contributions of Vav3 to AR activity in two CRPC cell lines that naturally express relatively high levels of Vav3 and AR3. Vav3 or AR3 knockdown greatly attenuated cell proliferation, soft agar growth, and ligand-independent AR activity. Vav3 potently enhanced the transcriptional activity of AR3 and another clinically relevant AR splice variant, ARv567es. Vav3 knockdown resulted in lowered nuclear AR3 levels, whereas total AR3 levels remained similar. Conversely, overexpression of Vav3 resulted in increased nuclear AR3. Coimmunoprecipitation revealed that AR3 and Vav3 interact. These novel data demonstrating physical and functional interactions between Vav3, a unique AR coactivator, and an AR splice variant provide insights into the mechanisms by which Vav3 exploits and enhances AR signaling in the progression to CRPC.
Despite advances in screening and therapeutic modalities, prostate cancer remains the second most common cancer worldwide and claims over a quarter of a million lives annually (1). Advanced or metastatic disease is responsible for the majority of prostate cancer-related deaths. Androgen-deprivation therapy, the gold standard of treatment for non-organ-confined disease, provides palliative relief with tumor regression and falling levels of prostate-specific antigen (PSA). However, the cancer inevitably recurs within 9–23 months in virtually all patients (2). At this stage, the disease is termed castration-resistant prostate cancer (CRPC) (3, 4) and indicates a rapidly progressing disease state for which treatment options are limited. The mechanistic processes underlying progression from androgen dependence to castration resistance are still not fully understood; however, continued or reactivated androgen receptor (AR) signaling is critical (3–10). Increased expression of constitutively active AR splice variants and AR coactivators are among the mechanisms that may promote AR transcriptional activity in CRPC (6, 9, 11–14).
AR is a steroid hormone receptor with an N-terminal transactivation domain, DNA-binding domain, hinge region, and C-terminal ligand-binding domain (LBD) (15, 16). AR splice variants were originally identified in CRPC cell lines derived from serially propagating the androgen-dependent human prostate cancer cell line CWR22 in castrated mice (17, 18). These variants retain the N-terminal transactivation domain of AR but lack various regions of the C terminus including the LBD (17–22). Most importantly, several AR splice variants are constitutively active in the absence of androgen (18–22). Recent clinical data have associated the constitutively active splice variant AR3 (also known as AR-V7) with poor clinical prognosis (19, 20, 23). Furthermore, AR3 confers castration resistance to androgen-dependent prostate cancer cell lines (20, 21). Given the importance of AR coactivators in promoting CRPC and that constitutively active AR splice variants are not bound by clinically used AR antagonists, it is imperative to evaluate the possible involvement of AR coactivators in modulating AR splice variant activity.
Several studies have highlighted the importance of the Rho GTPase guanine nucleotide exchange factor (GEF) Vav3 to CRPC progression. Previous work from our lab showed that Vav3 levels increase during progression to CRPC and that Vav3 enhances AR transcriptional activity in a GEF-independent manner even at subnanomolar levels of androgen (24–26). Additionally, Vav3 mRNA levels are up-regulated in the Nkx3.1;Pten mutant mouse model of prostate cancer progression (27) as well as in clinical samples of prostate cancer patients who have undergone androgen ablation therapy (from dataset of Ref. 28). Vav3 protein is elevated in a significant number of prostate cancer clinical specimens compared with benign tissue (29). Recently, it was found that not only is Vav3 elevated in late-stage and metastatic prostate cancer clinical samples but also that increased Vav3 expression correlates with decreased biochemical failure-free survival (30). Targeted expression of a constitutively active Vav3 allele to murine prostatic epithelium results in formation of high-grade prostatic intraepithelial neoplasia and prostate adenocarcinoma (31). Importantly, Vav3 confers castration resistance to an androgen-dependent cell line in vivo (26). Although Vav3 is a well-established enhancer of AR activity, its potential role in AR splice variant signaling is unknown and may play a crucial role in the progression to CRPC.
Materials and Methods
Cell culture and chemical reagents
The human prostate cancer cell lines LNCaP.FGC (ATCC catalog no. CRL 1740; batch F-11701), PC-3 (ATCC catalog no. CRL 1435; batch F-11154), and CWR-22Rv1 (CRL-2505, batch 4484055) were obtained from American Type Culture Collection (Manassas, VA). CWR-R1 and VCaP cells were generous gifts from Dr. Elizabeth M. Wilson (University of North Carolina, Chapel Hill, NC) and Dr. Kenneth Pienta (University of Michigan, Ann Arbor, MI), respectively. CWR-22Rv1-shGFP, -shAR, -shVav3, and -shAR3 knockdown cells were derived from 22Rv1 cells transduced with the corresponding plk0.1 short hairpin RNA (shRNA) construct and selected using 500 ng/ml puromycin. The same approach was followed with CWR-R1 cells and 22Rv1 cells transduced with the tet-plk0.1 system. LNCaP cells overexpressing green fluorescent protein (GFP) or Vav3 were isolated as described previously (26). Cell culture media (RPMI-1640 and DMEM) were obtained from Cellgro by Mediatech, Inc. (Manassas, VA). Fetal bovine serum (FBS) was obtained from Atlanta Biologicals, Inc. (Lawrenceville, GA). LNCaP, DU145, 22Rv1, CWR-R1, and PC-3 cell lines were cultured in RPMI supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin, 2 mm l-glutamine (Life Technologies, Inc., Carlsbad, CA) and 10% FBS. R1881 was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA), and doxycycline was purchased from Sigma Chemical Co. (St. Louis, MO).
Plasmids
The PSA luciferase (PSA-Luc) reporter plasmid (kindly provided by Dr. Carlos Perez-Stable, University of Miami) consists of the PSA promoter and 5′-flanking region, which contain both the distal (−5325 to −4023) and proximal (−542 to −12) androgen response element (ARE)-containing enhancer regions but lacks the intervening sequences. The ARE luciferase (ARE-Luc) was a generous gift from Dr. Zafar Nawaz (University of Miami). The following DNA constructs were generously provided: shAR (Dr. Paul Rennie, University of British Columbia, Vancouver, British Columbia, Canada), plk0.1 shGFP and tet-plk0.1 shGFP (Dr. Priya Rai, University of Miami), shAR3 and AR3 (Dr. Yun Qiu, University of Maryland School of Medicine, Baltimore, MD), shVav3 (Dr. Daniel Billadeau, Mayo Clinic, Rochester, MN), pcDNA3.1 ARv567es (Dr. Stephen Plymate, University of Washington, Seattle, WA). The nucleotide sequences used to target Vav3, AR, and AR3 mRNA were GCTTTGTCTAACATAAGAC, GAAAGCACTGCTACTCTTCAGCATTATTC, and GTAATAGTGGTTACCACTC respectively. Knockdown-resistant Vav3 was created through two stages of site-directed mutagenesis using the following primers: step 1 forward (5′-gaagaaatggctagaacagtttgaaatggctcttaccaatatacgcccagactatgcagactccaatttccacgac-3′), step 1 reverse (5′-gtcgtggaaattggagtctgcatagtctgggcgtatattggtaagagccatttcaaactgttctagccatttcttc-3′), step 2 forward (5′-gaagaaatggctagaacagtttgaaatggcgcttagcaatatccgcccagactatgcagactccaatttccacgac-3′), and step 2 reverse (5′-gtcgtggaaattggagtctgcatagtctgggcggatattgctaagcgccatttcaaactgttctagccatttcttc-3′). Vav3 ISOIII, W493L, and ΔPH mutants were cloned as previously described (24).
VCaP xenografts
All experiments involving animals were conducted in a manner approved by the University of Miami Animal Care and Use Committee (Miami, FL). VCaP cells stably expressing Vav3-Flag or GFP were generated and maintained as previously described (26). The mice were castrated when the tumors reached a volume of 300 mm3, and tumors were allowed to progress to castration resistance. When tumors reached approximately 1000 mm3 (6–9 wk after castration), tumors were then excised and flash frozen in liquid nitrogen for protein analysis.
Reporter gene assays and transfections
All transfections were carried out using the cationic lipid reagent Lipofectamine (Invitrogen/Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. For luciferase assays, cells were plated at a density of 3.0 × 105 cells in 35-mm dishes 16–20 h before transfection. Immediately before transfection, media were replaced with unsupplemented DMEM. For luciferase assays (PC3 cells), cells were transfected with 1.6 μg of reporter ARE luc; 83 ng CMV-hAR, pcDNA3.1ARv567es, or PQCXIP-AR3; and 83 ng of pIRES-egfp-Vav3 or empty vector. After a 5- to 6-h incubation with DNA/lipid complexes, cells were re-fed with RPMI supplemented with 2% charcoal-dextran-stripped serum (CSS) and treated with vehicle or 1 nm R1881. Cells were harvested 48 h after transfection, lysed, and assessed for luciferase activity using the Promega luciferase assay kit (Promega Corp., Madison, WI). Luciferase assays in CWR-R1 and 22Rv1 were performed with the addition of a 5-min DNA incubation with PLUS reagent (Invitrogen Life Technologies) as well as refeeding of the cells with 5% CSS with or without 1 μg of doxycycline (where applicable). Transfection of LNCaP Vav3/GFP cells was done with 250 ng PQCXIP or PQCXIP-AR3 per 60-mm plate.
RNA extraction and real-time RT-PCR
Total RNA was harvested using the Trizol method according to the manufacturer's protocol (Invitrogen Life Technologies). Total RNA (2 μg) was reverse transcribed using a cDNA archive kit (Applied Biosystems, Foster City, CA). Real-time PCR was performed using 100 ng cDNA and ABI StepOne. TaqMan probes from Applied Biosystems for HPRT1, UBE2C, GAPDH, FKBP5, IGFBP3, and KLK3 were used. Custom AB TaqMan probes were designed for AR3 using the following sequences: forward primer 5′-ttctgggtgtcactatggagctctc-3′, reverse primer 5′-TCAGGGTCTGGTCATTTTGAGATG-3′, and probe 5′-CTGGGAGAAAAATTCCGGGTTGG-3′. The comparative threshold cycle method was used to determine the relative expression level of mRNA.
Flow cytometry
The 22Rv1 cells (selected for expression of indicated constructs) were plated at 1 × 106 cells in 100-mm dishes. To synchronize the cell cycle, the following day, the cells were washed one time in PBS, incubated in unsupplemented RPMI for 3 h, and then incubated for an additional 21 h in 5% CSS at 37 C/5% CO2. Cells were then grown in 10% FBS media for 48 h. Cells were trypsinized, and approximately 1 × 106 were fixed in 70% ethanol at 4 C. Cells were counterstained with 50 ng/ml propidium iodide and RNA digested with 1 mg/ml ribonuclease (Roche Laboratories, Indianapolis, IN). To determine DNA content, propidium iodide-stained cells were analyzed by flow cytometry using the FACScan (Becton Dickinson, San Jose, CA) at the University of Miami, Sylvester Comprehensive Cancer Center, Flow Cytometry Core Facility. Analyses were done on 10,000 total gated cells.
Cellular fractionation
Cells were plated at 2 × 106 cells per 100-mm dish and grown in 5% CSS with or without 1 μg doxycycline for 72 h. Cellular fractionation was performed according to the manufacturer's protocol (Thermo Scientific, Waltham, MA), and 10 μg of each protein sample was loaded onto SDS-PAGE gels as described below.
Western blot analysis
Samples were run in 8–12% SDS-PAGE gels and transferred onto nitrocellulose membranes. Membranes were blocked with 5% nonfat dry milk in Tris-buffered saline [20 mm Tris base (pH 7.5), 50 mm NaCl, and 2.5 mm EDTA] containing 0.1% Tween 20 and then probed with anti-AR (N-20; 1:1000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-AR3 (1:1000; Precision Antibody, Columbia, MD), anti-histone (1:1000; Santa Cruz), anti-super oxide dismutase (1:1000; Santa Cruz); anti-cyclin A (1:1000; Santa Cruz); anti-cleaved poly ADP ribose polymerase (PARP) (1:1000; Cell Signaling), anti-Vav3 (1:1000, Cell Signaling), or anti-actin (1:1000; Santa Cruz) primary antibodies diluted in 5% nonfat dry milk in Tris-buffered saline with Tween 20. After washing in Tris-buffered saline with Tween 20, membranes were incubated with their appropriate horseradish peroxidase-conjugated secondary antibodies (Santa Cruz) and developed using an enhanced chemiluminescence detection system (Amersham Biosciences, Arlington Heights, IL) according to the instructions of the manufacturer. Densitometry was performed using ImageJ software (32).
Cell proliferation assay
Cells were plated at an initial density of 20,000 per well in 24-well dishes. The following day, cells were trypsinized and viable cells (excluded trypan blue) were counted using a hemocytometer. Experiments done in 5% CSS were subjected to an extra wash and 3-h incubation in unsupplemented RPMI to remove residual androgens. Experiments were performed in triplicate and were counted at a minimum of three time points.
Soft agar assays
The 22Rv1 cells were stably infected with shGFP, shAR, shAR3, or shVav3 as described above. Cells were counted and mixed with an equal volume of 20% FBS, 2× RPMI, and of 0.6% noble agar (final percentage, 10% FBS and 0.3% agar) and seeded at a density of 2500 cells in 35-mm petri plates containing a base layer of 0.5% noble agar. Plates were incubated for 4 wk. Colonies were stained with 0.005% crystal violet and counted using the Bio-Rad Geldoc system (Bio-Rad Laboratories, Inc., Hercules, CA).
Immunoprecipitation
HEK293 cells were plated at 3.5 × 106 cells per 100-mm dish. Cells were transfected with 5 μg of PQCXIP AR3 or AR and pIRES-egfp-Vav3-myc using a calcium-phosphate transfection kit according to manufacturer's instruction (Clontech, Mountain View, CA). After 48 h, the cells were lysed in Nonidet P-40 (NP-40) lysis buffer containing 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1% NP-40, and proteinase inhibitor cocktail (Sigma). Cell debris was removed by centrifuging at 16,000 × g for 10 min at 4 C. Cell lysates incubated with 30 μl of 25% protein G plus Sepharose (Santa Cruz Biotechnology) for 1 h to remove nonspecific bead binding. This was followed by immunoprecipitation with 2 μg nonspecific mouse IgG (Santa Cruz), 2 μg monoclonal mouse anti-myc (Invitrogen), 2 μg nonspecific rabbit IgG (Santa Cruz), and 2 μg rabbit polyclonal AR-N20 (Santa Cruz) overnight at 4 C, followed by adding 50 μl of 25% protein G plus Sepharose (Santa Cruz) for 2 h at 4 C. The precipitates were washed four times with immunoprecipitation wash buffer containing 0.1% NP-40, 50 mm Tris-HCl (pH 7.4) and denatured with Laemmli sodium dodecyl sulfate sample buffer and separated by SDS-PAGE as described above. The input lanes were loaded with 10 μg of the starting cell lysate.
Chromatin immunoprecipitation
The 22Rv1 cells stably expressing shGFP, shAR3, or shVav3 were grown in phenol red-free RPMI with 5% CSS for 3 d. Chromatin was then cross-linked with 1% formaldehyde for 10 min at room temperature. The cross-linked chromatin was then sonicated, diluted, and immunoprecipitated with AR3 antibody (Precision Antibody) or normal mouse IgG control at 4 C overnight. Protein G+ agarose beads with salmon sperm DNA were added and then washed with a low-salt buffer, followed by a high-salt buffer, then LiCl buffer, and finally Tris-EDTA buffer. The protein-DNA complexes were eluted and the cross-links reversed. The DNA fragments were purified with the QIAGEN PCR purification kit (QIAGEN, Chatsworth, CA). The fragments were then analyzed by real-time PCR. Real-time PCR was performed using an iCycler iQ PCR detection system (Bio-Rad) with iQ Sybr Green Supermix (Bio-Rad). The primers for UBE2C enhancer 2 were as follows: forward, 5′-CCACAAACTCTTCTCAGCTGGG-3′, and reverse, 5′-TTCTTTCCTTCCCTGTTACCCC-3′ (33). The primers for the PSA middle control region were as follows: forward, 5′-TTGGCTCAGACATCCTTG-3′, and reverse, 5′-CAACGCAACTTAACCTAAC-3′ (26).
Results
The naturally occurring AR splice variant AR3 (AR-V7) is up-regulated in castrate-resistant VCaP tumor xenografts
Using an AR antibody directed against the N terminus and an AR3 antibody that specifically recognizes the unique C-terminal tail of AR3, we confirmed the finding that AR3 protein (also known as AR-V7) was up-regulated after castration in xenografts generated by the androgen-dependent human prostate cancer cell line VCaP (Fig. 1A) (21). Additional lower-molecular-weight AR forms may correspond to other splice variants such as ARv567es. As expected, AR3 protein was negligible in VCaP tumors before castration and in the originating VCaP cell line (Fig. 1A). We previously showed that Vav3 confers castration-resistant growth to VCaP cells in vivo (26). We next examined whether castration-resistant VCaP tumor xenografts expressing Vav3 exhibited altered levels of AR3. Analysis of late-stage, castration-resistant VCaP xenografts (6–9 wk after castration) revealed considerable variability in AR3 levels. Nevertheless, there were significantly higher AR3 levels in Vav3-expressing, castration-resistant tumors compared with control (GFP) castration-resistant tumors (Fig. 1B). These data raised the possibility that one mechanism by which Vav3 confers castration-resistant growth is through effects on AR3.
Fig. 1.

The naturally occurring AR splice variant AR3 (AR-V7) is up-regulated in castration-resistant VCaP tumor xenografts. A, VCaP cells stably expressing GFP were inoculated into the hind flanks of SCID mice. When tumors reached a volume of approximately 300 mm3, three tumors were harvested and represent androgen-dependent tumors. The remaining mice were castrated, and the castration-resistant tumors were harvested when tumor volumes reached 1000 mm3 (approximately 6–9 wk after castration). Tumor lysates were immunoblotted for AR (N-terminal AR, AR-N20), AR3, and actin. The 22Rv1 and VCaP cell lines served as positive and negative controls, respectively. B, VCaP cells stably expressing GFP or Vav3-FLAG were injected into mice as described in A. AR3 levels in five GFP and six Vav3-FLAG VCaP tumors were analyzed via densitometry. Data (± sem) are graphed as AR3 levels normalized to actin. *, P < 0.05.
Vav3 enhances AR3 transcriptional activity
Because both Vav3 and AR3 increase during progression to CRPC, we investigated the ability of Vav3 to coactivate AR3. The activities of full-length AR and AR3 were initially examined using an ARE-driven luciferase reporter construct in the AR-null prostate cancer cell line PC3. Vav3 significantly enhanced the activities of both AR3 as well as full-length AR (Fig. 2A). As previously reported, Vav3 potentiated full-length AR only in response to hormone (Fig. 2A). Furthermore, we found that Vav3 enhanced the activity of another clinically relevant splice variant, ARv567es (Fig. 2B) (22). Because AR and AR3 are coexpressed in prostate cancer cells, we examined Vav3 effects on AR3 transfected into the human prostate cancer cell line LNCaP, which expresses full-length AR. Given that AR3 increases PSA and FKBP5 mRNA in LNCaP cells in androgen-depleted media (19, 20), we used PSA and FKBP5 gene expression as a measure of AR3 activity. Expression of AR3 in LNCaP cells stably expressing Vav3 resulted in a greater increase of both PSA and FKBP5 mRNA compared with control LNCaP cells expressing GFP in androgen-depleted media (Fig. 2C). Together these data indicate that under conditions of androgen depletion, Vav3 potentiated AR3 activity in the presence or absence of full-length AR.
Fig. 2.

Vav3 enhances AR3 (AR-V7) transcriptional activity. AR-negative human prostate cancer cell line PC3 was transfected with reporter plasmid ARE-Luc and Vav3 [or equivalent amounts of the corresponding empty vector (EV) and AR or AR3 (A)] or ARv567es (B). Cells were treated with either vehicle (−) or 1 nm R1881 (+). Luciferase activity (RLU) was determined 48 h after transfection. Data represent one of four experiments performed in triplicate, plotting the mean RLU/protein ± sem. Significance was determined using a two-tailed Student's t test. *, P < 0.05. C, LNCaP cells stably expressing either GFP or Vav3 were transfected with AR3 or equal amounts of EV. Cells were grown in 2% CSS for 48 h. RNA was extracted and reverse transcribed to cDNA, and real-time PCR was done with TaqMan probes for KLK3 (PSA), FKBP5, and HPRT1 (control). Data represent four independent experiments performed in triplicate. *, P < 0.05.
Vav3 interacts with AR3
We performed coimmunoprecipitations to determine whether AR3 and Vav3 were present in the same protein complexes in HEK293 cells expressing both proteins. These experiments revealed an interaction between Vav3-myc and AR3 proteins. This interaction occurred regardless of whether the AR- or Vav3-specific myc antibody was used for the immunoprecipitations (Fig. 3).
Fig. 3.
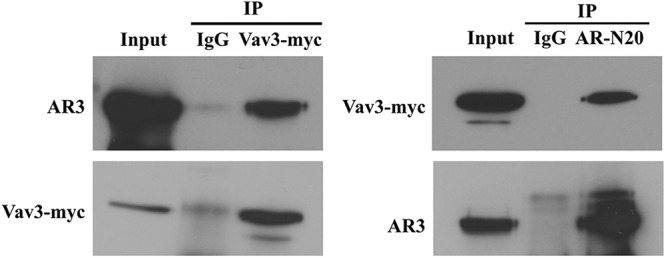
Vav3 interacts with AR3 (AR-V7). HEK293 cells were transfected with Vav3-myc and AR3 and harvested 48 h later. Coimmunoprecipitation (IP) was performed as described in Materials and Methods using antibodies to mouse or rabbit IgG (control), myc, or AR [N-terminal AR (AR-N20)]. Equivalent volumes were immunoblotted and probed for N-terminal AR and myc. A representative experiment of four independent experiments is shown.
Vav3 and AR3 are critical for anchorage-dependent and anchorage-independent growth
To define the contribution of Vav3 to CRPC growth, we used CWR-22Rv1 (heretofore referred to as 22Rv1), a CRPC cell line that naturally expresses Vav3, AR3, and full-length AR. Vav3 knockdown using Vav3 shRNA greatly inhibited cell proliferation as compared with the shGFP controls (plotted as doubling time, Fig. 4A, left panel). Proliferation was inhibited in 22Rv1 cells cultured in either complete media (10% FBS) or in media depleted of steroids and small growth factors (5% CSS). To specifically knock down AR3 in this cell line, we used an shRNA designed to target the 3′-untranslated region of AR3 but not full-length AR (20). Similar to the Vav3 knockdown experiments, AR3 depletion resulted in a large decrease in growth rate (Fig. 4A, middle panel). The doubling time of shVav3- and shAR3-expressing cells was roughly twice that of their shGFP counterparts in both 10% serum and androgen-depleted media. We observed little change in proliferation of 22Rv1 cells after depletion of full-length AR, thus revealing the strong reliance of these CRPC cells on AR3 (Fig. 4A, right panel). These observations were confirmed in CWR-R1, another CRPC cell line that expresses Vav3, AR3, and full-length AR (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). Because anchorage-independent growth often correlates with tumor aggressiveness, the growth of 22Rv1 cells in soft agar after depletion of Vav3, AR3, or full-length AR was investigated. The 22Rv1 cells expressing shVav3 showed a 20-fold reduction in soft agar colony formation as compared with the shGFP cell line control (Fig. 4B). A similar reduction was observed in 22Rv1 cells expressing shAR3, whereas knockdown of full-length AR resulted in no change in anchorage-independent growth compared with shGFP controls (Fig. 4B). These data further support our observations above that these CRPC cell models rely on AR splice variants to a greater extent than full-length AR and indicate that Vav3 is essential for anchorage-dependent and anchorage-independent proliferation of CRPC.
Fig. 4.

Vav3 and AR3 (AR-V7) are critical for anchorage-dependent and anchorage-independent growth. A, 22Rv1 cells stably expressing plk0.1-shGFP, -shVav3, -shAR3, or -shAR were plated at 20,000 cells per well in 24-well dishes in 10% FBS or 5% CSS. Cells were harvested and counted at three distinct time points. Data represent three independent experiments performed in triplicate and are plotted as mean doubling time (hours) ± sem as determined by the following equation: doubling time = ln(2)/growth rate. Cell lysates were immunoblotted with Vav3 or N-terminal AR and actin antibodies. Significance was determined using a Student's t test of comparison with shGFP controls. *, P < 0.05; **, P < 0.01. B, The growth in soft agar of 22Rv1 cells stably expressing the indicated shRNA vectors was assessed as described in Materials and Methods. Average colony number per plate ± sem is plotted. Data represent two independent experiments performed in triplicate.
Knockdown of Vav3 or AR3 in CRPC cells results in G0/G1 accumulation
Given the decreased growth rate of 22Rv1 cells upon either AR3 or Vav3 knockdown, the cell cycle profile of these cells was characterized. Knockdown of either AR3 or Vav3 resulted in an increased percentage of 22Rv1 cells in the G0/G1 phase (Table 1). Furthermore, there was an approximate 50% decrease in the amount of S-phase cells upon Vav3 or AR3 knockdown. Depletion of full-length AR resulted in no significant cell cycle change as compared with the shGFP control. These data suggest that Vav3 and AR3 are important for S-phase progression in 22Rv1 cells.
Table 1.
Knockdown of Vav3 or AR3 (AR-V7) in CRPC cells results in G0/G1 accumulation
| Phase |
|||
|---|---|---|---|
| G0/G1 | S | G2 | |
| shGFP | 68 (1.1) | 13 (1.1) | 19 (1.0) |
| shVav3 | 79b (0.6) | 7a (0.8) | 13a (1.4) |
| shAR3 | 81b (1.7) | 6b (0.2) | 13a (1.4) |
| shAR | 70 (1.9) | 12 (0.8) | 18 (1.5) |
The 22Rv1 cells stably expressing plk0.1-shGFP, -shVav3, -shAR3, or -shAR were plated at one million cells per 100-mm dish. Cells were grown in androgen-depleted media for 24 h, and then media were changed to 10% FBS for 48 h. Cells were fixed in 70% ethanol, and DNA content was determined with propidium iodide and flow cytometry. Data (sem) represent three independent experiments performed in triplicate. Significance was determined using a Student's t test of comparison with the shGFP-expressing controls.
P < 0.05.
P < 0.01.
Knockdown of Vav3 or AR3 promotes death of CRPC cells
We investigated the contributions of Vav3 and AR3 to cell survival. Depletion of Vav3 resulted in a greater than 3-fold increase in cell death as compared with the shGFP control cells (Fig. 5A). Knockdown of AR3 resulted in a greater than 2-fold increase in cell death, whereas depletion of full-length AR had no significant effect on cell death (Fig. 5A). To examine the role of apoptosis in this process, we examined PARP cleavage. PARP is cleaved by caspase 3 and is a marker of apoptosis (34). Knockdown of Vav3 or AR3 but not full-length AR led to increased levels of cleaved PARP, suggesting that depletion of either protein promotes apoptosis (Fig. 5B). These data are in accordance with our previous report that Vav3 decreases apoptosis in VCaP cells in a castration environment (26) and suggest that Vav3 and AR3 promote CRPC survival and avoidance of apoptosis.
Fig. 5.
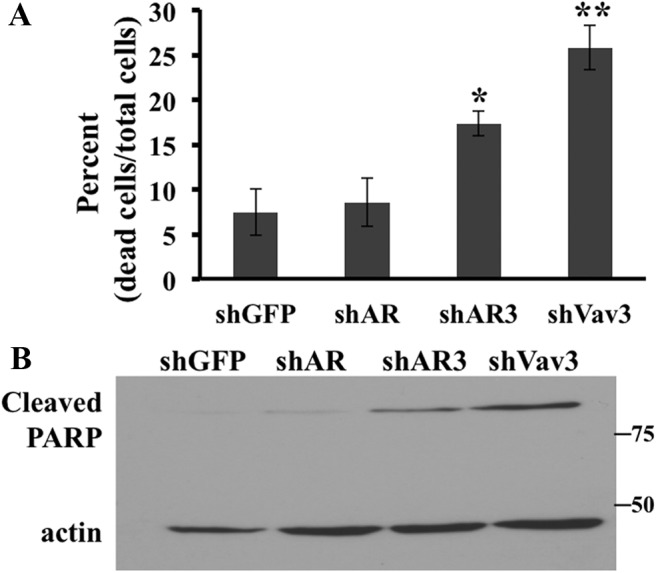
Knockdown of Vav3 or AR3 (AR-V7) promotes death of CRPC cells. A, 22Rv1 cells stably expressing plk0.1-shGFP, -shAR, -shAR3, or -shVav3 were plated and grown as in Table 1. Adherent and floating cells were harvested, and cells that stained with trypan blue or that excluded stain were counted. Data represent three independent experiments performed in triplicate. Significance was determined using a Student's t test of comparison with shGFP controls. *, P < 0.05; **, P < 0.01. B, Lysates from cells in A were immunoblotted with antibodies that recognize cleaved PARP or actin.
Vav3 and AR3 drive ligand-independent AR activity in CRPC
Ligand-independent AR activity may be an important mechanism for CRPC survival and progression (35). To determine the contribution of Vav3 to ligand-independent AR activity of CRPC, Vav3 was stably knocked down in 22Rv1 and CWR-R1 cells, and ligand-independent AR activity was examined by ARE-driven luciferase reporter gene experiments. The 22Rv1/shVav3 cells showed a greater than 85% decrease in luciferase activity (Supplemental Fig. 2A). This observation was confirmed in CWR-R1 cells (Supplemental Fig. 2B). To validate that this decrease in AR transcriptional activity was a result of Vav3-specific knockdown, a knockdown-resistant form of Vav3 was generated. The Vav3 knockdown-resistant form (Vav3KDres) altered nine nucleotides without changing the wild-type amino acid sequence, thereby rendering the protein resistant to shVav3-targeted knockdown (Fig. 6A, right panel). The Vav3 knockdown-resistant vector restored ligand-independent AR activity in the Vav3-depleted cell lines to the level seen in the control shGFP cells (Fig. 6A, left panel). This finding suggests that the effects of Vav3 knockdown were specific and not due to off-target actions of the shRNA construct. We also assessed whether acute Vav3, AR3, and AR knockdown altered ligand-independent AR activity using a tetracycline-inducible shRNA system. Knockdown of AR, AR3, and Vav3 was achieved within 3 d of doxycycline treatment (Fig. 6B, lower panel). Short-term knockdown of Vav3 resulted in an approximate 50% decrease in ligand-independent ARE-driven luciferase activity as compared with untreated tet-shVav3 control (Fig. 6B, upper panel). Furthermore, short-term knockdown of AR3 reduced luciferase activity by approximately 60%, whereas depletion of full-length AR resulted in no significant change in ligand-independent AR activity. Reporter gene assays were validated using real-time RT-PCR analysis. UBE2C is a marker of the castration-resistant AR transcriptome (33) and is increased in CRPC bone metastases concomitantly with AR splice variants (23). Recently, UBE2C was found to be higher in CRPC specimens that had increased levels of AR3 (36). UBE2C mRNA levels were significantly decreased upon depletion of Vav3 or AR3 but increased upon full-length AR depletion (Fig. 6C). We also examined UBE2C mRNA levels in the doxycycline-inducible system at a time point before any cell proliferation changes. Acute knockdown of AR3 and Vav3 led to decreased UBE2C mRNA levels (Fig. 6D). In addition, we examined the effects of Vav3 or AR3 knockdown upon the AR and AR3 negatively regulated gene IGFBP3 (20). Acute knockdown of either AR3 or Vav3 led to increased levels of IGFBP3 mRNA (Fig. 6D). These data further indicate that Vav3 and AR3 drive ligand-independent AR activity in these CRPC models.
Fig. 6.

Vav3 and AR3 (AR-V7) drive ligand-independent AR activity in CRPC. A, Left panel, 22Rv1 cells stably expressing either shGFP or shVav3 were transfected with reporter plasmid PSA-Luc and knockdown-resistant Vav3 (V3KDres) or equal amounts of empty vector. Luciferase activity (RLU) was determined 48 h after transfection. Data represent four experiments performed in triplicate and are plotted (fold induction ± sem) with shGFP empty vector set as one. Right panel, 22Rv1 cells stably expressing shGFP or shVav3 were transduced with WT Vav3-FLAG (Vav3) or knockdown-resistant Vav3-FLAG (Vav3KDres) or were not transduced (No tx). Cells were harvested 48 h later, and equivalent protein amounts were immunoblotted for FLAG and actin. B, 22Rv1 cells stably expressing tet-shGFP, tet-shVav3, tet-shAR3, or tet-shAR were transfected with the ARE-Luc reporter plasmid. Cells were grown for 72 h in 5% CSS with or without 1 μg doxycycline. Media were replaced every 48 h. Equivalent protein amounts were immunoblotted using antibodies AR (N-terminal), Vav3, and actin. Data represent three experiments done in triplicate. Fold induction is RLU/protein for doxycycline-treated cells divided by untreated cells. Significance was determined using a two-tailed Student's t test comparing the (treated/untreated) experimental group with the (treated/untreated) tet-shGFP. *, P < 0.05; **, P < 0.01. C, 22Rv1 cells stably expressing shGFP, shVav3, or shAR3 were grown in 5% CSS for 48 h. Real-time RT-PCR was done with TaqMan probes for UBE2C and HPRT1 (control). Data represent three to four independent experiments performed in triplicate with UBE2C mRNA levels for shGFP cells set as one (relative mRNA levels ± sem). *, P < 0.05; **, P < 0.01. D, 22Rv1 cells stably expressing tet-shVav3 or tet-shAR3 were grown for 72 h in 5% CSS with or without 1 μg doxycycline. Real-time RT-PCR was done with TaqMan probes for UBE2C, IGFBP3, and GAPDH (control). Data represent three independent experiments performed in triplicate with no-treatment control mRNA levels set as one (relative mRNA levels ± sem). *, P < 0.05; **, P < 0.01.
Vav3 mediates ligand-independent AR activity in a Vav3 GEF- and pleckstrin homology (PH) domain-independent manner
We investigated the effects of Vav3's GEF and PH domains on AR3 activity. Vav3 mutants deficient in GEF activity (ISOIII) and lacking a functional PH domain (W493L and ΔPH) were used. As previously reported (24), Vav3 enhanced full-length AR activity in a GEF-independent but PH domain-dependent manner (Fig. 7, A and B). However, Vav3 enhanced AR3 activity independently of both the GEF (Fig. 7A) and the PH domains (Fig. 7B and Supplemental Fig. 3). We found that a Vav3 PH domain-deficient knockdown-resistant mutant (Vav3-W493L KDres) rescued the loss of ligand-independent AR activity in 22Rv1 cells stably expressing shGFP or shVav3 (Fig. 7C). This finding further confirmed the dispensable nature of the PH domain in Vav3 enhancement of AR3 activity. Vav3 interacted preferentially with AR3 compared with full-length AR (Supplemental Fig. 4), further supporting a distinct mechanism by which Vav3 enhances the constitutively active AR splice variant.
Fig. 7.

Vav3 mediates ligand-independent AR activity in a Vav3 GEF- and PH domain-independent manner. AR-negative human prostate cancer cell line PC3 was transfected with AR or AR3, reporter plasmid ARE-Luc, and GEF-deficient Vav3 (ISOIII) (A), Vav3 PH domain mutant (W493L) (B) or equivalent amounts of the corresponding empty vector. Cells were treated with either vehicle (−) or 1 nm R1881 (+). Luciferase activity (RLU) was determined 48 h after transfection. Data represent one of three to six experiments performed in triplicate, plotting the mean RLU/protein ± sem. Significance was determined using a two-tailed Student's t test. *, P < 0.05. C, 22Rv1 cells stably expressing either shGFP or shVav3 were transfected with reporter plasmid PSA-Luc and a PH domain mutant knockdown-resistant Vav3 (Vav3-W493L KDres) or equal amounts of empty vector. Luciferase activity was determined 48 h after transfection. Data represent three experiments performed in triplicate and are plotted (fold induction ± sem) with shGFP (empty vector transfected) set as one.
Vav3 regulates nuclear levels of AR3
Due to the decrease of AR ligand-independent activity upon acute Vav3 depletion, AR3 levels were investigated. There was no significant difference in the total AR3 protein levels upon doxycycline-induced Vav3 knockdown (Fig. 8A). We also investigated whether Vav3 regulated AR3 mRNA levels. Acute knockdown of Vav3 had no effect on AR3 mRNA levels, whereas control experiments showed that acute knockdown of AR3 greatly decreased AR3 mRNA (Fig. 8B). Because nuclear AR3 is considered the active form, subcellular localization of AR3 was evaluated using an AR3-specific antibody. Vav3 knockdown resulted in a 65% decrease in AR3 nuclear levels (Fig. 8C). Using an antibody specific to the N-terminal portion of AR revealed a 75% decrease in nuclear AR splice variant levels with no significant change in input levels upon Vav3 depletion (Supplemental Fig. 5, A and B). These data were also verified in a non-tetracycline-inducible system in which Vav3 was stably depleted (data not shown). Note that the cytosolic fractions contain higher overall protein levels compared with nuclear fractions. Therefore, although the AR3 concentrations were proportionally lower in the cytosolic fractions compared with nuclear fractions, the total amount of AR3 was greater in the cytoplasm than in the nucleus. Thus, no significant change in total AR3 levels was observed after Vav3 knockdown despite a 65% decrease in AR3 nuclear levels. Furthermore, to determine whether Vav3 was involved in maintaining AR3 nuclear levels, either Vav3-FLAG or FLAG was stably overexpressed in 22Rv1 cells. The 22Rv1 cells expressing Vav3-FLAG showed only a minor increase in AR3 input levels (Fig. 8D) but a 75% increase in nuclear AR3 compared with the FLAG-expressing controls (Fig. 8E). Experiments using the N-terminal AR antibody showed more than a 2-fold increase in nuclear AR splice variants with no significant change in total AR splice variant levels (Supplemental Fig. 5, C and D). To test whether depletion of Vav3 led to decreased AR3 recruitment to chromatin-binding sites, we used 22Rv1 cells stably expressing shGFP, shAR3, or shVav3. In a ligand-independent fashion, AR binds to the ARE-enhancers of UBE2C in CRPC cells (33). Using primers to the second ARE-containing enhancer region of UBE2C, we found that AR3 recruitment to the UBE2C enhancer was decreased by approximately 2-fold upon either Vav3 or AR3 depletion compared with shGFP controls (Supplemental Fig. 6). We found no appreciable binding of AR3 to a non-ARE-containing site in the PSA promoter (data not shown). Together, these findings using two different antibodies to detect AR splice variants confirm that Vav3 increased nuclear AR3 levels and provide a plausible mechanism for enhancement of AR3 transcriptional activity by Vav3.
Fig. 8.

Vav3 regulates nuclear levels of AR3 (AR-V7). The 22Rv1 cells stably expressing tet-shGFP, tet-shVav3, or tet-shAR3 were grown in 5% CSS with or without 1 μg doxycycline (DOX) for 72 h. A, Equivalent amounts of total cellular protein were immunoblotted for AR3 and actin (left panel) and quantified (right panel). B, RNA was isolated after 48 and 72 h doxycycline treatment, and real-time RT-PCR was done with TaqMan probes for AR3 and HPRT1 (control). Data represent three independent experiments performed in triplicate (relative mRNA levels ± sem). ***, P < 0.001. C, Cells were fractionated and immunoblotted for AR3, histone, and super oxide dismutase (SOD) as described in Materials and Methods. Protein levels were normalized to SOD (cytoplasmic fraction) or histone (nuclear fraction). Protein levels were determined from three independent experiments and represent the ratios of doxycycline (DOX) to untreated cells and are compared with the tet-shGFP-treated/untreated values (set to one). Significance was determined using a two-tailed Student's t test comparing the (treated/untreated) experimental groups with (treated/untreated) tet-shGFP. *, P < 0.05. D, 22Rv1 cells stably expressing Vav3-FLAG or FLAG were grown in 5% CSS for 72 h. Equivalent amounts of total cellular protein were immunoblotted for AR3 and actin. E, Nuclear and cytosolic fractions from the same cells were immunoblotted for AR3, histone, and SOD. Densitometry was performed on blots from three independent experiments. *, P < 0.05.
Discussion
The progression from androgen dependence to castration resistance remains poorly understood; however, AR signaling is thought to be central in this process. We previously reported that Vav3 is a unique AR coactivator that is up-regulated during CRPC progression and confers castration resistance to androgen-dependent prostate cancer cells (24–26). Given the recent discovery of constitutively active AR splice variants, such as the prevalent variant AR3, that increase during progression to CRPC (21), and the potential for these variants to promote tumor growth and therapeutic resistance (20–22), we investigated the capacity of Vav3 to enhance the activity of AR3. We show that increased AR3 levels accompanied castration-resistant growth of Vav3-expressing tumors and that Vav3 strongly coactivated AR3 in a ligand-independent setting in either the presence or absence of full-length AR. We also found that Vav3 coactivated ARv567es, another clinically important AR splice variant (22). To our knowledge, this finding is the first reported evidence of AR splice variant coactivation. We found that Vav3 interacted preferentially with AR3 compared with full-length AR in coimmunoprecipitation studies (24) (Supplemental Fig. 4). Furthermore, in contrast to Vav3 coactivation of full-length AR, enhancement of AR3 by Vav3 was not dependent on the Vav3 PH domain. Thus, although Vav3 coactivates both full-length AR and AR3, this unique coactivator appears to use distinct mechanisms that commonly require the AR's active N terminus (24, 37, 38).
To pursue the impact of Vav3-AR3 interaction in a biologically relevant setting, we used an shRNA approach in two CRPC cell lines with relatively high endogenous levels of Vav3, AR3, and full-length AR. Knockdown of either Vav3 or AR3 resulted in greatly diminished anchorage-dependent and anchorage-independent growth, decreased G1/S-phase progression, and increased apoptosis. These data suggest that both Vav3 and AR3 drive proliferation and survival of CRPC. It is well established that AR transcriptional activity is key to maintaining prostate cancer growth at all clinical stages (39, 40). However, our data suggest that full-length AR may be dispensable in cells that coexpress the constitutively active splice variant AR3 and Vav3. Our findings also confirm that the AR3 variant is poised to drive CRPC growth and survival particularly in the setting of elevated Vav3.
In a castration-sensitive cell line such as LNCaP that lacks endogenous AR splice variants, addition of AR3 enhances the transcriptional activity of AR molecular targets in both an androgen-dependent and -independent setting (19, 20). In LNCaP cells expressing AR3, the addition of Vav3 further augmented the levels of PSA (KLK3) and FKBP5 mRNAs; both genes are validated molecular targets of AR3 (19, 20). The ubiquitinating enzyme UBE2C is a marker of the ligand-independent transcriptome whose levels are increased in castration-resistant disease (33). Knockdown of UBE2C mRNA in the CRPC line LNCaP-abl, but not in the parental LNCaP cells, leads to decreased ligand-independent cell growth (33). Importantly, depletion of AR3 or Vav3 in the CRPC cell line 22Rv1 resulted in decreased levels of UBE2C and decreased recruitment of AR3 to the UBE2C enhancer in chromatin as well as decreased cell proliferation in an androgen-depleted environment. It remains to be fully elucidated whether Vav3 enhances AR3 transcriptional activity for all or a subset of AR3 target genes and thereby influences cell survival and proliferation. Previous studies demonstrated that AKT1 is an AR3 target gene in 22Rv1 cells grown in serum (20). However, we did not observe AR3 regulation of AKT1 in 22Rv1 cells cultured in androgen-depleted media (data not shown). These findings reinforce the theory that AR has differential transcriptional targets and uses distinct drivers of growth and survival in an androgen-replete vs. ligand-independent setting (33). The collection of AR3 transcriptional targets that drive malignancy may depend on Vav3 and other yet to be identified coactivators.
The clinical implications of our findings are that current therapeutic modalities that target the AR LBD may be ineffective against CRPC driven by Vav3 and AR splice variants. Indeed, castration-resistant xenografts that are also resistant to abiraterone, a clinically promising androgen synthesis inhibitor, display increased levels of AR and AR splice variants (41). Furthermore, MDV3100, a potent AR antagonist, stimulates increased levels of constitutively active AR splice variants (36, 42). Given that Vav3 augments the activity of full-length AR in subnanomolar levels of androgen (24), ARv567es (this study), and AR3 (this study), Vav3 may promote resistance to drugs that target AR signaling and serve as a prognostic indicator for therapeutic failure.
Our data support a model in which constitutive AR splice variant activity shifts reliance away from full-length AR when Vav3 is expressed. Data presented here suggest that Vav3 controls AR splice variant transcriptional activity through a mechanism novel to this AR coactivator. Strikingly, doxycycline-induced depletion of Vav3 specifically decreased AR3 nuclear levels within 72 h, whereas Vav3 overexpression increased nuclear AR3 levels. Taken together, these data suggest that Vav3 is integral in the maintenance of nuclear AR3 levels in a castrate environment and is a key determinant of cell proliferation and survival. It is unclear whether Vav3-AR3 interaction is required for AR3 nuclear translocation or whether Vav3 indirectly regulates nuclear AR3 levels. Little is known about the regulation of nuclear-cytoplasmic trafficking of AR3. Both of the classical bipartite nuclear localization signals present in full-length AR are absent in AR3. A recent report demonstrated that the ability of AR3 to localize to the nucleus is independent of canonical AR nuclear import pathways such as importin-β and heat shock protein 90 chaperone activity (43). Microtubules were shown to participate in shuttling full-length AR into the nucleus upon androgen stimulation (44). Vav3, which participates in the maintenance of the microtubule cytoskeleton (45), may influence AR3 nuclear entry conceivably via microtubule cytoskeleton dynamics. Together, these data support efforts to therapeutically target Vav3-AR3 interaction in CRPC because Vav3 and AR3 actions may underlie CRPC evasion of androgen deprivation and AR antagonist therapies. A benefit of such an approach would be that the oncogenic nuclear form of AR3 would be selectively targeted and might produce fewer undesirable effects.
Supplementary Material
Acknowledgments
As cited in the text, the following individuals generously provided reagents and/or cells: Dr. Yun Qiu, Dr. Priya Rai, Dr. Elizabeth M. Wilson, Dr. Stephen Plymate, Dr. Kenneth Pienta, Dr. Carlos Perez-Stable, Dr. Paul Rennie, Dr. Daniel Billadeau, and Dr. Zafar Nawaz. We are grateful to Drs. Wayne Balkan (University of Miami), Maria Mudryi (University of California, Davis), Fayi Wu (University of Miami), and Leah Lyons (Nova Southeastern University) for helpful advice. We are also grateful for Mr. James Phillips' and Dr. Shannon Opiela's assistance with flow cytometry.
Research performed in this manuscript was supported by National Institutes of Health (NIH) Grant CA132200 (to K.L.B.). S.O.P. and C.D.F. were supported by NIH predoctoral fellowships F30AG038275 and T32HL007188, respectively.
Disclosure Summary: The authors have nothing to disclose.
NURSA Molecule Pages:
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- AR
- Androgen receptor
- ARE
- androgen response element
- CRPC
- castration-resistant prostate cancer
- CSS
- charcoal-dextran-stripped serum
- FBS
- fetal bovine serum
- GEF
- guanine nucleotide exchange factor
- GFP
- green fluorescent protein
- LBD
- ligand-binding domain
- NP-40
- Nonidet P-40
- PARP
- poly ADP ribose polymerase
- PH
- pleckstrin homology
- PSA
- prostate-specific antigen
- shRNA
- short hairpin RNA.
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. 2011. Global cancer statistics. CA Cancer J Clin 61:69–90 [DOI] [PubMed] [Google Scholar]
- 2. Halabi S, Small EJ, Kantoff PW, Kattan MW, Kaplan EB, Dawson NA, Levine EG, Blumenstein BA, Vogelzang NJ. 2003. Prognostic model for predicting survival in men with hormone-refractory metastatic prostate cancer. J Clin Oncol 21:1232–1237 [DOI] [PubMed] [Google Scholar]
- 3. Dehm SM, Tindall DJ. 2006. Molecular regulation of androgen action in prostate cancer. J Cell Biochem 99:333–344 [DOI] [PubMed] [Google Scholar]
- 4. Agoulnik IU, Weigel NL. 2006. Androgen receptor action in hormone-dependent and recurrent prostate cancer. J Cell Biochem 99:362–372 [DOI] [PubMed] [Google Scholar]
- 5. Hååg P, Bektic J, Bartsch G, Klocker H, Eder IE. 2005. Androgen receptor down regulation by small interference RNA induces cell growth inhibition in androgen sensitive as well as in androgen independent prostate cancer cells. J Steroid Biochem Mol Biol 96:251–258 [DOI] [PubMed] [Google Scholar]
- 6. Knudsen KE, Penning TM. 2010. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab 21:315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. 2002. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res 62:1008–1013 [PubMed] [Google Scholar]
- 8. Cheng H, Snoek R, Ghaidi F, Cox ME, Rennie PS. 2006. Short hairpin RNA knockdown of the androgen receptor attenuates ligand-independent activation and delays tumor progression. Cancer Res 66:10613–10620 [DOI] [PubMed] [Google Scholar]
- 9. Taplin ME, Balk SP. 2004. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. J Cell Biochem 91:483–490 [DOI] [PubMed] [Google Scholar]
- 10. Snoek R, Cheng H, Margiotti K, Wafa LA, Wong CA, Wong EC, Fazli L, Nelson CC, Gleave ME, Rennie PS. 2009. In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castration-resistant prostate tumors. Clin Cancer Res 15:39–47 [DOI] [PubMed] [Google Scholar]
- 11. Debes JD, Tindall DJ. 2004. Mechanisms of androgen-refractory prostate cancer. N Engl J Med 351:1488–1490 [DOI] [PubMed] [Google Scholar]
- 12. Dehm SM, Tindall DJ. 2011. Alternatively spliced androgen receptor variants. Endocr Relat Cancer 18:R183–R196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo Z, Qiu Y. 2011. A new trick of an old molecule: androgen receptor splice variants taking the stage?! Int J Biol Sci 7:815–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feldman BJ, Feldman D. 2001. The development of androgen-independent prostate cancer. Nat Rev Cancer 1:34–45 [DOI] [PubMed] [Google Scholar]
- 15. Jänne OA, Palvimo JJ, Kallio P, Mehto M. 1993. Androgen receptor and mechanism of androgen action. Ann Med 25:83–89 [DOI] [PubMed] [Google Scholar]
- 16. Gelmann EP. 2002. Molecular biology of the androgen receptor. J Clin Oncol 20:3001–3015 [DOI] [PubMed] [Google Scholar]
- 17. Tepper CG, Boucher DL, Ryan PE, Ma AH, Xia L, Lee LF, Pretlow TG, Kung HJ. 2002. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res 62:6606–6614 [PubMed] [Google Scholar]
- 18. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. 2008. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res 68:5469–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. 2009. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 69:16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AM, Edwards J, Qiu Y. 2009. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 69:2305–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. 2010. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci USA 107:16759–16765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, Nelson PS, Plymate SR. 2010. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest 120:2715–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hörnberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A, Wikström P. 2011. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One 6:e19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lyons LS, Burnstein KL. 2006. Vav3, a Rho GTPase guanine nucleotide exchange factor, increases during progression to androgen independence in prostate cancer cells and potentiates androgen receptor transcriptional activity. Mol Endocrinol 20:1061–1072 [DOI] [PubMed] [Google Scholar]
- 25. Lyons LS, Rao S, Balkan W, Faysal J, Maiorino CA, Burnstein KL. 2008. Ligand-independent activation of androgen receptors by Rho GTPase signaling in prostate cancer. Mol Endocrinol 22:597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rao S, Lyons LS, Fahrenholtz CD, Wu F, Farooq A, Balkan W, Burnstein KL. 2012. A novel nuclear role for the Vav3 nucleotide exchange factor in androgen receptor coactivation in prostate cancer. Oncogene 31:716–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Banach-Petrosky W, Jessen WJ, Ouyang X, Gao H, Rao J, Quinn J, Aronow BJ, Abate-Shen C. 2007. Prolonged exposure to reduced levels of androgen accelerates prostate cancer progression in Nkx3.1; Pten mutant mice. Cancer Res 67:9089–9096 [DOI] [PubMed] [Google Scholar]
- 28. Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, Ryan C, Smith S, Scher H, Scardino P, Reuter V, Gerald WL. 2004. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol 164:217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dong Z, Liu Y, Lu S, Wang A, Lee K, Wang LH, Revelo M, Lu S. 2006. Vav3 oncogene is overexpressed and regulates cell growth and androgen receptor activity in human prostate cancer. Mol Endocrinol 20:2315–2325 [DOI] [PubMed] [Google Scholar]
- 30. Lin KT, Gong J, Li CF, Jang TH, Chen WL, Chen HJ, Wang LH. 2012. Vav3-rac1 signaling regulates prostate cancer metastasis with elevated vav3 expression correlating with prostate cancer progression and posttreatment recurrence. Cancer Res 72:3000–3009 [DOI] [PubMed] [Google Scholar]
- 31. Liu Y, Mo JQ, Hu Q, Boivin G, Levin L, Lu S, Yang D, Dong Z, Lu S. 2008. Targeted overexpression of vav3 oncogene in prostatic epithelium induces nonbacterial prostatitis and prostate cancer. Cancer Res 68:6396–6406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abramoff MD, Magalhaes PJ, Ram SJ. 2004. Image processing with ImageJ. Biophoton Int 11:36–42 [Google Scholar]
- 33. Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, Wu T, Regan MM, Meyer CA, Carroll JS, Manrai AK, Jänne OA, Balk SP, Mehra R, Han B, Chinnaiyan AM, Rubin MA, True L, Fiorentino M, Fiore C, Loda M, et al. 2009. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. 1993. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res 53:3976–3985 [PubMed] [Google Scholar]
- 35. Lamont KR, Tindall DJ. 2011. Alternative activation pathways for the androgen receptor in prostate cancer. Mol Endocrinol 25:897–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, Edwards J, Isaacs W, Nelson PS, Bluemn E, Plymate S, Luo J. 2012. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res 72:3457–3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simental JA, Sar M, Lane MV, French FS, Wilson EM. 1991. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem 266:510–518 [PubMed] [Google Scholar]
- 38. MacLean HE, Warne GL, Zajac JD. 1997. Localization of functional domains in the androgen receptor. J Steroid Biochem Mol Biol 62:233–242 [DOI] [PubMed] [Google Scholar]
- 39. Chen Y, Sawyers CL, Scher HI. 2008. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 8:440–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Knudsen KE, Scher HI. 2009. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res 15:4792–4798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, Nelson PS, Montgomery RB. 2011. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res 17:5913–5925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, Vakar-Lopez F, Vessella RL, Plymate SR. 2011. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One 6:e27970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chan SC, Li Y, Dehm SM. 2012. Androgen receptor splice variants activate AR target genes and support aberrant prostate cancer cell growth independent of the canonical AR nuclear localization signal. J Biol Chem 287:19736–19749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Darshan MS, Loftus MS, Thadani-Mulero M, Levy BP, Escuin D, Zhou XK, Gjyrezi A, Chanel-Vos C, Shen R, Tagawa ST, Bander NH, Nanus DM, Giannakakou P. 2011. Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res 71:6019–6029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu JY, Seno H, Miletic AV, Mills JC, Swat W, Stappenbeck TS. 2009. Vav proteins are necessary for correct differentiation of mouse cecal and colonic enterocytes. J Cell Sci 122:324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.