

Abstract
Radon and radon progeny inhalation exposure are recognized to induce lung cancer. To explore the role of mitochondria in radon-induced carcinogenesis in humans, an in vitro partially depleted mitochondrial DNA (mtDNA) cell line (ρ−) was generated by treatment of human bronchial epithelial (HBE) cells (ρ+) with ethidium bromide (EB). The characterization of ρ− cells indicated the presence of dysfunctional mitochondria and might thus serve a reliable model to investigate the role of mitochondria. In a gas inhalation chamber, ρ− and ρ+ cells were exposed to radon gas produced by a radium source. Results showed that apoptosis was significantly increased both in ρ− and ρ+ cells irradiated by radon. Moreover, apoptosis in ρ− cells showed a lower level than in ρ+ cells. Radon was further found to depress mitochondrial membrane potential (MMP) of HBE cells with knock-down mtDNA. Production of reactive oxygen species (ROS) was markedly elevated both in ρ− and ρ+ cells exposed to radon. The distribution of phases of cell cycle was different in ρ− compared to ρ+ cells. Radon-irradiation induced a rise in G2/M and decrease in S phase in ρ+ cells. In ρ− cells, G1, G2/M and S populations remained similar to cells exposed to radon. In conclusion, radon-induced changes in ROS generation, MMP and cell cycle are all attributed to reduction of apoptosis which may trigger and promote cell transformation leading to carcinogenesis. Our study indicates that the use of the ρ− knock-down mtDNA HBE cells may serve as a reliable model to study the role played by mitochondria in carcinogenic diseases.
Introduction
Radon gas is formed during the radioactive decay of uranium-238, which occurs naturally in rocks and soils in the environment. In 1988, the International Agency for Research on Cancer (IARC) classified radon as a human lung cancer, based on studies of underground miners historically exposed to high levels of radon gas (IARC, 1988; Krewski et al, 2006). Radon gas enters homes through cracks and other openings in the foundation and accumulates largely in the basement and lower living areas (Marcinowski et al., 1994). Recent efforts to combine data from individual case–control studies showed positive associations between ecological indicators of residential radon and occurrence of lung cancer (Michelle et al., 2011; Tse et al., 2011). Despite the fact that radon exposure has been generally acknowledged as the second leading cause of human lung cancer after smoking, the cellular mechanisms underlying radon-induced carcinogenesis remain to be determined.
It is well known that nuclear DNA (nDNA) is the direct target of radiation, in which oncogenes and tumor suppressor genes are responsible for initiation and development of cancer in case of gene mutations (Huang et al., 2003; Kaufmann et al., 2007; Li et al 2007). Among the organelles in the cell, mitochondria are unique as these cells contain their own DNA, mitochondrial DNA (mtDNA). Human mitochondria possess their own double-stranded circular DNA encoding 13 protein components of 4 enzyme complexes including I, II, IV and V involved in electron transport and oxidative phosphorylation (Fernandez-Silva et al. 2003).
In the present study, a cell model of human bronchial epithelial (HBE) cells with mtDNA knock-down of mitochondria DNA (zero potential; ρ−) was established and used to examine the role of mitochondria in radon- and its progeny-induced cellular apoptotic mediated pathways that may be involved in cell transformation and malignancy.
Materials and Methods
Cell culture and generation of HBE cells with partially depleted mtDNA ρ− cells
The immortalized human bronchial epithelial cell line (HBE) was a gift from Professor Wen Cheng (School of Public Health, Zhongshan University). The parental HBE cells (ρ+) were maintained in growth medium containing a 4.5 g/L glucose DMEM, 2 mmol/L L-glutamine, 10% fetal bovine serum (FBS), 100 IU/mL penicillin and 100 μg/ml streptomycin. The partially depleted mtDNA cells (ρ−) were generated by treatment of ρ+ HBE cells with 50 ng/ml ethidium bromide (EB) in growth medium supplemented with 50 μg/ml uridine (Sigma) and 100 μg/ml sodium pyruvate. The ρ− cells were cultured using the growth medium plus 12.5 ng/ml EB and 50 μg/ml uridine, which provides an alternative source of energy through glycolysis to ensure optimal growth. Cell viability was assessed by the MTT assay and found to be 85%.
Determination of mtDNA copies
The mitochondrial DNA (mtDNA) copy number was tested by quantitative real time PCR using the 2−ΔΔCt method with GAPDH as a reference as described previously (Evdokimovsky et al., 2011). Using the ρ+ HBE cells as the standard, whose mtDNA copy number (mtDNA amount / nDNA amount) was defined as one, the relative mtDNA copy numbers of the ρ− cells were calculated. ND1 and 16S rRNA was expressed as mean mtDNA, while 18S rRNA was expressed as mean nDNA. Briefly, total cell DNA was extracted with E.Z.N.A.® Tissue DNA Kit (OMEGA, USA) and quantified by spectrometry. Fifty ng DNA was used to amplify the primers (Table. 1) using SYBR Green detection on an 7500 Real-time PCR System (Applied Biosystem, Carlsbad, CA, USA). All reactions were conducted in triplicate in 96-well MicroAmp® optical tubes (Applied Biosystems, USA). PCR conditions were set as follows: an initial step of 2 min at 50 °C and 10 min at 95 °C, followed by 40 cycles of 15 sec at 95 °C for 30 sec, 55 °C for 30 sec, and 72 °C.
Table 1.
Mitochondrial DNA primer sequence
| Gene name | Primer sequence (5′-3′) |
|---|---|
| 16S rRNA | CTTAGCCAAACCATTTACCCAAA |
| CATCTTTCCCTTGCGGTACTATATC | |
| ND1 | CCCTAAAACCCGCCACATCT |
| GAGCGATGGTGAGAGCTAAGGT | |
| 18S rRNA | ACGACCCATTCGAACGTCTG |
| CCGTTTCTCAGGCTCCCTC | |
| GAPDH | CGACCACTTTGTCAAGCTCA |
| AGAGTTGTCAGGGCCCTTTT |
Radon and its progeny Irradiation
Exponentially growing ρ− and ρ+ HBE cells were plated onto Transwell membrane (Corning, USA) with a 6 μm mylar bottom at a density of 1 × 105 cells 2 days before irradiation. Cells were placed in a gas inhalation chamber (Chinese Academy of Military Medical Sciences, Beijing, China). The gas chamber was connected with a multifunctional radon chamber purchased from Donghua University in China. Randon and its progeny was produced by a radium source using a Changhe pump machine (model BT00-300, China) and pumped into the gas chamber. Cells were directly exposed to radon and its progeny at concentration of 20, 000 Bq/m3 for 20 min every other day. During exposure, the cells were kept at 37 °C in medium bath. This procedure was continued for 10 passages and each passage was exposed twice. The optimal time and concentration were determined by MTT assay.
Cell staining with fluorescent probe
All cell staining was performed with cells grown in 35 mm dish with 0.17 mm glass bottom. mtDNA staining was achieved by diluting stock PicoGreen solution at 3 μl/ml directly into cell culture medium. (Molecular Probes Inc., Oregon, USA), which specifically visualizes only mitochondrial nucleic acid. The cells were then incubated for 1 hr, and washed twice with medium. Mitochondrion of cells were co-stained with mitochondrion selective dye Mitotracker Red CM-H2XRos (Molecular Probes) by adding 50 nM directly to the culture medium and incubating cells for 30 min.. The cells were then rinsed thrice in pre-warmed PBS and visualized with living cell system (Cell^ R, Olympus, Japan).
Apoptosis analysis with flow cytometry
Apoptosis was determined using annexin V-FITC/propidium iodide (PI) staining assay with an apoptosis assay kit (Invitrogen, USA). Cells with confluence of 70-80% were trypsinized and stained for apoptosis detection through FACS5000 flow cytometry (BECKMAN, CA, USA) according to the manufacturer’s instructions. For analysis, early apoptotic cells showed a positive staining of annexin V-FITC. Late apoptotic and dead cells display both membrane positive staining with annexin V-FITC and nuclear positive staining with PI.
Measurement of mitochondrial transmembrane potential
Mitochondrial transmembrane potential (MMP) was investigated using JC-1 (5,5V,6,6V -tetrachloro-1,1V,3,3V-tetraethylbenzimidazolyl-carbocyanine iodide), which was reported to provide more accurate estimates of MMP than other fluorescent dyes (Mathur et al., 2000). This dye accumulates in the mitochondrial matrix under the influence of MMP, where it reversibly forms monomers (green) and J aggregates (red) with characteristic absorption and emission spectra (Reers et al., 1995).
Five×105 cells were incubated with JC-1 (5 μg/ml final concentration) for 1 hr at 4°C, washed, and immediately analyzed by flow cytometry using 488 as excitation and 530 or 590 nm as emission wavelengths. Bivariate plots of FL1 versus FL2 were used to analyze MMP (Smiley et al, 1991) Flow cytometric data presented are representative of three experiments. The ratio of fluorescent identity at FL2/FL1 was considered as the relative MMP value.
Measurement of intracellular ROS levels
Intracellular reactive oxygen species (ROS) levels were measured by a cell-permeating probe CM-H2DCFDA (Invitrogen, USA). CM-H2DCFDA is a non-polar compound which is hydrolyzed after entering the cell to form a non-fluorescent derivative, subsequently converted into a fluorescent product in the presence of an oxidant. Briefly, cells were washed, and then loaded with 10 μM CM-H2DCFDA for 1 hr. Excitation was set at 488 nm, and emission was recorded on an FL1 detector (525 nm). Data presented here are representative of three experiments. The fluorescent intensity was taken as the relative intracellular ROS level.
Cell cycle analysis
The cells were harvested with a trypsin-EDTA solution to produce a single-cell suspension, pelleted by centrifugation at 600 g for 10 min, and washed with PBS. The cells were then resuspended in a solution of PBS (pH 7.4) containing 25 mg/ml PI (Sigma Chemical Co., St. Louis, MO, USA), 0.1 mM EDTA and 0.01 mg/ml DNase-free RNase. The samples were incubated for 15 min at room temperature and followed with analysis on a FACS5000 flow cytometer.
Statistic analysis
Data are presented as mean ± standard deviations. Data were subjected to ANOVA and comparisons between irradiation groups (radon) and controls were determined by the Student’s t-test. A p <0.05 was considered to be significant.
Results
Characterization of mitochondrial DNA–knockdown (ρ−) and parental (ρ+) cells
In our initial attempts, HBE cells were treated with 50 ng/ml EB for 9 days, and subsequently maintained in the presence of 12.5 μg/ml EB for 30 days. Quantitative real time PCR of total cell DNA showed that mitochondrial DNA knockdown (ρ−) cells contained reduced mtDNA in approximately 20% of the parental ρ+ cells (Table. 1). Subsequently, a series of experiments were conducted to characterize the differences between ρ− cells and their parental ρ+ cells. After removal of uridine for 5 days, there was almost complete loss of viability of ρ− cells (Figure. 1A).
Figure 1.

Characterization of mitochondrial DNA–knockdown (ρ−) cells and their parental (ρ+) cells. HBE cells were treated with 50 ng/ml EB for 9 days, and then maintained in the presence of 12.5 μg/ml EB for 30 days. (A) The ρ+ cells were grew well in culture (left, × 400), while ρ− HBE cells died after withdrawal of uridine in the medium for 5 days (right, × 400). (B) ρ− and ρ+ HBE cells were labeled with PicoGreen and Mitotracker Red CM-H2XRos. Merging the red and green signals shows a yellow color in regions that overlap. The ρ− HBE cells stained with PicoGreen show bright nuclear and punctuate cytoplasmic staining, and ρ+ cells only nuclear staining is apparent. (C) Cells were stained with the mitochondrial membrane potential (MMP) probe JC-1. In ρ+ cells, normal MMP was noted where red J-aggregates was detected. In ρ− cells, depressed MMP resulted in few J-aggregates and staining was mostly green.
In order to visualize mtDNA in situ within viable cells, cells were stained with PicoGreen. Although nuclear staining was visible in both ρ− and ρ+ HBE cells, ρ− cells were observed with a faint cytoplasmic speckling, while ρ+ displayed bright punctuate cytoplasmic staining. The morphology of mitochondria was not apparently different between ρ− and ρ+ HBE cell (Figure. 1B). Subsequently, mitochondrial function was examined using probe JC-1. In ρ+ cells normal mitochondrial membrane potential was noted as evidenced by red J-aggregates. In ρ− cells, the depressed mitochondrial membrane potential resulted in few J-aggregates and staining was mostly green (Figure 1C). Data thus indicate that ρ− cells possess dysfunctional mitochondria and that this cell line is a reliable model to investigate the role of mitochondria in in vitro in radon and its progeny-induced cell transformations
Radon-irradiation induced apoptosis in ρ− and ρ+ HBE cells
To explore the role of mitochondria in apoptosis of HBE cells irradiated by radon and its progeny, the % cells undergoing apoptosis and necrosis (post-apoptosis) were determined by staining with annexin V-FITC and PI. Cells in the bottom right quadrant indicated apoptosis, whereas cells in the top right quadrant represented post-apoptotic necrotic population (Figure. 2A). Flow cytometric analysis showed significant increased apoptosis % both in ρ− and ρ+ HBE cells following exposure to radon and its progeny. However, when compared with ρ+ cells, ρ− cells demonstrated a significantly lower level of apoptosis in Figure 2B. Although the early apoptotic rate in ρ− cell is higher than in ρ+ cells, it is noteworthy that post-apoptosis was almost absent in ρ− cells (Figure. 2B). Therefore, data indicate that partial mitochondria deletions as present in knock-down HBE cells may decrease apoptosis induced by radon especially in post-apoptosis in this cellular model.
Figure 2.

Radon-irradiation induces apoptosis in ρ− and ρ+ HBE cells. Apoptotic cells were detected by flow cytometric analysis using annexin V and PI in FL-1 versus FL-2 channels. (A) The cells in the bottom right quadrant indicated apoptosis, whereas cells in the top right quadrant represented post-apoptotic necrotic population. (B) The % apoptotic and post-apoptotic cells were determined by fluorescence-activated cell sorting analysis. Data presented are representative from three experiments.
Data are expressed as means ± S.D.
*, p<0.05, compared to control cells;
Δ, p<0.05, compared to ρ+ cell.
Radon-irradiation effect on mitochondrial member potential in ρ− and ρ+ HBE cells
The contribution of mitochondria in mediating apoptosis of HBE cells exposed to radon and its progeny was determined by measurement of mitochondrial member potential (MMP) in ρ− and ρ+ using JC-1 fluorescence dye. In cells with normal mitochondrial function, membrane potential–driven accumulation of these dyes results in formation of red fluorescent J-aggregates as shown in control HBE cells (Figure 3A). In ρ− cells with lower MMP mitochondria appear green when stained with JC-1 dye. After exposure to radon, the lower MMP in ρ− HBE cells resulted in few J-aggregates and staining was predominantly green (Figure. 3A). The analysis by flow cytometry also verified that MMP was significantly decreased in ρ− cells compared to ρ+ HBE cells (Figure. 3B).
Figure 3.
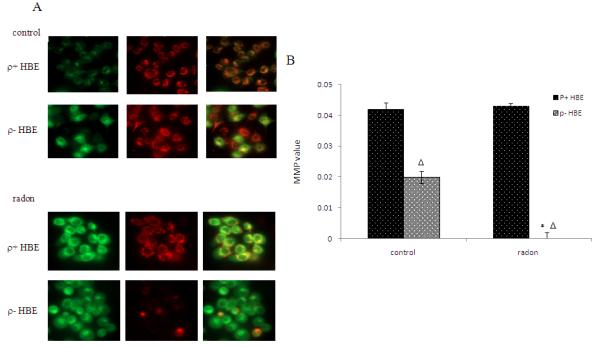
Radon-irradiation effect on mitochondrial member potential in ρ− cells. Mitochondrial membrane potential (MMP) was detected using JC-1 dye. (A) The normal MMP–driven accumulation of JC-1 dyes results in the formation of red fluorescent J-aggregates in ρ+ HBE cells. In ρ− cells with depressed MMP, the mitochondria appear green. After exposure to radon, depressed MMP in ρ− HBE cells resulted in few J-aggregates and stained green. (B) MMP was determined by flow cytometry. The ratio of the fluorescent identity at FL2/FL1 was considered as relative MMP value.
Data presented are representative from three experiments. Data are expressed as means ± S.D.
*, p<0.05, compared to control cells
Δ, p<0.05, compared to ρ+ cell.
Radon-irradiation influence on ROS levels in ρ− and ρ+ HBE cells
To determine the role of mitochondria in radon-induced oxidative burst, the intracellular ROS levels were examined in ρ− and ρ+ HBE cells. After irradiation by radon and its progeny, production of ROS was markedly increased in ρ− and ρ+ HBE cells (Figure 4). Further, intracellular ROS was significantly lower in ρ− compared to ρ+ HBE cells, suggesting that reduction of ROS occurs in mtDNA knock-down HBE cells.
Figure 4.
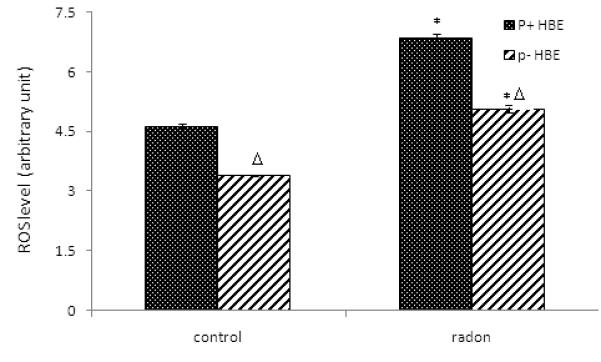
Radon-irradiation influence on ROS levels. Cells were loaded with 10 μM CM-H2DCFDA for 1 hr. The intracellular ROS levels were examined by flow cytometry. The fluorescent intensity was taken as the relative intracellular ROS level. Data presented here are representative of three experiments.
Data are expressed as means ± S.D.
*, p<0.05, compared to control cells
Δ, p<0.05, compared to ρ+ cell.
Radon-irradiation effect on cell cycle in ρ+ and ρ− HBE cells
The cell cycle distribution was analyzed by flow cytometry after exposure to radon and its progeny (Figure 5A). In ρ+ cells, a significant finding was the decrease in S and increase in the G2/M phase fraction produced by radon and its progeny. In ρ− cells, G1, G2/M and S populations remained similar to cells exposed to radon (Figure 5B). It is of interest that S phase fraction changed from a rise to a fall following radon radiation in ρ− compared to ρ+ cells. A significant elevation in G1 phase fraction was observed in control and radon-irradiated ρ− cells compared to ρ+ HBE cells. These results indicate that the distribution of phases of cell cycle induced by radon irradiation may be different in ρ− and ρ+ HBE cells.
Figure 5.
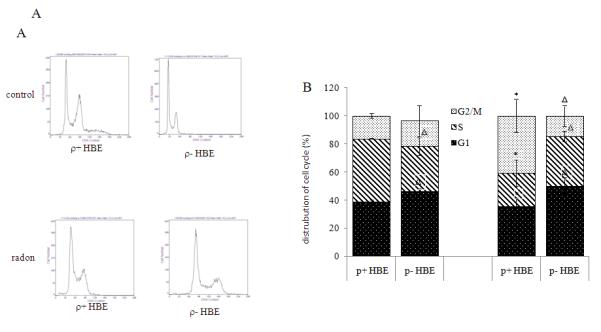
Radon-irradiation effect on cell cycle in ρ+ HBE cells. The cells were labeled with a solution of PBS (pH 7.4) containing 25 mg/ml PI, 0.1 mM EDTA and 0.01 mg/ml DNase-free RNase. The samples were incubated for 15 min at room temperature. (A) Histograms of distribution of DNA content with flow cytometry in ρ− and ρ+ HBE cells. (B) The % cells within different cells stages were analyzed by flow cytometry. Data presented are representative of three experiments.
Data are expressed as means ± S.D.
*, p<0.05, compared to control cells
Δ, p<0.05, compared to ρ+ cell.
Discussion
Chronic exposure of human bronchial epithelial cells to EB (50 ng/ml), a HBE ρ− cell line was developed to investigate the effects of depleting mtDNA on cellular processes, with a focus on mitochondrial function and apoptosis following radon exposure. After several weeks of culture in EB, cells became dependant on glycolysis for ATP production and required supplementation with uridine and pyruvate. Thus the presence of EB in the culture is necessary to preserve 80% knock-down of mtDNA. Thus in our model the cell line (ρ−) was defined as knock-down rather than knockout or depleted as reported in other studies (King and Attardi, 1996; Ray et al., 2004 )
Mitochondria play a key role in the process of cell death. One of the prominent examples of apoptosis inhibition acting at the mitochondrial level is the over-expression of anti-apoptotic proteins of the Bcl-2 family such as Bcl-2 or its close homologues Bcl-XL and Mcl-1 (Adams and Cory, 2007). In this study, the apoptosis % in ρ− cells was found to be significantly lower than in ρ+ cells. Although radon exposure increased the apoptotic rate to a higher level, a reduction in the rate in ρ− cells was still clearly seen when compared to ρ+ cells. These observations indicated a key role that mitochondria play in inhibition of cellular apoptosis. In agreement with this finding several investigators demonstrated that mitochondria play an important role in apoptotic pathways (Simone, 2010).
Mitochondria produce reactive oxygen species (ROS), which are involved in the regulation of many physiological processes, but which might also be harmful to the cell if produced in excessive amounts. An increased mitochondrial formation of ROS triggers the intrinsic pathway by opening permeability transmission pores with enhanced permeability of the outer mitochondrial membrane (Palmer et al., 2000). A number of conditions may lead to the intra-mitochondrial excessive generation of ROS, which favor oxidative damage-dependent mutagenesis and hence promote tumorigenesis (Zhou et al., 2007). Dasgupta et al.(2008) found that enhanced electron leakage and ROS overproduction resulted in mutations of the nuclear or mitochondrial DNA may affect components of the respiratory chain such as cytochrome b and favor uncoupling. It is of interest that such mutations are also noted in a wide variety of cancers (Modica-Napolitano and Singh, 2004). In the present study, intracellular ROS levels in ρ− cells were markedly lower than in ρ+ cells. This is not surprising since knock-down of mtDNA may lead to significant reduction in number of mitochondria and then diminished ROS production. As in the case of apoptosis, radon exposure induced a significant elevation of ROS levels in both cell types but the level of ROS in ρ− cells was only 48% of that in ρ+ cells. Data thus suggest that less radon-induced apoptosis in ρ− cells may be associated with absence of mitochondria as evidenced by reduced ROS generation.
The dissipation of the mitochondrial electrochemical potential gradient (ΔΨm) is considered an early event in apoptosis which is inhibited in tumor cells due to decreased release of apoptotic factors. Enhanced mitochondrial gene expression may attenuate apoptosis in cell transformation (Shidara et al., 2005). In our study MMP was decreased by radon irradiation. However, the association of this observation with apoptosis requires further investigation.
Singh (2006) postulated that mitochondrial damage checkpoint (mitocheckpoint) might be activated by mitochondrial derangement, thereby preventing cell cycle progression until restoration of mitochondrial functions. In the present study, the distribution of phases of cell cycle was altered in both control ρ− and radon exposed cells with a prolonged G1 phase. After radon exposure, the fraction of S phase increased while that of G2/M phase decreased in ρ− compared to ρ+ cells. These results indicate a checkpoint arrest of G1 phase in cell cycle due to either mtDNA knockdown or radon irradiation, which may be a consequence or a parallel event occurring in apoptosis.
In conclusion, a mtDNA knockdown cell model was established to study the role of mitochondria in cell apoptosis induced by radon and its progeny. Changes in ROS generation, MMP and cell cycle are all attributed to a reduction in apoptosis that may trigger and promote cell transformation leading to radon-induced carcinogenesis.
Table 2.
mtDNA copy number in HBE cells treated with EB
| cells | mtDNA copy number |
|
|---|---|---|
| 16S/18S | ND1/18S | |
| ρ+ HBE | 1±0.16 | 1±0.16 |
| ρ− HBE | 0.22.8±0.12 | 0.22.7±0.12 |
Acknowledgements
The work was supported by the National Natural Science Foundation grants 81020108028 and 81072286, and in part funding from the National Institutes of Health grants ES05786, CA49062, Superfund grant P42 ES10349, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
References
- Adams J, M., Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Hoque MO, Upadhyay S, Sidransky D. Mitochondrial cytochrome B gene mutation promotes tumor growth in bladder cancer. Cancer Res. 2008;68:700–706. doi: 10.1158/0008-5472.CAN-07-5532. [DOI] [PubMed] [Google Scholar]
- Evdokimovsky EV, Ushakova TE, Kudriavtcev AA, Gaziev AI. Alteration of mtDNA copy number, mitochondrial gene expression and extracellular DNA content in mice after irradiation at lethal dose. Radiat Environ Biophys. 2011;50:181–188. doi: 10.1007/s00411-010-0329-6. [DOI] [PubMed] [Google Scholar]
- Fernandez-Silva P, Enriquez JA, Montoya J. Replication and transcription of mammalian mitochondrial DNA. Exp Physiol. 2003;88:41–56. doi: 10.1113/eph8802514. [DOI] [PubMed] [Google Scholar]
- Huang L, Snyder AR, Morgan WF. Radiation-induced genomic instability and its implications for radiation carcinogenesis. Oncogene. 2003;22:5848–5854. doi: 10.1038/sj.onc.1206697. [DOI] [PubMed] [Google Scholar]
- International Agency for Research on Cancer . Monographs on the evaluation of carcinogenic risk to humans: manmade fibres and radon. International Agency for Research on Cancer; Lyon, France: 1988. [Google Scholar]
- Kaufmann WK. Initiating the uninitiated replication of damaged DNA and carcinogenesis. Cell Cycle. 2007;6:1460–1467. [PubMed] [Google Scholar]
- King M, Attardi G. Isolation of human cell lines lacking mitochondrial DNA. Meth Enzymol. 1996;264:304–313. doi: 10.1016/s0076-6879(96)64029-4. [DOI] [PubMed] [Google Scholar]
- Krewski D, Lubin JH, Zielinski JM, Alavanja M, Catalan VS, Field RW, Klotz JB, Letourneau EG, Lynch CF, Lyon JL, Sandler DP, Schoenberg JB, Steck DJ, Stolwijk JA, Weinberg C, Wilcox HB. A combined analysis of North American case-control studies of residential radon and lung cancer. J. Toxicol. Environ. Health A. 2006;69:533–597. doi: 10.1080/15287390500260945. [DOI] [PubMed] [Google Scholar]
- Li JX, Fu CL, Chen R, Sun J, Nie JH, Tong J. Screening of differential expression genes in bone marrow cells of radon-exposed mice. J. Toxicol. Environ. Health A. 2007;70:964–969. doi: 10.1080/15287390701290766. [DOI] [PubMed] [Google Scholar]
- Marcinowski F, Lucas RM, Yeager WM. National and regional distributions of airborne radon concentrations in U.S. homes. Health Phys. 1994;66:699–706. doi: 10.1097/00004032-199406000-00009. [DOI] [PubMed] [Google Scholar]
- Michelle CT, Daniel K, Chen Y, Arden P, III, Gapstur S, Thun MJ. Radon and lung cancer in the American Cancer Society cohort. Cancer Epidemiol. Biomarkers Prev. 2011;20:438–448. doi: 10.1158/1055-9965.EPI-10-1153. [DOI] [PubMed] [Google Scholar]
- Modica-Napolitano JS, Singh KK. Mitochondrial dysfunction in cancer. Mitochondrion. 2004;4:755–762. doi: 10.1016/j.mito.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Mathur A, Hong Y, Kemp BK, Barrientos AA, Erusalimskya JD. Evaluation of fluorescent dyes for the detection of MMP changes in cultured ardiomyocytes. Cardiovasc Res. 2000;46:126–138. doi: 10.1016/s0008-6363(00)00002-x. [DOI] [PubMed] [Google Scholar]
- Ray A, Jacqueline YC, Jennifer K, Kristina AW, Kristin AM, Raymond PP, Lionel DL. The effects of ethidium bromide induced loss of mitochondrial DNA on mitochondrial phenotype and ultrastructure in a human leukemia T-cell line (MOLT-4 cells) Toxicol Appl Pharmacol. 2004;196:68–79. doi: 10.1016/j.taap.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC-1 dye. Meth Enzymol. 1995;260:406–417. doi: 10.1016/0076-6879(95)60154-6. [DOI] [PubMed] [Google Scholar]
- Palmer AM, Greengrass PM, Cavalla D. The role of mitochondria in apoptosis. Drug News Persp. 2000;13:398–384. [PubMed] [Google Scholar]
- Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, Oda H, Ohta S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65:1655–1663. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- Simone F. Evasion of apoptosis as a cellular stress response in cancer. Int. J Cell Biol. 2010 doi: 10.1155/2010/370835. Article ID 370835, doi:10.1155/2010/370835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KK. Mitochondria damage checkpoint, aging, and cancer. Ann. NY Acad. Sci. 2006;1067:182–190. doi: 10.1196/annals.1354.022. [DOI] [PubMed] [Google Scholar]
- Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD, Jr, Chen LB. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse LA, Yu IT, Qiu H, Joseph Siu Kai Au, Wang XR. A case–referent study of lung cancer and incense smoke, smoking, and residential radon in Chinese men. Environ. Health Persp. 2011;119:1641–1646. doi: 10.1289/ehp.1002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Kachhap S, Sun W, Wu G, Chuang A, Poeta L, Grumbine L, Mithani SK, Chatterjee A, Koch W, Westra WH, Maitra A, Glazer C, Carducci M, Sidransky D, McFate T, Verma A, Califano JA. Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc. Natl. Acad. Sci. USA. 2007;104:7540–7545. doi: 10.1073/pnas.0610818104. [DOI] [PMC free article] [PubMed] [Google Scholar]