

Abstract
Orphan nuclear receptors regulate diverse biological processes. These important molecules are ligand-activated transcription factors that act as natural sensors for a wide range of steroid hormones and xenobiotic ligands. Because of their importance in regulating various novel signaling pathways, recent research has focused on identifying xenobiotics targeting these receptors for the treatment of multiple human diseases. In this review, we will highlight these receptors in several physiologic and pathophysiologic actions and demonstrate how their functions can be exploited for the successful development of newer drugs.
Keywords: agonist, antagonist, ligand binding domain, orphan nuclear receptor
INTRODUCTION
Nuclear receptors (NRs) define the largest superfamily of ligand-dependent transcription factors. They are involved in a wide variety of biological functions, including cell proliferation, differentiation, development and homeostasis (1, 2). Since the discovery of the first NR in the 1970s (3), several more structurally similar receptors were discovered, defining a new class of NRs called the “orphan nuclear receptors” (ONRs) (Fig. 1) (1). ONRs are defined by a lack of identifiable ligands controlling their physiological functions in vivo. In recent years, low affinity ligands have been discovered for some of the orphans and were subsequently classified as “adopted” ONRs (1, 2). The ligand-binding pocket of these adopted receptors (e.g. pregnane X receptor (PXR; NR1I2), liver X receptor (LXR; NR1H2 and NR1H3), farnesoid X receptor (FXR; NR1H4), constitutive androstane receptor (CAR; NR1I3 and NR1I4), peroxisome proliferator activated receptor (PPAR; NR1C2, NR1C3, and NR1C4), etc.) are larger than classical NRs and bind to a large diversity of molecules with lower affinity (1).
Fig. 1.

Classification of NR superfamily based on the identification of their respective ligands. This includes the classical NRs with known high affinity hormonal ligands, orphan receptors with no known identifiable ligands and adopted orphan receptors with low affinity dietary ligands.
Functionally, ONRs are very similar to classical NRs. The classical function of NRs is to transcriptionally regulate expression of target genes by the recruitment of co-activators or co-repressors. Ligand binding to these receptors recruits the co-activators (activation) or co-repressors (repression), thereby regulating the coordinate expression of their target genes (Fig. 2) (4, 5). Generally, NRs bind to co-repressors in their un-liganded apo-form with histone deacetylase (HDAC) activity and act as a transcriptional suppressor (6, 7). With agonist ligand binding, conformational change in the helix 12 (H12) of the ligand-binding domain creates an open conformation of the NR holo-form favoring co-activator binding with histone acetyltransferase (HAT) activity for its activation properties (7). Discovery of antagonist molecules has been targeted to this H12 structure with the hypothesis that molecules that inhibit folding of H12 can act as antagonist for the corresponding NR (7). Though the classical mechanism of NR action is called transactivation, alternative mechanisms of NR action have also been reported (e.g., transrepression, where NRs, instead of binding directly to DNA, interact with other promoter-specific transcription factors to deactivate the target gene (8, 9), and non-genomic mechanisms, such as the very fast actions of NRs via membrane-associated signal transduction machineries (10, 11)).
Fig. 2.

Schematic diagram describing classical NR function: NR binds to its ligand in the cytoplasm, which leads to its translocation into the nucleus. Ligand-bound NR dimerizes with its obligate partner to bind to the target gene regulatory element, with further recruitment of coactivators and RNA polymerase complex in the nucleus. This leads to target gene transcription with more protein production in the cytoplasm for gene specific activity.
Structurally, classical NRs contain four distinct functional domains: (1) AF1 or ligand-independent activation domain or A/B domain at the amino-terminal end, (2) DBD or DNA-binding domain containing two conserved C4-type zinc-finger motifs, (3) a highly variable flexible hinge region connecting the DBD with the (4) LBD or ligand-binding domain that is associated with second activation domain (AF2) at the extreme carboxy-terminal end (Fig. 3) (12). NRs bind to the target gene DNA response element using their conserved DBD. These response elements contain conserved hexameric sequences that can be arranged in several configurations, such as inverted or direct repeats (13, 14). Though the majority of ONRs possess all the functional domains common to the classical NRs, diversity is present in some ONR structures (1, 15). The structures of ONRs within the LBD in general and highly diverse (e.g. the ligand-binding pocket of PXR is very large and flexible due to the presence of two additional strands of β sheet, which explains the promiscuous nature of PXR binding to diverse range of compounds) (16–18). Some orphan receptors contain only one of the two characteristic domains (DBD or LBD) of the NR superfamily. In vertebrates, DAX-1 (NR0B1) and small heterodimer partner (SHP/SHP-1; NR0B2) contain only a LBD and lack a classic DBD (19, 20). In other species, such as Drosophila Knirps, KNRL and EGON (NR0A1, 2, 3) lack either of these domains (21, 22). The size of the domains in ONRs also varies, e.g., the A/B domain of some receptors is short (e.g., RORβ (NR2F2) and TLX (NR2E1)), whereas the same domain is quite large for NGFI-B/NR4A group members (15). The diversity of ONRs is also present in the modes of their DNA binding. While most of them bind to DNA as homodimers on direct repeat elements (e.g. HNF4 (NR2A1, A2, A3), COUP-TFs (NR2F1, F2, F3), TLX, and TR2/4 (NR2C1, C2)), some bind to DNA by interacting with retinoid X receptor (RXR) as a heterodimer partner (e.g. PXR, CAR, FXR, LXR and PPAR) (23), some oligomerize (e.g. GCNF (NR6A1)) on binding to a direct repeat (24), whereas several other orphan receptors (e.g. Rev-erbs (NR1D1, D2), RORs (NR2F1, F2, F3), SF-1 (NR5A1), NURR1 (NR4A2), NOR1 (NR4A3) and ERRs (NR3B1, B2, B3)) have been shown to bind DNA as a monomer to a half-site sequence (25). Additionally, estrogen receptor α, a ligand-activated NR, contains an additional carboxy-terminal F domain with unknown function (26). Furthermore, the NR diversity is also evident in their expressions in different species both vertebrates and invertebrates, such that there are 21 and more than 270 NR-like genes identified in Drosophila melanogaster and in Caenorhabditis elegans, respectively (27, 28). A list of vertebrate ONRs is presented in Table I, information that will be very useful to study individual receptor in these research animals for drug design and in vivo testing (1).
Fig. 3.

Structural domain of the NR superfamily: extreme amino terminal domain is called the A/B domain that contains the AF1 region, followed by the conserved DNA binding domain (DBD), and linked by a hinge region with the ligand binding domain (LBD). The extreme carboxy terminal end of LBD is called the AF2 region.
Table I.
Known Vertebrate Orphan Nuclear Receptors
| NR | Subtypes | Nomenclature | Species | References |
|---|---|---|---|---|
| PXR | NR1I2 | h, m, x | (81,88,298) | |
| CAR | α, β | NR1I3 (α), | h, m | (299,300) |
| NR1I4 (β) | ||||
| FXR | NR1H4 | h, m, r | (301,302) | |
| LXR | α, β | NR1H3 (α), | h, m, r | (301,303–306) |
| NR1H2 (β) | ||||
| PPAR | α, β/δ, γ | NR1C1 (α), | h, m, r, k, l, | (166,307–314) |
| NR1C2 (β/δ), | b, p, g, x | |||
| NR1C3 (γ) | ||||
| NURR1 | NR4A2 | h, m, r | (196,315,316) | |
| Nur77 | NR4A1 | h, m, r, x | (317–319) | |
| TLX | NR2E1 | h, m, c, x, f | (212,320,321) | |
| DAX-1 | NR0B1 | h, p, m, r | (19,322) | |
| ROR | α, β, γ | NR2F1 (α), | h, m, r | (323–326) |
| NR2F2 (β), | ||||
| NR2F3 (γ) | ||||
| SHP | NR0B2 | h, m, r | (20,327) | |
| SF-1 | NR5A1 | h, m, c, b | (328–331) | |
| ERR | α, β, γ | NR3B1 (α), | h, m, r | (257,259,332) |
| NR3B2 (β), | ||||
| NR3B3 (γ) | ||||
| HNF4 | α, β, γ | NR2A1 (α), | h, m, r, x | (272,333,334) |
| NR2A3 (β), | ||||
| NR2A2 (γ) | ||||
| GCNF | NR6A1 | h, m, x | (19,322) | |
| LRH-1 | NR5A2 | h, m, r, c | (335–338) | |
| PNR | NR2E3 | h, m, r, c | (211,292,339,340) |
Because of this structural and functional diversity, promiscuous nature of DNA and ligand-binding properties (e.g. CAR and PXR) and their ability to bind to a broad range of molecules, thereby regulating a vast array of target genes, ONRs have become an attractive target for drug development. In this review, we will discuss how individual ONRs can be exploited (using agonist and antagonist molecules of these receptors) for a successful drug discovery in various human diseases (Table II).
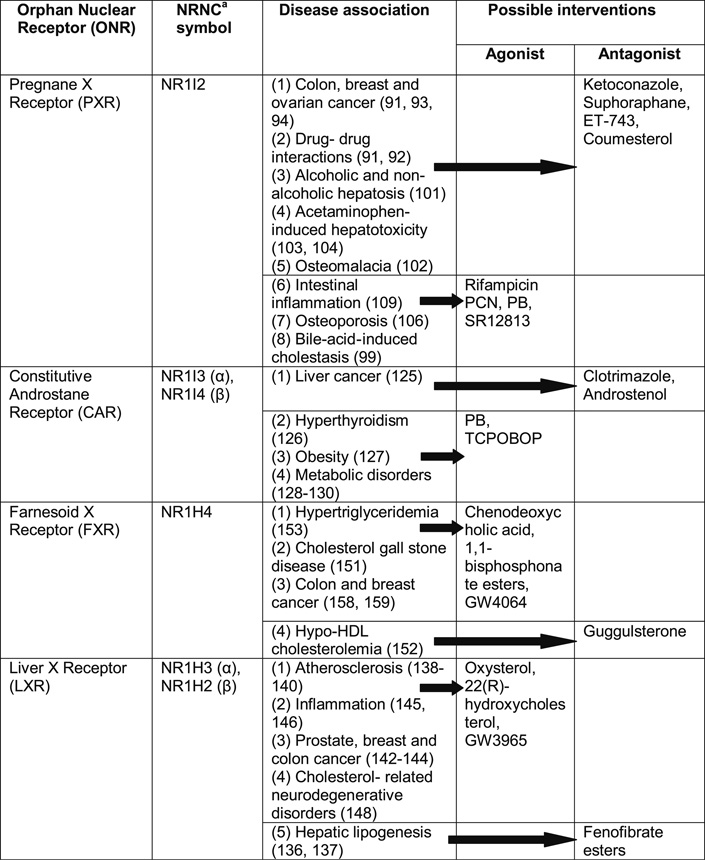
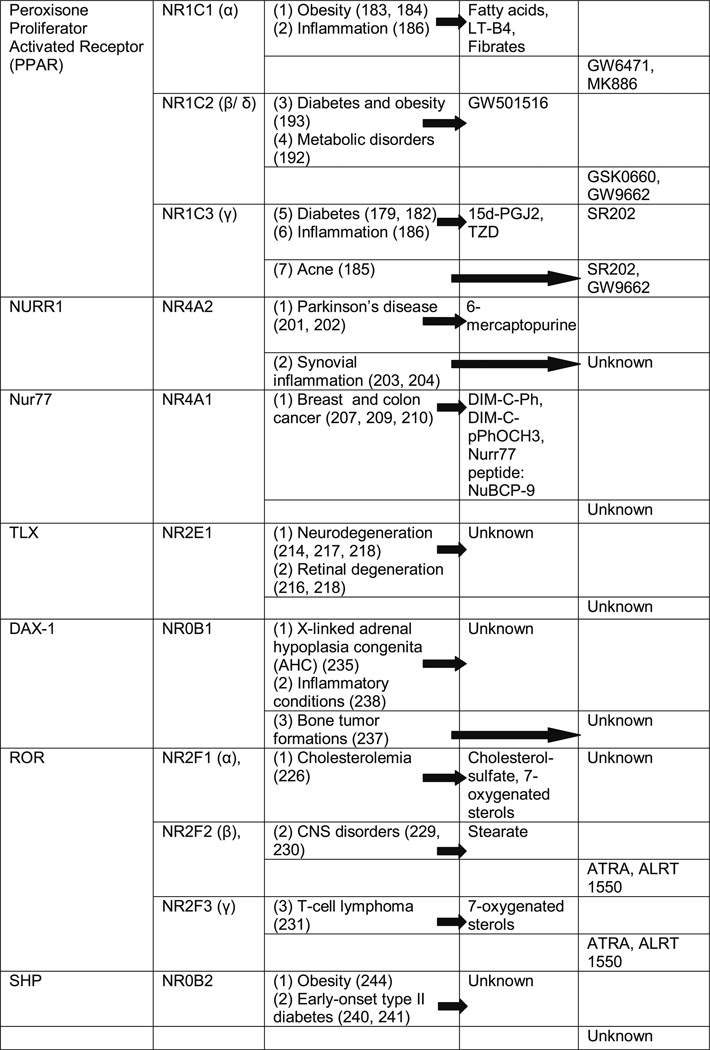
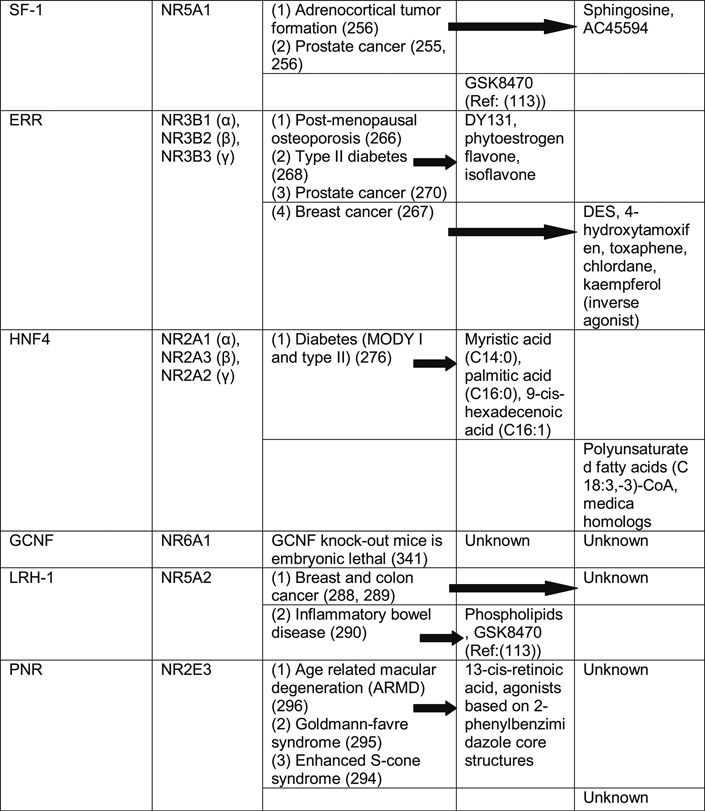
Table II. Orphan Nuclear Receptor Phenotypes and Possible Therapeutic Interventions with Known Agonist and Antagonist Compounds for Various Human Diseases.
 |
 |
 |
NUCLEAR RECEPTOR DRUG DISCOVERY TOOLS
Identification of potential drug candidates for NRs traditionally has been done by using several drug development strategies (Fig. 4). The various strategies are described below.
Fig. 4.

Schematic diagrams of various drug discovery tools. Detailed descriptions of each can be found in the text.
In cell-based reporter assays where NR LBD is fused with DBD of yeast transcription factor Gal4, cells are transfected with this construct along with a reporter construct containing Gal4 upstream activation sequences (UAS) upstream of a reporter gene (e.g., luciferase, β-galactosidase). The activation and/or inactivation of the NRs by binding of a ligand is monitored by the expression of the reporter gene. Multiple modifications of this assay have been made since its discovery to allow for the measurement of receptor activation or inhibition by compounds and also to determine compound selectivity and potency (29).
In vitro ligand binding assays (e.g., scintillation proximity and fluorescent polarization assays) screen potential ligands by their competition with radiolabeled known ligands for the LBD of respective NR (30, 31).
Yeast and mammalian are two hybrid assays in which ligand-dependent NR-coactivator interactions are determined to identify potential compounds that augment/inhibit NR-coactivator interactions (32, 33).
Similarly, NR-coactivator interaction properties are exploited for more specific, sensitive and high-throughput assays, such as the fluorescence resonance energy transfer (FRET, or more specifically time-resolved FRET or TR-FRET) (34), ligand-sensing assay (LiSA) (35) and AlphaScreen assay (Amplified Luminescent Proximity Homogenous assay) (36). In FRET assays, NR and coactivator are labeled with proper fluorescent donor-acceptor probes (e.g., europian cryptate [Eu (K)]-cross linked conjugate of allophycocyanin (XL665) or cyan fluorescent protein (CFP)-yellow fluorescent protein (YFP)), and ligand-induced NR-coactivator interactions are measured by the energy transfer between the fluorophores. In TR-FRET assay, this energy transfer is time-gated in order to reduce short-lived fluorescent background, thereby increasing the sensitivity of the detection (37, 38). Additionally, the rapid fashion in which TR-FRET assay is accomplished gives the advantage of screening large drug samples in high-throughput format. In LiSA, similar to FRET assay, biotin-streptavidin interaction properties are used for fluorophore labeling of NR and coactivator. AlphaScreen is a bead-based assay where NR is coupled to donor beads and coactivator to acceptor beads. Ligand-induced conformation change in NR and hence its binding to co-activator triggers the donor-acceptor beads to come in proximity with subsequent generation of a signal.
Another assay, known as amide hydrogen/deuterium exchange, coupled with proteolysis and liquid chromatography-mass spectrometry (H/D-Ex), is gaining popularity as a drug development tool for the analysis of NR-ligand interactions (39). In this assay, the rate of amide hydrogen exchange depends on local fluctuations in the protein structure, and, for this reason, the rate of H/D-Ex exchange is a good indicator of protein conformational change. Hence, H/D-Ex is used to detect differences of protein dynamics in apo and holo forms of NR LBD.
Structural detailing of NR LBD has triggered the development of in silico approaches, like virtual ligand screening (VLS) for NR drug development. VLS is a knowledge-driven approach based on the structural information of either ligand (ligand-based approach) or NR target (receptor-or target-based approach). This method expands the concept of central similarity-property principle, which depicts that similar molecules exhibit similar properties. Based on this, similarity calculations can be performed, and molecules from large chemical libraries can be screened and subsequently scored and ranked using computational methods (40).
Computational methods have become an extremely powerful tool not only to provide insights into novel agonist-antagonist interactions with NRs but also have facilitated the identifications of off-target interactions of NRs responsible for undesirable side effects (41). Results from computational methods have shown that NRs undergo significant conformational changes upon ligand binding or release, and the key factor of such NR-ligand interaction depends on the quaternary state of the receptor (41, 42). Computational studies have also been helpful to understand and characterize the structural domains of NR required for the recognition and specificity of interacting partners for its transcriptional activities (43, 44). Similarly, computational analysis of the three-dimensional coordinates of NR protein structure has helped to identify newer functional residues using the evolutionary trace computational methods, which are important for NR-coregulator interactions (45, 46).
Combined in silico-in vitro approaches have also shown their importance in studying evolutionary change in the LBD of different NRs responsible for their varying ligand specification across species (47). Finally, docking studies with structural similarity comparison methods have been helpful to understand the molecular mechanisms of adverse drug effects. This in silico approach, therefore, can be used to identify early off-target side effects of newly discovered drugs. This approach can also be expanded to drug repurposing or repositioning, where newer related therapeutic applications of older marketed drugs are exploited while eliminating the activity at the original target, thereby reducing the cost and time associated with the drug development processes (48, 49).
LIMITATIONS OF ONR-BASED PHARMACOLOGIC THERAPY
Drug adverse effects are a common cause of morbidity and mortality world-wide, and it is estimated that 20% of all adverse drug effects are due to drug-drug interactions (50, 51). Drug-drug interactions are a process by which administration of one drug alters the systemic drug levels (i.e. increasing or decreasing effective concentration) of another co-administered drug. Among the mechanisms of drug-drug interactions, drug metabolism and transport are considered to be most important, since they directly affect the therapeutic plasma concentrations of drugs administered, thereby determining drug toxicity or loss of drug response (52, 53).
Tissues that are mostly responsible for drug metabolism and transport are liver and the intestine. In these tissues, oxidative metabolism of drugs occurs through CYP450 group of enzymes, and drug efflux transport is mediated via MDR1 (also known as p-glycoprotein, ABCB1, ATP binding cassette subfamily B member 1) (52, 54). ONRs are critical in regulating these important mediators. Arguably, the most important ONR involved in drug-drug interaction is PXR, since it can be activated by a wide array of chemicals because of its promiscuous nature of ligand binding. Moreover, PXR regulates expression of the two most important drug metabolizing proteins—CYP3A4 (cytochrome P450 family 3 subfamily A member 4, the most abundant of the CYP450 isozymes in the liver and intestine) and MDR1—and is thereby responsible for the metabolism of ~50% of marketed drugs (55, 56). CAR is activated by fewer compounds than PXR but is responsible for similar effects on drug interactions via CYP3A4 and MDR1 (57). Additionally, FXR has also become an important ONR involved in drug interactions, since functional FXR response elements have been identified in CYP3A4 and another drug transporter MRP2 (58, 59). Since any given drug can activate more than one receptor and NR themselves regulate each other, this complex drug-drug interaction network regulated by ONRs should be very carefully exploited for safer pharmaceutical applications (60, 61).
While the numbers of compounds that can activate orphans have rapidly increased over the last decade, a few prescription drugs also cause clinically relevant drug-interactions. Drugs in these categories are anticonvulsants (e.g. phenobarbital, carbamazepine, phenytoin, valproic acid), antibiotics (e.g. rifampicin, nafcillin, rifabutin), human immunodeficiency virus protease inhibitors (ritonavir, nelfinavir, tipranavir) and non-nucleoside reverse transcriptase inhibitors (e.g. nevirapine, efavirenz) (62–66). Additionally, several herbal medicines (e.g. St. John’s Wort, Ginkgo biloba) similarly affect drug-drug interactions through ONR signaling pathways (67, 68). In several disease conditions (e.g. tuberculosis, HIV infection, epilepsy) where chronic treatment is required, drugs used for these purposes can also activate ONRs as an off-target effect causing multiple side effects. For example, anticonvulsants and rifampicin (anti-tuberculosis drug) can cause hypothyroidism by increasing thyroid hormone turnover in liver via induction of glucuronidation, sulfation and biliary excretion (69, 70). Rifampicin treatment results in bone-loss and osteomalacia by interfering with vitamin D signaling via PXR (71). Additionally, HIV protease inhibitors by activating PXR and CAR are associated with the development of fatty liver (72).
Classical estrogen receptor exerts its effect on various tissues, and synthetic estrogens have shown its potential to act in tissue-specific manner (e.g. selective estrogen receptor modulators) (73, 74). While this effect is beneficial for receptor drug targeting to prevent off-target adverse effects, unfortunately, no tissue-specific pharmacologic agents targeting ONR have been described to date.
Since ONRs are very crucial as a drug development target, predictions of in vivo drug interactions have become very important for successful drug discovery. Several experimental systems have been employed that include cultured primary human hepatocytes, humanized mouse models, transformed cell lines (e.g. DPX-2, a derivative of HepG2 cells, that harbors human PXR and luciferase-linked CYP3A4 promoter and Fa2N-4 immortalized human hepatocyte clone), reporter gene assays, coactivator recruitment assays and receptor binding assays (55, 75–78).
Therefore, it is evident that both ONR agonist/antagonist therapies and prescription drugs causing off-target ONR effects could affect the therapeutic outcomes of treatment. Hence, serious considerations should be given while designing ONR-based pharmaceutics to avoid drug adverse effects.
PREGNANE X RECEPTOR (PXR): AGONIST AND ANTAGONIST—IMPLICATION FOR MULTITUDE OF DISEASES
Pregnane X Receptor (PXR) is an ONR encoded by the NR1I2 gene. It is involved in drug metabolism, bile acid transport, cancer, cholesterol metabolism and inflammation (79–81). While it is highly expressed in the liver and proximal small intestine, reduced levels of expression are seen in the large intestine (80, 82). PXR ligands, such as rifampicin, pregnenolone and phenobarbitone, are typically activators, although a small number of antagonists have also been identified, such as the ketoconazole (and related azoles), suphoraphane, ecteinascidin (ET-743) and coumesterol (83, 84).
Crystal structure of PXR LBD has shown that the ligand binding pocket of PXR is highly unrestricted because of its larger volume (more than 1,600 Å). Hence, it is able to function as a broad-specificity sensor of lipophilic xenobiotics and therefore regulate a vast array of target genes (16).
PXR has a substantial cross-species difference in terms of ligand binding. PXR LBD amino acid sequence identity has shown only 75% identity among human and rodents and 50% among human and chicken or zebrafish sequences (85). Since PXR and CAR (discussed later in the article) play a major role in drug metabolism and they show interspecies sequence variation in terms of ligand-mediated gene transcription, drug bioavailability testing, efficacy and toxicity evaluations become difficult in animal models of human disease. To address these issues better, humanized mouse models of PXR, CAR and double humanized (both PXR and CAR) models have been created, where mouse PXR and CAR genes have been exchanged with their human counterparts (75, 86, 87).
PXR Functions and Therapeutic Implications
The metabolism of xenobiotics via CYP3A4 and MDR1 pathways are regulated by PXR (88, 89). By binding to the promoter region of CYP3A4 gene, PXR brings about its activation, leading to enhanced xenobiotic metabolism. Possible roles of PXR antagonist (e.g., ketoconazole) are that they may bind to PXR LBD and inhibit its interaction with its coactivator (SRC-1, steroid receptor coactivator 1), thereby inhibiting target gene activation (90). Inhibition of PXR causes decreased metabolism and, hence, increased bioavailability of bioactive compounds. This provides a novel mechanism (PXR antagonist) to increase bioavailability of the chemotherapeutic drugs, while induction of PXR by agonists can lead to drug resistance (91, 92).
Recent investigations have shown that PXR has an anti-apoptotic role in colon cancer cells, implicating its role in tumorigenesis. Anti-apoptotic role of PXR is independent of the xenobiotic metabolizing role. Instead, it is associated with up-regulation of multiple anti-apoptotic genes, including BAG3 (BAG family molecular chaperone regulator 3), BIRC2 (Baculoviral IAP repeat-containing protein 2) and MCL-1 (Induced myeloid leukemia cell differentiation protein), and down-regulation of pro-apoptotic genes, such as BAK1 (Bcl-2 homologous antagonist/killer) and TP53/p53 (Tumor protein 53/protein 53) (93). This antiapoptotic role is also seen in normal colonic epithelial cells. For the treatment of colon cancer, this information will be very useful in developing PXR antagonist. PXR transcriptionally activates organic anion transporter OATP1A2 (mediates cellular uptake of estrogen metabolites), and this effect leads to increased proliferative potential of estrogen in breast tissues. Specific PXR antagonists have been shown to inhibit proliferative effects of estrogen (94). Additionally, PXR activation has been shown to increase proliferative potentials of ovarian cancer cell-lines, further strengthening the basis for finding novel non-toxic inhibitors of PXR activation to control cell growth (91).
PXR transcriptionally activates dehydroepiandrosterone (DHEA) sulfotransferase (SULT2A1; phase II drug conjugating enzyme) and regulates bile acid metabolism by facilitating elimination of lithocholic acid from the body (95). Additionally, a complex network of ONRs (PXR, FXR, CAR, LRH-1(liver receptor homolog/NR5A2), HNFs, SHP) regulates genes involved in organic anion uptake (NTCP, OATPs), bile canalicular export (BSEP, MRP2) and alternative basolateral export (MRP3, MRP4) in liver (96–98). Furthermore, PXR has also been shown to inhibit cholesterol 7α hydroxylase (CYP7A1, rate-limiting enzyme in bile acid biosynthetic pathway) transcription via a complex regulatory mechanism involving HNF4α (hepatocyte nuclear factor α) and PGC-1α (peroxisome proliferators-activated receptor γ coactivator) (99). This describes a novel protective mechanism of PXR activation against bile acid-induced cholestasis. SULT2A1 also has a role in energy and lipid homeostasis, and this involvement may highlight treatment potentials for many metabolic disorders targeting PXR.
PXR also activates fatty acid uptake transporter CD36 (in a complex interregulatory network involving PXR/LXR/PPARγ) and several accessory lipogenic enzymes, such as stearoyl CoA desaturase-1 (SCD-1) and long-chain free fatty acid elongase (FAE) (100). This is related to fat accumulation in the liver cells. It indicates that antagonism of PXR-related pathways may be utilized to treat alcoholic and non-alcoholic hepatic steatosis (101).
Other examples where PXR anatagonism would be beneficial are in disease conditions such as osteomalacia and acetaminophen-induced hepatotoxicity. PXR activators lead to osteomalacia by increased clearance of 1, 25 dihydroxyvitamin D3 (102). Similarly, activation of hepatic PXR increases conversion of acetaminophen to hepatotoxic metabolites (103, 104).
Intestine-specific PXR/CYP27A1/LXRα pathway regulates intestinal cholesterol efflux and high-density lipoprotein (HDL) assembly, targeting mitochondrial sterol 27-hydroxylase (CYP27A1), which catalyzes oxidative cleavage of the sterol side chain in the bile acid biosynthetic Orphan Nuclear Receptor and Drug Development 1447 pathway in the liver and 27-hydroxylation of cholesterol in most tissues. PXR transcriptionally activates CYP27A1 to produce 27-hydroxycholesterol in intestine, which in turn activates liver X receptor α (LXRα) to induce cholesterol efflux transporters ABCA1 and ABCG1 in macrophages (105). Therefore, this provides the evidence of PXR as a target for regulation of cholesterol efflux and HDL assembly, indicating its role in hyperlipidemia.
Vitamin K2 plays an important role in bone formation. It has been found that vitamin K2 binds to and transcriptionally activates PXR. PXR mRNA is expressed in osteosarcoma cell lines, and vitamin K2 with known PXR agonists induces the expression of the prototypical PXR target gene CYP3A4 in these cells (106). Thus, PXR is likely to be involved in the maintenance of bone homeostasis. This reveals a novel biological function of PXR and indicates that PXR agonists can function as effective therapeutic agents in the treatment of osteoporosis.
An inverse relationship was found between the PXR level and estrogen receptor (ER) status in breast cancer cells. It has been shown that PXR level is lower in ER+ breast cancer cells than in ER− cells. However, the level is the same in ER− cells as in normal cells. But there is no relation between the progesterone receptors. This may point to the fact that PXR has a role in breast cancer and may be utilized for the treatment for ER− tumor cells (107).
Promoter region of inducible nitric oxide synthase (iNOS) gene contains responsive elements for PXR. Since an iNOS-induced production of nitric oxide (NO) is known to influence inflammation and apoptosis, a PXR-regulated iNOS activity may explain a modulatory effect of steroids and xenobiotics on these cellular processes (108).
Besides that, PXR has also been shown to down-regulate NF-κB activation to inhibit inflammatory processes (109). Thus, potent PXR agonists can be used for the treatment of inflammatory diseases.
CONSTITUTIVE ANDROSTANE RECEPTOR (CAR): DRUG TREATMENT FOR LIVER CANCER AND METABOLISM
Constitutive androstane/active receptor is another NR from the same subfamily of NR1I as PXR. Apart from its lower levels of expression in heart, muscle, kidney and lung, it is predominantly expressed in the liver and small intestine. CAR can be activated by a wide variety of xenobiotics and is involved in phase I and II detoxification of drugs, steroid hormones, thyroid hormones and bilirubin. What sets it apart from classical NRs is that it is constitutively active even without any ligand, and ligand binding modulates its activity (12, 110). Other ONRs that show similar constitutive activities are RORα, LXRα, LRH-1, HNF4, NR4A subfamily of receptors and ERR (111–116). CAR remains sequestered in the cytoplasm by binding to chaperone proteins, and after binding to its agonist, it translocates to the nucleus to bind (as a dimer with RXR) to its target gene regulatory element (62).
Discovery of the crystal structure of CAR LBD provides information about its constitutive activity and the molecular basis for inverse agonism. Additionally, it was also found that CAR ligand binding pocket is smaller (~675 Å) and less flexible than in PXR, making it less promiscuous (117). The structure of CAR bound to androstenol (inverse agonist) showed that this androstenol binding sterically blocks the constitutive active position of helix 12 (110, 118). While phenobarbitone (PB) and TCPOBOP (1, 4-Bis [2-(3, 5-dichloropyridyloxy)] benzene) are well known activators of CAR, there is a good number of CAR antagonists also available, including clotrimazole and androstenol (57, 110, 119). PB is not an agonist but a well-known activator of CAR. It facilitates nuclear translocation of CAR with subsequent activation but without directly binding to CAR (120). PB exerts its tumorigenic effects by causing sustained activation of CAR (121, 122). Besides PB and TCPOBOP, steroid hormones also modulate the CAR-mediated regulation of target gene transcription. Estrogens activate mouse CAR and induce the CYP2B10 gene in mouse liver, whereas androgens and progesterone repress estrogen-activated mouse CAR (123).
Similar to PXR, CAR also shows species specificity in regards to some of its agonists. For example, human CAR, unlike mouse CAR, does not respond to steroid hormones, and a single residue difference in the C-terminal region of the mouse versus human CAR (T350M) has been shown to be responsible for this species specificity (110, 117).
CAR receptor associates itself with many other NRs to exert its effects via many cross-regulatory pathways, some of which are discussed as follows with possible implications for drug discovery: PB causes liver cell growth and tumor promotion and regulates glucose metabolism, steroid and thyroid hormone metabolism, drug metabolism and bile acid synthesis. Upon activation by PB and numerous PB-type inducers, the orphan receptor CAR mediates those pleiotropic actions by regulating various target genes, utilizing multiple regulatory mechanisms. This provides an idea about the pleiotropic effects of CAR (124). Tumorigenesis by PB is induced by the DNA methylation, which is mediated by CAR as well. This methylation may have a role in the carcinogenesis. Hence, similar to PXR, CAR is also a potential candidate for tumor drug development, especially for hepatocarcinogenesis (125).
CAR induces metabolism of the thyroid hormones T4 and T3. With CAR agonist therapy, it was found that both T4 and T3 levels dropped. CAR participates in the molecular mechanisms contributing to homeostatic resistance to weight loss by regulating lipid and glucose metabolism (126–128).
Activation of CAR suppresses lipid metabolism by reducing lipogenic transcription factor SREBP-1 (sterol regulatory element binding protein 1) protein levels (129). CAR induces Insig-1, a protein with anti-lipogenic properties, which lead to reduced levels of active SREBP-1 with subsequent reduction in target gene expression involved in triglyceride synthesis (130). Hence, this information implies that CAR represents a novel therapeutic target to uncouple metabolic rate from food intake and has implications in obesity and its associated disorders. Through a multiplex promoter spanning 218 kb, the phase II UDP-glucurono-syltransferase 1A (UGT1A1) gene encodes at least eight differently regulated mRNAs whose protein products function as the principal means to eliminate a vast array of steroids, heme metabolites, environmental toxins, and drugs. It was found that CAR, in association with PXR, activates this pathway of metabolism as well, in addition to the P450 system (131). UGT1A1 is also associated with many genetic diseases associated with bilirubin clearance, like Crigler Najjar syndrome and Gilbert’s disease. These findings may provide new dimensions to the understanding of these disorders with newer treatment options.
LIVER X RECEPTOR (LXR): ROLE IN CHOLESTEROL HOMEOSTASIS AND INFLAMMATION
LXR is a member of the NR1H NR subfamily involved in the regulation of cholesterol, fatty acid and glucose homeostasis (132). Two isoforms of LXR have been described as LXRα and β. LXRα is expressed in the liver, kidney, intestine, fat tissue, macrophage, lung and spleen, while LXRβ is expressed in almost all tissues (hence, earlier it was called as ubiquitous receptor) (132, 133). Both LXRα and LXRβ can be activated by endogenous oxidized derivatives of cholesterol, oxysterols (132).
X-ray crystal structures of both isoforms of LXR are reported in complex with synthetic LXR agonist T0901317 (can also activate FXR, PXR and CAR). LXRβ LBD shows that T0901317 can adopt two distinct conformations in the ligand binding pocket because of the pockets larger size (~830Å). This explains the molecular basis of LXR activation by a wide range of endogenous ligands. The conservation of amino acid sequence among human LXR isoforms is also very high, which makes it difficult to design isoform-selective agonists (134, 135).
LXR Functions and Pharmacological Implications
LXR activates fatty acid synthase (FAS) gene expression through binding to a direct-repeat 4 (DR-4) element in the promoter. Another orphan receptor, LRH-1 binds to a distinct element of FAS gene 21 bases downstream to DR-4 element, which is critical for the maximal response of LXR towards FAS expression, and this binding of LRH-1 is blocked by SHP (136). Hence, LXR plays a role in fatty acid synthesis along with two other orphan receptors, LRH-1 and SHP. Fatty acids serve many specialized functions, including cholesterol esterification, lung surfactant production, mammary gland secretions, signaling molecules and many others, including energy storage. Fatty acid biosynthesis is regulated mainly through two enzymes: acetyl-CoA carboxylase (ACC, the rate-limiting enzyme in fatty acid biosynthesis) and FAS. Besides being regulated by LXR, FAS can also be independently regulated by a large number of signals, including insulin, fatty acids, thyroid hormone, sterols, oxysterols, glucocorticoids, growth factors and cyclic AMP. These signals exert effects via FAS promoter with binding sites for E-box binding proteins USF1 & 2, sterol regulatory element binding proteins (SREBPs), thyroid hormone receptor (TR), LXR and carbohydrate response element binding protein. Additionally, LXR activation by oxysterols has been shown to up-regulate transcription of SREBPs (particularly SREBP-1c), which explains a mechanism of coordinate regulation of homeostatic balance between fatty acids and sterols (137).
It has been found that LXR promotes reverse cholesterol transport and inhibits atherosclerosis. In the presence of LXR agonists T0901317 and 22(R) hydroxycholesterol, fluid phase pinocytosis of low-density lipoprotein (LDL) by the macrophages are suppressed (138). Recently, it has also been observed that NAD-dependent deacetylase SIRT1 (sirtuin 1, silent mating type information regulation 2 homolog) deacetylates and activates LXR, thereby potentially regulating reverse cholesterol transport (139). This shows that there are mechanisms to inhibit macrophage cholesterol accumulation and atherosclerosis, by inhibiting macrophage uptake of LDL by activated LXR. Hence, as a transcriptional regulator of genes, such as the ABC transport proteins and genes involved in fatty acid and cholesterol metabolism, LXR has become an attractive target for the development of drugs for atherosclerotic disorders.
However, though the desirable therapeutic effects of increasing reverse cholesterol transport and cholesterol catabolism on activating LXR-mediated gene transcription can be achieved, the undesirable side effect of increasing hepatic lipogenesis can occur in the presence of LXR agonists. Therefore, searching for a LXR antagonist has identified fenofibrate esters (not fenofibric acid), which represses LXR activation-mediated lipogenesis but without negating the beneficial roles of LXR in increasing reverse cholesterol transport and cholesterol secretion (140).
Androgen ablation therapy is the mainstay therapy for certain prostate cancers. But many patients who receive the ablation therapy develop androgen-independent tumors. It has been found that androgen paradoxically inhibits the Orphan Nuclear Receptor and Drug Development 1449 proliferation of these cells partially by down-regulating c-myc and inducing the CDK-inhibitor p27kip1, which causes cell-cycle growth arrest (141). It was also found that LXR agonists decrease proliferation of both androgen-dependent and -independent prostate cancer cells. This effect of LXR is mediated by the ATP binding cassette transporter A1 (ABCA1) as well as by LXR signaling (LXR/ABCA1/27-hydroxylase) in the retardation of progression of prostate cancer growth (142). This indicates that LXR may be involved in the prostate cancer progression in vivo and suggests LXR signaling may be a useful target for prostate cancer treatment.
Similarly, LXR may also play a role in breast cancer. LXR controls estrogen homeostasis by regulating the basal and inducible hepatic expression of estrogen sulfotransferase (EST or SULT1E1), which is critical for metabolic estrogen deactivation. Estrogen has an important role in normal physiology, as well as in breast cancer and other hormonal disturbances. Therefore, this represents LXR to be a novel target for drug development in the treatment of breast cancer (143). Recently, it has been shown that LXR activation suppresses colon cancer proliferation in a β-catenin-dependent pathway, further implicating its role in the regulation of cancer development (144).
Macrophages have a central role in innate immunity, by functioning as a scavenger for pathogens and apoptotic cells as well as by coordinating inflammatory response through production of cytokines and inflammatory mediators. It has been found that LXR inhibits genes involved in innate immune response and stimulate those involved in lipid metabolism, serving as a link between these two pathways. Additionally, LXR in macrophages can be transcriptionally activated by oxidized lipoproteins. LXR then acts to promote cholesterol efflux (via ATP-binding cassette transporters ABCA1 and ABCG1) from the cell to prevent lipid overload and limit the production of inflammatory mediators (145, 146). Thus, LXR may function to integrate metabolic and immune signaling. Many of the genes inhibited by LXR are established targets of NF-κB signaling (e.g., iNOS, COX-2 (Cycloxygenase-2), MMP-9 (Matrix metallopeptidase 9) etc.) (145). This role may be important in atherogenesis. Oxidized lipids may act as inducers or inhibitors of inflammatory gene expression, depending upon the context—the inhibitory effects being mediated via LXR. Since the anti-inflammatory effect of LXR is not limited to promoting cholesterol efflux (which has a direct implication in atherosclerosis) but to decrease the inflammatory mediators as well, LXR agonists may have the utility in the treatment of other chronic inflammatory disease where macrophages play an important role (e.g., osteoarthritis, contact dermatitis, etc.).
Cholesterol is an important constituent of mammalian cell membranes and is a major constituent of myelin. It plays an important role in central nervous system (CNS) synapto-genesis and is essential for optimal neurotransmitter release. Most of the cholesterol is synthesized in situ, and to maintain homeostasis, cholesterol leaves CNS in the form of 24(S)-hydroxycholesterol, which is secreted across blood-brain barrier in high concentrations. It was found that patients with Alzheimer’s disease have high circulating levels of 24(S)-hydroxycholesterol, which might be a reflection of ongoing neurodegeneration. LXR, which has a close relationship with cholesterol metabolism, is expressed in the brain and is related to cholesterol efflux from CNS. Therefore, it has been suggested that LXR may have an implication in neurodegenerative diseases as well by modulating CNS cholesterol content, cholesterol efflux and inflammation. In fact, it was found that astrocytes treated with synthetic LXR ligands exhibit enhanced cholesterol efflux and increased expression of LXR target genes (ABCA1, ABCG1 and apoE) (147), whereas loss of the two isoforms of LXR in mice led to degenerative processes in brain characterized by enhanced lipid accumulation, astrocyte proliferation and disorganized myelination (148).
FARNESOID X RECEPTOR (FXR): IMPLICATING TREATMENT OPTIONS FOR HYPERTRIGLYCERIDEMIA
Farnesoid X Receptor is a nuclear hormone receptor encoded by the NR1H4 gene. It is similar in form to PPAR, LXR and RXR. When activated by its ligands, such as chenodeoxycholic acid, it translocates to the cell nucleus. One important function of FXR is to suppress the expression of cholesterol alpha hydroxylase (CYP7A1), which is a rate-limiting enzyme in bile acid synthesis (37).
Crystal structure of FXR LBD (~720 Å) has been reported in complex with coactivator peptide (GRIP-1) and two different bile acids. With the availability of FXR crystal structures, newer potent therapeutic bile acids and non-steroidal FXR modulators can be designed for the treatment of hyperlipidemia and cholestasis, where FXR plays a major regulatory role (149). FXR is highly expressed in the liver, gut, kidney, and adrenal cortex and at low levels in heart, lung, stomach, and adipose tissue.
FXR Functions and Therapeutic Implications
It has been found that bile acids repress the transcription of CYP7A1, which catalyzes the rate-limiting step in bile acid biosynthesis. It was shown that bile acids activate FXR, which leads to the expression of orphan receptor SHP-1. SHP-1 in turn inhibits LRH-1, which is another orphan receptor known to regulate CYP7A1 expression positively. Therefore, FXR plays an important role in the coordinated regulation of cholesterol and bile acid synthesis in FXR/SHP-1/LRH-1 cascade (150).
FXR is a good therapeutic target for cholesterol gall stone disease, and this effect is mediated by the FXRmediated expression of bile acid transporter ABCB11 and ABCB4 (151). A new class of FXR agonist, 1, 1-bis-phosphonate esters has been discovered that up-regulates the bile acid transporter intestinal bile acid binding protein (I-BABP), which, like other agonists, increases the degradation of HMG-CoA reductase leading to hypocholesterolemia in normal animals (37).
In another study, FXR has been found to negatively regulate serum HDL and apolipoprotein A-I (apoA-I) levels. Ligand-activated FXR directly binds to apoA-I promoter and decreases its expression (152). As serum levels of HDL and risk of coronary heart disease (CHD) are inversely correlated, potent FXR antagonists could be promising in the treatment of CHD by raising serum HDL levels.
Syndecan-1 (SDC1) is a member of the family of transmembrane heparan sulfate proteoglycans, which are widely expressed in many cell types and tissues and has a significant contribution to the lipoprotein metabolism. Their principal function is to modulate the ligand-dependent activation of primary signaling receptors at the cell surface, leading to an increase in binding and/or internalization of extracellular ligands. It is highly expressed in the liver. In liver heparan sulfate proteoglycans (HSPGs) bind to lipoproteins via several accessory proteins, such as the lipoprotein-lipase, apoE, and hepatic lipase, which are crucial for binding and sequestering of lipoprotein remnants before transfer to specific receptors (such as the LDL receptor) with subsequent endocytosis. It has been found that hepatic SDC1 is induced in a FXR-dependent manner. Therefore, increased expression of SDC1 may be one mechanism by which administration of chenodeoxycholic acid (naturally occurring FXR ligand) and synthetic FXR ligand GW4064 leads to hypotriglyceridemic effect (153).
The plant sterol guggulsterone, which has been used to treat hyperlipidemia, has been found to be an antagonist of FXR and to decrease expression of FXR target genes (154). Although acting as an antagonist, it enhances FXR agonist-induced transcription of bile salt export pump (BSEP), which is a major hepatic bile acid transporter. It has been proposed that it mediates its effects via SHP, a known FXR target. Therefore, guggulsterone defines a novel class of FXR ligands characterized by antagonistic activities by coactivator association assays but at the same time can enhance the action of agonists on BSEP expression in vivo.
Human kininogen belongs to plasma kallikrein-kinin system. High molecular weight kininogen is the precursor of two-chain kinin-free kininogen and bradykinin. The former has properties of anti-adhesion, anti-platelet aggregation, anti-thrombosis, while the latter is a potent vasodilator and mediator of inflammation. It has been found that human kininogen gene is strongly up-regulated by agonists of the FXR. FXR response element (inverse repeat, IR-1) was also found in the promoter of kininogen at −66/−54, where FXR-RXR heterodimer binds. Hence, it means that FXR and its agonists (e.g., bile acids) may play a role in vasodilatation and anti-coagulation process (155).
FXR directly regulates the expression of FGF-19 (FGF-15, mouse ortholog), which is a member of the fibroblast growth factor family of signaling molecules (156). FGFs bind to the extracellular domain of their cognate cell surface receptor (FGFRs) and induce receptor dimerization and tyrosine kinase phosphorylation, which, in turn, leads to the activation of a number of intracellular pathways. This FXR/FGF-19 pathway is also involved in the bile acid homeostasis. In mice lacking apical ileal bile acid transporter (Asbt −/− mice, animal model of bile acid malabsorption), it was found that the FXR/FGF-15 pathway is disrupted. With the treatment of Asbt −/− mice with either FXR agonist or FGF-15 peptide, CYP7A1 expression becomes less. This suggests that FXR agonist (and/or FGF-15 peptide) could be used in the treatment of patients of bile acid malabsorption to decrease excessive bile acid synthesis (157). It is understood that the FGFs regulate cell growth, differentiation, and morphogenesis; however, it is now apparent that some of these proteins are also important components of specific homeostatic pathways (e.g., bile acid homeostasis). It means that FXR has more implications than previously thought which expands its importance in drug discovery targeting FXR/FGF-19 pathway. FXR has also been shown to play a role in tumor formation. It was also observed that FXR deficiency results in increased colon cell proliferation and tumorigenesis, which is accompanied by the up-regulation of the genes involved in cell-cycle progression and inflammation (e.g. cyclin D1 and interleukin-6) (158). Additionally, in breast cancer cells, FXR plays a pro-apoptotic role by regulating the expression of genes involved in the transport of bile acids, amino acids and xenobiotics (159). Hence, activation of FXR could be a novel intervention strategy for the protection of intestinal and breast carcinogenesis.
FXR also regulates the expression of various transport proteins and biosynthetic enzymes crucial to the physiological maintenance of lipids, cholesterol and bile acid homeostasis (38).
PEROXISOMAL PROLIFERATOR-ACTIVATED RECEPTOR (PPAR): DRUG TREATMENT FOR DIABETES, LIPID DISORDERS, ACNE AND INFLAMMATION
PPAR is another class of ONR which has been very well studied. It was initially described in Xenopus frogs, where it leads to the proliferation of peroxisomes (160). PPAR was shown to increase peroxisomes in the rodent liver apart Orphan Nuclear Receptor and Drug Development 1451 from improving insulin sensitivity; hence, they were called peroxisome proliferator activator receptors. There are three types of PPAR described so far: PPARα, γ and δ/β, the latter two being closely related (161).
X-ray crystal structures of the PPARα, γ and δ ligand binding domains (LBDs) have revealed that the receptors contain a much larger ligand binding pocket (~1,300 Å) compared to other NRs (162–165).
The size of this pocket may explain the ability of the PPARs to bind a variety of naturally occurring and synthetic lipophilic acids. Known endogenous ligands for PPARs are free fatty acids and eicosanoids (166). Leukotrienes B4 is specific for PPARα, while prostaglandin J2 (PG-J2) is specific for PPARγ (167). Each class of PPAR has its own modulators targeting their functions. PPARα is targeted by the fibrates, such as clofibrate, fenofibrates, gemfibrozil, etc., which are used in the treatment of hyperlipidemias. PPARγ modulators include thiazolidinediones (TZD), which are used as anti-diabetic drugs, although some interactions with NSAIDs have been demonstrated as well (168–170). PPARδ modulators, still in the experimental stage, include GW501516 (171). A new experimental class of PPAR modulators that affect more than one class of PPARs includes muraglitazar and tesaglitazar, which are being developed for metabolic syndromes (172). PPARα is distributed mainly in the liver, kidney, heart, muscle, and adipose tissue. PPARδ/β are mainly expressed in the brain, adipose tissue and skin. PPARγ has a very wide distribution in the body and is found in three forms formed by alternate splicing in the same gene. PPARγ1 is expressed in heart, muscle, kidney, colon, pancreas and spleen, while PPARγ2 is expressed mainly in adipose tissue, and PPARγ3 is expressed in macrophages and adipose tissue (161).
PPAR Functions and Therapeutic Implications
Cannabinoids (CB) are a group of terpenophenolic compound present naturally in nervous and immune systems of mammals (173). CB activate G-protein coupled receptor CB1 (cannabinoid receptor) to inhibit calcium-induced neurotoxicity (174). Studies in CB receptor knock-out mice have revealed non-CB receptor-mediated responses both in CNS and periphery (175). These non-CB responses are shown to be mediated via PPARs (176). The monounsaturated analog of the endocannabinoid anandamide oleoylethanolamide regulates feeding and body weight, stimulates fat utilization and has neuroprotective effects mediated through activation of PPARα. Other CBs, like palmitoylethanolamide, anandamide, virodhamine and noladin, also act via PPARα to regulate lipid metabolism. Few (anandamide and 2-arachidonoylglycerol) act on PPARγ to exert anti-inflammatory activities. This opens a new domain of application of PPARs as a target of naturally occurring CB. This also supports the importance of PPARs as a target for lipid disorders and also for neuroprotective and cardioprotective tretaments (177).
PPARγ agonist TZDs are used clinically to treat insulin resistance and diabetes, disease conditions strongly associated with obesity (178). It is believed that elevated fatty acids produced by adipose tissues promote insulin resistance, resulting in increased hepatic gluconeogenesis and decreased glucose utilization in the periphery (179). TZDs induce the expression of genes involved in adipocyte differentiation and lipogenesis through PPARγ activation, and these mechanisms are responsible for the insulin-sensitizing actions of these drugs in the treatment of type II diabetes (180, 181). Additional mechanisms of TZD action in diabetes have also been discovered, where TZDs target adipocyte gluconeogenesis (182). Gluconeogenesis is a process, where glycerol-3-phosphate is produced from precursors other than glycerol or glucose in adipose tissue when glucose utilization is reduced during fasting. Hence, gluconeogenesis in adipocytes acts as a fatty acid homeostatic pathway that allows the re-esterification of fatty acids (FAs) from glycerol-3-phosphate for triacylglycerol synthesis at the time when their breakdown is occurring through lipolysis. This action therefore prevents the release of FAs into the blood. Phosphoenol pyruvate carboxy kinase enzyme (PEPCK) is the central regulator of gluconeogenesis and is being closely coupled with cytosolic aspartate transaminase (cAspAT) in the liver. It was found that TZD responsiveness of cAspAT is dependent on PPARγ and protein synthesis. This effect of TZD on PEPCK and cAspAT leads to hypolipidemia by inducing gluconeogenesis, which is a novel mechanism of anti-diabetic drug therapy by TZDs (179, 182).
Recently, many NRs expressed in skeletal muscle have been shown to improve glucose tolerance, insulin resistance, and dyslipidemia. Skeletal muscle and NRs are rapidly emerging as critical targets in the battle against cardiovascular disease risk factors. It has also been found that estrogen receptor-related receptor (ERR) stimulates PPARα-mediated energy metabolism in cardiac and skeletal muscle cells via activation of medium chain acyl-CoA dehydrogenase (183). Understanding the function of NRs in skeletal muscle has enormous pharmacological utility for the treatment of cardiovascular disease and for obesity (184).
Activation of orphan receptors, in particular activation of PPARα and PPARγ, can regulate lipogenesis in human sebaceous glands. PPARγ activation induces COX-2 expression in sebocytes, which leads to sebocyte proliferation and/or lipogenesis (185). As suppression of sebum secretion is associated with reduced acne activity, the NRs involved may open new avenues in the development of novel acne treatments.
Both PPARα and PPARγ receptor subtypes have been reported to regulate inflammatory responses, both in vivo and in vitro. Leukotriene B4 (LTB4) is an endogenous ligand for PPARα, which leads to transcription of genes of the ω- and β-oxidation pathways that can catabolize LTB4 itself. Activation of PPARα by NSAIDs contributes to the anti-inflammatory, antipyretic, and analgesic properties of these drugs through stimulation of oxidative pathways involved in the catabolism of eicosanoids. PPARγ regulates the activity of iNOS, and activation of this PPAR subtype controls inflammation by diminishing nitric oxide production. PPARγ has shown involvement in other inflammatory pathways as well. This provides a new avenue to develop treatment for autoimmune diseases, which has not been well explored so far (186).
All-trans retinoic acid (ATRA) leads to activation of apoptotic pathways via its interaction with RAR/RXR dimers. But it was also found to bind to PPARβ/δ with nanomolar affinity, modulating the conformation of the receptor, promoting interaction with the coactivator SRC-1, and efficiently activating PPARβ/δ-mediated transcription, leading to activation of growth promoting and antiapoptotic pathways. While the current use of retinoids as chemotherapeutic agents is due to its growth-inhibitory effects mediated via RAR, it can also promote growth via activation of PPARβ/δ (187).
PPARδ has a wide expression pattern in adult animal and is expressed very early during embryogenesis (188). PPARδ gene disruption is lethal due to placental defect. The surviving knock-out mice are smaller than their control, and they have reduced body fat mass, skin defects and alterations in myelinisation (189).
PPARδ also plays important role in lipid absorption in the intestine by directly regulating genes involved in lipid uptake, such as fatty acid binding protein and fatty acid translocase (190). Furthermore, activation of PPARδ has been shown to regulate lipid metabolism by activating fatty acid oxidation (191). Consistent with this role, it was observed that PPARδ expression increases in muscle during physical exercise with subsequent increase in fatty acid burning (192). Hence, PPARδ agonist treatment could have therapeutic usefulness in metabolic syndromes to increase insulin sensitivity and obesity (193).
Human immunodeficiency virus long terminal repeat (HIV-1 LTR)-driven transcription is regulated by NR-responsive element (NRRE) located in its promoter. This NRRE contains tightly clustered binding sites for RXRα, RARα, apolipoprotein AI regulatory protein, HNF-4, NGFI-B and PPAR. These findings suggest that a complex network of NR signaling pathways that include 9-cis- and all-trans-retinoic acid, fatty acids, peroxisome proliferators, growth factors, membrane depolarization, and possibly other signals, converge onto the HIV-1 NRRE and may participate in modulation of viral gene expression (194). PPARγ activation has also been shown to suppress HIV-1 replication in an animal model of encephalitis, further implicating the role of PPARs in anti-viral drug treatment (195).
NURR1: ROLE IN DOPAMINERGIC DYSFUNCTION AND SYNOVIAL INFLAMMATION
NURR1 belongs to the NR4A subfamily of ONRs and is expressed predominantly in the CNS, especially in the substantia nigra, the ventral tegmental area, the midbrain and limbic areas (196, 197). Recent reports have indicated its essential role in the development and survival of dopaminergic neurons (198). The purine anti-metabolite 6-mercaptopurine is reported to act as an agonist of NURR1, and it activates NURR1 transcription (199). Crystal structure of the NURR1 LBD has been reported and shows its two distinctive features. First, the NURR1 lacks a classic ligand binding pocket because of the tight packing of side chains. Second, NURR1 lacks a classical binding site for coactivators. Despite these features, the structure shows that NURR1 can be transcriptionally activated in a ligand-independent fashion (200).
Biological Importance of NURR1 as a Drug Target
Studies with NURR1 knock-out mice have shown that NURR1 deficiency results in impaired dopaminergic activity and apoptosis in midbrain dopaminergic neurons, which degenerate in Parkinson’s disease (PD). Mutations in the gene encoding NURR1 and decreased NURR1 expression have been associated with disorders related to dopaminergic dysfunction, including PD, schizophrenia and manic depression (201, 202). Therefore, selective NURR1 agonist with high potency could be exploited for the prevention and treatment of PD.
Additionally, NURR1 is found to be expressed in inflamed synovial tissue. It was shown that enhanced binding of NF-κB and cAMP response element binding protein (CREB) to NURR1 promoter by inflammatory mediators (e. g., IL-1β, TNF-α, PG-E2) increases local production of NURR1 in the inflamed joints. NURR1 in turn acts as the mediator of an autocrine regulatory inflammatory cascade to amplify the inflammatory response by increasing synovial corticotrophin-releasing hormone (CRH) expression (203, 204). Hence, NURR1 transcriptional regulation can be selectively modulated using antagonist compounds for the prevention of inflammatory joint diseases.
NUR77: CANCER TREATMENT OPTIONS
Another member of the NR4A superfamily of ONRs, nerve growth factor IB (NGFIB or Nur77), is involved in many Orphan Nuclear Receptor and Drug Development 1453 biological processes, such as cell-cycle regulation, apoptosis and inflammation (205). However, a physiological ligand for Nur77 has not been identified. The octaketide cyto-sporone B (Csn-B) has been found to be a naturally occurring agonist for Nur77. Csn-B specifically binds to the LBD of Nur77 (modeling is based on the crystal structure of Nur77, PDB code 2QW4) and stimulates Nur77-dependent transactivation (206).
Csn-B has been found to elevate blood glucose levels in fasting mice by inducing multiple genes involved in gluconeogenesis pathway. These biological effects were not observed in Nur77-null mice, indicating that Csn-B regulates gluconeogenesis through Nur77 (206).
Nur77 has been shown to induce cytochrome c release and apoptosis through interaction with anti-apoptotic protein Bcl-2. Nur77 binding to Bcl-2 induces a conformational change in Bcl-2, resulting in conversion of Bcl-2 from a protector (anti-apoptotic) to killer (apoptotic) protein (207, 208). This suggests that Nur77 could be a new target for novel therapeutic applications for cancer.
Very recently it was also shown that a short Nurr77-derived peptide NuBCP-9 has a protective role in drug-resistant breast tumors (209). Novel compounds with Nur77 agonistic activity, such as 1, 1-bis (3′-indolyl)-1-(phenyl) methane (DIM-C-Ph) and 1, 1-bis (3′-indolyl)-1-(p-anisyl) methane (DIM-C-pPhOCH3) have been discovered recently and have been shown to act as anti-colon-cancer drugs (210).
TLX (HUMAN HOMOLOG OF DROSOPHILA TAILLESS GENE): INVOLVEMENT IN NEURODEGENERATION AND RETINAL DEGENERATION
Orphan receptor TLX, encoded by the NR2E1 gene, is specifically expressed in the brain and has an important role in vertebrate brain and eye functions (211–213). It is the human homolog of Drosophila tailless gene. It plays an essential role in the maintenance of neural stem-cell proliferation and self-renewal in the adult mouse brain (214, 215). Additionally, TLX is a key component of retinal development and is essential for vision (216). Though the crystal structure of TLX is not available to date, identification of endogenous and synthetic TLX ligands are under investigation using novel affinity/GC-MS technology, chemical screening and cell-based assays.
Targeting TLX for Neuro- and Retino-degenerative Therapeutics
TLX is important for the formation of superficial cortical layers in embryonic brains, regulation of neurogenesis and patterning of lateral telencephalic progenitor domains during development. TLX-expressing neural cells can proliferate, self-renew and differentiate into all neural cell types, whereas TLX-null neural cells show a significant reduction of cell proliferation (217). This shows the importance of TLX in neural development (214).
TLX is also found to be expressed in retinal progenitor cells in the neuroblastic layer during the period of retinal layer formation and is crucial for controlling the generation of appropriate numbers of retinal progenies. The TLX knock-out neural retinas were significantly thinner than controls (218). It is well known that malformations in the eye can be caused by either an excess or deficiency of retinoids, by regulating the expression of its early target gene, retinoic acid receptor β (RARβ). A TLX response element has been identified in RARβ promoter, which is important for retinoic acid-mediated induction of RARβ. These results show an important role for TLX in autologous regulation of the RARβ gene in the eye, critical for its development (219). TLX-Pax2 (paired box homeodomain transcription factor) regulatory network has also been identified as involved in vertebrate eye development. It has been shown that TLX by binding to Pax2 gene promoter represses its expression, which is essential for retinal development and vision (216).
RETINOIC ACID RECEPTOR-RELATED ORPHAN RECEPTOR (ROR): MAINTENANCE OF CHOLESTEROL HOMEOSTASIS
ROR is a member of NR1F subfamily of ONRs. The RORs are somewhat unusual in that they appear to bind as monomers to hormone response elements as opposed to the majority of other NRs which bind as dimers (1, 220). There are three subtypes of ROR: RORα, β and γ. RORα is expressed in specific areas of the brain, including purkinje cells in the cerebellum and the suprachiasmatic nucleus of the hypothalamus. It is also expressed in the spleen, thymus and macrophages (220, 221).
Crystal structure of RORα has been solved in complex with cholesterol-3-O-sulphate, suggesting that cholesterol sulphate could regulate RORα functions in vivo (222). Despite the high homology between RORα and RORβ LBD, cholesterol is not a ligand of RORβ; instead, RORβ LBD has been shown to bind all-trans retinoic acid (ATRA) in crystal structure (223). ATRA has been shown to work as an antagonist for the constitutively active RORβ.
ROR Disease Associations
RORα knock-out mice, which show a stagger phenotype, exhibit ataxia resulting from neurodegeneration in the cerebellum involving the purkinje cells (224). Furthermore, the staggerer mice display lowered plasma apoAI/II levels, decreased plasma HDL cholesterol and triglycerides, and develop hypo-α-lipoproteinemia and atherosclerosis (225).
It has been shown that the muscle carnitine palmitoyltransferase-1 and caveolin-3 promoters are directly regulated by RORα, implying that RORα could play an important role in the control of lipid homeostasis in skeletal muscle (226). It has also been reported that cholesterol and its sulfonated derivatives might function as RORα ligands. Additionally, it was shown that oxysterol 7α-hydroxylase (CYP7B1), which plays a critical role in cholesterol homeostasis, is a RORα target gene. Studies in RORα- and LXR-deficient mice have revealed an interesting functional crosstalk between them in endobiotic metabolic gene regulation (227). These data suggest that RORα could have an important role in the maintenance of cholesterol homeostasis and thus could be used for the treatment of cholesterol-related diseases (222, 228).
RORβ is expressed in areas of CNS that are involved in the processing of sensory information and the circadian rhythm. Therefore, it could be possible that target genes of RORβ play an important role in sensory input integration and maintenance of biological clock. Besides, RORβ knock-out mice display a duck-like gait, disrupted reproduction in males, blindness and abnormal circadian rhythm (229, 230). These observations suggest that RORβ ligands can become very useful for RORβ-related CNS disorders.
In contrast to other ROR genes, RORγ is not expressed in the CNS. Instead, RORγ is found at high levels in skeletal muscle and thymocytes. RORγ knock-out mice lack peripheral and mesenteric lymph nodes and peyer’s patches (231). Though the functional role of RORγ is not fully characterized, its role in lymphoid organogenesis could be exploited for thymopoiesis and the maintenance of T cell homeostasis.
DOSAGE-SENSITIVE SEX REVERSAL-ADRENAL HYPOPLASIA CONGENITA CRITICAL REGION ON THE X CHROMOSOME GENE 1 (DAX-1): BONE DEVELOPMENT AND GLUCOCORTICOID RECEPTOR MODULATOR
DAX-1 is a member of the NR0B subfamily of ONRs. It acts by inhibiting the activity of other NRs, such as steroidogenic factor 1 (SF-1), estrogen receptor and androgen receptor by heterodimerization, thereby acting as a negative regulator of steroidogenesis. It is involved in controlling the development of the hypothalamic-pituitary axis, as well as in gonadal development and sex determination (232). DAX-1 is expressed primarily in reproductive tissues (ovary, testis, and uterus), endocrine tissues (adrenal gland) and the CNS (pituitary and hypothalamus) (232). While the crystal structure of DAX-1 is unavailable, DAX-1 LBD homology model have been constructed to get an idea about the position of mutations and deletions in DAX-1 gene that are responsible for gonadal dysfunction (233). It was found that missense mutations and codon deletion in DAX-1 all mapped to the predicted hydrophobic core of its LBD (233, 234).
DAX-1 is involved in various disease states, such as X-linked adrenal hypoplasia congenita (AHC), which is caused by mutations in the NR0B1 gene. More than 90 NR0B1 mutations that cause AHC have been identified, and several of these mutations delete all or part of the NR0B1 gene, preventing the production of DAX-1 protein. Loss of DAX-1 function leads to adrenal insufficiency and hypogonadotropic hypogonadism, which are the main characteristics of this disorder (235). DAX-1 knock-out mice are associated with delayed testis development and male sterility.
Additionally, DAX-1 expression has been detected in totipotent murine embryonic stem cells, which suggests an important function of DAX-1 in early embryonic development, which is independent of its role in steroidogenesis (236).
DAX-1 expression was found to increase with osteoblast differentiation and in a variety of tumor tissues (e.g. adrenal and pituitary adenomas, breast, ovarian and prostate cancer), implicating its potential role in bone cell development and malignancy (237).
Another important aspect of DAX-1 function is that it has been shown to physically interact with glucocorticoid receptor (GR) and thus acts as a novel selective GR modulator. It specifically inhibits ligand-dependent GR transactivation with little effect on GR-mediated transrepression. Clinically, glucocorticoids are extensively prescribed for their anti-inflammatory or immune-suppressive effects (i.e. transrepression). However, long-term use of steroids is often associated with a wide range of adverse effects. It is well known that GR-mediated transrepression of target genes, particularly pro-inflammatory cytokines and cytokine receptors are responsible for the beneficial effects of glucocorticoids in preventing inflammation, whereas the side effects are associated with GR-mediated transactivation. Therefore, selective GR modulators, like DAX-1 that enhances or has no effect on GR-mediated transrepression but reduce transactivation, are expected to have great a therapeutic value due to improved benefit-torisk ratio in various inflammatory conditions (238).
SMALL HETERODIMER PARTNER (SHP): DRUG TREATMENT FOR OBESITY
SHP belongs to the NR0B subfamily of ONRs. It is expressed primarily in endocrine organs (adrenal, pancreas), gastrointestinal organs (stomach, duodenum, ileum, colon and gall bladder), metabolic organs (liver, kidney), Orphan Nuclear Receptor and Drug Development 1455 reproductive organs (ovary and testis), cardiopulmonary organs (heart and lung), and CNS (cerebrum) (239). No crystal structure of SHP has been determined yet.
SHP dysfunction is associated with mild early-onset obesity, and early-onset type II diabetes (240, 241). Loss of SHP in mice causes abnormal accumulation and increased synthesis of bile acids due to de-repression of rate-limiting CYP7A1 and CYP8B1 hydroxylase enzymes in the bile acid biosynthetic pathway (242, 243).
SHP has also been shown to act as a transcriptional inhibitor of adipogenesis through inhibition of adipogenic transcription factors C/EBP alpha and PPARγ2 and other adipogenic stimulators Ebf3 and Stat5a (244). This property of SHP could be exploited (SHP agonists) for the treatment for obesity.
STEROIDOGENIC FACTOR 1 (SF-1): ROLE IN PROSTATE CANCER AND ADRENOCORTICAL TUMOR FORMATION
SF-1 is a member of the NR5A subfamily of ONR transcription factors that is essential for the development of adrenals and gonads and plays a role in sexual development. SF-1 is expressed primarily in the adrenal gland, hypothalamus, ovary and testes and acts by regulating the secretion of steroid hormones (245, 246). Crystal structure of SF-1 LBD has been solved in complex with non-bacterial phospholipids, and these phospholipids were shown to readily exchange bacterial phospholipids fortuitously bound to SF-1 ligand binding pocket (247–250).
SF-1 knock-out mice develop loss of pituitary gonadotrope function and fail to develop adrenal gland and gonads (245). Abnormalities in SF-1 function have been implicated in adrenocortical insufficiency, sex reversal, cryptorchidism, insulin resistance and type II diabetes (251, 252). Tandem Mass spectrometry and cell-based receptor selection and amplification technology (R-SAT) assays have identified sphingosine and 4-(heptyloxy) phenol (AC45594) as negative regulators of SF-1 activity, respectively (248, 253, 254). These properties of SF-1 modulators can be exploited to suppress both adrenal androgen and gonadal testosterone synthesis in the treatment of prostate cancer and in adrenocortical tumors (255, 256).
ESTROGEN RECEPTOR-RELATED RECEPTOR (ERR): TREATMENT OPTIONS FOR POSTMENOPAUSAL OSTEOPOROSIS, BREAST TUMOR AND TYPE II DIABETES
ERR is a member of the NR3B orphan subfamily of the NR superfamily of transcription factors. While ERR is structurally homologous to estrogen receptors and binds estrogen response elements, it is not activated by estrogens. It functions as a metabolic regulator by modulating the expression of enzymes involved in adipogenesis, energy metabolism, and lipid, eicosanoid, and steroid synthesis (257). It has three subtypes: ERRα, ERRβ and ERRγ. ERRα is expressed robustly in tissues in all major physiological systems (CNS, endocrine, metabolic, gastrointestinal, immune, reproductive, cardiovascular and respiratory) with particularly high levels in the olfactory bulb, jejunum, ileum, kidney, brown adipose tissue, heart and skeletal muscle. ERRβ is expressed specifically in the placenta, and ERRγ is expressed in brain, kidney, testis, lung, adrenal gland, pancreas, placenta and bone marrow (258–261).
Crystal structure of apo ERRα has revealed that it has a very small ligand binding pocket (~100Å) and shows constitutive activity (222, 262). Searches for ERRα ligand have mostly identified inverse agonists, namely 4-hydroxytamoxifen and diethylstilbestrol (263). Crystal structures of the ERRγ LBD were determined in three distinct states: unliganded, inverse agonist bound (with 4-hydroxytamoxifen), and agonist bound (with GSK4716) (264). For ERRβ, also, crystal structure of ERRβ LBD with 4-hydroxytamoxifen has been reported (265).
Disease Conditions and Possible Clinical Interventions
Postmenopausal osteoporosis is a condition where serum estrogen levels decrease, resulting in decreased bone mineralization. Estrogen receptors (ERs) are expressed in osteoblasts. Strikingly, mice lacking ER show only minor skeletal deformities, suggesting other mechanisms or receptors (additional to ER) are involved in this process. ERRα is more widely distributed in osteoblast and osteoblast-like cells than ERs. It has also been shown to positively regulate osteopontin gene (extracellular matrix molecule secreted by osteblast) expression and thereby regulate bone remodeling (266).
ERR transcriptionally regulates estrogen-responsive breast cancer marker pS2, and it has been shown that ERR-responsive element in pS2 promoter is required for both estrogen and ERR response on pS2 expression. Transcriptional response of pS2 is completely abolished by diethylstilbestrol (DES) treatment, which is an inhibitor of ERR function, showing that DES treatment completely abolishes both ER+ and ER− breast tumor growth through ERR pathway (267). Several reports have shown that diabetics as well as individuals with a family history of diabetes have reduced mitochondrial oxidative phosphorylation (OXPHOS) capacity in muscle. OXPHOS genes are downstream targets of the transcriptional coactivator, PGC-1α. ERRα is an early target gene of PGC-1α. When activated by PGC-1α, ERRα expression is induced in an autocrine loop leading to increased OXPHOS target gene expression, as both ERRα and PGC-1α (they directly interact with one another) bind to promoter of OXPHOS target genes. Thus, as an ONR, targeting of ERRα with small molecules is an attractive strategy to increase mitochondrial OXPHOS function in type II diabetic patients. Moreover, because ERRα is involved in the regulation of fatty acid β-oxidation, activating ERRα can ameliorate the lipid accumulation in skeletal muscle, which is believed to contribute to insulin resistance (268).
ERRβ is specifically expressed in the chorion, suggesting that ERRβ may play a role in early placental development. The ERRβ knockout embryos have severely impaired placental formation and die owing to a lack of nutrients. The synthetic estrogen diethylstilbestrol (DES) has been shown to act as an inverse agonist of ERRβ, thereby affecting normal placental development (269).
ERRγ has been shown to play an anti-proliferative role in both androgen-sensitive and -insensitive prostate cancer by directly inducing cyclin-dependent kinase inhibitors p21 and p27, which results in cell-cycle arrest at G1-S transition. Therefore, by selectively activating ERRγ by its synthetic agonist DY131, growth proliferation of prostate cancer cells can be prevented (270).
HEPATOCYTE NUCLEAR FACTOR 4 (HNF4): TARGET FOR ANTI-DIABETIC DRUG DISCOVERY
HNF4 is a member of the NR2A orphan subfamily of the NR transcription factors that is required for the development of liver (271, 272). Three subtypes of HNF4 are known: α, β and γ. Crystal structure of HNF4 LBD shows that it is structurally similar to other ONR LBD and can bind fatty acids (273). HNF4 is highly expressed in the gastrointestinal tract, liver, and kidney, with lower levels in adipose tissue and pancreas (274, 275).
While little is known about HNF4β and HNF4γ, HNF4α has been shown to constitutively bind fatty acids, and its dysfunction has been implicated in the development of maturity onset diabetes of the young (MODY) type I, adult onset type II diabetes mellitus, high serum lipid levels and chronic kidney failure (276). HNF4α knock-out mice show defects in embryonic, liver, biliary system and CNS development. It has been previously published that missense mutations of HNF4α LBD result in MODY-1, which can be rescued by fatty acid agonist activation of HNF4α (276). Hence, this property of HNF4α could be utilized for the treatment of MODY-1 patients by selective HNF4α agonist.
HNF4α also regulates coordinate nuclear-receptormediated response to xenobiotics and is involved in the PXR and CAR-mediated gene activation of CYP3A4, a drug-metabolizing enzyme with possible implications in drug metabolism (277).
GERM CELL NUCLEAR FACTOR (GCNF) AND LIVER RECEPTOR HOMOLOG (LRH-1)
GCNF (NR6A1) is a member of the NR6A subfamily of NR transcription factors and acts as a transcriptional repressor. It plays an important function in vertebrate embryogenesis and is highly expressed in oocytes and spermatogenic cells (278). In the absence of a GCNF crystal structure, very little is known about its ligand or any heterodimerization partner and cofactor.
It is predicted that GCNF regulates protamine gene expression in response to an unknown ligand, which is critical for testicular development. In the absence of a ligand, it is a repressor of transcription, and part of its repression is mediated by the corepressor N-CoR (279). Germ-cell-specific expression of GCNF, thereby regulating gametogenesis, could be exploited as a contraceptive target (280). The complex temporal and spatial expression pattern of GCNF suggests its involvement in different developmental processes in addition to its role in gametogenesis (281). Functional gene targeting studies have shown that GCNF knock-out mice are embryonic-lethal, and death is found to be due to severe cardiovascular and posterior developmental defects, suggesting its role in embryogenesis (282).
LRH-1 is a member of the NR5A subfamily of ONRs. It is expressed in liver, intestine and pancreas, where it is involved in bile acid metabolism, cholesterol homeostasis and liver development. LRH-1 transcriptionally up-regulates ATP binding cassette half-transporters ABCG-5 and ABCG-8 to facilitate biliary and intestinal removal of neutral sterols (283). It is also expressed in the preadipocytes, adrenal and sex glands including ovaries and placenta (284). Crystal structure of LRH-1 LBD has shown that it can exist in active conformation as a monomer without a ligand (285). But later findings from different groups have shown that ligand binding pocket of LRH-1 can be occupied by phospholipids (247, 248).
This receptor plays a role in cell proliferation by inducing the expression of cyclins D1 and E1 through both direct and indirect interactions with β-catenin (286). LRH-1 also regulates the transcription of genes responsible for hormone synthesis, including the expression of the estrogen-synthesis enzyme CYP19 in the pre-adipocytes of cancerous breast tissue (287).
LRH-1 has also been identified to play a role in colon cancer. It was shown that LRH-1 synergizes with β-catenin/T-cell factor 4 signaling to stimulate intestinal crypt cell renewal. LRH-1 happloinsifficiency thereby blunts intestinal tumorigenesis in APCmin/+ mouse and chemical carcinogen (azoxymethane) models of intestinal cancer (288). LRH-1 has also been shown to promote breast tumorigenesis by activating aromatase II promoter (CYP19 gene) activity. Hence, selective inhibition of LRH-1 Orphan Nuclear Receptor and Drug Development 1457 may represent a novel strategy for the discovery of selective aromatase modulators (289).
Additionally, it has been observed that LRH-1 has a protective role in mouse models of inflammatory bowel disease by triggering local production of glucocorticoids (290). Structural and functional studies have shown that phospholipids bind to LRH-1 and regulate its downstream transcriptional events. GSK8470, a small-molecule high-affinity ligand for LRH-1, and SF-1 have been discovered by FRET-based biochemical assays to increase LRH-1 target gene SHP expression (113).
It has been found that both GCNF and LRH-1 compete for the same Oct-4 (member of POU homeodomain family of transcription factors) promoter elements during embryonic cell (ES) differentiation. LRH-1 induces expression of Oct-4 for the induction of ES cell differentiation, whereas GCNF represses Oct-4 to maintain the pluripotency of ES cells. Hence, agonist for LRH-1 and antagonist for GCNF could be used to maintain ES cell pluripotency and selfr-enewal for large scale culture of ES cells; similarly, LRH-1 antagonist and GCNF agonist could be used to silence pluripotency genes like Oct-4 for differentiation of ES cells to target cells for therapeutic purposes (291).
PHOTORECEPTOR-SPECIFIC NUCLEAR RECEPTOR (PNR): INVOLVEMENT IN AGE-RELATED MACULAR DEGENERATION (AMD)
PNR (NR2E3) is a member of the NR2E subfamily of ONRs which plays a role in retinal differentiation and degeneration. It is exclusively expressed in the rod photoreceptor cells of retina (not expressed in other tissues, including brain) (292). PNR gene was first identified through a search for genes related to TLX, which is involved in the development of midbrain and eye. In vitro assays have shown that PNR is capable of binding to a subset of TLX target sequences. TLX gene knock-out experiments in mouse revealed specific defects in forebrain derivatives; however, other regions, such as eye, develop normally, thus implicating compensation of loss of TLX function by other proteins in mice (211). Analysis of human PNR gene at the gene locus 15q24, a region susceptible for retinal degeneration, has supported a role for this receptor in retinal cell function (292, 293). Mutation of PNR gene gives rise to retinal degeneration in mice that resulted in abnormal development of rods and cones, enhanced S-cone and Goldmann-favre syndromes (inherited vitreoretinal dystrophy) (294, 295). The rd7 mice containing a sporadic deletion in PNR gene have abnormal development of rods and cones, leading to the development of age-related macular degeneration (AMD), the leading cause of blindness world-wide (296). Similarly, PNR gene mutations in humans have been correlated with various retinal diseases.
Based on the evidence regarding the involvement of PNR in retinal degeneration, agonist of PNR has become an attractive drug target for the treatment and prevention of AMD. It is noteworthy that 13-cis-retinoic acid, a receptor in retinal epithelium, has been found to be a weak PNR agonist in cell-based assays. While in the absence of a crystal structure of PNR LBD, recent high throughput screening has identified a new class of PNR agonist based on 2-phenylbenzimidazole core. This identification thus opens the door for future research into the development of PNR ligands (297).
CONCLUSIONS
ONRs are a group of important biological molecules, whose functions can be modified by the application of potent receptor-specific agonist and antagonist to regulate diverse biological processes. Discoveries of various ligands to these receptors will help us to modulate various important physiological processes for therapeutic purposes for various human disease states. Thus, drug targeting of these ONRs is an important and viable means of treatment for various human disorders, and this will act as a challenge for better development of therapeutics to the pharmacological and pharmaceutical research.
ACKNOWLEDGEMENTS
This work was supported by the Damon Runyon Clinical Investigator Award (CI015-02), 1R01CA127231-01 (NCI) and Miriam Mandel Cancer Research Scholar Fund, Albert Einstein College of Medicine, Bronx, NY. We sincerely apologize to those, whose works have not been mentioned due to space limitations.
REFERENCES
- 1.Giguere V. Orphan nuclear receptors: from gene to function. Endocr Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- 2.Laudet V. Evolution of the nuclear receptor superfamily: early diversification from an ancestral orphan receptor. J Mol Endocrino. 1997;19:207–226. doi: 10.1677/jme.0.0190207. [DOI] [PubMed] [Google Scholar]
- 3.Jensen EV. Estrogen receptor: ambiguities in the use of this term. Science. 1968;159:1261. [PubMed] [Google Scholar]
- 4.Horwitz KB, Jackson TA, Bain DL, Richer JK, Takimoto GS, Tung L. Nuclear receptor coactivators and corepressors. Mol Endocrinol. 1996;10:1167–1177. doi: 10.1210/mend.10.10.9121485. [DOI] [PubMed] [Google Scholar]
- 5.McKenna NJ, Lanz RB, O’Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 6.Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, et al. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373–380. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 7.Hashimoto Y, Miyachi H. Nuclear receptor antagonists designed based on the helix-folding inhibition hypothesis. Bioorg Med Chem. 2005;13:5080–5093. doi: 10.1016/j.bmc.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 8.Pascual G, Glass CK. Nuclear receptors versus inflammation: mechanisms of transrepression. Trends Endocrinol Metab. 2006;17:321–327. doi: 10.1016/j.tem.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bjornstrom L, Sjoberg M. Estrogen receptor-dependent activation of AP-1 via non-genomic signalling. Nucl Recept. 2004;2:3. doi: 10.1186/1478-1336-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zivadinovic D, Gametchu B, Watson CS. Membrane estrogen receptor-alpha levels in MCF-7 breast cancer cells predict cAMP and proliferation responses. Breast Cancer Res. 2005;7:R101–R112. doi: 10.1186/bcr958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olefsky JM. Nuclear receptor minireview series. J Biol Chem. 2001;276:36863–36864. doi: 10.1074/jbc.R100047200. [DOI] [PubMed] [Google Scholar]
- 13.Umesono K, Murakami KK, Thompson CC, Evans RM. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell. 1991;65:1255–1266. doi: 10.1016/0092-8674(91)90020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naar AM, Boutin JM, Lipkin SM, Yu VC, Holloway JM, Glass CK, et al. The orientation and spacing of core DNA-binding motifs dictate selective transcriptional responses to three nuclear receptors. Cell. 1991;65:1267–1279. doi: 10.1016/0092-8674(91)90021-p. [DOI] [PubMed] [Google Scholar]
- 15.Benoit G, Cooney A, Giguere V, Ingraham H, Lazar M, Muscat G, et al. International Union of Pharmacology. LXVI. Orphan nuclear receptors. Pharmacol Rev. 2006;58:798–836. doi: 10.1124/pr.58.4.10. [DOI] [PubMed] [Google Scholar]
- 16.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, et al. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 17.Watkins RE, Noble SM, Redinbo MR. Structural insights into the promiscuity and function of the human pregnane X receptor. Curr Opin Drug Discov Dev. 2002;5:150–158. [PubMed] [Google Scholar]
- 18.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–828. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 19.Zanaria E, Muscatelli F, Bardoni B, Strom TM, Guioli S, Guo W, et al. An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita. Nature. 1994;372:635–641. doi: 10.1038/372635a0. [DOI] [PubMed] [Google Scholar]
- 20.Seol W, Choi HS, Moore DD. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science. 1996;272:1336–1339. doi: 10.1126/science.272.5266.1336. [DOI] [PubMed] [Google Scholar]
- 21.Oro AE, Ong ES, Margolis JS, Posakony JW, McKeown M, Evans RM. The Drosophila gene knirps-related is a member of the steroid-receptor gene superfamily. Nature. 1988;336:493–496. doi: 10.1038/336493a0. [DOI] [PubMed] [Google Scholar]
- 22.Nauber U, Pankratz MJ, Kienlin A, Seifert E, Klemm U, Jackle H. Abdominal segmentation of the Drosophila embryo requires a hormone receptor-like protein encoded by the gap gene knirps. Nature. 1988;336:489–492. doi: 10.1038/336489a0. [DOI] [PubMed] [Google Scholar]
- 23.Perlmann T, Jansson L. A novel pathway for vitamin A signaling mediated by RXR heterodimerization with NGFI-B and NURR1. Genes Dev. 1995;9:769–782. doi: 10.1101/gad.9.7.769. [DOI] [PubMed] [Google Scholar]
- 24.Gu P, Morgan DH, Sattar M, Xu X, Wagner R, Raviscioni M, et al. Evolutionary trace-based peptides identify a novel asymmetric interaction that mediates oligomerization in nuclear receptors. J Biol Chem. 2005;280:31818–31829. doi: 10.1074/jbc.M501924200. [DOI] [PubMed] [Google Scholar]
- 25.Wilson TE, Fahrner TJ, Milbrandt J. The orphan receptors NGFI-B and steroidogenic factor 1 establish monomer binding as a third paradigm of nuclear receptor-DNA interaction. Mol Cell Biol. 1993;13:5794–5804. doi: 10.1128/mcb.13.9.5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bain DL, Heneghan AF, Connaghan-Jones KD, Miura MT. Nuclear receptor structure: implications for function. Annu Rev Physiol. 2007;69:201–220. doi: 10.1146/annurev.physiol.69.031905.160308. [DOI] [PubMed] [Google Scholar]
- 27.Maglich JM, Sluder A, Guan X, Shi Y, McKee DD, Carrick K, et al. Comparison of complete nuclear receptor sets from the human, Caenorhabditis elegans and Drosophila genomes. Genome Biol. 2001;2:RESEARCH0029. doi: 10.1186/gb-2001-2-8-research0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sluder AE, Mathews SW, Hough D, Yin VP, Maina CV. The nuclear receptor superfamily has undergone extensive proliferation and diversification in nematodes. Genome Res. 1999;9:103–120. [PubMed] [Google Scholar]
- 29.Blumberg B, Evans RM. Orphan nuclear receptors—new ligands and new possibilities. Genes Dev. 1998;12:3149–3155. doi: 10.1101/gad.12.20.3149. [DOI] [PubMed] [Google Scholar]
- 30.Nichols JS, Parks DJ, Consler TG, Blanchard SG. Development of a scintillation proximity assay for peroxisome proliferator-activated receptor gamma ligand binding domain. Anal Biochem. 1998;257:112–119. doi: 10.1006/abio.1997.2557. [DOI] [PubMed] [Google Scholar]
- 31.Lin S, Bock CL, Gardner DB, Webster JC, Favata MF, Trzaskos JM, et al. A high-throughput fluorescent polarization assay for nuclear receptor binding utilizing crude receptor extract. Anal Biochem. 2002;300:15–21. doi: 10.1006/abio.2001.5437. [DOI] [PubMed] [Google Scholar]
- 32.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 33.Fearon ER, Finkel T, Gillison ML, Kennedy SP, Casella JF, Tomaselli GF, et al. Karyoplasmic interaction selection strategy: a general strategy to detect protein-protein interactions in mammalian cells. Proc Natl Acad Sci USA. 1992;89:7958–7962. doi: 10.1073/pnas.89.17.7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boute N, Jockers R, Issad T. The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends Pharmacol Sci. 2002;23:351–354. doi: 10.1016/s0165-6147(02)02062-x. [DOI] [PubMed] [Google Scholar]
- 35.Spencer TA, Li D, Russel JS, Collins JL, Bledsoe RK, Consler TG, et al. Pharmacophore analysis of the nuclear oxysterol receptor LXRalpha. J Med Chem. 2001;44:886–897. doi: 10.1021/jm0004749. [DOI] [PubMed] [Google Scholar]
- 36.Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature. 2002;415:813–817. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- 37.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 38.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 39.Hamuro Y, Coales SJ, Morrow JA, Molnar KS, Tuske SJ, Southern MR, et al. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 2006;15:1883–1892. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol. 2007;152:9–20. doi: 10.1038/sj.bjp.0707305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ai N, Krasowski MD, Welsh WJ, Ekins S. Understanding nuclear receptors using computational methods. Drug Discov Today. 2009;14:486–494. doi: 10.1016/j.drudis.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sonoda MT, Martinez L, Webb P, Skaf MS, Polikarpov I. Ligand dissociation from estrogen receptor is mediated by receptor dimerization: evidence from molecular dynamics simulations. Mol Endocrinol. 2008;22:1565–1578. doi: 10.1210/me.2007-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang CY, Li CW, Chen JD, Welsh WJ. Structural model reveals key interactions in the assembly of the pregnane X receptor/corepressor complex. Mol Pharmacol. 2006;69:1513–1517. doi: 10.1124/mol.106.022368. [DOI] [PubMed] [Google Scholar]
- 44.Teotico DG, Frazier ML, Ding F, Dokholyan NV, Temple BR, Redinbo MR. Active nuclear receptors exhibit highly correlated AF-2 domain motions. PLoS Comput Biol. 2008;4:e1000111. doi: 10.1371/journal.pcbi.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Folkertsma S, van Noort PI, Brandt RF, Bettler E, Vriend G, de Vlieg J. The nuclear receptor ligand-binding domain: a family-based structure analysis. Curr Med Chem. 2005;12:1001–1016. doi: 10.2174/0929867053764699. [DOI] [PubMed] [Google Scholar]
- 46.Raviscioni M, He Q, Salicru EM, Smith CL, Lichtarge O. Evolutionary identification of a subtype specific functional site in the ligand binding domain of steroid receptors. Proteins. 2006;64:1046–1057. doi: 10.1002/prot.21074. [DOI] [PubMed] [Google Scholar]
- 47.Reschly EJ, Ai N, Ekins S, Welsh WJ, Hagey LR, Hofmann AF, et al. Evolution of the bile salt nuclear receptor FXR in vertebrates. J Lipid Res. 2008;49:1577–1587. doi: 10.1194/jlr.M800138-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chong CR, Sullivan DJ., Jr New uses for old drugs. Nature. 2007;448:645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 49.Bisson WH, Cheltsov AV, Bruey-Sedano N, Lin B, Chen J, Goldberger N, et al. Discovery of antiandrogen activity of nonsteroidal scaffolds of marketed drugs. Proc Natl Acad Sci USA. 2007;104:11927–11932. doi: 10.1073/pnas.0609752104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.White TJ, Arakelian A, Rho JP. Counting the costs of drug-related adverse events. Pharmacoeconomics. 1999;15:445–458. doi: 10.2165/00019053-199915050-00003. [DOI] [PubMed] [Google Scholar]
- 51.Kohler GI, Bode-Boger SM, Busse R, Hoopmann M, Welte T, Boger RH. Drug-drug interactions in medical patients: effects of in-hospital treatment and relation to multiple drug use. Int J Clin Pharmacol Ther. 2000;38:504–513. doi: 10.5414/cpp38504. [DOI] [PubMed] [Google Scholar]
- 52.Nebert DW, Gonzalez FJ. P450 genes: structure, evolution, and regulation. Annu Rev Biochem. 1987;56:945–993. doi: 10.1146/annurev.bi.56.070187.004501. [DOI] [PubMed] [Google Scholar]
- 53.Balayssac D, Authier N, Cayre A, Coudore F. Does inhibition of P-glycoprotein lead to drug-drug interactions? Toxicol Lett. 2005;156:319–329. doi: 10.1016/j.toxlet.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 54.Varadi A, Szakacs G, Bakos E, Sarkadi B. P glycoprotein and the mechanism of multidrug resistance. Novartis Found Symp. 2002;243:54–65. discussion 65–58, 180–185. [PubMed] [Google Scholar]
- 55.Goodwin B, Hodgson E, Liddle C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol Pharmacol. 1999;56:1329–1339. doi: 10.1124/mol.56.6.1329. [DOI] [PubMed] [Google Scholar]
- 56.Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem. 2001;276:14581–14587. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- 57.Moore LB, Parks DJ, Jones SA, Bledsoe RK, Consler TG, Stimmel JB, et al. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem. 2000;275:15122–15127. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- 58.Gnerre C, Blattler S, Kaufmann MR, Looser R, Meyer UA. Regulation of CYP3A4 by the bile acid receptor FXR: evidence for functional binding sites in the CYP3A4 gene. Pharmacogenetics. 2004;14:635–645. doi: 10.1097/00008571-200410000-00001. [DOI] [PubMed] [Google Scholar]
- 59.Kast HR, Goodwin B, Tarr PT, Jones SA, Anisfeld AM, Stoltz CM, et al. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem. 2002;277:2908–2915. doi: 10.1074/jbc.M109326200. [DOI] [PubMed] [Google Scholar]
- 60.Jung D, Mangelsdorf DJ, Meyer UA. Pregnane X receptor is a target of farnesoid X receptor. J Biol Chem. 2006;281:19081–19091. doi: 10.1074/jbc.M600116200. [DOI] [PubMed] [Google Scholar]
- 61.Maglich JM, Stoltz CM, Goodwin B, Hawkins-Brown D, Moore JT, Kliewer SA. Nuclear pregnane×receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol Pharmacol. 2002;62:638–646. doi: 10.1124/mol.62.3.638. [DOI] [PubMed] [Google Scholar]
- 62.Kawamoto T, Sueyoshi T, Zelko I, Moore R, Washburn K, Negishi M. Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol Cell Biol. 1999;19:6318–6322. doi: 10.1128/mcb.19.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Acocella G. Clinical pharmacokinetics of rifampicin. Clin Pharmacokinet. 1978;3:108–127. doi: 10.2165/00003088-197803020-00002. [DOI] [PubMed] [Google Scholar]
- 64.Qureshi GD, Reinders TP, Somori GJ, Evans HJ. Warfarin resistance with nafcillin therapy. Ann Intern Med. 1984;100:527–529. doi: 10.7326/0003-4819-100-4-527. [DOI] [PubMed] [Google Scholar]
- 65.King JR, Acosta EP. Tipranavir: a novel nonpeptidic protease inhibitor of HIV. Clin Pharmacokinet. 2006;45:665–682. doi: 10.2165/00003088-200645070-00003. [DOI] [PubMed] [Google Scholar]
- 66.Mouly S, Lown KS, Kornhauser D, Joseph JL, Fiske WD, Benedek IH, et al. Hepatic but not intestinal CYP3A4 displays dose-dependent induction by efavirenz in humans. Clin Pharmacol Ther. 2002;72:1–9. doi: 10.1067/mcp.2002.124519. [DOI] [PubMed] [Google Scholar]
- 67.Ruschitzka F, Meier PJ, Turina M, Luscher TF, Noll G. Acute heart transplant rejection due to Saint John’s wort. Lancet. 2000;355:548–549. doi: 10.1016/S0140-6736(99)05467-7. [DOI] [PubMed] [Google Scholar]
- 68.Yin OQ, Tomlinson B, Waye MM, Chow AH, Chow MS. Pharmacogenetics and herb-drug interactions: experience with Ginkgo biloba and omeprazole. Pharmacogenetics. 2004;14:841–850. doi: 10.1097/00008571-200412000-00007. [DOI] [PubMed] [Google Scholar]
- 69.Kodama S, Tanaka K, Konishi H, Momota K, Nakasako H, Nakayama S, et al. Supplementary thyroxine therapy in patients with hypothyroidism induced by long-term anticonvulsant therapy. Acta Paediatr Jpn. 1989;31:555–562. doi: 10.1111/j.1442-200x.1989.tb01354.x. [DOI] [PubMed] [Google Scholar]
- 70.Qatanani M, Zhang J, Moore DD. Role of the constitutive androstane receptor in xenobiotic-induced thyroid hormone metabolism. Endocrinology. 2005;146:995–1002. doi: 10.1210/en.2004-1350. [DOI] [PubMed] [Google Scholar]
- 71.Shah SC, Sharma RK, Hemangini, Chitle AR. Rifampicin induced osteomalacia. Tubercle. 1981;62:207–209. doi: 10.1016/0041-3879(81)90008-8. [DOI] [PubMed] [Google Scholar]
- 72.Riddle TM, Kuhel DG, Woollett LA, Fichtenbaum CJ, Hui DY. HIV protease inhibitor induces fatty acid and sterol biosynthesis in liver and adipose tissues due to the accumulation of activated sterol regulatory element-binding proteins in the nucleus. J Biol Chem. 2001;276:37514–37519. doi: 10.1074/jbc.M104557200. [DOI] [PubMed] [Google Scholar]
- 73.Osborne CK, Zhao H, Fuqua SA. Selective estrogen receptor modulators: structure, function, and clinical use. J Clin Oncol. 2000;18:3172–3186. doi: 10.1200/JCO.2000.18.17.3172. [DOI] [PubMed] [Google Scholar]
- 74.Levenson AS, Jordan VC. Selective oestrogen receptor modulation: molecular pharmacology for the millennium. Eur J Cancer. 1999;35:1628–1639. doi: 10.1016/s0959-8049(99)00183-5. [DOI] [PubMed] [Google Scholar]
- 75.Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, Nelson MC, et al. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature. 2000;406:435–439. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- 76.Ma X, Cheung C, Krausz KW, Shah YM, Wang T, Idle JR, et al. A double transgenic mouse model expressing human pregnane X receptor and cytochrome P450 3A4. Drug Metab Dispos. 2008;36:2506–2512. doi: 10.1124/dmd.108.022723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mills JB, Rose KA, Sadagopan N, Sahi J, de Morais SM. Induction of drug metabolism enzymes and MDR1 using a novel human hepatocyte cell line. J Pharmacol Exp Ther. 2004;309:303–309. doi: 10.1124/jpet.103.061713. [DOI] [PubMed] [Google Scholar]
- 78.Trubetskoy O, Marks B, Zielinski T, Yueh MF, Raucy J. A simultaneous assessment of CYP3A4 metabolism and induction in the DPX-2 cell line. AAPS J. 2005;7:E6–E13. doi: 10.1208/aapsj070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Biswas A, Mani S, Redinbo MR, Krasowski MD, Li H, Ekins S. Elucidating the ‘Jekyll and Hyde’ nature of PXR: the case for discovering antagonists or allosteric antagonists. Pharm Res. 2009;26:1807–1815. doi: 10.1007/s11095-009-9901-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blumberg B, Sabbagh W, Jr, Juguilon H, Bolado J, Jr, van Meter CM, Ong ES, et al. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 1998;12:3195–3205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 82.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23:687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- 84.Kliewer SA. The nuclear pregnane X receptor regulates xenobiotic detoxification. J Nutr. 2003;133:2444S–2447S. doi: 10.1093/jn/133.7.2444S. [DOI] [PubMed] [Google Scholar]
- 85.Krasowski MD, Yasuda K, Hagey LR, Schuetz EG. Evolution of the pregnane×receptor: adaptation to cross-species differences in biliary bile salts. Mol Endocrinol. 2005;19:1720–1739. doi: 10.1210/me.2004-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang J, Huang W, Chua SS, Wei P, Moore DD. Modulation of acetaminophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science. 2002;298:422–424. doi: 10.1126/science.1073502. [DOI] [PubMed] [Google Scholar]
- 87.Scheer N, Ross J, Rode A, Zevnik B, Niehaves S, Faust N, et al. A novel panel of mouse models to evaluate the role of human pregnane X receptor and constitutive androstane receptor in drug response. J Clin Invest. 2008;118:3228–3239. doi: 10.1172/JCI35483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Synold TW, Dussault I, Forman BM. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med. 2001;7:584–590. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- 90.Wang H, Huang H, Li H, Teotico DG, Sinz M, Baker SD, et al. Activated pregnenolone X-receptor is a target for ketoconazole and its analogs. Clin Cancer Res. 2007;13:2488–2495. doi: 10.1158/1078-0432.CCR-06-1592. [DOI] [PubMed] [Google Scholar]
- 91.Gupta D, Venkatesh M, Wang H, Kim S, Sinz M, Goldberg GL, et al. Expanding the roles for pregnane X receptor in cancer: proliferation and drug resistance in ovarian cancer. Clin Cancer Res. 2008;14:5332–5340. doi: 10.1158/1078-0432.CCR-08-1033. [DOI] [PubMed] [Google Scholar]
- 92.Zhou C, Poulton EJ, Grun F, Bammler TK, Blumberg B, Thummel KE, et al. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol Pharmacol. 2007;71:220–229. doi: 10.1124/mol.106.029264. [DOI] [PubMed] [Google Scholar]
- 93.Zhou J, Liu M, Zhai Y, Xie W. The antiapoptotic role of pregnane X receptor in human colon cancer cells. Mol Endocrinol. 2008;22:868–880. doi: 10.1210/me.2007-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Meyer zu Schwabedissen HE, Tirona RG, Yip CS, Ho RH, Kim RB. Interplay between the nuclear receptor pregnane X receptor and the uptake transporter organic anion transporter polypeptide 1A2 selectively enhances estrogen effects in breast cancer. Cancer Res. 2008;68:9338–9347. doi: 10.1158/0008-5472.CAN-08-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fang HL, Strom SC, Ellis E, Duanmu Z, Fu J, Duniec-Dmuchowski Z, et al. Positive and negative regulation of human hepatic hydroxysteroid sulfotransferase (SULT2A1) gene transcription by rifampicin: roles of hepatocyte nuclear factor 4alpha and pregnane X receptor. J Pharmacol Exp Ther. 2007;323:586–598. doi: 10.1124/jpet.107.124610. [DOI] [PubMed] [Google Scholar]
- 96.Uppal H, Toma D, Saini SP, Ren S, Jones TJ, Xie W. Combined loss of orphan receptors PXR and CAR heightens sensitivity to toxic bile acids in mice. Hepatology. 2005;41:168–176. doi: 10.1002/hep.20512. [DOI] [PubMed] [Google Scholar]
- 97.Zollner G, Fickert P, Fuchsbichler A, Silbert D, Wagner M, Arbeiter S, et al. Role of nuclear bile acid receptor, FXR, in adaptive ABC transporter regulation by cholic and ursodeoxycholic acid in mouse liver, kidney and intestine. J Hepatol. 2003;39:480–488. doi: 10.1016/s0168-8278(03)00228-9. [DOI] [PubMed] [Google Scholar]
- 98.Teng S, Piquette-Miller M. The involvement of the pregnane X receptor in hepatic gene regulation during inflammation in mice. J Pharmacol Exp Ther. 2005;312:841–848. doi: 10.1124/jpet.104.076141. [DOI] [PubMed] [Google Scholar]
- 99.Li T, Chiang JY. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7 alpha-hydroxylase gene transcription. Am J Physiol Gastrointest Liver Physiol. 2005;288:G74–G84. doi: 10.1152/ajpgi.00258.2004. [DOI] [PubMed] [Google Scholar]
- 100.Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, Toma D, et al. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem. 2006;281:15013–15020. doi: 10.1074/jbc.M511116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hoekstra M, Lammers B, Out R, Li Z, Van Eck M, Van Berkel TJ. Activation of the nuclear receptor PXR decreases plasma LDL-cholesterol levels and induces hepatic steatosis in LDL receptor knockout mice. Mol Pharmacol. 2009;6:182–189. doi: 10.1021/mp800131d. [DOI] [PubMed] [Google Scholar]
- 102.Pascussi JM, Robert A, Nguyen M, Walrant-Debray O, Garabedian M, Martin P, et al. Possible involvement of pregnane X receptor-enhanced CYP24 expression in drug-induced osteomalacia. J Clin Invest. 2005;115:177–186. doi: 10.1172/JCI21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wolf KK, Wood SG, Hunt JA, Walton-Strong BW, Yasuda K, Lan L, et al. Role of the nuclear receptor pregnane X receptor in acetaminophen hepatotoxicity. Drug Metab Dispos. 2005;33:1827–1836. doi: 10.1124/dmd.105.005256. [DOI] [PubMed] [Google Scholar]
- 104.Guo GL, Moffit JS, Nicol CJ, Ward JM, Aleksunes LA, Slitt AL, et al. Enhanced acetaminophen toxicity by activation of the pregnane X receptor. Toxicol Sci. 2004;82:374–380. doi: 10.1093/toxsci/kfh286. [DOI] [PubMed] [Google Scholar]
- 105.Li T, Chen W, Chiang JY. PXR induces CYP27A1 and regulates cholesterol metabolism in the intestine. J Lipid Res. 2007;48:373–384. doi: 10.1194/jlr.M600282-JLR200. [DOI] [PubMed] [Google Scholar]
- 106.Tabb MM, Sun A, Zhou C, Grun F, Errandi J, Romero K, et al. Vitamin K2 regulation of bone homeostasis is mediated by the steroid and xenobiotic receptor SXR. J Biol Chem. 2003;278:43919–43927. doi: 10.1074/jbc.M303136200. [DOI] [PubMed] [Google Scholar]
- 107.Dotzlaw H, Leygue E, Watson P, Murphy LC. The human orphan receptor PXR messenger RNA is expressed in both normal and neoplastic breast tissue. Clin Cancer Res. 1999;5:2103–2107. [PubMed] [Google Scholar]
- 108.Toell A, Kroncke KD, Kleinert H, Carlberg C. Orphan nuclear receptor binding site in the human inducible nitric oxide synthase promoter mediates responsiveness to steroid and xenobiotic ligands. J Cell Biochem. 2002;85:72–82. [PubMed] [Google Scholar]
- 109.Zhou C, Tabb MM, Nelson EL, Grun F, Verma S, Sadatrafiei A, et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest. 2006;116:2280–2289. doi: 10.1172/JCI26283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Suino K, Peng L, Reynolds R, Li Y, Cha JY, Repa JJ, et al. The nuclear xenobiotic receptor CAR: structural determinants of constitutive activation and heterodimerization. Mol Cell. 2004;16:893–905. doi: 10.1016/j.molcel.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 111.Schrader M, Danielsson C, Wiesenberg I, Carlberg C. Identification of natural monomeric response elements of the nuclear receptor RZR/ROR. They also bind COUP-TF homodimers. J Biol Chem. 1996;271:19732–19736. doi: 10.1074/jbc.271.33.19732. [DOI] [PubMed] [Google Scholar]
- 112.Forman BM, Ruan B, Chen J, Schroepfer GJ, Jr, Evans RM. The orphan nuclear receptor LXRalpha is positively and negatively regulated by distinct products of mevalonate metabolism. Proc Natl Acad Sci USA. 1997;94:10588–10593. doi: 10.1073/pnas.94.20.10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Whitby RJ, Dixon S, Maloney PR, Delerive P, Goodwin BJ, Parks DJ, et al. Identification of small molecule agonists of the orphan nuclear receptors liver receptor homolog-1 and steroidogenic factor-1. J Med Chem. 2006;49:6652–6655. doi: 10.1021/jm060990k. [DOI] [PubMed] [Google Scholar]
- 114.Wisely GB, Miller AB, Davis RG, Thornquest AD, Jr, Johnson R, Spitzer T, et al. Hepatocyte nuclear factor 4 is a transcription factor that constitutively binds fatty acids. Structure. 2002;10:1225–1234. doi: 10.1016/s0969-2126(02)00829-8. [DOI] [PubMed] [Google Scholar]
- 115.Maxwell MA, Muscat GE. The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl Recept Signal. 2006;4:e002. doi: 10.1621/nrs.04002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tremblay GB, Kunath T, Bergeron D, Lapointe L, Champigny C, Bader JA, et al. Diethylstilbestrol regulates trophoblast stem cell differentiation as a ligand of orphan nuclear receptor ERR beta. Genes Dev. 2001;15:833–838. doi: 10.1101/gad.873401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xu RX, Lambert MH, Wisely BB, Warren EN, Weinert EE, Waitt GM, et al. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol Cell. 2004;16:919–928. doi: 10.1016/j.molcel.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 118.Shan L, Vincent J, Brunzelle JS, Dussault I, Lin M, Ianculescu I, et al. Structure of the murine constitutive androstane receptor complexed to androstenol: a molecular basis for inverse agonism. Mol Cell. 2004;16:907–917. doi: 10.1016/j.molcel.2004.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kelley M, Lambert I, Merrill J, Safe S. 1, 4-Bis[2-(3, 5-dichloropyridyloxy)]benzene (TCPOBOP) and related compounds as inducers of hepatic monooxygenases. Structure-activity effects. Biochem Pharmacol. 1985;34:3489–3494. doi: 10.1016/0006-2952(85)90722-1. [DOI] [PubMed] [Google Scholar]
- 120.Zelko I, Negishi M. Phenobarbital-elicited activation of nuclear receptor CAR in induction of cytochrome P450 genes. Biochem Biophys Res Commun. 2000;277:1–6. doi: 10.1006/bbrc.2000.3557. [DOI] [PubMed] [Google Scholar]
- 121.Honkakoski P, Moore R, Washburn KA, Negishi M. Activation by diverse xenochemicals of the 51-base pair phenobarbital-responsive enhancer module in the CYP2B10 gene. Mol Pharmacol. 1998;53:597–601. doi: 10.1124/mol.53.4.597. [DOI] [PubMed] [Google Scholar]
- 122.Honkakoski P, Zelko I, Sueyoshi T, Negishi M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol Cell Biol. 1998;18:5652–5658. doi: 10.1128/mcb.18.10.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kawamoto T, Kakizaki S, Yoshinari K, Negishi M. Estrogen activation of the nuclear orphan receptor CAR (constitutive active receptor) in induction of the mouse Cyp2b10 gene. Mol Endocrinol. 2000;14:1897–1905. doi: 10.1210/mend.14.11.0547. [DOI] [PubMed] [Google Scholar]
- 124.Kodama S, Negishi M. Phenobarbital confers its diverse effects by activating the orphan nuclear receptor car. Drug Metab Rev. 2006;38:75–87. doi: 10.1080/03602530600569851. [DOI] [PubMed] [Google Scholar]
- 125.Huang W, Zhang J, Washington M, Liu J, Parant JM, Lozano G, et al. Xenobiotic stress induces hepatomegaly and liver tumors via the nuclear receptor constitutive androstane receptor. Mol Endocrinol. 2005;19:1646–1653. doi: 10.1210/me.2004-0520. [DOI] [PubMed] [Google Scholar]
- 126.Maglich JM, Watson J, McMillen PJ, Goodwin B, Willson TM, Moore JT. The nuclear receptor CAR is a regulator of thyroid hormone metabolism during caloric restriction. J Biol Chem. 2004;279:19832–19838. doi: 10.1074/jbc.M313601200. [DOI] [PubMed] [Google Scholar]
- 127.Gao J, He J, Zhai Y, Wada T, Xie W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J Biol Chem. 2009;284:25984–25992. doi: 10.1074/jbc.M109.016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dong B, Saha PK, Huang W, Chen W, Abu-Elheiga LA, Wakil SJ, et al. Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc Natl Acad Sci USA. 2009;106:18831–18836. doi: 10.1073/pnas.0909731106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Roth A, Looser R, Kaufmann M, Meyer UA. Sterol regulatory element binding protein 1 interacts with pregnane X receptor and constitutive androstane receptor and represses their target genes. Pharmacogenet Genomics. 2008;18:325–337. doi: 10.1097/FPC.0b013e3282f706e0. [DOI] [PubMed] [Google Scholar]
- 130.Roth A, Looser R, Kaufmann M, Blattler SM, Rencurel F, Huang W, et al. Regulatory cross-talk between drug metabolism and lipid homeostasis: constitutive androstane receptor and pregnane X receptor increase Insig-1 expression. Mol Pharmacol. 2008;73:1282–1289. doi: 10.1124/mol.107.041012. [DOI] [PubMed] [Google Scholar]
- 131.Sugatani J, Nishitani S, Yamakawa K, Yoshinari K, Sueyoshi T, Negishi M, et al. Transcriptional regulation of human UGT1A1 gene expression: activated glucocorticoid receptor enhances constitutive androstane receptor/pregnane X receptor-mediated UDP-glucuronosyltransferase 1A1 regulation with glucocorticoid receptor-interacting protein 1. Mol Pharmacol. 2005;67:845–855. doi: 10.1124/mol.104.007161. [DOI] [PubMed] [Google Scholar]
- 132.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 133.Chuu CP, Kokontis JM, Hiipakka RA, Liao S. Modulation of liver X receptor signaling as novel therapy for prostate cancer. J Biomed Sci. 2007;14:543–553. doi: 10.1007/s11373-007-9160-8. [DOI] [PubMed] [Google Scholar]
- 134.Svensson S, Ostberg T, Jacobsson M, Norstrom C, Stefansson K, Hallen D, et al. Crystal structure of the heterodimeric complex of LXRalpha and RXRbeta ligand-binding domains in a fully agonistic conformation. EMBO J. 2003;22:4625–4633. doi: 10.1093/emboj/cdg456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Williams S, Bledsoe RK, Collins JL, Boggs S, Lambert MH, Miller AB, et al. X-ray crystal structure of the liver X receptor beta ligand binding domain: regulation by a histidine-tryptophan switch. J Biol Chem. 2003;278:27138–27143. doi: 10.1074/jbc.M302260200. [DOI] [PubMed] [Google Scholar]
- 136.Matsukuma KE, Wang L, Bennett MK, Osborne TF. A key role for orphan nuclear receptor liver receptor homologue-1 in activation of fatty acid synthase promoter by liver X receptor. J Biol Chem. 2007;282:20164–20171. doi: 10.1074/jbc.M702895200. [DOI] [PubMed] [Google Scholar]
- 137.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors. LXRalpha and LXRbeta. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Beaven SW, Tontonoz P. Nuclear receptors in lipid metabolism: targeting the heart of dyslipidemia. Annu RevMed. 2006;57:313–329. doi: 10.1146/annurev.med.57.121304.131428. [DOI] [PubMed] [Google Scholar]
- 139.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 140.Thomas J, Bramlett KS, Montrose C, Foxworthy P, Eacho PI, McCann D, et al. A chemical switch regulates fibrate specificity for peroxisome proliferator-activated receptor alpha (PPARalpha) versus liver X receptor. J Biol Chem. 2003;278:2403–2410. doi: 10.1074/jbc.M209629200. [DOI] [PubMed] [Google Scholar]
- 141.Kokontis JM, Hay N, Liao S. Progression of LNCaP prostate tumor cells during androgen deprivation: hormone-independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Mol Endocrinol. 1998;12:941–953. doi: 10.1210/mend.12.7.0136. [DOI] [PubMed] [Google Scholar]
- 142.Chuu CP, Hiipakka RA, Kokontis JM, Fukuchi J, Chen RY, Liao S. Inhibition of tumor growth and progression of LNCaP prostate cancer cells in athymic mice by androgen and liver X receptor agonist. Cancer Res. 2006;66:6482–6486. doi: 10.1158/0008-5472.CAN-06-0632. [DOI] [PubMed] [Google Scholar]
- 143.Gong H, Guo P, Zhai Y, Zhou J, Uppal H, Jarzynka MJ, et al. Estrogen deprivation and inhibition of breast cancer growth in vivo through activation of the orphan nuclear receptor liver X receptor. Mol Endocrinol. 2007;21:1781–1790. doi: 10.1210/me.2007-0187. [DOI] [PubMed] [Google Scholar]
- 144.Uno S, Endo K, Jeong Y, Kawana K, Miyachi H, Hashimoto Y, et al. Suppression of beta-catenin signaling by liver X receptor ligands. Biochem Pharmacol. 2009;77:186–195. doi: 10.1016/j.bcp.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 145.Hong C, Tontonoz P. Coordination of inflammation and metabolism by PPAR and LXR nuclear receptors. Curr Opin Genet Dev. 2008;18:461–467. doi: 10.1016/j.gde.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, et al. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc Natl Acad Sci USA. 2000;97:12097–12102. doi: 10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Patel NV, Forman BM. Linking lipids, Alzheimer’s and LXRs? Nucl Recept Signal. 2004;2:e001. doi: 10.1621/nrs.02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci USA. 2002;99:7604–7609. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mi LZ, Devarakonda S, Harp JM, Han Q, Pellicciari R, Willson TM, et al. Structural basis for bile acid binding and activation of the nuclear receptor FXR. Mol Cell. 2003;11:1093–1100. doi: 10.1016/s1097-2765(03)00112-6. [DOI] [PubMed] [Google Scholar]
- 150.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors, FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 151.Moschetta A, Bookout AL, Mangelsdorf DJ. Prevention of cholesterol gallstone disease by FXR agonists in a mouse model. Nat Med. 2004;10:1352–1358. doi: 10.1038/nm1138. [DOI] [PubMed] [Google Scholar]
- 152.Claudel T, Sturm E, Duez H, Torra IP, Sirvent A, Kosykh V, et al. Bile acid-activated nuclear receptor FXR suppresses apolipo-protein A-I transcription via a negative FXR response element. J Clin Invest. 2002;109:961–971. doi: 10.1172/JCI14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Anisfeld AM, Kast-Woelbern HR, Meyer ME, Jones SA, Zhang Y, Williams KJ, et al. Syndecan-1 expression is regulated in an isoform-specific manner by the farnesoid-X receptor. J Biol Chem. 2003;278:20420–20428. doi: 10.1074/jbc.M302505200. [DOI] [PubMed] [Google Scholar]
- 154.Cui J, Huang L, Zhao A, Lew JL, Yu J, Sahoo S, et al. Guggulsterone is a farnesoid X receptor antagonist in coactivator association assays but acts to enhance transcription of bile salt export pump. J Biol Chem. 2003;278:10214–10220. doi: 10.1074/jbc.M209323200. [DOI] [PubMed] [Google Scholar]
- 155.Zhao A, Lew JL, Huang L, Yu J, Zhang T, Hrywna Y, et al. Human kininogen gene is transactivated by the farnesoid X receptor. J Biol Chem. 2003;278:28765–28770. doi: 10.1074/jbc.M304568200. [DOI] [PubMed] [Google Scholar]
- 156.Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jung D, Inagaki T, Gerard RD, Dawson PA, Kliewer SA, Mangelsdorf DJ, et al. FXR agonists and FGF15 reduce fecal bile acid excretion in a mouse model of bile acid malabsorption. J Lipid Res. 2007;48:2693–2700. doi: 10.1194/jlr.M700351-JLR200. [DOI] [PubMed] [Google Scholar]
- 158.Maran RR, Thomas A, Roth M, Sheng Z, Esterly N, Pinson D, et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J Pharmacol Exp Ther. 2009;328:469–477. doi: 10.1124/jpet.108.145409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Swales KE, Korbonits M, Carpenter R, Walsh DT, Warner TD, Bishop-Bailey D. The farnesoid X receptor is expressed in breast cancer and regulates apoptosis and aromatase expression. Cancer Res. 2006;66:10120–10126. doi: 10.1158/0008-5472.CAN-06-2399. [DOI] [PubMed] [Google Scholar]
- 160.Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68:879–887. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- 161.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 162.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 163.Uppenberg J, Svensson C, Jaki M, Bertilsson G, Jendeberg L, Berkenstam A. Crystal structure of the ligand binding domain of the human nuclear receptor PPARgamma. J Biol Chem. 1998;273:31108–31112. doi: 10.1074/jbc.273.47.31108. [DOI] [PubMed] [Google Scholar]
- 164.Gampe RT, Jr, Montana VG, Lambert MH, Miller AB, Bledsoe RK, Milburn MV, et al. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol Cell. 2000;5:545–555. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- 165.Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999;3:397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- 166.Schmidt A, Endo N, Rutledge SJ, Vogel R, Shinar D, Rodan GA. Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids. Mol Endocrinol. 1992;6:1634–1641. doi: 10.1210/mend.6.10.1333051. [DOI] [PubMed] [Google Scholar]
- 167.Lin Q, Ruuska SE, Shaw NS, Dong D, Noy N. Ligand selectivity of the peroxisome proliferator-activated receptor alpha. Biochemistry. 1999;38:185–190. doi: 10.1021/bi9816094. [DOI] [PubMed] [Google Scholar]
- 168.Barter PJ, Rye KA. Cardioprotective properties of fibrates: which fibrate, which patients, what mechanism? Circulation. 2006;113:1553–1555. doi: 10.1161/CIRCULATIONAHA.105.620450. [DOI] [PubMed] [Google Scholar]
- 169.Panigrahy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, et al. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest. 2002;110:923–932. doi: 10.1172/JCI15634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Bishop-Bailey D, Warner TD. PPARgamma ligands induce prostaglandin production in vascular smooth muscle cells: indomethacin acts as a peroxisome proliferator-activated receptor-gamma antagonist. FASEB J. 2003;17:1925–1927. doi: 10.1096/fj.02-1075fje. [DOI] [PubMed] [Google Scholar]
- 171.Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, et al. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 172.Buse JB, Rubin CJ, Frederich R, Viraswami-Appanna K, Lin KC, Montoro R, et al. Muraglitazar, a dual (alpha/gamma) PPAR activator: a randomized, double-blind, placebo-controlled: 24-week monotherapy trial in adult patients with type 2 diabetes. Clin Ther. 2005;27:1181–1195. doi: 10.1016/j.clinthera.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 173.Lambert DM, Fowler CJ. The endocannabinoid system: drug targets, lead compounds, and potential therapeutic applications. J Med Chem. 2005;48:5059–5087. doi: 10.1021/jm058183t. [DOI] [PubMed] [Google Scholar]
- 174.Liu J, Li H, Burstein SH, Zurier RB, Chen JD. Activation and binding of peroxisome proliferator-activated receptor gamma by synthetic cannabinoid ajulemic acid. Mol Pharmacol. 2003;63:983–992. doi: 10.1124/mol.63.5.983. [DOI] [PubMed] [Google Scholar]
- 175.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 176.Sun Y, Alexander SP, Kendall DA, Bennett AJ. Cannabinoids and PPARalpha signalling. Biochem Soc Trans. 2006;34:1095–1097. doi: 10.1042/BST0341095. [DOI] [PubMed] [Google Scholar]
- 177.O’Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 179.Tobin JF, Freedman LP. Nuclear receptors as drug targets in metabolic diseases: new approaches to therapy. Trends Endocrinol Metab. 2006;17:284–290. doi: 10.1016/j.tem.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 180.Jiang G, Dallas-Yang Q, Li Z, Szalkowski D, Liu F, Shen X, et al. Potentiation of insulin signaling in tissues of Zucker obese rats after acute and long-term treatment with PPARgamma agonists. Diabetes. 2002;51:2412–2419. doi: 10.2337/diabetes.51.8.2412. [DOI] [PubMed] [Google Scholar]
- 181.Way JM, Harrington WW, Brown KK, Gottschalk WK, Sundseth SS, Mansfield TA, et al. Comprehensive messenger ribonucleic acid profiling reveals that peroxisome proliferator-activated receptor gamma activation has coordinate effects on gene expression in multiple insulin-sensitive tissues. Endocrinology. 2001;142:1269–1277. doi: 10.1210/endo.142.3.8037. [DOI] [PubMed] [Google Scholar]
- 182.Tordjman J, Leroyer S, Chauvet G, Quette J, Chauvet C, Tomkiewicz C, et al. Cytosolic aspartate aminotransferase, a new partner in adipocyte glyceroneogenesis and an atypical target of thiazolidinedione. J Biol Chem. 2007;282:23591–23602. doi: 10.1074/jbc.M611111200. [DOI] [PubMed] [Google Scholar]
- 183.Kim MS, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Suppression of estrogen-related receptor alpha and medium-chain acyl-coenzyme A dehydrogenase in the acute-phase response. J Lipid Res. 2005;46:2282–2288. doi: 10.1194/jlr.M500217-JLR200. [DOI] [PubMed] [Google Scholar]
- 184.Gilde AJ, van der Lee KA, Willemsen PH, Chinetti G, van der Leij FR, van der Vusse GJ, et al. Peroxisome proliferator-activated receptor (PPAR) alpha and PPARbeta/delta, but not PPARgamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res. 2003;92:518–524. doi: 10.1161/01.RES.0000060700.55247.7C. [DOI] [PubMed] [Google Scholar]
- 185.Zhang Q, Seltmann H, Zouboulis CC, Konger RL. Involvement of PPARgamma in oxidative stress-mediated prostaglandin E(2) production in SZ95 human sebaceous gland cells. J Invest Dermatol. 2006;126:42–48. doi: 10.1038/sj.jid.5700028. [DOI] [PubMed] [Google Scholar]
- 186.Bailey ST, Ghosh S. ‘PPAR’ting ways with inflammation. Nat Immunol. 2005;6:966–967. doi: 10.1038/ni1005-966. [DOI] [PubMed] [Google Scholar]
- 187.Shaw N, Elholm M, Noy N. Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor beta/delta. J Biol Chem. 2003;278:41589–41592. doi: 10.1074/jbc.C300368200. [DOI] [PubMed] [Google Scholar]
- 188.Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 189.Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, et al. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta) Mol Cell Biol. 2000;20:5119–5128. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Poirier H, Niot I, Monnot MC, Braissant O, Meunier-Durmort C, Costet P, et al. Differential involvement of peroxisome-proliferator-activated receptors alpha and delta in fibrate and fatty-acid-mediated inductions of the gene encoding liver fatty-acid-binding protein in the liver and the small intestine. Biochem J. 2001;355:481–488. doi: 10.1042/0264-6021:3550481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci USA. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, et al. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 2003;17:2299–2301. doi: 10.1096/fj.03-0269fje. [DOI] [PubMed] [Google Scholar]
- 193.Luquet S, Gaudel C, Holst D, Lopez-Soriano J, Jehl-Pietri C, Fredenrich A, et al. Roles of PPAR delta in lipid absorption and metabolism: a new target for the treatment of type 2 diabetes. Biochim Biophys Acta. 2005;1740:313–317. doi: 10.1016/j.bbadis.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 194.Ladias JA. Convergence of multiple nuclear receptor signaling pathways onto the long terminal repeat of human immunodeficiency virus-1. J Biol Chem. 1994;269:5944–5951. [PubMed] [Google Scholar]
- 195.Potula R, Ramirez SH, Knipe B, Leibhart J, Schall K, Heilman D, et al. Peroxisome proliferator-activated receptor-gamma activation suppresses HIV-1 replication in an animal model of encephalitis. AIDS. 2008;22:1539–1549. doi: 10.1097/QAD.0b013e3283081e08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Law SW, Conneely OM, DeMayo FJ, O’Malley BW. Identification of a new brain-specific transcription factor, NURR1. Mol Endocrinol. 1992;6:2129–2135. doi: 10.1210/mend.6.12.1491694. [DOI] [PubMed] [Google Scholar]
- 197.Zetterstrom RH, Williams R, Perlmann T, Olson L. Cellular expression of the immediate early transcription factors Nurr1 and NGFI-B suggests a gene regulatory role in several brain regions including the nigrostriatal dopamine system. Brain Res Mol Brain Res. 1996;41:111–120. doi: 10.1016/0169-328x(96)00074-5. [DOI] [PubMed] [Google Scholar]
- 198.Zetterstrom RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- 199.Ordentlich P, Yan Y, Zhou S, Heyman RA. Identification of the antineoplastic agent 6-mercaptopurine as an activator of the orphan nuclear hormone receptor Nurr1. J Biol Chem. 2003;278:24791–24799. doi: 10.1074/jbc.M302167200. [DOI] [PubMed] [Google Scholar]
- 200.Wang Z, Benoit G, Liu J, Prasad S, Aarnisalo P, Liu X, et al. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature. 2003;423:555–560. doi: 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- 201.Le WD, Xu P, Jankovic J, Jiang H, Appel SH, Smith RG, et al. Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet. 2003;33:85–89. doi: 10.1038/ng1066. [DOI] [PubMed] [Google Scholar]
- 202.Buervenich S, Carmine A, Arvidsson M, Xiang F, Zhang Z, Sydow O, et al. NURR1 mutations in cases of schizophrenia and manic-depressive disorder. Am J Med Genet. 2000;96:808–813. doi: 10.1002/1096-8628(20001204)96:6<808::aid-ajmg23>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 203.McEvoy AN, Murphy EA, Ponnio T, Conneely OM, Bresnihan B, FitzGerald O, et al. Activation of nuclear orphan receptor NURR1 transcription by NF-kappa B and cyclic adenosine 5′-monophosphate response element-binding protein in rheumatoid arthritis synovial tissue. J Immunol. 2002;168:2979–2987. doi: 10.4049/jimmunol.168.6.2979. [DOI] [PubMed] [Google Scholar]
- 204.Murphy EP, McEvoy A, Conneely OM, Bresnihan B, FitzGerald O. Involvement of the nuclear orphan receptor NURR1 in the regulation of corticotropin-releasing hormone expression and actions in human inflammatory arthritis. Arthritis Rheum. 2001;44:782–793. doi: 10.1002/1529-0131(200104)44:4<782::AID-ANR134>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 205.Pei L, Castrillo A, Tontonoz P. Regulation of macrophage inflammatory gene expression by the orphan nuclear receptor Nur77. Mol Endocrinol. 2006;20:786–794. doi: 10.1210/me.2005-0331. [DOI] [PubMed] [Google Scholar]
- 206.Zhan Y, Du X, Chen H, Liu J, Zhao B, Huang D, et al. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat Chem Biol. 2008;4:548–556. doi: 10.1038/nchembio.106. [DOI] [PubMed] [Google Scholar]
- 207.Lin B, Kolluri SK, Lin F, Liu W, Han YH, Cao X, et al. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell. 2004;116:527–540. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]
- 208.Li H, Kolluri SK, Gu J, Dawson MI, Cao X, Hobbs PD, et al. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science. 2000;289:1159–1164. doi: 10.1126/science.289.5482.1159. [DOI] [PubMed] [Google Scholar]
- 209.Kolluri SK, Zhu X, Zhou X, Lin B, Chen Y, Sun K, et al. A short Nur77-derived peptide converts Bcl-2 from a protector to a killer. Cancer Cell. 2008;14:285–298. doi: 10.1016/j.ccr.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Cho SD, Yoon K, Chintharlapalli S, Abdelrahim M, Lei P, Hamilton S, et al. Nur77 agonists induce proapoptotic genes and responses in colon cancer cells through nuclear receptor-dependent and nuclear receptor-independent pathways. Cancer Res. 2007;67:674–683. doi: 10.1158/0008-5472.CAN-06-2907. [DOI] [PubMed] [Google Scholar]
- 211.Monaghan AP, Bock D, Gass P, Schwager A, Wolfer DP, Lipp HP, et al. Defective limbic system in mice lacking the tailless gene. Nature. 1997;390:515–517. doi: 10.1038/37364. [DOI] [PubMed] [Google Scholar]
- 212.Yu RT, McKeown M, Evans RM, Umesono K. Relationship between Drosophila gap gene tailless and a vertebrate nuclear receptor Tlx. Nature. 1994;370:375–379. doi: 10.1038/370375a0. [DOI] [PubMed] [Google Scholar]
- 213.Miyawaki T, Uemura A, Dezawa M, Yu RT, Ide C, Nishikawa S, et al. Tlx, an orphan nuclear receptor, regulates cell numbers and astrocyte development in the developing retina. J Neurosci. 2004;24:8124–8134. doi: 10.1523/JNEUROSCI.2235-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Shi Y, Chichung Lie D, Taupin P, Nakashima K, Ray J, Yu RT, et al. Expression and function of orphan nuclear receptor TLX in adult neural stem cells. Nature. 2004;427:78–83. doi: 10.1038/nature02211. [DOI] [PubMed] [Google Scholar]
- 215.Shi Y. Orphan nuclear receptors in drug discovery. Drug Discov Today. 2007;12:440–445. doi: 10.1016/j.drudis.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Yu RT, Chiang MY, Tanabe T, Kobayashi M, Yasuda K, Evans RM, et al. The orphan nuclear receptor Tlx regulates Pax2 and is essential for vision. Proc Natl Acad Sci USA. 2000;97:2621–2625. doi: 10.1073/pnas.050566897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Zhang CL, Zou Y, He W, Gage FH, Evans RM. A role for adult TLX-positive neural stem cells in learning and behaviour. Nature. 2008;451:1004–1007. doi: 10.1038/nature06562. [DOI] [PubMed] [Google Scholar]
- 218.Zhang CL, Zou Y, Yu RT, Gage FH, Evans RM. Nuclear receptor TLX prevents retinal dystrophy and recruits the corepressor atrophin1. Genes Dev. 2006;20:1308–1320. doi: 10.1101/gad.1413606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kobayashi M, Yu RT, Yasuda K, Umesono K. Cell-type-specific regulation of the retinoic acid receptor mediated by the orphan nuclear receptor TLX. Mol Cell Biol. 2000;20:8731–8739. doi: 10.1128/mcb.20.23.8731-8739.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Jetten AM, Kurebayashi S, Ueda E. The ROR nuclear orphan receptor subfamily: critical regulators of multiple biological processes. Prog Nucleic Acid Res Mol Biol. 2001;69:205–247. doi: 10.1016/s0079-6603(01)69048-2. [DOI] [PubMed] [Google Scholar]
- 221.Jetten AM. Recent advances in the mechanisms of action and physiological functions of the retinoid-related orphan receptors (RORs) Curr Drug Targets Inflamm Allergy. 2004;3:395–412. doi: 10.2174/1568010042634497. [DOI] [PubMed] [Google Scholar]
- 222.Kallen J, Schlaeppi JM, Bitsch F, Delhon I, Fournier B. Crystal structure of the human RORalpha Ligand binding domain in complex with cholesterol sulfate at 2.2 A. J Biol Chem. 2004;279:14033–14038. doi: 10.1074/jbc.M400302200. [DOI] [PubMed] [Google Scholar]
- 223.Stehlin-Gaon C, Willmann D, Zeyer D, Sanglier S, Van Dorsselaer A, Renaud JP, et al. All-trans retinoic acid is a ligand for the orphan nuclear receptor ROR beta. Nat Struct Biol. 2003;10:820–825. doi: 10.1038/nsb979. [DOI] [PubMed] [Google Scholar]
- 224.Dussault I, Fawcett D, Matthyssen A, Bader JA, Giguere V. Orphan nuclear receptor ROR alpha-deficient mice display the cerebellar defects of staggerer. Mech Dev. 1998;70:147–153. doi: 10.1016/s0925-4773(97)00187-1. [DOI] [PubMed] [Google Scholar]
- 225.Mamontova A, Seguret-Mace S, Esposito B, Chaniale C, Bouly M, Delhaye-Bouchaud N, et al. Severe atherosclerosis and hypoalphalipoproteinemia in the staggerer mouse, a mutant of the nuclear receptor RORalpha. Circulation. 1998;98:2738–2743. doi: 10.1161/01.cir.98.24.2738. [DOI] [PubMed] [Google Scholar]
- 226.Lau P, Nixon SJ, Parton RG, Muscat GE. RORalpha regulates the expression of genes involved in lipid homeostasis in skeletal muscle cells: caveolin-3 and CPT-1 are direct targets of ROR. J Biol Chem. 2004;279:36828–36840. doi: 10.1074/jbc.M404927200. [DOI] [PubMed] [Google Scholar]
- 227.Wada T, Kang HS, Jetten AM, Xie W. The emerging role of nuclear receptor RORalpha and its crosstalk with LXR in xeno - and endobiotic gene regulation. Exp Biol Med (Maywood) 2008;233:1191–1201. doi: 10.3181/0802-MR-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wiesenberg I, Missbach M, Kahlen JP, Schrader M, Carlberg C. Transcriptional activation of the nuclear receptor RZR alpha by the pineal gland hormone melatonin and identification of CGP 52608 as a synthetic ligand. Nucleic Acids Res. 1995;23:327–333. doi: 10.1093/nar/23.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sumi Y, Yagita K, Yamaguchi S, Ishida Y, Kuroda Y, Okamura H. Rhythmic expression of ROR beta mRNA in the mice suprachiasmatic nucleus. Neurosci Lett. 2002;320:13–16. doi: 10.1016/s0304-3940(02)00011-3. [DOI] [PubMed] [Google Scholar]
- 230.Schaeren-Wiemers N, Andre E, Kapfhammer JP, Becker-Andre M. The expression pattern of the orphan nuclear receptor RORbeta in the developing and adult rat nervous system suggests a role in the processing of sensory information and in circadian rhythm. Eur J NeuroSci. 1997;9:2687–2701. doi: 10.1111/j.1460-9568.1997.tb01698.x. [DOI] [PubMed] [Google Scholar]
- 231.Kurebayashi S, Ueda E, Sakaue M, Patel DD, Medvedev A, Zhang F, et al. Retinoid-related orphan receptor gamma (RORgamma) is essential for lymphoid organogenesis and controls apoptosis during thymopoiesis. Proc Natl Acad Sci USA. 2000;97:10132–10137. doi: 10.1073/pnas.97.18.10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Iyer AK, McCabe ER. Molecular mechanisms of DAX1 action. Mol Genet Metab. 2004;83:60–73. doi: 10.1016/j.ymgme.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 233.Zhang YH, Guo W, Wagner RL, Huang BL, McCabe L, Vilain E, et al. DAX1 mutations map to putative structural domains in a deduced three-dimensional model. Am J Hum Genet. 1998;62:855–864. doi: 10.1086/301782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Goodfellow PN, Camerino G. DAX-1, an ‘antitestis’ gene. Cell Mol Life Sci. 1999;55:857–863. doi: 10.1007/PL00013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Muscatelli F, Strom TM, Walker AP, Zanaria E, Recan D, Meindl A, et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. 1994;372:672–676. doi: 10.1038/372672a0. [DOI] [PubMed] [Google Scholar]
- 236.Swain A, Narvaez V, Burgoyne P, Camerino G, Lovell-Badge R. Dax1 antagonizes Sry action in mammalian sex determination. Nature. 1998;391:761–767. doi: 10.1038/35799. [DOI] [PubMed] [Google Scholar]
- 237.Billiard J, Moran RA, Whitley MZ, Chatterjee-Kishore M, Gillis K, Brown EL, et al. Transcriptional profiling of human osteoblast differentiation. J Cell Biochem. 2003;89:389–400. doi: 10.1002/jcb.10514. [DOI] [PubMed] [Google Scholar]
- 238.Zhou J, Oakley RH, Cidlowski JA. DAX-1 (dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X-chromosome, gene 1) selectively inhibits transactivation but not transrepression mediated by the glucocorticoid receptor in a LXXLL-dependent manner. Mol Endocrinol. 2008;22:1521–1534. doi: 10.1210/me.2007-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lee HK, Lee YK, Park SH, Kim YS, Lee JW, Kwon HB, et al. Structure and expression of the orphan nuclear receptor SHP gene. J Biol Chem. 1998;273:14398–14402. doi: 10.1074/jbc.273.23.14398. [DOI] [PubMed] [Google Scholar]
- 240.Nishigori H, Tomura H, Tonooka N, Kanamori M, Yamada S, Sho K, et al. Mutations in the small heterodimer partner gene are associated with mild obesity in Japanese subjects. Proc Natl Acad Sci USA. 2001;98:575–580. doi: 10.1073/pnas.021544398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Hung CC, Farooqi IS, Ong K, Luan J, Keogh JM, Pembrey M, et al. Contribution of variants in the small heterodimer partner gene to birthweight, adiposity, and insulin levels: mutational analysis and association studies in multiple populations. Diabetes. 2003;52:1288–1291. doi: 10.2337/diabetes.52.5.1288. [DOI] [PubMed] [Google Scholar]
- 242.Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell. 2002;2:721–731. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 243.Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, et al. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell. 2002;2:713–720. doi: 10.1016/s1534-5807(02)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Song G, Park K, Wang L. Gene expression profiling reveals a diverse array of pathways inhibited by nuclear receptor SHP during adipogenesis. Int J Clin Exp Pathol. 2009;2:275–285. [PMC free article] [PubMed] [Google Scholar]
- 245.Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77:481–490. doi: 10.1016/0092-8674(94)90211-9. [DOI] [PubMed] [Google Scholar]
- 246.Ikeda Y, Luo X, Abbud R, Nilson JH, Parker KL. The nuclear receptor steroidogenic factor 1 is essential for the formation of the ventromedial hypothalamic nucleus. Mol Endocrinol. 1995;9:478–486. doi: 10.1210/mend.9.4.7659091. [DOI] [PubMed] [Google Scholar]
- 247.Li Y, Choi M, Cavey G, Daugherty J, Suino K, Kovach A, et al. Crystallographic identification and functional characterization of phospholipids as ligands for the orphan nuclear receptor steroidogenic factor-1. Mol Cell. 2005;17:491–502. doi: 10.1016/j.molcel.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 248.Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, et al. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. 2005;120:343–355. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 249.Wang W, Zhang C, Marimuthu A, Krupka HI, Tabrizizad M, Shelloe R, et al. The crystal structures of human steroidogenic factor-1 and liver receptor homologue-1. Proc Natl Acad Sci USA. 2005;102:7505–7510. doi: 10.1073/pnas.0409482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Sablin EP, Blind RD, Krylova IN, Ingraham JG, Cai F, Williams JD, et al. Structure of SF-1 bound by different phospholipids: evidence for regulatory ligands. Mol Endocrinol. 2009;23:25–34. doi: 10.1210/me.2007-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Liu W, Liu M, Fan W, Nawata H, Yanase T. The Gly146Ala variation in human SF-1 gene: its association with insulin resistance and type 2 diabetes in Chinese. Diabetes Res Clin Pract. 2006;73:322–328. doi: 10.1016/j.diabres.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 252.Majdic G, Young M, Gomez-Sanchez E, Anderson P, Szczepaniak LS, Dobbins RL, et al. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology. 2002;143:607–614. doi: 10.1210/endo.143.2.8652. [DOI] [PubMed] [Google Scholar]
- 253.Del Tredici AL, Andersen CB, Currier EA, Ohrmund SR, Fairbain LC, Lund BW, et al. Identification of the first synthetic steroidogenic factor 1 inverse agonists: pharmacological modulation of steroidogenic enzymes. Mol Pharmacol. 2008;73:900–908. doi: 10.1124/mol.107.040089. [DOI] [PubMed] [Google Scholar]
- 254.Urs AN, Dammer E, Sewer MB. Sphingosine regulates the transcription of CYP17 by binding to steroidogenic factor-1. Endocrinology. 2006;147:5249–5258. doi: 10.1210/en.2006-0355. [DOI] [PubMed] [Google Scholar]
- 255.Fan W, Yanase T, Morinaga H, Gondo S, Okabe T, Nomura M, et al. Atrazine-induced aromatase expression is SF-1 dependent: implications for endocrine disruption in wildlife and reproductive cancers in humans. Environ Health Perspect. 2007;115:720–727. doi: 10.1289/ehp.9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Doghman M, Cazareth J, Douguet D, Madoux F, Hodder P, Lalli E. Inhibition of adrenocortical carcinoma cell proliferation by steroidogenic factor-1 inverse agonists. J Clin Endocrinol Metab. 2009;94:2178–2183. doi: 10.1210/jc.2008-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Giguere V, Yang N, Segui P, Evans RM. Identification of a new class of steroid hormone receptors. Nature. 1988;331:91–94. doi: 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- 258.Bonnelye E, Vanacker JM, Spruyt N, Alric S, Fournier B, Desbiens X, et al. Expression of the estrogen-related receptor 1 (ERR-1) orphan receptor during mouse development. Mech Dev. 1997;65:71–85. doi: 10.1016/s0925-4773(97)00059-2. [DOI] [PubMed] [Google Scholar]
- 259.Pettersson K, Svensson K, Mattsson R, Carlsson B, Ohlsson R, Berkenstam A. Expression of a novel member of estrogen response element-binding nuclear receptors is restricted to the early stages of chorion formation during mouse embryogenesis. Mech Dev. 1996;54:211–223. doi: 10.1016/0925-4773(95)00479-3. [DOI] [PubMed] [Google Scholar]
- 260.Hong H, Yang L, Stallcup MR. Hormone-independent transcriptional activation and coactivator binding by novel orphan nuclear receptor ERR3. J Biol Chem. 1999;274:22618–22626. doi: 10.1074/jbc.274.32.22618. [DOI] [PubMed] [Google Scholar]
- 261.Heard DJ, Norby PL, Holloway J, Vissing H. Human ERR-gamma, a third member of the estrogen receptor-related receptor (ERR) subfamily of orphan nuclear receptors: tissue-specific isoforms are expressed during development and in the adult. Mol Endocrinol. 2000;14:382–392. doi: 10.1210/mend.14.3.0431. [DOI] [PubMed] [Google Scholar]
- 262.Kallen J, Schlaeppi JM, Bitsch F, Filipuzzi I, Schilb A, Riou V, et al. Evidence for ligand-independent transcriptional activation of the human estrogen-related receptor alpha (ERRalpha): crystal structure of ERRalpha ligand binding domain in complex with peroxisome proliferator-activated receptor coactivator-1alpha. J Biol Chem. 2004;279:49330–49337. doi: 10.1074/jbc.M407999200. [DOI] [PubMed] [Google Scholar]
- 263.Horard B, Vanacker JM. Estrogen receptor-related receptors: orphan receptors desperately seeking a ligand. J Mol Endocrinol. 2003;31:349–357. doi: 10.1677/jme.0.0310349. [DOI] [PubMed] [Google Scholar]
- 264.Wang L, Zuercher WJ, Consler TG, Lambert MH, Miller AB, Orband-Miller LA, et al. X-ray crystal structures of the estrogen-related receptor-gamma ligand binding domain in three functional states reveal the molecular basis of small molecule regulation. J Biol Chem. 2006;281:37773–37781. doi: 10.1074/jbc.M608410200. [DOI] [PubMed] [Google Scholar]
- 265.Wang Y, Chirgadze NY, Briggs SL, Khan S, Jensen EV, Burris TP. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor beta. Proc Natl Acad Sci USA. 2006;103:9908–9911. doi: 10.1073/pnas.0510596103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Bonnelye E, Merdad L, Kung V, Aubin JE. The orphan nuclear estrogen receptor-related receptor alpha (ERRalpha) is expressed throughout osteoblast differentiation and regulates bone formation in vitro. J Cell Biol. 2001;153:971–984. doi: 10.1083/jcb.153.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Lu D, Kiriyama Y, Lee KY, Giguere V. Transcriptional regulation of the estrogen-inducible pS2 breast cancer marker gene by the ERR family of orphan nuclear receptors. Cancer Res. 2001;61:6755–6761. [PubMed] [Google Scholar]
- 268.Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci USA. 2004;101:6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Chung AC, Cooney AJ. The varied roles of nuclear receptors during vertebrate embryonic development. Nucl Recept Signal. 2003;1:e007. doi: 10.1621/nrs.01007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Yu S, Wang X, Ng CF, Chen S, Chan FL. ERRgamma suppresses cell proliferation and tumor growth of androgen-sensitive and androgen-insensitive prostate cancer cells and its implication as a therapeutic target for prostate cancer. Cancer Res. 2007;67:4904–4914. doi: 10.1158/0008-5472.CAN-06-3855. [DOI] [PubMed] [Google Scholar]
- 271.Sladek FM. Orphan receptor HNF-4 and liver-specific gene expression. Receptor. 1994;4:64. [PubMed] [Google Scholar]
- 272.Chartier FL, Bossu JP, Laudet V, Fruchart JC, Laine B. Cloning and sequencing of cDNAs encoding the human hepatocyte nuclear factor 4 indicate the presence of two isoforms in human liver. Gene. 1994;147:269–272. doi: 10.1016/0378-1119(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 273.Dhe-Paganon S, Duda K, Iwamoto M, Chi YI, Shoelson SE. Crystal structure of the HNF4 alpha ligand binding domain in complex with endogenous fatty acid ligand. J Biol Chem. 2002;277:37973–37976. doi: 10.1074/jbc.C200420200. [DOI] [PubMed] [Google Scholar]
- 274.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol. 2001;21:1393–1403. doi: 10.1128/MCB.21.4.1393-1403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Stegmann A, Hansen M, Wang Y, Larsen JB, Lund LR, Ritie L, et al. Metabolome, transcriptome, and bioinformatic cis-element analyses point to HNF-4 as a central regulator of gene expression during enterocyte differentiation. Physiol Genomics. 2006;27:141–155. doi: 10.1152/physiolgenomics.00314.2005. [DOI] [PubMed] [Google Scholar]
- 276.Hertz R, Ben-Haim N, Petrescu AD, Kalderon B, Berman I, Eldad N, et al. Rescue of MODY-1 by agonist ligands of hepatocyte nuclear factor-4alpha. J Biol Chem. 2003;278:22578–22585. doi: 10.1074/jbc.M212138200. [DOI] [PubMed] [Google Scholar]
- 277.Li T, Chiang JY. Rifampicin induction of CYP3A4 requires pregnane X receptor cross talk with hepatocyte nuclear factor 4alpha and coactivators, and suppression of small heterodimer partner gene expression. Drug Metab Dispos. 2006;34:756–764. doi: 10.1124/dmd.105.007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Cooney AJ, Lee CT, Lin SC, Tsai SY, Tsai MJ. Physiological function of the orphans GCNF and COUP-TF. Trends Endocrinol Metab. 2001;12:247–251. doi: 10.1016/s1043-2760(01)00424-6. [DOI] [PubMed] [Google Scholar]
- 279.Yan Z, Jetten AM. Characterization of the repressor function of the nuclear orphan receptor retinoid receptor-related testis-associated receptor/germ cell nuclear factor. J Biol Chem. 2000;275:35077–35085. doi: 10.1074/jbc.M005566200. [DOI] [PubMed] [Google Scholar]
- 280.Katz D, Niederberger C, Slaughter GR, Cooney AJ. Characterization of germ cell-specific expression of the orphan nuclear receptor, germ cell nuclear factor. Endocrinology. 1997;138:4364–4372. doi: 10.1210/endo.138.10.5444. [DOI] [PubMed] [Google Scholar]
- 281.Susens U, Aguiluz JB, Evans RM, Borgmeyer U. The germ cell nuclear factor mGCNF is expressed in the developing nervous system. Dev Neurosci. 1997;19:410–420. doi: 10.1159/000111238. [DOI] [PubMed] [Google Scholar]
- 282.Chung AC, Katz D, Pereira FA, Jackson KJ, DeMayo FJ, Cooney AJ, et al. Loss of orphan receptor germ cell nuclear factor function results in ectopic development of the tail bud and a novel posterior truncation. Mol Cell Biol. 2001;21:663–677. doi: 10.1128/MCB.21.2.663-677.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Freeman LA, Kennedy A, Wu J, Bark S, Remaley AT, Santamarina-Fojo S, et al. The orphan nuclear receptor LRH-1 activates the ABCG5/ABCG8 intergenic promoter. J Lipid Res. 2004;45:1197–1206. doi: 10.1194/jlr.C400002-JLR200. [DOI] [PubMed] [Google Scholar]
- 284.Wang ZN, Bassett M, Rainey WE. Liver receptor homologue-1 is expressed in the adrenal and can regulate transcription of 11 beta-hydroxylase. J Mol Endocrinol. 2001;27:255–258. doi: 10.1677/jme.0.0270255. [DOI] [PubMed] [Google Scholar]
- 285.Sablin EP, Krylova IN, Fletterick RJ, Ingraham HA. Structural basis for ligand-independent activation of the orphan nuclear receptor LRH-1. Mol Cell. 2003;11:1575–1585. doi: 10.1016/s1097-2765(03)00236-3. [DOI] [PubMed] [Google Scholar]
- 286.Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, et al. Synergy between LRH-1 and beta-catenin induces G1 cyclin-mediated cell proliferation. Mol Cell. 2004;15:499–509. doi: 10.1016/j.molcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 287.Clyne CD, Speed CJ, Zhou J, Simpson ER. Liver receptor homologue-1 (LRH-1) regulates expression of aromatase in preadipocytes. J Biol Chem. 2002;277:20591–20597. doi: 10.1074/jbc.M201117200. [DOI] [PubMed] [Google Scholar]
- 288.Schoonjans K, Dubuquoy L, Mebis J, Fayard E, Wendling O, Haby C, et al. Liver receptor homolog 1 contributes to intestinal tumor formation through effects on cell cycle and inflammation. Proc Natl Acad Sci USA. 2005;102:2058–2062. doi: 10.1073/pnas.0409756102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Clyne CD, Kovacic A, Speed CJ, Zhou J, Pezzi V, Simpson ER. Regulation of aromatase expression by the nuclear receptor LRH-1 in adipose tissue. Mol Cell Endocrinol. 2004;215:39–44. doi: 10.1016/j.mce.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 290.Coste A, Dubuquoy L, Barnouin R, Annicotte JS, Magnier B, Notti M, et al. LRH-1-mediated glucocorticoid synthesis in enterocytes protects against inflammatory bowel disease. Proc Natl Acad Sci USA. 2007;104:13098–13103. doi: 10.1073/pnas.0702440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Mullen EM, Gu P, Cooney AJ. Nuclear receptors in regulation of mouse ES cell pluripotency and differentiation. PPAR Res. 2007;2007:61563. doi: 10.1155/2007/61563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Kobayashi M, Takezawa S, Hara K, Yu RT, Umesono Y, Agata K, et al. Identification of a photoreceptor cell-specific nuclear receptor. Proc Natl Acad Sci USA. 1999;96:4814–4819. doi: 10.1073/pnas.96.9.4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Mitchell SJ, McHale DP, Campbell DA, Lench NJ, Mueller RF, Bundey SE, et al. A syndrome of severe mental retardation, spasticity, and tapetoretinal degeneration linked to chromosome 15q24. Am J Hum Genet. 1998;62:1070–1076. doi: 10.1086/301821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Haider NB, Jacobson SG, Cideciyan AV, Swiderski R, Streb LM, Searby C, et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat Genet. 2000;24:127–131. doi: 10.1038/72777. [DOI] [PubMed] [Google Scholar]
- 295.Chavala SH, Sari A, Lewis H, Pauer GJ, Simpson E, Hagstrom SA, et al. An Arg311Gln NR2E3 mutation in a family with classic Goldmann-Favre syndrome. Br J Ophthalmol. 2005;89:1065–1066. doi: 10.1136/bjo.2005.068130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Akhmedov NB, Piriev NI, Chang B, Rapoport AL, Hawes NL, Nishina PM, et al. A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc Natl Acad Sci USA. 2000;97:5551–5556. doi: 10.1073/pnas.97.10.5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Wolkenberg SE, Zhao Z, Kapitskaya M, Webber AL, Petrukhin K, Tang YS, et al. Identification of potent agonists of photoreceptor-specific nuclear receptor (NR2E3) and preparation of a radioligand. Bioorg Med Chem Lett. 2006;16:5001–5004. doi: 10.1016/j.bmcl.2006.07.056. [DOI] [PubMed] [Google Scholar]
- 298.Blumberg B, Kang H, Bolado J, Jr, Chen H, Craig AG, Moreno TA, et al. BXR, an embryonic orphan nuclear receptor activated by a novel class of endogenous benzoate metabolites. Genes Dev. 1998;12:1269–1277. doi: 10.1101/gad.12.9.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Baes M, Gulick T, Choi HS, Martinoli MG, Simha D, Moore DD. A new orphan member of the nuclear hormone receptor superfamily that interacts with a subset of retinoic acid response elements. Mol Cell Biol. 1994;14:1544–1552. doi: 10.1128/mcb.14.3.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Choi HS, Chung M, Tzameli I, Simha D, Lee YK, Seol W, et al. Differential transactivation by two isoforms of the orphan nuclear hormone receptor CAR. J Biol Chem. 1997;272:23565–23571. doi: 10.1074/jbc.272.38.23565. [DOI] [PubMed] [Google Scholar]
- 301.Seol W, Choi HS, Moore DD. Isolation of proteins that interact specifically with the retinoid X receptor: two novel orphan receptors. Mol Endocrinol. 1995;9:72–85. doi: 10.1210/mend.9.1.7760852. [DOI] [PubMed] [Google Scholar]
- 302.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 303.Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995;9:1033–1045. doi: 10.1101/gad.9.9.1033. [DOI] [PubMed] [Google Scholar]
- 304.Song C, Kokontis JM, Hiipakka RA, Liao S. Ubiquitous receptor: a receptor that modulates gene activation by retinoic acid and thyroid hormone receptors. Proc Natl Acad Sci USA. 1994;91:10809–10813. doi: 10.1073/pnas.91.23.10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Shinar DM, Endo N, Rutledge SJ, Vogel R, Rodan GA, Schmidt A. NER, a new member of the gene family encoding the human steroid hormone nuclear receptor. Gene. 1994;147:273–276. doi: 10.1016/0378-1119(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 306.Teboul M, Enmark E, Li Q, Wikstrom AC, Pelto-Huikko M, Gustafsson JA. OR-1, a member of the nuclear receptor superfamily that interacts with the 9-cis-retinoic acid receptor. Proc Natl Acad Sci USA. 1995;92:2096–2100. doi: 10.1073/pnas.92.6.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Sher T, Yi HF, McBride OW, Gonzalez FJ. cDNA cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry. 1993;32:5598–5604. doi: 10.1021/bi00072a015. [DOI] [PubMed] [Google Scholar]
- 308.Mukherjee R, Jow L, Noonan D, McDonnell DP. Human and rat peroxisome proliferator activated receptors (PPARs) demonstrate similar tissue distribution but different responsiveness to PPAR activators. J Steroid Biochem Mol Biol. 1994;51:157–166. doi: 10.1016/0960-0760(94)90089-2. [DOI] [PubMed] [Google Scholar]
- 309.Tugwood JD, Aldridge TC, Lambe KG, Macdonald N, Woodyatt NJ. Peroxisome proliferator-activated receptors: structures and function. Ann N Y Acad Sci. 1996;804:252–265. doi: 10.1111/j.1749-6632.1996.tb18620.x. [DOI] [PubMed] [Google Scholar]
- 310.Guan Y, Zhang Y, Davis L, Breyer MD. Expression of peroxisome proliferator-activated receptors in urinary tract of rabbits and humans. Am J Physiol. 1997;273:F1013–F1022. doi: 10.1152/ajprenal.1997.273.6.F1013. [DOI] [PubMed] [Google Scholar]
- 311.Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Greene ME, Blumberg B, McBride OW, Yi HF, Kronquist K, Kwan K, et al. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: expression in hematopoietic cells and chromosomal mapping. Gene Expr. 1995;4:281–299. [PMC free article] [PubMed] [Google Scholar]
- 313.Zhu Y, Alvares K, Huang Q, Rao MS, Reddy JK. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J Biol Chem. 1993;268:26817–26820. [PubMed] [Google Scholar]
- 314.Elbrecht A, Chen Y, Cullinan CA, Hayes N, Leibowitz M, Moller DE, et al. Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors gamma 1 and gamma 2. Biochem Biophys Res Commun. 1996;224:431–437. doi: 10.1006/bbrc.1996.1044. [DOI] [PubMed] [Google Scholar]
- 315.Scearce LM, Laz TM, Hazel TG, Lau LF, Taub R. RNR-1, a nuclear receptor in the NGFI-B/Nur77 family that is rapidly induced in regenerating liver. J Biol Chem. 1993;268:8855–8861. [PubMed] [Google Scholar]
- 316.Mages HW, Rilke O, Bravo R, Senger G, Kroczek RA. NOT, a human immediate-early response gene closely related to the steroid/thyroid hormone receptor NAK1/TR3. Mol Endocrinol. 1994;8:1583–1591. doi: 10.1210/mend.8.11.7877627. [DOI] [PubMed] [Google Scholar]
- 317.Milbrandt J. Nerve growth factor induces a gene homologous to the glucocorticoid receptor gene. Neuron. 1988;1:183–188. doi: 10.1016/0896-6273(88)90138-9. [DOI] [PubMed] [Google Scholar]
- 318.Hazel TG, Nathans D, Lau LF. A gene inducible by serum growth factors encodes a member of the steroid and thyroid hormone receptor superfamily. Proc Natl Acad Sci USA. 1988;85:8444–8448. doi: 10.1073/pnas.85.22.8444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Ryseck RP, Macdonald-Bravo H, Mattei MG, Ruppert S, Bravo R. Structure, mapping and expression of a growth factor inducible gene encoding a putative nuclear hormonal binding receptor. EMBO J. 1989;8:3327–3335. doi: 10.1002/j.1460-2075.1989.tb08494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Jackson A, Panayiotidis P, Foroni L. The human homologue of the Drosophila tailless gene (TLX): characterization and mapping to a region of common deletion in human lymphoid leukemia on chromosome 6q21. Genomics. 1998;50:34–43. doi: 10.1006/geno.1998.5270. [DOI] [PubMed] [Google Scholar]
- 321.Hollemann T, Bellefroid E, Pieler T. The Xenopus homologue of the Drosophila gene tailless has a function in early eye development. Development. 1998;125:2425–2432. doi: 10.1242/dev.125.13.2425. [DOI] [PubMed] [Google Scholar]
- 322.Guo W, Lovell RS, Zhang YH, Huang BL, Burris TP, Craigen WJ, et al. Ahch, the mouse homologue of DAX1: cloning, characterization and synteny with GyK, the glycerol kinase locus. Gene. 1996;178:31–34. doi: 10.1016/0378-1119(96)00320-4. [DOI] [PubMed] [Google Scholar]
- 323.Giguere V, Tini M, Flock G, Ong E, Evans RM, Otulakowski G. Isoform-specific amino-terminal domains dictate DNA-binding properties of ROR alpha, a novel family of orphan hormone nuclear receptors. Genes Dev. 1994;8:538–553. doi: 10.1101/gad.8.5.538. [DOI] [PubMed] [Google Scholar]
- 324.Becker-Andre M, Andre E, DeLamarter JF. Identification of nuclear receptor mRNAs by RT-PCR amplification of conserved zinc-finger motif sequences. Biochem Biophys Res Commun. 1993;194:1371–1379. doi: 10.1006/bbrc.1993.1976. [DOI] [PubMed] [Google Scholar]
- 325.Matsui T, Sashihara S, Oh Y, Waxman SG. An orphan nuclear receptor, mROR alpha, and its spatial expression in adult mouse brain. Brain Res Mol Brain Res. 1995;33:217–226. doi: 10.1016/0169-328x(95)00126-d. [DOI] [PubMed] [Google Scholar]
- 326.Matysiak-Scholze U, Nehls M. The structural integrity of ROR alpha isoforms is mutated in staggerer mice: cerebellar coexpression of ROR alpha1 and ROR alpha4. Genomics. 1997;43:78–84. doi: 10.1006/geno.1997.4757. [DOI] [PubMed] [Google Scholar]
- 327.Masuda N, Yasumo H, Tamura T, Hashiguchi N, Furusawa T, Tsukamoto T, et al. An orphan nuclear receptor lacking a zinc-finger DNA-binding domain: interaction with several nuclear receptors. Biochim Biophys Acta. 1997;1350:27–32. doi: 10.1016/s0167-4781(96)00196-0. [DOI] [PubMed] [Google Scholar]
- 328.Oba K, Yanase T, Nomura M, Morohashi K, Takayanagi R, Nawata H. Structural characterization of human Ad4bp (SF-1) gene. Biochem Biophys Res Commun. 1996;226:261–267. doi: 10.1006/bbrc.1996.1343. [DOI] [PubMed] [Google Scholar]
- 329.Yu RN, Ito M, Jameson JL. The murine Dax-1 promoter is stimulated by SF-1 (steroidogenic factor-1) and inhibited by COUP-TF (chicken ovalbumin upstream promoter-transcription factor) via a composite nuclear receptor-regulatory element. Mol Endocrinol. 1998;12:1010–1022. doi: 10.1210/mend.12.7.0131. [DOI] [PubMed] [Google Scholar]
- 330.Rust W, Stedronsky K, Tillmann G, Morley S, Walther N, Ivell R. The role of SF-1/Ad4BP in the control of the bovine gene for the steroidogenic acute regulatory (StAR) protein. J Mol Endocrinol. 1998;21:189–200. doi: 10.1677/jme.0.0210189. [DOI] [PubMed] [Google Scholar]
- 331.Val P, Lefrancois-Martinez AM, Veyssiere G, Martinez A. SF-1 a key player in the development and differentiation of steroidogenic tissues. Nucl Recept. 2003;1:8. doi: 10.1186/1478-1336-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Yang N, Shigeta H, Shi H, Teng CT. Estrogen-related receptor, hERR1, modulates estrogen receptor-mediated response of human lactoferrin gene promoter. J Biol Chem. 1996;271:5795–5804. doi: 10.1074/jbc.271.10.5795. [DOI] [PubMed] [Google Scholar]
- 333.Sladek FM, Zhong WM, Lai E, Darnell Jr JE. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990;4:2353–2365. doi: 10.1101/gad.4.12b.2353. [DOI] [PubMed] [Google Scholar]
- 334.Holewa B, Zapp D, Drewes T, Senkel S, Ryffel GU. HNF4beta, a new gene of the HNF4 family with distinct activation and expression profiles in oogenesis and embryogenesis of Xenopus laevis. Mol Cell Biol. 1997;17:687–694. doi: 10.1128/mcb.17.2.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Ortlund EA, Lee Y, Solomon IH, Hager JM, Safi R, Choi Y, et al. Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nat Struct Mol Biol. 2005;12:357–363. doi: 10.1038/nsmb910. [DOI] [PubMed] [Google Scholar]
- 336.Higashiyama H, Billin AN, Okamoto Y, Kinoshita M, Asano S. Expression profiling of peroxisome proliferator-activated receptor-delta (PPAR-delta) in mouse tissues using tissue microarray. Histochem Cell Biol. 2007;127:485–494. doi: 10.1007/s00418-007-0279-5. [DOI] [PubMed] [Google Scholar]
- 337.Falender AE, Lanz R, Malenfant D, Belanger L, Richards JS. Differential expression of steroidogenic factor-1 and FTF/LRH-1 in the rodent ovary. Endocrinology. 2003;144:3598–3610. doi: 10.1210/en.2002-0137. [DOI] [PubMed] [Google Scholar]
- 338.Kudo T, Sutou S. Chicken LRH-1 gene is transcribed from multiple promoters in steroidogenic organs. Gene. 2006;367:38–45. doi: 10.1016/j.gene.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 339.Milam AH, Rose L, Cideciyan AV, Barakat MR, Tang WX, Gupta N, et al. The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc Natl Acad Sci USA. 2002;99:473–478. doi: 10.1073/pnas.022533099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Yanagi Y, Takezawa S, Kato S. Distinct functions of photoreceptor cell-specific nuclear receptor, thyroid hormone receptor beta2 and CRX in one photoreceptor development. Invest Ophthalmol Vis Sci. 2002;43:3489–3494. [PubMed] [Google Scholar]
- 341.Zechel C. The germ cell nuclear factor (GCNF) Mol Reprod Dev. 2005;72:550–556. doi: 10.1002/mrd.20377. [DOI] [PubMed] [Google Scholar]