

Abstract
Interferon-alpha (IFN-α) is an immunomodulatory cytokine that is used clinically for the treatment of melanoma in the adjuvant setting. The cellular actions of IFN-α are regulated by the suppressors of cytokine signaling (SOCS) family of proteins. We hypothesized that the anti-tumor activity of exogenous IFN-α would be enhanced in SOCS1-deficient mice. SOCS1-deficient (SOCS1−/−) or control (SOCS1+/+) mice on an IFN-γ−/− C57BL/6 background bearing intraperitoneal (i.p.) JB/MS murine melanoma cells were treated for 30 days with i.p. injections of IFN-A/D or PBS (vehicle). Log-rank Kaplan-Meier survival curves were used to evaluate survival. Tumor-bearing control SOCS1+/+ mice receiving IFN-A/D had significantly enhanced survival versus PBS–treated mice (P = 0.0048). The anti-tumor effects of IFN-A/D therapy were significantly enhanced in tumor-bearing SOCS1−/− mice; 75% of these mice survived tumor challenge, whereas PBS-treated SOCS1−/− mice all died at 13-16 days (P = 0.00038). Antibody (Ab) depletion of CD8+ T cells abrogated the anti-tumor effects of IFN-A/D in SOCS1−/− mice as compared with mice receiving a control antibody (P = 0.0021). CD4+ T-cell depletion from SOCS1−/− mice also inhibited the effects of IFN-A/D (P = 0.0003). IFN-A/D did not alter expression of CD80 or CD86 on splenocytes of SOCS1+/+ or SOCS1−/− mice, or the proportion of T regulatory cells or myeloid-derived suppressor cells in SOCS1+/+ or SOCS1−/− mice. An analysis of T-cell function did reveal increased proliferation of SOCS1-deficient splenocytes at baseline and in response to mitogenic stimuli. These data suggest that modulation of SOCS1 function in T-cell subsets could enhance the anti-tumor effects of IFN-α in the setting of melanoma.
Keywords: Interferon-alpha, Suppressors of cytokine signaling, T cell, Interferon
Introduction
Malignant melanoma is the most aggressive form of skin cancer and its worldwide incidence is rising faster than that of any other cancer [1]. Few treatment options are available for patients with metastatic disease, and standard chemotherapeutic agents are generally ineffective. Immunotherapy with cytokines such as interleukin-2 (IL-2) or high-dose interferon-alpha (IFN-α) has been used extensively as a treatment option for patients with metastatic melanoma. Collectively, this approach has produced clinical responses in approximately 10% of patients [2, 3]. However, in patients who show therapeutic benefit, responses can be quite durable. Although widespread use of high-dose IFN-α for therapy of metastatic melanoma has declined in recent years, exogenous administration of this cytokine remains the only standard FDA-approved therapy for patients in the post-operative setting who have undergone complete excision of a primary tumor but are at high-risk for recurrence. Given the ability of IFN-α therapy to alter the disease course of melanoma, novel strategies to enhance its therapeutic efficacy deserve further investigation.
The Janus kinase and signal transducer and activator of transcription (Jak-STAT) signal transduction pathway is activated in both immune cells and tumor cells in response to IFN-α stimulation [4]. Upon binding to its receptor, IFN-α initiates a series of phosphorylation events in the upstream Jak1 and Tyk2 tyrosine kinases, which ultimately lead to phosphorylation of the STAT family of transcription factors [4, 5]. Upon activation, STAT proteins translocate to the cell nucleus together with other chaperone/adapter proteins to drive the expression of genes that mediate the biologic effects of the IFNs [6, 7]. We have previously shown that STAT1 signal transduction within immune cells, but not tumor cells, was critical for the anti-tumor activity of exogenously administered IFN-α [8, 9]. Therefore, factors that modulate STAT1-mediated signal transduction in immune cells might influence the anti-tumor activity of IFN-α.
The Suppressors of Cytokine Signaling (SOCS) proteins function as negative regulators of cytokine-induced Jak-STAT signal transduction [10]. The SOCS family of proteins has eight members, all of which have a central SH2 domain, an amino terminal domain, and a 40–amino acid motif at the carboxy terminus that is known as the SOCS box. SOCS1, in particular, is a potent inhibitor of signal transduction induced by both Type I (i.e. IFN-α, IFN-β) and Type II (IFN-γ) interferons, though its ability to bind the catalytic groove of Jak2 and inhibit its kinase activity [11, 12]. Although recombinant IFN-γ is not used clinically for immunotherapy of melanoma, its endogenous production is instrumental in regulating inflammation and various other aspects of immunologic function. The importance of SOCS proteins in normal physiologic processes is exemplified by the observation that homozygous SOCS1-deficient mice die of inflammatory sequelae unless the endogenous release of interferon-gamma (IFN-γ) is eliminated [13, 14]. SOCS1-deficient mice can be maintained without excess inflammation on an IFN-γ-deficient background.
We hypothesized that SOCS1-deficient mice would have an enhanced responsiveness to the anti-tumor effects of IFN-α. We now report that administration of IFN-α to melanoma-bearing SOCS1-deficient mice on the C57BL/6 background led to enhanced anti-tumor activity as compared with wild-type mice. We further demonstrate that both the CD8+ and CD4+ T-cell compartments were required for the augmented anti-tumor activity of IFN-α in this murine melanoma model and that splenocytes from SOCS1-deficient mice had increased in vitro proliferation at baseline and in response to standard mitogenic stimuli. These data suggest that targeting SOCS1 in the T-cell compartment could represent a novel means to enhance the anti-tumor activity of exogenously administered IFN-α.
Materials and methods
Reagents and cell lines
IFN-A/D (specific activity 1.1 × 108 U/mg; PBL Biomedical Laboratories, Piscataway, NJ) was used in all murine tumor challenge experiments and was administered via the intraperitoneal (i.p.) route at a dose of 2 × 104 U/day. IFN-A/D, or universal type I IFN, is a human hybrid recombinant type I IFN constructed from recombinant hu-IFN-αA and hu-IFN-αD that shows activity in a wide variety of mammalian cells [16]. The JB/MS murine melanoma cell line was obtained from Dr. Vincent Hearing (National Cancer Institute, Bethesda, MD) and cultured as previously described [15].
Antibodies for cell depletion experiments
Rat anti-mouse CD8 (clone 2.43) and rat anti-mouse CD4 (clone GK1.5) antibodies were purchased from the National Cell Culture Center (NCCC; Minneapolis, MN). For depletion experiments, 100 μg of antibody was injected i.p. on days −3, −1, +1, +3 and every 4 days thereafter in relation to the tumor challenge. Purified Rat IgG was used as a control (Sigma, St. Louis, MO). Depletion of CD8+ and CD4+ cells was confirmed by flow cytometric analysis of lymphocytes obtained from venous blood.
Animals
Homozygous SOCS1-deficient animals are susceptible to lethal inflammation in the presence of endogenous IFN-γ. Therefore, SOCS1-deficient animals must be maintained on an IFN-γ-deficient background (IFN-γ−/−). SOCS1−/−IFN-γ−/− mice were originally obtained on a mixed 129 Sv, C57BL/6 background from Dr. J. Ihle (St. Jude Children’s Research Institute, Memphis, TN). These animals were then backcrossed onto a C57BL/6 background as described [16]. This murine strain was generously provided to us by Dr. S. Ilangumaran (University of Sherbrooke, Sherbrooke, QC, Canada). Genotyping of SOCS1-deficient mice and confirmation of SOCS1 protein deficiencies were performed as previously described [13, 17].
Murine tumor models
An i.p. model of murine melanoma was used to evaluate the in vivo efficacy of IFN-A/D as previously described [18]. Briefly, mice were injected i.p. with 106 JB/MS murine melanoma cells and received daily i.p. injections of either PBS (negative control) or IFN-A/D (2 × 104 U). Mice were examined daily, and those exhibiting signs of progressive disease were euthanized via CO2 inhalation. For a subset of mechanistic experiments, tumor-bearing mice were euthanized via CO2 inhalation following 10–14 consecutive days of treatment with either IFN-A/D or PBS (vehicle). Blood was obtained by cardiac puncture immediately following euthanasia. Spleens were removed aseptically, and mechanically dispersed through 70 μM cell strainers. Splenocytes were washed with PBS and 5% FBS, pelleted by centrifugation and resuspended in RPMI-1640 and 10% FBS. All experiments were conducted in compliance with the guidelines of the Institutional Laboratory Animal Care and Use Committee of The Ohio State University.
Flow cytometric analysis
Freshly isolated splenocytes were evaluated using extracellular flow cytometry with fluorochrome-conjugated antibodies specific for various cellular markers (CD4, CD8, Gr1, CD11b, CD80, CD86) (BD Bioscience, San Jose, CA). All samples were stained on ice for 30 min, washed in flow buffer (PBS + 5% fetal bovine serum), and fixed in 1% formalin prior to flow cytometric analysis. To detect the presence of CD4+CD25+FoxP3+ T regulatory cells within splenocytes, a commercially available murine regulatory T-cell staining kit was utilized according to manufacturer’s instructions (eBioscience, San Deigo, CA). A minimum of 10,000 events gated on the lymphocyte population was used for analysis by WinMDI (created by Joseph Trotter; available at: http://pingu.salk.edu/software.html) [18].
Measurement of cell proliferation by carboxyfluorescein diacetate, succinimidyl ester (CFSE) labeling
Splenocyte proliferation was measured by the CFSE Cell Proliferation assay and flow cytometry according to the manufacturer’s instructions (Invitrogen). Briefly, splenocytes were isolated from SOCS1−/− or IFN-γ−/− mice and T cells were negatively selected using a murine T-cell negative isolation kit (Invitrogen). T cells were then labeled with 10 μM CFSE and cultured in complete media with or without antibodies against CD3 and CD28 (10 μg/mL; eBioscience) for 72 h. At this time point, cells were harvested, fixed, and analyzed on a BD LSR II flow cytometer.
Statistical analysis
Overall survival was determined from the date of the first treatment to death or to the date of last observation. Kaplan-Meier survival curves were generated and compared using the log-rank test. Student’s t test was used to compare the percentage of T-cell proliferation between groups. Results were considered statistically significant if P < 0.05.
Results
SOCS1-deficiency enhances the anti-tumor effects of exogenous IFN-α in vivo. The anti-tumor effects of exogenous IFN-α were studied in a murine model of malignant melanoma in which SOCS1−/−IFN-γ−/− [SOCS1−/−] or SOCS1+/+IFN-γ−/− [GKO (gamma knock out) background] mice bearing JB/MS tumors received i.p. injections of IFN-A/D or PBS (vehicle) for 30 days. All animals in these studies were on the C57BL/6 background. This i.p. tumor model represents a scenario of widespread metastatic disease. This model was chosen as our group has previously shown exogenous IFN-α administration leads to a modest, but statistically significant survival advantage in wild-type, C57BL/6 mice [8]. As expected, treatment of tumor-bearing, SOCS1+/+IFN-γ−/− C57Bl/6 mice with IFN-A/D led to a statistically significant improvement in survival (approximately 7 days) as compared with treatment with PBS alone (Fig. 1). The anti-tumor effects of IFN-A/D were markedly enhanced in SOCS1−/−IFN-γ−/− mice, as 75% of these animals exhibited long-term survival as compared with IFN-A/D-treated SOCS1+/+IFN-γ−/− mice (Fig. 1). There was no evidence that mice with SOCS1 deficiency experienced increased systemic toxicity in response to IFN-A/D treatments [18]. SOCS1-deficient mice that survived following IFN-A/D treatment were observed for an additional 3 weeks following the completion of IFN-A/D treatment. At this time point, surviving mice were euthanized and exhibited no evidence of disease on necropsy.
Fig. 1.

SOCS1 deficiency enhances the anti-tumor effects of IFN-A/D in a murine model of malignant melanoma. Survival analysis of SOCS1+/+ IFN-γ−/− (GKO) and SOCS1−/− IFN-γ−/− mice (n = 5 per group) bearing intraperitoneal (i.p.) murine JB/MS melanoma cells. Mice were challenged with tumor on day 0. Beginning on day 1, mice received daily injections of PBS or 2 × 104 U of IFN-A/D and were monitored for survival. IFN-A/D significantly prolonged the survival of GKO mice (P = 0.0048) as compared with PBS-treated GKO mice. Survival of SOCS1−/− IFN-γ−/− mice was further prolonged as compared with IFN-A/D-treated GKO mice (P = 0.0046)
The enhanced anti-tumor activity of IFN-α in SOCS1-deficient mice is dependent on CD8+ T cells
The role of the CD8+ T-cell compartment in mediating the anti-tumor activity of IFN-A/D in this model was next examined. SOCS1-competent and SOCS1-deficient mice were depleted of CD8+ T cells via i.p. injection of a rat anti-mouse CD8 Ab and challenged with i.p. JB/MS tumors. Control mice received normal rat IgG. CD8+ T-cell depletion led to a modest reduction in the survival of tumor-bearing, SOCS-competent GKO (background) mice treated with IFN-A/D as compared with IFN-treated GKO mice that had received an isotype control Ab, but these results were not statistically significant (Fig. 2a). As expected, SOCS1-deficient mice receiving isotype control Ab displayed significantly prolonged survival in response to IFN-A/D therapy as compared with SOCS1 competent mice that received a control Ab and IFN therapy (Fig. 2b). However, depletion of CD8+ T cells abrogated the anti-tumor effects of IFN-A/D in SOCS1−/−IFN-γ−/− mice (Fig. 2b).
Fig. 2.

The anti-tumor effects of IFN-A/D in SOCS1-deficient mice is dependent on CD8+ T cells. (a) Tumor-bearing SOCS1+/+ IFN-γ−/− mice (n = 5 per group) were injected i.p. with CD8 depleting antibodies or isotype control on days −3, −1, +1, +3, and every 4 days thereafter. Mice were challenged with JB/MS tumors (i.p.) on day 0 and received daily i.p. injections of PBS or IFN-A/D as described above. There was no significant difference in survival of IFN-A/D-treated GKO mice receiving either CD8-depleting or control antibodies (P = 0.5782). (b) Survival of JB/MS bearing, SOCS1−/− IFN-γ−/− mice receiving an isotype control antibody (n = 5 per group) was significantly enhanced after IFN-A/D treatment (P = 0.0018). Treatment with CD8-depleting antibodies significantly reduced the anti-tumor effect of IFN-A/D injections (P = 0.0021)
The enhanced anti-tumor activity of IFN-α in SOCS1-deficient mice is also dependent upon the CD4+ T-cell compartment
The role for CD4+ T cell help in mediating the enhanced anti-tumor activity of IFN-A/D in SOCS1-deficient mice has not been explored (18). Using the JB/MS tumor model, we further demonstrated that Ab-mediated depletion of CD4+ T cells significantly reduced the anti-tumor effects of IFN-A/D in SOCS1−/−IFN-γ−/− mice as compared with mice treated with isotype control antibody (Fig. 3). These data suggest that CD4+ T cell help contributes to the enhanced anti-tumor action of IFN-A/D in SOCS1-deficient mice.
Fig. 3.

The anti-tumor effects of IFN-A/D in SOCS1-deficient mice are dependent on CD4+ T cells. SOCS1−/− IFN-γ−/− mice (n = 5 per group) were injected i.p. with CD4+ depleting antibodies or isotype control on days −3, −1, +1, +3, and every 4 days thereafter. Mice were challenged with JB/MS tumors (i.p.) on day 0 and received daily i.p. injections of PBS or IFN-A/D as described above. IFN-A/D-treated mice receiving CD4-depleting antibody had significantly reduced survival as compared with mice receiving isotype control antibody (P = 0.0003)
Prevalence of immunosuppressive cell subsets and immune cell expression of co-stimulatory molecules in SOCS1-deficient mice
It was hypothesized that the efficient tumor clearance in SOCS1-deficient animals may be the result of fewer numbers of cells with an immunosuppressive phenotype or higher expression of co-stimulatory molecules on T lymphocytes. We therefore conducted an immunophenotyping study in tumor-bearing, SOCS1−/−IFN-γ−/− and SOCS1+/+IFN-γ−/− mice following 14 consecutive days of treatment with IFN-A/D. The 14-day time point was chosen for the analysis of splenocytes because it would provide an opportunity to monitor immune subset differences across all treatment groups prior to death of control animals. There was no evidence for a significant difference in the CD4/CD8 ratio across strain or treatment groups (range 0.35–0.82, data not shown). In addition, there was no evidence that CD3+ T cells from tumor-bearing, SOCS1-deficient mice receiving IFN-A/D treatment displayed an increase in CD80 or CD86 co-stimulatory markers as compared with IFN-A/D-treated controls (Fig. 4a, b). The overall level of splenic T regulatory cells and MDSC was relatively low in all animals, consistent with an IFN-γ-deficient phenotype [19, 20]. Furthermore, treatment of tumor-bearing SOCS1−/− mice with IFN-A/D did not appreciably alter the percentage of these cell types as compared with GKO mice treated with IFN-A/D (Fig. 4c, d).
Fig. 4.
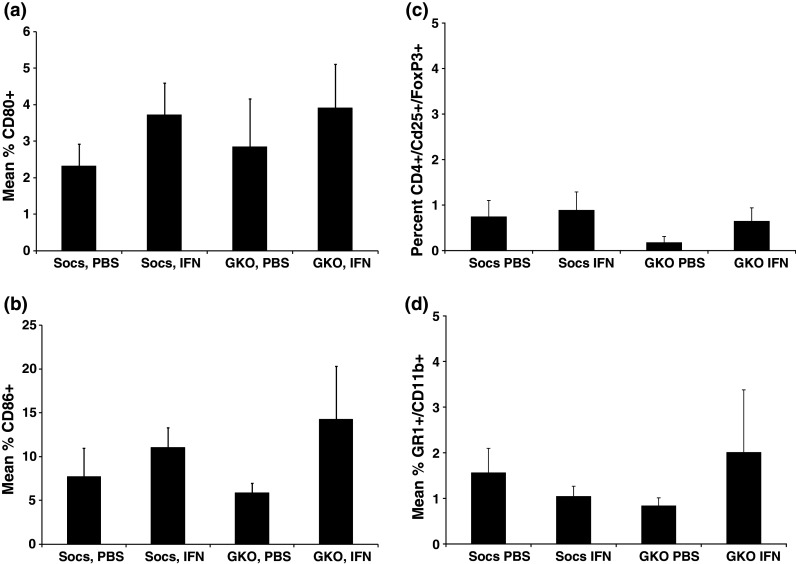
Expression of T regulatory cells and myeloid-derived suppressor cells (MDSC). The level of a CD80 and b CD86 expression was evaluated in splenocytes by flow cytometric analysis. Mice bearing i.p. JB/MS tumors were treated with PBS or IFN-A/D for 14 days and euthanized to obtain splenocytes for analysis. The number of c CD4+CD25+FoxP3+ T regulatory cells and d CD11b+Gr1+ myeloid-derived suppressor cells was evaluated in splenocytes by flow cytometric analysis. All flow cytometric data are presented as the mean total % of positive cells within splenocytes. Error bars represent the standard deviation from n = 4 mice per group. All flow cytometry data were collected using a minimum of 10,000 events gated on the lymphocyte population based on forward and side scatter properties, and all voltages were set using appropriate isotype control antibodies
Proliferation of splenocytes from SOCS1-deficient mice
We next tested whether splenocytes from SOCS1-deficient mice might display altered cell proliferation in response to CD3/CD28 ligation as a standard mitogenic stimulus. As expected, T-cell proliferation was enhanced by CD3/CD28 ligation in both SOCS1-deficient and GKO mice (Fig. 5). However, cell proliferation at baseline was significantly greater in SOCS1-deficient splenocytes as compared with splenoctyes from GKO mice (Fig. 5). These data suggest that SOCS1-deficient immune cells have altered baseline activity that impacts their responsiveness to CD3-mediated signaling pathways.
Fig. 5.

SOCS1-deficient splenocytes exhibit elevated in vitro proliferative activity T cells from naïve, non-tumor-bearing SOCS1+/+ IFN-γ−/− (GKO) and SOCS1−/− IFN-γ−/− (SOCS1) were assessed for cell proliferation as measured by CFSE labeling and flow cytometric analysis. CFSE proliferation was assessed following 72 h of culture with complete media or anti-CD3 and anti-CD28 stimulation. Significantly enhanced proliferation of SOCS1-deficient splenocytes was observed as compared with splenocytes from GKO mice (P = 0.01). Data are presented as the mean percent of proliferating T cells, and error bars represent the standard deviation from n = 3 mice per strain (GKO or SOCS1)
Discussion
We have demonstrated that administration of IFN-A/D to tumor-bearing, SOCS1-deficient mice on a C57BL/6 background enhanced survival as compared with IFN-A/D-treated SOCS1-competent mice. The enhanced anti-tumor effects of IFN-A/D were dependent upon CD8+ T cells and required CD4+ T cell help as well. There was no evidence for a difference in the percentage of CD4+CD25+FoxP3+ T regulatory cells or MDSC in splenocytes from IFN-treated SOCS1-deficient or GKO mice. IFN-A/D treatment also did not appear to augment the proliferative response of SOCS1-deficient splenocytes in vitro. However, the proliferation of SOCS1-deficient splenocytes was elevated both at baseline and in response to CD3/CD28 ligation, as compared with splenocytes from control animals. These data indicate that SOCS1 protein expression in T-lymphocyte compartments could function to limit the therapeutic efficacy of IFN-α.
We acknowledge that the immunologic phenotype of these animals is likely influenced by a systemic lack of IFN-γ. However, it remains noteworthy that exogenous IFN-A/D can elicit a potent anti-tumor effect even in the absence of this important immunoregulatory cytokine. The findings of the present study highlight a unique ability of IFN-α to promote anti-tumor activity by mechanisms involving the host immune system [8, 21, 22].
The present study shows that type I IFN can mediate dramatic anti-tumor activity in a SOCS1-deficient host [18]. These experiments were performed in a well-characterized murine strain and provide novel evidence for a role of the CD8+ and CD4+ T-cell compartment in contributing to the anti-tumor responses in this model. Although the exact mechanisms by which SOCS1-deficient T-cell compartments augment the cellular actions of exogenous IFN-α are unknown, our data are in agreement with prior reports documenting a hyper-reactive phenotype of T cells from SOCS1-deficient mice [24]. In addition, it has been demonstrated that SOCS1−/− mice develop increased numbers of mature CD8+ T cells relative to CD4+ T cells [23, 24]. Interestingly, the tumor-bearing animals used in the present study did not have this phenotype. It remains to be determined how SOCS1 inhibition might alter other aspects of T-cell function that are influenced by IFN-α treatment. For example, it is possible that SOCS1-deficient T cells may display a differential degree of cytotoxicity against tumor cell lines after IFN-α stimulation. This could potentially contribute to the enhanced anti-tumor activity of IFN-α in SOCS1-deficient mice. It will be important to test whether or not SOCS1 alters antigen-specific CD8+ T-cell-mediated cytotoxicity in future studies.
Although the present study highlights the potential for modulating SOCS1 expression in CD4+ and CD8+ T cells, reduced expression of SOCS1 in other cellular compartments may influence anti-tumor immunity. For example, Tanaka et al. have previously reported that mice harboring a conditional knockout of SOCS1 in the T-cell compartment fail to develop the Th17 subset of immune cells [25]. Despite these interesting findings, analysis of plasma from mice in the present study did not reveal any detectable IL-17 within any treatment group (data not shown). Evel-Kabler et al. have also demonstrated that adoptive transfer of dendritic cells transduced with a SOCS1-targeting siRNA construct can overcome self-tolerance and elicit enhanced anti-tumor responses against murine B16 melanoma tumors. These effects were dependent on the presence of IL-12 and were abrogated in the absence of CD4+ or CD8+ T cells [26]. These observations suggest that lymphocytes from SOCS1-deficient mice not only display enhanced signaling in response to IFN-α but may also be capable of enhanced uptake and cross-presentation of tumor-associated antigen by DCs. In addition, a recent study by Hashimoto et al. showed that conditional silencing of SOCS1 in macrophages can suppress carcinogen-induced colon tumor development and rejection of B16 melanoma [27].
Targeting negative regulatory proteins such as SOCS1 represents a potential approach for enhancing the anti-tumor effects of cytokine immunotherapy with IFN-α. However, pursuit of this approach should be exercised with caution. SOCS1 plays an important role in the regulation of cytokine pathways involved in normal inflammatory responses. This indispensable role for SOCS1 is emphasized by the potent and lethal inflammatory effects of IFN-γ in homozygous, SOCS1-deficient mice [13]. In addition, SOCS1 could also be involved in regulating cancer development in response to chronic inflammation. This possibility is illustrated by the observation that SOCS1−/− transgenic mice exhibit a high incidence of spontaneous gastrointestinal carcinomas [28]. One approach for minimizing potential inflammatory side effects associated with SOCS1 inhibition would be to target SOCS1 function in the precise cellular compartments necessary for mediating the enhanced anti-tumor efficacy of IFNs in SOCS1-deficient mice treated with IFN-α. Modulation of SOCS proteins specifically within CD8+ T cells or CD4+ T cells could be a valuable tool enhancing the activity of IFN-α in patients with advanced stage melanoma. This approach has the potential to minimize the putative off-target effects of SOCS1 inhibition on normal tissues.
Acknowledgments
We thank the OSU CCC Analytical Cytometry and Biostatistics Shared Resoucres. This work was supported by The Melanoma Research Foundation, The Valvano Foundation for Cancer Research Award (to G.B. Lesinski), National Institutes of Health (NIH) Grants CA84402, K24 CA93670 (to W.E. Carson), K22 CA134551 (to G.B. Lesinski), P30-CA16058, P01-CA95426 (to M.A. Caligiuri), T32 CA009338.
Footnotes
K. D. Guenterberg and G. B. Lesinski have contributed equally to this manuscript.
References
- 1.Lens MB, Dawes M. Global perspectives of contemporary epidemiological trends of cutaneous malignant melanoma. Br J Dermatol. 2004;150(2):179–185. doi: 10.1111/j.1365-2133.2004.05708.x. [DOI] [PubMed] [Google Scholar]
- 2.Atkins MB. Immunotherapy and experimental approaches for metastatic melanoma. Hematol Oncol Clin North Am. 1998;12(4):877–902. doi: 10.1016/S0889-8588(05)70029-0. [DOI] [PubMed] [Google Scholar]
- 3.Saleh FH, Crotty KA, Hersey P, Menzies SW, Rahman W. Autonomous histopathological regression of primary tumours associated with specific immune responses to cancer antigens. J Pathol. 2003;200(3):383–395. doi: 10.1002/path.1369. [DOI] [PubMed] [Google Scholar]
- 4.Darnell JE., Jr Studies of IFN-induced transcriptional activation uncover the Jak-Stat pathway. J Interferon Cytokine Res. 1998;18(8):549–554. doi: 10.1089/jir.1998.18.549. [DOI] [PubMed] [Google Scholar]
- 5.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 6.Haque SJ, Williams BR. Signal transduction in the interferon system. Semin Oncol. 1998;25(Suppl 1):14–22. [PubMed] [Google Scholar]
- 7.Ransohoff RM. Cellular responses to interferon’s and other cytokines: the JAK-STAT paradigm. N Engl J Med. 1998;338(9):616–618. doi: 10.1056/NEJM199802263380911. [DOI] [PubMed] [Google Scholar]
- 8.Lesinski GB, Anghelina M, Zimmerer J, Bakalakos T, Badgwell B, Parihar R, Hu Y, Becknell B, Abood G, Chaudhury AR, Magro C, Durbin J, Carson WE., III The antitumor effects of IFN-alpha are abrogated in a STAT1-deficient mouse. J Clin Invest. 2003;112(2):170–180. doi: 10.1172/JCI16603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lesinski GB, Valentino D, Hade EM, Jones S, Magro C, Chaudhury AR, Walker MJ, Carson WE., III Expression of STAT1 and STAT2 in malignant melanoma does not correlate with response to interferon-alpha adjuvant therapy. Cancer Immunol Immunother. 2005;54(9):815–825. doi: 10.1007/s00262-004-0649-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol. 2002;2(6):410–416. doi: 10.1038/nri818. [DOI] [PubMed] [Google Scholar]
- 11.Nicholson SE, Willson TA, Farley A, Starr R, Zhang JG, Baca M, Alexander WS, Metcalf D, Hilton DJ, Nicola NA. Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J. 1999;18(2):375–385. doi: 10.1093/emboj/18.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, Ohtsuka S, Imaizumi T, Matsuda T, Ihle JN, Yoshimura A. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999;18(5):1309–1320. doi: 10.1093/emboj/18.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, Yoshimura A, Ihle JN. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. 1999;98(5):609–616. doi: 10.1016/S0092-8674(00)80048-3. [DOI] [PubMed] [Google Scholar]
- 14.Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, Hilton DJ, Alexander WS. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci U S A. 1998;95(24):14395–14399. doi: 10.1073/pnas.95.24.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berkelhammer J, Oxenhandler RW, Hook RR, Jr, Hennessy JM. Development of a new melanoma model in C57BL/6 mice. Cancer Res. 1982;42(8):3157–3163. [PubMed] [Google Scholar]
- 16.Gagnon J, Ramanathan S, Leblanc C, Ilangumaran S. Regulation of IL-21 signaling by suppressor of cytokine signaling-1 (SOCS1) in CD8(+) T lymphocytes. Cell Signal. 2007;19(4):806–816. doi: 10.1016/j.cellsig.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 17.Zimmerer JM, Lesinski GB, Kondadasula SV, Karpa VI, Lehman A, Raychaudhury A, Becknell B, Carson WE., III IFN-alpha-induced signal transduction, gene expression, and antitumor activity of immune effector cells are negatively regulated by suppressor of cytokine signaling proteins. J Immunol. 2007;178(8):4832–4845. doi: 10.4049/jimmunol.178.8.4832. [DOI] [PubMed] [Google Scholar]
- 18.Lesinski GB, Kondadasula SV, Crespin T, Shen L, Kendra K, Walker M, Carson WE., III Multiparametric flow cytometric analysis of inter-patient variation in STAT1 phosphorylation following interferon Alfa immunotherapy. J Natl Cancer Inst. 2004;96(17):1331–1342. doi: 10.1093/jnci/djh252. [DOI] [PubMed] [Google Scholar]
- 19.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111(8):4233–4244. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 20.Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168(2):689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 21.Gogas H, Ioannovich J, Dafni U, Stavropoulou-Giokas C, Frangia K, Tsoutsos D, Panagiotou P, Polyzos A, Papadopoulos O, Stratigos A, Markopoulos C, Bafaloukos D, Pectasides D, Fountzilas G, Kirkwood JM. Prognostic significance of autoimmunity during treatment of melanoma with interferon. N Engl J Med. 2006;354(7):709–718. doi: 10.1056/NEJMoa053007. [DOI] [PubMed] [Google Scholar]
- 22.Moschos SJ, Kirkwood JM, Konstantinopoulos PA. Present status and future prospects for adjuvant therapy of melanoma: time to build upon the foundation of high-dose interferon alfa-2b. J Clin Oncol. 2004;22(1):11–14. doi: 10.1200/JCO.2004.10.952. [DOI] [PubMed] [Google Scholar]
- 23.Catlett IM, Hedrick SM. Suppressor of cytokine signaling 1 is required for the differentiation of CD4 + T cells. Nat Immunol. 2005;6(7):715–721. doi: 10.1038/ni1211. [DOI] [PubMed] [Google Scholar]
- 24.Chong MM, Cornish AL, Darwiche R, Stanley EG, Purton JF, Godfrey DI, Hilton DJ, Starr R, Alexander WS, Kay TW. Suppressor of cytokine signaling-1 is a critical regulator of interleukin-7-dependent CD8 + T cell differentiation. Immunity. 2003;18(4):475–487. doi: 10.1016/S1074-7613(03)00078-5. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka K, Ichiyama K, Hashimoto M, Yoshida H, Takimoto T, Takaesu G, Torisu T, Hanada T, Yasukawa H, Fukuyama S, Inoue H, Nakanishi Y, Kobayashi T, Yoshimura A. Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN-gamma on STAT3 and Smads. J Immunol. 2008;180(6):3746–3756. doi: 10.4049/jimmunol.180.6.3746. [DOI] [PubMed] [Google Scholar]
- 26.Evel-Kabler K, Song XT, Aldrich M, Huang XF, Chen SY. SOCS1 restricts dendritic cells’ ability to break self tolerance and induce antitumor immunity by regulating IL-12 production and signaling. J Clin Invest. 2006;116(1):90–100. doi: 10.1172/JCI26169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashimoto M, Ayada T, Kinjyo I, Hiwatashi K, Yoshida H, Okada Y, Kobayashi T, Yoshimura A. Silencing of SOCS1 in macrophages suppresses tumor development by enhancing antitumor inflammation. Cancer Sci. 2009;100(4):730–736. doi: 10.1111/j.1349-7006.2009.01098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, Koga K, Minoda Y, Sanada T, Yoshioka T, Mimata H, Kato S, Yoshimura A. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med. 2006;203(6):1391–1397. doi: 10.1084/jem.20060436. [DOI] [PMC free article] [PubMed] [Google Scholar]