

Abstract
Molecular dynamics simulations of unprecedented duration now can provide new insights into biomolecular mechanisms. Analysis of a 1-ms molecular dynamics simulation of the small protein bovine pancreatic trypsin inhibitor reveals that its main conformations have different thermodynamic profiles and that perturbation of a single geometric variable, such as a torsion angle or interresidue distance, can select for occupancy of one or another conformational state. These results establish the basis for a mechanism that we term entropy–enthalpy transduction (EET), in which the thermodynamic character of a local perturbation, such as enthalpic binding of a small molecule, is camouflaged by the thermodynamics of a global conformational change induced by the perturbation, such as a switch into a high-entropy conformational state. It is noted that EET could occur in many systems, making measured entropies and enthalpies of folding and binding unreliable indicators of actual thermodynamic driving forces. The same mechanism might also account for the high experimental variance of measured enthalpies and entropies relative to free energies in some calorimetric studies. Finally, EET may be the physical mechanism underlying many cases of entropy–enthalpy compensation.
Keywords: calorimetry, heat capacity, control, correlation, causation
The entropic and enthalpic components of free energy can be informative regarding the mechanisms of biophysical processes, like protein folding and protein–ligand binding. Moreover, information on changes in entropy and enthalpy can usefully guide the design of improved drug molecules (1), with advantageous specificity (2) and physical properties (3). However, calorimetric studies of biomolecular binding and folding often reveal unexpected changes in entropy and enthalpy that are difficult to interpret in terms of physical driving forces (4–8). Some of these puzzling results are instances of entropy–enthalpy compensation (9), a common but not universal (10, 11) phenomenon in which perturbations of a system that increase the enthalpy also increase the entropy or vice versa; therefore, the net change in the free energy remains small. Experimental error in measured enthalpies, particularly when obtained by the relatively imprecise van ’t Hoff method, can generate spurious entropy–enthalpy compensation (12); the nonphysical operation of Berkson's paradox (selection bias) (13) can also generate this compensation. However, analysis of collected calorimetric data for protein–ligand binding indicates that entropy–enthalpy compensation is a genuine and common physical phenomenon (13).
Today, novel computational technologies, such as graphical processor units (14–17) and the specialized Anton computer (18), are enabling dramatically longer molecular dynamics (MD) simulations than hitherto feasible. The enormous conformational sampling power of these technologies has the potential to open a new window on the molecular mechanisms underlying the thermodynamics of biomolecular systems. For example, it can provide increasingly accurate estimates of thermodynamic quantities that are notoriously difficult to converge, such as configurational entropy (19–21). Such thermodynamic results, coupled with atomistic structural and dynamical data, hold the potential to address some of the conundrums in calorimetric data outlined above. Recently, a 1-ms MD simulation of a relatively simple protein, bovine pancreatic trypsin inhibitor (BPTI), was released to the scientific community (22). This simulation provides a level of sampling unparalleled by any other biomolecular simulation to date, making it an ideal dataset for gaining a deeper understanding of the thermodynamics of proteins.
Here, we present a statistical thermodynamic analysis of the main conformational clusters observed in the 1-ms BPTI simulation using the maximum information spanning tree (MIST) (23, 24) approach to estimate configurational entropy. A central finding is that even this simple protein is delicately poised between states of similar free energy but very different entropy and enthalpy. We also introduce a method, based on the Jensen–Shannon divergence (25, 26), to show that there exists structural degrees of freedom that can easily be perturbed to drive the system into any of the major conformational states. These observations combine to yield a concept of entropy–enthalpy transduction (EET), in which local thermodynamic driving forces are transduced into different global thermodynamics through conformational selection. Finally, we provide a statistical thermodynamic description of EET that does not rely on the idealization of conformational selection. The methods and results presented here bear on the interpretation of simulation and calorimetry data and the physical basis for entropy–enthalpy compensation.
Results
We carried out a thermodynamic analysis of the trajectory of the longest MD simulation of a protein published to date (22), a 1-ms simulation of the small trypsin inhibitor BPTI. The simulation results are available to the scientific community and agree reasonably well with NMR data (27). The previously defined conformational clusters (22) 0, 1, and 2 (C0, C1, and C2) account for 92.3% of the simulation time and include the required number of simulation snapshots for thermodynamic analysis. Cluster C1 is the most rigid and similar to crystal structures of BPTI bound to trypsin (22), whereas C2 is the most mobile (Fig. 1, Upper).
Fig. 1.

(Upper) Representative backbone traces of the main conformational clusters C0, C1, and C2 as labeled, and their percent occupancies during the simulation; color and thickness of the traces indicate rms fluctuation of the backbone (narrow blue, 0.4 Å; thick red, 3.7 Å) after structural superposition. (Lower) Bar graph of the computed thermodynamics (kilocalories per mole) of clusters C0 (red) and C2 (green) relative to cluster C1. ΔE, total potential energy; ΔEprot-prot, potential energy of protein; ΔEprot-solv, interaction energy of protein and solvent; ΔEsolv-solv, potential energy of solvent; ΔG, free energy; −TΔS, total entropic free energy contribution; −TΔSconfig, configurational entropy estimated by MIST at second order and corrected by subtraction of matched results with block-shuffled data; −TΔSsolv, solvent entropy computed as −T(ΔS − ΔSconfig).
Although the occupancies (probabilities) of the three conformational clusters vary about fivefold, they are of similar stability, with free energies ranging over only ∼0.95 kcal/mol (1 kcal = 4.18 kJ) because of, essentially, the logarithmic relationship between probability and free energy (Methods). However, considerably larger entropy and enthalpy differences underlie these small free energy differences. For example, although clusters C1 and C2 are equistable to within 0.5 kcal/mol, their entropy (−TS) and enthalpy differences are, respectively, −3.2 and 3.7 kcal/mol; their configurational entropy differences are estimated at 19 kcal/mol, and the more mobile C2 has weaker intraprotein (25 kcal/mol) and intrasolvent (22 kcal/mol) interactions, partly balanced by stronger protein-solvent interactions (−42 kcal/mol). The magnitudes of these thermodynamic components reveal an underlying potential for much larger swings in energy and entropy: even slightly imbalanced perturbation of these terms, such as by mutations or changes in solvent composition, could generate larger net entropy and enthalpy differences between the clusters.
Because the three conformational clusters are so closely balanced in free energy, small perturbations of the system might easily shift their probabilities. Such shifts would then be expected to yield significant changes in the entropy–enthalpy balance at minimal cost in free energy. In particular, we hypothesized the existence of control variables, here defined as geometric variables xi, whose perturbations could select for one of three main conformational clusters and therefore, influence the global entropy and enthalpy. In searching for such variables, one may use the fact that a canonical MD simulation contains information on how the equilibrium distribution of conformations will respond to local perturbations, because perturbing the marginal probability distribution function (pdf) of a subset of variables xc from p(xc) to p′(xc) will cause the equilibrium joint pdf over all variables x to change from p(x) to p′(x) = p(x|xc)p′(xc). The conditional pdf thus represents a response function of the system to shifts in the pdf of the control variables (SI Text shows the derivation). We focus here on perturbations in which the accessible range of a single variable is reduced, because these perturbations are simple to analyze; also, good statistics are available for the associated conditional probabilities. Thus, for each cluster Ca, where a = 0, 1, or 2, we sought a geometric variable xi, either an interatomic distance or a torsion angle, for which a range R could be identified such that, to good approximation, 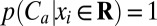 and
and 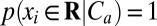 . These conditions are best met when there is little overlap of the pdfs of xi conditioned on the three clusters, p(xi|Ca). Because the Jensen–Shannon divergence (25, 26) is zero for identical pdfs and reaches its maximum (
. These conditions are best met when there is little overlap of the pdfs of xi conditioned on the three clusters, p(xi|Ca). Because the Jensen–Shannon divergence (25, 26) is zero for identical pdfs and reaches its maximum (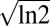 ) for nonoverlapping pdfs, we used it to define a metric Mia,
) for nonoverlapping pdfs, we used it to define a metric Mia,
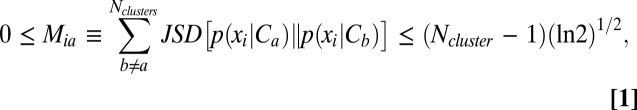 |
which scores the ability of variable xi to select cluster Ca from all other clusters Cb.
For each cluster, we found multiple carbon α-carbon α distances (xi) whose Mia scores are near the theoretical maximum of 1.67 (Fig. 2, row 1) and hence, had low overlap of the pdfs p(xi|C0), p(xi|C1), and p(xi|C2) (Fig. 2, row 3). For example, restricting the Thr11–Tyr35 distance to be greater than 6 Å (Fig. 2, row 2) drives the protein almost entirely into cluster C2. Interestingly, experiments have shown that the Tyr35Gly mutation makes the loop region of BPTI more flexible (28). Note that the significance of certain carbon α-carbon α distances does not derive from direct energetic interactions between the two corresponding residues, but rather from a complex set of couplings of each distance with the structure as a whole. We also used the same analysis to seek other classes of geometric variable that could yield similarly large values of the Mia metric and indeed, found a number of torsional pdfs with similarly low overlaps, as shown in Fig. 2, row 4. This result amplifies the observation that many control variables are available to shift the conformational state of this protein.
Fig. 2.

Identification and analysis of control variables. (Row 1) Heat maps of the Mia values for each cluster a, where the possible control variables xi are Cα–Cα distances among all 58 residues. The brightest pixels correspond to distances that can best isolate the respective cluster. (Row 2) Representative structure of the respective clusters, with the Cα–Cα distance, xi, corresponding to the largest Mia value shown as a dashed line. (Row 3) 1D pdfs of the Cα–Cα distances labeled in the row 2 for all three clusters; the pdf corresponding to the largest Mia value for a particular cluster is shaded. (Row 4) Analogous pdfs corresponding to the largest Mia value for each cluster when torsions are used as the xi instead of Cα–Cα distances.
We validated the ability of the control variables to isolate thermodynamically distinct clusters as follows. For each cluster, we chose the control variable with the greatest value of Mia (1.55, 1.52, and 1.59 for clusters C0, C1, and C2, respectively). We then reweighted the entire trajectory by imposing on the associated control variable an infinitely high-walled square-well potential positioned to select the bulk of the conformations associated with the desired cluster, while avoiding values of the control variable shared by any other cluster. The thermodynamic quantities of the reweighted trajectory were compared with those quantities associated with the original cluster. The results for the reweighted trajectories agree with the original cluster thermodynamics to within 0.3 kcal/mol for all ΔGab, 0.4 kcal/mol for all ΔEab, and 0.6 kcal/mol for all −TΔSab (Table S1). Thus, the control variables select the intended conformational substates with their respective thermodynamic signatures.
Discussion
Thermodynamic and Structural Analysis of the BPTI Simulation.
The present analysis of a long MD simulation indicates that BPTI can exist in multiple conformational states with distinct thermodynamic signatures, and that a variety of control variables are available to drive it into selected conformational states. Given that BPTI is a small protein rigidified by three disulfide bonds and not normally regarded as allosteric, it seems likely that larger, more flexible proteins have even greater scope to shift among global states of different entropy and enthalpy. If so, then various perturbations of biomolecules, such as ligand binding and mutation, should often be accompanied by changes in entropy and entropy that derive from global population shifts rather than local interactions. Even relatively subtle changes, such as chemical changes across a congeneric series of ligands, could produce large changes in entropy and enthalpy of binding if the global states are of similar free energy. Furthermore, because the local forces driving, for example, ligand binding need not be the same as the global thermodynamic consequences of binding, one may view the biomolecule as transducing the local driving force into a quite different global thermodynamic signature. This phenomenon, essentially a manifestation of thermodynamic linkage (29), may be termed entropy–enthalpy transduction (EET).
Illustration and Implications of EET.
Some of the implications of EET may be illustrated quantitatively by considering a protein P, which can occupy either a low-entropy state, A, or a high-entropy state, B, where the intrinsic free energy difference between the two states is x kcal/mol (Fig. 3, Upper). The ligand L binds exclusively to state B through mainly enthalpic forces. (Note that, as in the BPTI analysis above, the changes in entropy and enthalpy in this model are considered to include contributions from the protein, ligand, and solvent.) When x exceeds thermal energy (RT = 0.6 kcal/mol, and R is the gas constant), the free protein exists primarily in state B, and the overall binding thermodynamics are simply the thermodynamics of the ligand's binding to B and hence are seen to be enthalpy-driven (Fig. 3, Lower). When x < −RT, the free protein exists primarily in state A, but it can switch to the high-entropy state B when L binds; therefore, the binding process now seems entropy-driven. In this way, the protein's ability to switch states on binding enables transduction of the ligand's binding enthalpy into entropy. Conversely, the Tyr35Gly mutant of BPTI, which makes its loops more flexible without visibly perturbing the trypsin-bound conformation (30), causes trypsin binding to become more favorable enthalpically but less favorable entropically (30), which was expected for a mutation that selectively stabilizes a conformational state similar in character to cluster C2. The EET mechanism can also reinforce or amplify the driving forces of binding and other reactions. In the present example, this would occur if state B, rather than state A, were more stable enthalpically and less stable entropically. The enthalpic driving force for binding would then seem much stronger than the actual enthalpic forces driving ligand binding to the protein. In general, EET can generate shifts in enthalpy and entropy that are opposite to or far larger than expected based on local considerations. This finding implies that calorimetric data alone cannot be relied on to report the actual thermodynamic forces driving binding.
Fig. 3.

Two-state model system illustrating EET. (Upper) The protein P has two states A and B with a free energy difference of ΔGAB. By construction, state B is higher in entropy and lower in enthalpy than A. Ligand L binds only state B and hence, drives P from a two- to one-state system. The overall binding free energy is ΔGPL. (Lower) Overall thermodynamics of ligand–protein binding as a function of a small free energy bias, x, added to ΔGAB to shift the preferred unbound state of P from state A (x < 0) to state B (x > 0). Here, ΔHBL = −7.5, −TΔSBL = 2.5, ΔHAB = 10.0, −TΔSAB = −10.0, and RT = 0.6, where ΔHBL and −TΔSBL correspond to the enthalpy and entropy of the ligand L binding to state B. All quantities are in kilocalories per mole.
Especially when unexpected shifts are observed, a structure-based search for substantial and potentially functionally relevant conformational changes may be fruitful, as illustrated by prior studies of drug binding to various sequences of DNA (31) and the binding of agonists and antagonists to the A2B adenosine receptor (32). Changes in NMR-order parameters (33) can be informative in this regard. In some cases, they indicate increases in protein flexibility—and hence, presumably in configurational entropy—caused by protein–ligand binding (34–36). However, an increase in configurational entropy identified in this way may be balanced by a decrease in solvent entropy, as in the present calculations and further considered below, leading to either a positive or negative change in the net entropy. It is also worth noting that, although likely cases of EET amounting to 10 kcal/mol or more have been uncovered experimentally (31, 32, 35, 37, 38), identifying them required a combination of calorimetric and noncalorimetric experiments. Thus, there are presumably many systems where EET affects calorimetric results but has not been brought to light. Similarly, although measured changes in heat capacity are often used to elucidate the polar or nonpolar character of a protein–protein binding interface (39), if binding induces a significant conformational change in either protein, then the measured heat capacity may not accurately indicate the nature of the binding interface. The potential significance of this scenario is supported by the present calculations, because the computed heat capacity of conformational cluster C2 is ∼0.09 and 0.13 kcal/mol per Kelvin greater than the computed heat capacities of clusters C0 and C1, respectively (Table S2).
A biomolecular system may access a high-entropy state not only through increased protein motion, but also through a conformational shift that leads to release of water or other solvent molecules far from the site of the inducing perturbation. The potential role of solvent in EET is illustrated by thrombin, whose affinity for dissolved Na+ increases when substrate binds (40), leading to possible cobinding of substrate and Na+. Because binding of Na+ to the enzyme–substrate complex at 300 K is enthalpically favorable (−18 kcal/mol) and entropically unfavorable (15.6 kcal/mol) (40), cobinding of substrate with Na+ could cause a substrate with intrinsic binding that is entropy-driven to seem enthalpy-driven. More generally, subtle changes in solvent, or indeed, in other experimental conditions, could lead to marked changes in measured entropy and enthalpy, with minimal change in free energy. Thus, in the scenario of Fig. 3, small changes in experimental conditions may produce small changes in the balance between states A and B (i.e., changes in x) and hence, large variations in the apparent thermodynamics of binding. Such sensitivity to experimental conditions could help explain cases where the variance of calorimetric enthalpies exceeds the variance of free energy (41), although other factors can also contribute (42).
Finally, the EET mechanism could underlie many cases of entropy–enthalpy compensation, a phenomenon in which related processes, such as a drug-like molecule binding various similar biomolecules, are associated with similar free energy changes but seemingly disproportionately large entropy and enthalpy changes that correlate near-linearly with a slope or compensation temperature, 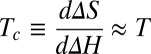 . Entropy–enthalpy compensation is often explained on physical grounds by arguing that deeper energy wells are narrower and hence lower in entropy than shallow ones (43, 44). Although this mechanism may indeed play a role, it is not known whether the depths and widths of energy wells in fact anticorrelate or show no correlation at all (45). Moreover, this mechanism would not explain why compensation is usually near-linear with slope
. Entropy–enthalpy compensation is often explained on physical grounds by arguing that deeper energy wells are narrower and hence lower in entropy than shallow ones (43, 44). Although this mechanism may indeed play a role, it is not known whether the depths and widths of energy wells in fact anticorrelate or show no correlation at all (45). Moreover, this mechanism would not explain why compensation is usually near-linear with slope 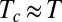 . The present analysis of BPTI suggests that a series of related proteins with similar binding sites could readily access different conformations of similar free energy but different enthalpy and entropy. As a consequence, the same ligand could bind the series of proteins with similar free energies but quite different entropies and enthalpies. In this way, the EET mechanism can lead to highly linear entropy–enthalpy compensation. This observation is broadly consistent with prior suggestions, derived from plausible mathematical models rather than detailed simulation, regarding the importance of delicately poised equilibria (46), large fluctuations, and a high density of states (47–51). Although the EET mechanism can, in this manner, generate a highly linear correlation between entropy and enthalpy, it can also generate other thermodynamic patterns. For example, if the various drug-like molecules that bind a protein drive it into global conformational states with free energies that vary substantially, one may still observe a degree of entropy–enthalpy compensation, but it will not be as clear cut as in a system where the protein instead moves among states of very similar free energy.
. The present analysis of BPTI suggests that a series of related proteins with similar binding sites could readily access different conformations of similar free energy but different enthalpy and entropy. As a consequence, the same ligand could bind the series of proteins with similar free energies but quite different entropies and enthalpies. In this way, the EET mechanism can lead to highly linear entropy–enthalpy compensation. This observation is broadly consistent with prior suggestions, derived from plausible mathematical models rather than detailed simulation, regarding the importance of delicately poised equilibria (46), large fluctuations, and a high density of states (47–51). Although the EET mechanism can, in this manner, generate a highly linear correlation between entropy and enthalpy, it can also generate other thermodynamic patterns. For example, if the various drug-like molecules that bind a protein drive it into global conformational states with free energies that vary substantially, one may still observe a degree of entropy–enthalpy compensation, but it will not be as clear cut as in a system where the protein instead moves among states of very similar free energy.
General Formalism for EET.
To unify and generalize the ideas above, we describe here how the concepts of control variables and response functions allow partitioning of the thermodynamics of protein–ligand binding into an intrinsic part, due to the ligand's direct interactions with the protein, and a transduced part, due to the long-ranged conformational response to the ligand's manipulation of control variables in the binding site: 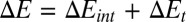 ,
, 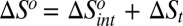 , and
, and 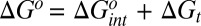 . (A detailed derivation is provided in SI Text.) Given the full set of spatial coordinates, x, the coordinates of the atoms local to the binding site are defined as the control variables, xc, which will be manipulated by the ligand when it binds. The coordinates of the remaining atoms are termed the transducing variables, xt, which will respond to the manipulation of the control variables. For the free protein, the potential energy function of the whole system
. (A detailed derivation is provided in SI Text.) Given the full set of spatial coordinates, x, the coordinates of the atoms local to the binding site are defined as the control variables, xc, which will be manipulated by the ligand when it binds. The coordinates of the remaining atoms are termed the transducing variables, xt, which will respond to the manipulation of the control variables. For the free protein, the potential energy function of the whole system 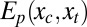 is expressed as the sum of a binding site part that depends only on the control variables and a transducing part that depends on the transducing variables as well. For the energy of the ligand–protein complex,
is expressed as the sum of a binding site part that depends only on the control variables and a transducing part that depends on the transducing variables as well. For the energy of the ligand–protein complex, 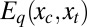 , the binding site part also depends on the conformation, position, and orientation of the ligand,
, the binding site part also depends on the conformation, position, and orientation of the ligand, 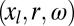 , whereas the ligand's long-ranged energetic interactions with atoms beyond the binding site may be approximated as a small constant value, Elt. Thus, binding of the ligand is approximated as directly affecting only the control variables, while the energy landscape of the transducing variables remains unchanged for any conformation of the control variables. This approximation should be good in a number of cases, such as when a small ligand binds a large protein. Hence,
, whereas the ligand's long-ranged energetic interactions with atoms beyond the binding site may be approximated as a small constant value, Elt. Thus, binding of the ligand is approximated as directly affecting only the control variables, while the energy landscape of the transducing variables remains unchanged for any conformation of the control variables. This approximation should be good in a number of cases, such as when a small ligand binds a large protein. Hence,
and
The energetic and entropic contributions of the transducing part of the system may now be expressed as functions of the control variables, 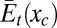 and
and 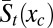 , respectively, and these functions can, furthermore, be combined to yield the contribution of the transducing variables to the potential of mean force over the control variable Wt(xc):
, respectively, and these functions can, furthermore, be combined to yield the contribution of the transducing variables to the potential of mean force over the control variable Wt(xc):
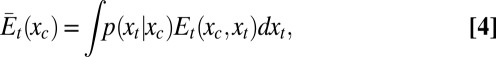 |
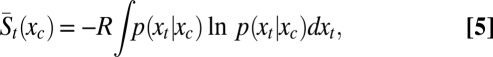 |
where R is the gas constant, and
These thermodynamic response functions, which apply to both the free protein and the complex, are mediated by p(xt|xc), the pdf of the transduced variables conditioned on the control variables. The net contribution of the transducing variables to the binding thermodynamics may now be written as
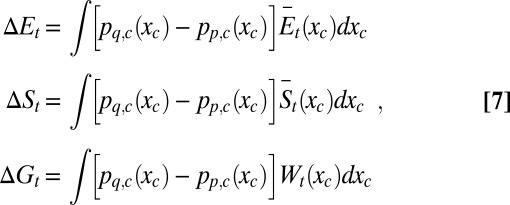 |
where pq,c(xc) and pp,c(xc) are the marginal pdfs over the control variables for the complex and free protein, respectively, at equilibrium.
The intrinsic binding terms, which make up the remainder of the binding thermodynamics, are as follows:
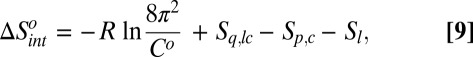 |
and
Here, the angle brackets signify Boltzmann averages for the complex, free protein, and free ligand, respectively; El is the potential function of the free ligand, and Sx signifies the Gibbs–Shannon entropies of the subscripted variables for the complex, free protein, and free ligand, respectively. Note that, although the intrinsic and transduced quantities are formally separated, the control and transducing variables are linked in the sense that the potential of mean force of the control variables, which influences both the intrinsic and transduced quantities, is determined jointly by both parts of the system. Thus, the control variables may be viewed as a boundary between two parts of the system, the ligand and transducing part of the protein. The present analysis, thus, is reminiscent of the fluctuating boundary ensemble (48).
The transduction mechanism, then, operates as follows. When a ligand binds, it changes the pdf of the control variables from pp,c(xc) to pq,c(xc), driving the transducing variables to adopt a new pdf dictated by the response function p(xt|xc) and thus, altering the thermodynamic contributions of the transducing part of the protein according to the above expressions. Because there is no requirement that this conformational distribution match any state of the free protein, conformational selection is not an issue. Also note that 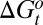 can be negative in sign. For example, stabilizing interactions internal to the binding site of the free protein might hold it in a closed conformation that keeps the transducing part in an unstable state; then, binding of the ligand could open up the binding site and allow the transducing part to relax.
can be negative in sign. For example, stabilizing interactions internal to the binding site of the free protein might hold it in a closed conformation that keeps the transducing part in an unstable state; then, binding of the ligand could open up the binding site and allow the transducing part to relax.
Entropy–enthalpy compensation occurs when 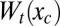 , the transducing variables’ contribution to the potential of mean force over the control variables, varies little (e.g., ∼RT or less) over some conformational region R. In this case, the transducing variables permit the conformation of the binding site to rearrange freely within the bounds of R in response to a variety of ligands. However, saying that
, the transducing variables’ contribution to the potential of mean force over the control variables, varies little (e.g., ∼RT or less) over some conformational region R. In this case, the transducing variables permit the conformation of the binding site to rearrange freely within the bounds of R in response to a variety of ligands. However, saying that 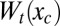 varies little with xc places no constraint on either
varies little with xc places no constraint on either 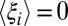 or
or 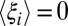 individually; therefore, these components can still vary substantially over R. As a consequence, there can be many possible changes in p(xc) that are associated with small changes in
individually; therefore, these components can still vary substantially over R. As a consequence, there can be many possible changes in p(xc) that are associated with small changes in 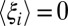 but large changes in
but large changes in 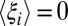 and
and 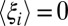 . Such changes will lead directly to entropy–enthalpy compensation across the series of ligands, with compensation temperatures, Tc, near unity, as commonly observed experimentally. Deviations from such perfect (or strong) (50) entropy–enthalpy compensation will occur, however, if some ligands drive the binding site conformation (i.e., the control variables) into regions of conformational space associated with large changes in
. Such changes will lead directly to entropy–enthalpy compensation across the series of ligands, with compensation temperatures, Tc, near unity, as commonly observed experimentally. Deviations from such perfect (or strong) (50) entropy–enthalpy compensation will occur, however, if some ligands drive the binding site conformation (i.e., the control variables) into regions of conformational space associated with large changes in 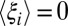 and hence, generate different values of
and hence, generate different values of 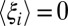 or if the intrinsic energy and entropy changes vary substantially across the set of ligands.
or if the intrinsic energy and entropy changes vary substantially across the set of ligands.
Methods
The numerical thermodynamic analysis presented above and detailed in SI Text is based on the recent 1-ms BPTI simulation (22). Because the simulation yields a canonical distribution, the relative free energy ΔGab of two clusters a and b can be estimated as −RT ln(na/nb), where ni is the number of simulation snapshots in cluster i. By the same token, the Boltzmann-averaged potential energy of cluster a, Ea, can be computed as a simple mean over the snapshots in each cluster. The entropy difference between clusters a and b then is given by −TΔSab = ΔGab − ΔEab. Furthermore, we decomposed the total energy into protein–protein, water–water, and protein–water contributions and estimated the protein configurational (19, 20) entropy of each cluster by means of the MIST approach (23, 24). Issues with noise and convergence were minimized by subtracting out the results for block-permuted data (SI Text). Solvation entropy differences were then estimated as differences between total entropy differences and configurational entropy differences (52). Finally, the heat capacity of each conformational cluster was computed by application of existing formulae (53). Fig. 1 and Table S2 summarize the results, and Figs. S1, S2, and S3 provide convergence plots of the various thermodynamic quantities.
Supplementary Material
Acknowledgments
We thank Drs. C. McClendon and L. Pierce for comments, and D. E. Shaw Research for providing the BPTI trajectory and their assignment of snapshots to conformational clusters. This publication was supported by National Institute of General Medical Sciences of the National Institutes of Health Grant GM61300.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213180109/-/DCSupplemental.
References
- 1.Freire E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov Today. 2008;13(19–20):869–874. doi: 10.1016/j.drudis2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohtaka H, et al. Thermodynamic rules for the design of high affinity HIV-1 protease inhibitors with adaptability to mutations and high selectivity towards unwanted targets. Int J Biochem Cell Biol. 2004;36(9):1787–1799. doi: 10.1016/j.biocel.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 3.Ferenczy GG, Keserũ GM. Thermodynamics guided lead discovery and optimization. Drug Discov Today. 2010;15(21–22):919–932. doi: 10.1016/j.drudis.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 4.Talhout R, Villa A, Mark AE, Engberts JBFN. Understanding binding affinity: A combined isothermal titration calorimetry/molecular dynamics study of the binding of a series of hydrophobically modified benzamidinium chloride inhibitors to trypsin. J Am Chem Soc. 2003;125(35):10570–10579. doi: 10.1021/ja034676g. [DOI] [PubMed] [Google Scholar]
- 5.Krishnamurthy VM, Bohall BR, Semetey V, Whitesides GM. The paradoxical thermodynamic basis for the interaction of ethylene glycol, glycine, and sarcosine chains with bovine carbonic anhydrase II: An unexpected manifestation of enthalpy/entropy compensation. J Am Chem Soc. 2006;128(17):5802–5812. doi: 10.1021/ja060070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010;53(14):5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeLorbe JE, et al. Thermodynamic and structural effects of conformational constraints in protein-ligand interactions. Entropic paradoxy associated with ligand preorganization. J Am Chem Soc. 2009;131(46):16758–16770. doi: 10.1021/ja904698q. [DOI] [PubMed] [Google Scholar]
- 8.Reynolds CH, Holloway MK. Thermodynamics of ligand binding and efficiency. ACS Med Chem Lett. 2011;2(6):433–437. doi: 10.1021/ml200010k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lumry R, Rajender S. Enthalpy-entropy compensation phenomena in water solutions of proteins and small molecules: A ubiquitous property of water. Biopolymers. 1970;9(10):1125–1227. doi: 10.1002/bip.1970.360091002. [DOI] [PubMed] [Google Scholar]
- 10.Gallicchio E, Kubo MM, Levy RM. Entropy-enthalpy compensation in solvation and ligand binding revisited. J Am Chem Soc. 1998;120(18):4526–4527. [Google Scholar]
- 11.Rekharsky MV, et al. A synthetic host-guest system achieves avidin-biotin affinity by overcoming enthalpy-entropy compensation. Proc Natl Acad Sci USA. 2007;104(52):20737–20742. doi: 10.1073/pnas.0706407105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krug RR, Hunter WG, Grieger RA. Statistical interpretation of enthalpy-entropy compensation. Nature. 1976;261(5561):566–567. [Google Scholar]
- 13.Olsson TSG, Ladbury JE, Pitt WR, Williams MA. Extent of enthalpy-entropy compensation in protein-ligand interactions. Protein Sci. 2011;20(9):1607–1618. doi: 10.1002/pro.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedrichs MS, et al. Accelerating molecular dynamic simulation on graphics processing units. J Comput Chem. 2009;30(6):864–872. doi: 10.1002/jcc.21209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Götz AW, et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J Chem Theory Comput. 2012;8(5):1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierce LCT, Salomon-Ferrer R, Augusto F de Oliveira C, McCammon JA, Walker RC. Routine access to millisecond time scale events with accelerated molecular dynamics. J Chem Theory Comput. 2012;8(9):2997–3002. doi: 10.1021/ct300284c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stone JE, Hardy DJ, Ufimtsev IS, Schulten K. GPU-accelerated molecular modeling coming of age. J Mol Graph Model. 2010;29(2):116–125. doi: 10.1016/j.jmgm.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw DE, et al. Anton, a special-purpose machine for molecular dynamics simulation. SIGARCH Comput Archit News. 2007;35(2):1–12. [Google Scholar]
- 19.Killian BJ, Yundenfreund Kravitz J, Gilson MK. Extraction of configurational entropy from molecular simulations via an expansion approximation. J Chem Phys. 2007;127(2):024107. doi: 10.1063/1.2746329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hnizdo V, Tan J, Killian BJ, Gilson MK. Efficient calculation of configurational entropy from molecular simulations by combining the mutual-information expansion and nearest-neighbor methods. J Comput Chem. 2008;29(10):1605–1614. doi: 10.1002/jcc.20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Numata J, Knapp E-W. Balanced and bias-corrected computation of conformational entropy differences for molecular trajectories. J Chem Theory Comput. 2012;8(4):1235–1245. doi: 10.1021/ct200910z. [DOI] [PubMed] [Google Scholar]
- 22.Shaw DE, et al. Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330(6002):341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 23.King BM, Tidor B. MIST: Maximum Information Spanning Trees for dimension reduction of biological data sets. Bioinformatics. 2009;25(9):1165–1172. doi: 10.1093/bioinformatics/btp109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King BM, Silver NW, Tidor B. Efficient calculation of molecular configurational entropies using an information theoretic approximation. J Phys Chem B. 2012;116(9):2891–2904. doi: 10.1021/jp2068123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin J. Divergence measures based on the Shannon entropy. IEEE Trans Inf Theory. 1991;37(1):145–151. [Google Scholar]
- 26.Endres DM, Schindelin JE. A new metric for probability distributions. IEEE Trans Inf Theory. 2003;49(7):1858–1860. [Google Scholar]
- 27.Xue Y, Ward JM, Yuwen T, Podkorytov IS, Skrynnikov NR. Microsecond time-scale conformational exchange in proteins: Using long molecular dynamics trajectory to simulate NMR relaxation dispersion data. J Am Chem Soc. 2012;134(5):2555–2562. doi: 10.1021/ja206442c. [DOI] [PubMed] [Google Scholar]
- 28.Hanson WM, Beeser SA, Oas TG, Goldenberg DP. Identification of a residue critical for maintaining the functional conformation of BPTI. J Mol Biol. 2003;333(2):425–441. doi: 10.1016/j.jmb.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 29.Wyman JJ., Jr Heme proteins. Adv Protein Chem. 1948;4:407–531. doi: 10.1016/s0065-3233(08)60011-x. [DOI] [PubMed] [Google Scholar]
- 30.Hanson WM, Domek GJ, Horvath MP, Goldenberg DP. Rigidification of a flexible protease inhibitor variant upon binding to trypsin. J Mol Biol. 2007;366(1):230–243. doi: 10.1016/j.jmb.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breslauer KJ, et al. Enthalpy-entropy compensations in drug-DNA binding studies. Proc Natl Acad Sci USA. 1987;84(24):8922–8926. doi: 10.1073/pnas.84.24.8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gessi S, et al. Thermodynamics of A2B adenosine receptor binding discriminates agonistic from antagonistic behaviour. Biochem Pharmacol. 2008;75(2):562–569. doi: 10.1016/j.bcp.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104(17):4546–4559. [Google Scholar]
- 34.Zídek L, Novotny MV, Stone MJ. Increased protein backbone conformational entropy upon hydrophobic ligand binding. Nat Struct Biol. 1999;6(12):1118–1121. doi: 10.1038/70057. [DOI] [PubMed] [Google Scholar]
- 35.Arumugam S, et al. Increased backbone mobility in β-barrel enhances entropy gain driving binding of N-TIMP-1 to MMP-3. J Mol Biol. 2003;327(3):719–734. doi: 10.1016/s0022-2836(03)00180-3. [DOI] [PubMed] [Google Scholar]
- 36.Diehl C, et al. Protein flexibility and conformational entropy in ligand design targeting the carbohydrate recognition domain of galectin-3. J Am Chem Soc. 2010;132(41):14577–14589. doi: 10.1021/ja105852y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edink E, Jansen C, Leurs R, de Esch IJP. The heat is on: Thermodynamic analysis in fragment-based drug discovery. Drug Discov Today Technol. 2010;7(3):189–201. doi: 10.1016/j.ddtec.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 38.Tzeng S-R, Kalodimos CG. Protein activity regulation by conformational entropy. Nature. 2012;488(7410):236–240. doi: 10.1038/nature11271. [DOI] [PubMed] [Google Scholar]
- 39.D’Aquino JA, et al. The magnitude of the backbone conformational entropy change in protein folding. Proteins. 1996;25(2):143–156. doi: 10.1002/(SICI)1097-0134(199606)25:2<143::AID-PROT1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 40.Wells CM, Di Cera E. Thrombin is a Na(+)-activated enzyme. Biochemistry. 1992;31(47):11721–11730. doi: 10.1021/bi00162a008. [DOI] [PubMed] [Google Scholar]
- 41.Myszka DG, et al. The ABRF-MIRG’02 study: Assembly state, thermodynamic, and kinetic analysis of an enzyme/inhibitor interaction. J Biomol Tech. 2003;14(4):247–269. [PMC free article] [PubMed] [Google Scholar]
- 42.Tellinghuisen J, Chodera JD. Systematic errors in isothermal titration calorimetry: Concentrations and baselines. Anal Biochem. 2011;414(2):297–299. doi: 10.1016/j.ab.2011.03.024. [DOI] [PubMed] [Google Scholar]
- 43.Dunitz JD. Win some, lose some: Enthalpy-entropy compensation in weak intermolecular interactions. Chem Biol. 1995;2(11):709–712. doi: 10.1016/1074-5521(95)90097-7. [DOI] [PubMed] [Google Scholar]
- 44.Searle MS, Westwell MS, Williams DH. Application of a generalised enthalpy-entropy relationship to binding cooperativity and weak associations in solution. J Chem Soc, Perkin Trans. 1995;2(1):141–151. [Google Scholar]
- 45.Ford DM. Enthalpy-entropy compensation is not a general feature of weak association. J Am Chem Soc. 2005;127(46):16167–16170. doi: 10.1021/ja054519c. [DOI] [PubMed] [Google Scholar]
- 46.Eftink MR, Anusiem AC, Biltonen RL. Enthalpy-entropy compensation and heat capacity changes for protein-ligand interactions: General thermodynamic models and data for the binding of nucleotides to ribonuclease A. Biochemistry. 1983;22(16):3884–3896. doi: 10.1021/bi00285a025. [DOI] [PubMed] [Google Scholar]
- 47.Weber G. van 't Hoff revisited: Enthalpy of association of protein subunits. J Phys Chem. 1995;99(3):1052–1059. [Google Scholar]
- 48.Qian H. Entropy-enthalpy compensation: Conformational fluctuation and induced-fit. J Chem Phys. 1998;109(22):10015–10017. [Google Scholar]
- 49.Qian H, Hopfield JJ. Entropy-enthalpy compensation: Perturbation and relaxation in thermodynamic systems. J Chem Phys. 1996;105(20):9292–9298. [Google Scholar]
- 50.Sharp K. Entropy-enthalpy compensation: Fact or artifact? Protein Sci. 2001;10(3):661–667. doi: 10.1110/ps.37801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Starikov EB, Nordén B. Enthalpy-entropy compensation: A phantom or something useful? J Phys Chem B. 2007;111(51):14431–14435. doi: 10.1021/jp075784i. [DOI] [PubMed] [Google Scholar]
- 52.Zhou H-X, Gilson MK. Theory of free energy and entropy in noncovalent binding. Chem Rev. 2009;109(9):4092–4107. doi: 10.1021/cr800551w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prabhu NV, Sharp KA. Heat capacity in proteins. Annu Rev Phys Chem. 2005;56:521–548. doi: 10.1146/annurev.physchem.56.092503.141202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.