

The major molecular mechanism of non-steroidal anti-inflammatory drugs (NSAIDs) is the inhibition of cyclooxygenases (COX) that leads to suppression of prostanoid formation.[1] Among the prostanoids, prostaglandin (PG) E2 is involved in several pathologic conditions such as fever, inflammation, pain, and cancer, but also in key physiological functions.[2] Accordingly, NSAIDs possess high efficacy against the aforementioned pathological states but also give rise to target-related side effects due to suppression of constitutively generated PGE2 with gastro-protective function and of PGs with other house-keeping properties.[1,3] Hence, novel pharmacological concepts are required that more selectively interfere with the generation of pathologically relevant PGs while sparing the suppression of prostanoids with homeostatic functionalities. Among the three mammalian PGE2 synthases, microsomal prostaglandin E2 synthase-1 (mPGES-1)[4] is an inducible isoform that is upregulated at various pathophysiological stages and, in conjunction with the inducible COX-2, produces massive PGE2 from PGH2 formed by COX enzymes from arachidonic acid.[5] Thus, mPGES-1 inhibitors could potentially possess high anti-inflammatory efficacy, while lacking NSAID-related toxicity.[5,6] Experiments with mPGES-1-deficient mice and preclinical studies with mPGES-1 inhibitors support the therapeutic potential of mPGES-1 blockers.[6,7]
The three-dimensional structure of mPGES-1,[8] site-directed mutagenesis data,[9] and computational pharmacophore models[10] might facilitate the successful discovery of potent and selective mPGES-1 inhibitors. We recently established two pharmacophore models to identify mPGES-1 inhibitors, and nine chemically diverse compounds from the US National Cancer Institute (NCI) and the commercial compound provider SPECS (www.specs.net) were identified in a virtual screening campaign as novel mPGES-1 inhibitors with IC50 values in the low micromolar range.[10b] The first model (M1) consists of a negatively ionizable feature, one aromatic ring feature, four hydrophobic features, and a shape restriction to limit the size of fitting compounds. The second model (M2) is a partial query of M1, in which the aromatic feature or one of the hydrophobic features is allowed to be omitted during the screening. While M1 achieved a more favorable enrichment of active compounds in a virtual screening, M2 correctly recognized chemically diverse mPGES-1 inhibitors for which M1 was too restrictive.[10b]
Here, we demonstrate the usefulness of these pharmacophore models and the success of the virtual screening for identification of lichen constituents as potent mPGES-1 inhibitors. In silico screening using the two pharmacophore models (M1 and M2) of a three-dimensional, multi-conformational natural product library, the Chinese herbal medicine (CHM) database, consisting of 10216 compounds found in medicinal preparations applied in traditional Chinese medicine,[11] resulted in hit rates of 0.04% with M1 and 0.6% with M2. Intriguingly, more than 10% of the 61 virtual hits (M1: perlatolic acid (8); M2: baeomycesic acid, barbatinic acid, diffrataic acid, evernic acid (7), gyrophoric acid, ramalic acid, and squamatic acid) were identified as constituents of lichen species, belonging to the chemical class of depsides. A set of ten compounds containing previously isolated lichen depsides (e.g., virtual hits 7 and 8) and a related group of depsidones was virtually screened with the established pharmacophore models (Figure 1). This resulted in the identification of two further virtual hits from M2, that is, physodic acid (2) and olivetoric acid (9). Six compounds (1, 3–6, and 10) did not map the features of M1 or M2 (Table 1).
Figure 1.

General structures of both the depside and depsidone scaffolds with numbering, and the chemical structures of lichen constituents 1–10 and reference mPGES-1 inhibitor 11.
Table 1.
Virtual prediction and the biological effects of lichen depsides and depsidones on the activity of mPGES-1 determined in a cell-free assay.
| Comp | Model[a] | Activity[b] [%] | IC50[c] [νm] |
|---|---|---|---|
| 1 | – | 74.6±10.7 | >10 |
| 2 | M2 | 14.9±4.9 | 0.43 |
| 3 | – | 60.8±10.7 | >10 |
| 4 | – | 75.5±12.9 | >10 |
| 5 | – | 73.3±9.7 | >10 |
| 6 | – | 89.1±12.5 | >10 |
| 7 | M2 | 53.9±2.7 | >10 |
| 8 | M1 | 12.8±3.7 | 0.4 |
| 9 | M2 | 22.1±5.6 | 1.15 |
| 10 | – | 87.5±13.2 | >10 |
Pharmacophore models: “M1”= retrieved by pharmacophore model M1; “M2”= retrieved by pharmacophore model M2; “–”= not found by M1 or M2.
mPGES-1 % remaining activity at 10 μm. Data represent the mean ±SE of n=3–4 independent experiments.
IC50 against mPGES-1. Values quoted were determined by extrapolation. See the Experimental Section for details.
Lichen secondary metabolites were proposed to exhibit antiinflammatory, antipyretic, and analgesic properties.[12] However, little is known about the bioactivities of lichen compounds on the PG biosynthetic pathway, and only one study has reported that lichen depsides and depsidones interfere with COX activity in a cell-free assay using rabbit renal microsomes.[13] To the best of our knowledge, inhibition of mPGES-1 by depsides and depsidones has not yet been reported in the literature, and any interference of these substances with PGE2 biosynthesis in isolated cells or in vivo is thus far unknown. In order to investigate whether or not lichen constituents 1–10 inhibit mPGES-1 activity, the compounds were analyzed in a well-established cell-free activity assay.[4,6] Inhibition of mPGES-1 activity (transformation of PGH2 to PGE2) was assessed using the microsomal fraction of IL-1β-stimulated A549 cells as an enzyme source and PGH2 as the substrate (20 μm).[14] This assay allows the definitive assessment of the enzymatic transformation of PGH2 to PGE2 by mPGES-1 and as such enables the direct identification of mPGES-1 inhibitors. Known mPGES-1 inhibitor MK-886 (11; Figure 1)[15] was used as a reference compound.
In a first screening round, all compounds were tested at a concentration of 10 μm. As shown in Table 1, depsidone 2 and the depsides perlatolic acid (8) and olivetoric acid (9) strongly suppressed mPGES-1 activity. Note that these three active compounds all contain at least one lipophilic alkyl chain with five or more C-atoms, which are absent in the other seven molecules. However, the depsidone salazinic acid (3) and the depside evernic acid (7), both of which lack extended lipophilic alkyl residues, also inhibited mPGES-1, although greater than 50% enzyme activity still remained. In contrast, fumarprotocetraric acid (1), variolaric acid (4), scensidin (5), methyl-betaorcinol-carboxylate (6) and atranorin (10) were inactive or only showed very weak inhibition of mPGES-1 (Table 1).
A more detailed analysis of compounds 2, 8, and 9 revealed potent and concentration-dependent inhibition of mPGES-1 with IC50 values of 0.43, 0.4, and 1.15 μm, respectively (Figure 2). For comparison, known mPGES-1 inhibitor 11 was analyzed under the same assay conditions, and an IC50 value of 2.4 μm was determined. Compounds 2 and 8 are remarkably potent inhibitors of mPGES-1, exhibiting a ten-fold greater activity over reference compound 11, with IC50 values of 0.43 and 0.4 μm, respectively. To the best of our knowledge, among natural products, only curcumin isolated from Curcuma longa (IC50 = 0.3 μm)[16] is comparably potent, whereas all other natural compounds, such as hyperforin from Hypericum perforatum (commonly known as St. John’s wort),[17] arzanol from Helichrysum italicum,[18] myrtucommulone from Myrtus communis,[19] boswellic acids from Boswellia species,[20] epigallocatechin-3-gallate from Camellia sinensis (commonly known as green tea),[21] and garcinol from Garcinia indica,[22] were less efficient mPGES-1 inhibitors. Interestingly, compounds 2, 8, and 9 caused only moderate inhibition of COX-1 activity (IC50> 30 μm) and failed to significantly inhibit COX-2 at a concentration of up to 30 μm, whereas the NSAIDs indomethacin and celecoxib both efficiently inhibited COX-1/2 activity (data not shown). Together, lichen compounds 2, 8, and 9 could represent new lead structures for the development of novel anti-inflammatory agents targeting mPGES-1. Moreover, the suppressive effect on PGE2 generation exhibited by lichen depsides and depsidones could provide a reasonable explanation for the anti-inflammatory, analgesic and antipyretic properties of lichen extracts observed in folk medicine and in scientific studies.[23]
Figure 2.
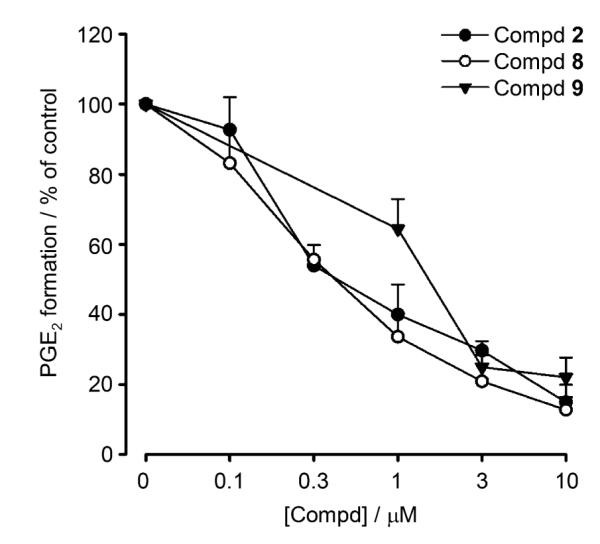
Inhibition of mPGES-1 by depsides and depsidones from lichen. Concentration–response curves of physodic acid (2), perlatolic acid (8), and olivetoric acid (9) for inhibition of mPGES-1 activity in microsomal preparations of IL-1β-stimulated A549 cells. Experiments were performed as described in the Experimental Section. Data represent the mean ± SE of n=3–4 independent experiments.
Among the depsidones, only compound 2 exhibited strong mPGES-1 inhibition. The free carboxylic moiety at position 1′ (numbering according to Figure 1) seems crucial for activity, as inactive compounds 3, 4, and 5 lack such a substituent at that position. Within the depsides evaluated, the importance of the acidic group was also confirmed. Inactive depsides 6 and 10 both lack a free carboxylic acid group. In both scaffolds, depsides and depsidones, the size and length of two hydrophobic substituents at positions 6 and 6′ seem to be crucial for mPGES-1 inhibition. The three highly active compounds (2, 8, and 9) all bear alkyl chains at these positions. Compound 7 with its small substituents (both methyl groups) at positions 6 and 6′ is a weak mPGES-1 inhibitor. Based on the experimental data, it can be concluded that both depsides and depsidones require a free acidic group for interference with mPGES-1 and hydrophobic substituents at positions 6 and 6′ for increased potency.
Fitting of compounds 2, 8, and 9 into pharmacophore model M2 confirmed the importance of their n-pentyl substituents. All three compounds mapped two of the hydrophobic features with their n-pentyl residues (Figure 3). Within our data set, the optimal chain length of the hydrophobic substituents was five carbon atoms; however, a systematic investigation of different chain lengths and introducing unsaturated bonds within these chains might yield even more active compounds. For a refinement of the pharmacophore model M1, it would be plausible to delete the aromatic ring feature as compounds 2 and 9 cannot map this feature. Compound 2 is as potent as compound 8, which maps all features of the M1 model. Thus, mapping of the aromatic ring feature does not seem to be important for mPGES-1 inhibition.
Figure 3.
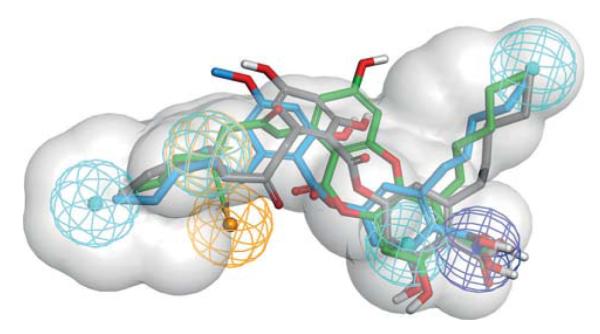
Pharmacophore model for acidic mPGES-1 inhibitors. Chemical features are color-coded: hydrophobic (cyan), aromatic ring (gold), negatively ionizable (blue), spatial shape restriction (grey). The screening model M1 required a compound to map all of these features to be considered a virtual hit. During the screening with M2, one hydrophobic feature or the aromatic ring feature was allowed to be omitted. The mPGES-1 inhibitors found in this study (compound 2 in green, 8 in blue, and 9 in grey) all map two of the hydrophobic features with their alkyl chains.
There is accumulating evidence that mPGES-1 might represent a druggable target. Recent mPGES-1 gene deletion studies and pharmacological approaches with select mPGES-1 inhibitors support a role for mPGES-1 in inflammatory reactions, fever, pain, neurological disorders, and tumorigenesis.[6,7,25] In view of the considerable gastrointestinal, renal and cardiovascular side effects of COX inhibitors, the class of drugs widely used to treat inflammation, fever and pain, mPGES-1 might be a promising alternative target to COX. Accordingly, extensive efforts are underway to develop mPGES-1 inhibitors, however, only a few synthetic agents show sufficient efficacy in preclinical models of inflammation and pain and thus could potentially enter clinical trials.[6,7] Inhibition of mPGES-1 by the lichen depsides or depsidones described here is unprecedented and unexpected. Of interest, several known lichen depsides and depsidones suppressed COX activity in rabbit renal microsomes,[13] and the lichen depside 4-O-methylcryptochlorophaeic acid was found to interact with COX-1.[26] In our study, compounds 2, 8, and 9 that potently inhibited mPGES-1 failed to efficiently inhibit COX-1/2. It was recently shown that abdication of the carboxylic group might be suitable to obtain more selective mPGES-1 inhibitors lacking inhibition of COX enzymes.[10a] Accordingly, the design of nonacidic derivatives of compounds 2, 8, or 9 could yield even more selective mPGES-1 inhibitors.
To the best of our knowledge, no other studies are available that addressed the interference of lichen compounds with PG biosynthesis, COX enzymes or PGES. More than 1000 secondary lichen metabolites have been identified, and aside from depsides and depsidones, a variety of bioactive phenolic compounds including hydroxybenzoic acid derivatives, dibenzofuran usnic acid, anthraquinones, and naphthoquinones are produced by lichen.[12] However, even for this large collection of diverse compounds, little is known about the anti-inflammatory activity (in vitro or in vivo) of lichen phenolic compounds, and their therapeutic potential, in particular that of depsides and depsidones, as anti-inflammatory agents is largely unexplored. In conclusion, the discovery of compounds 2, 8, and 9 as potent mPGES-1 inhibitors through the use of two pharmacophore models and virtual screening is a crucial result providing new lead structures for the development of novel anti-inflammatory agents. These results also offer broader perspectives and possibilities for the application of depsides and depsidones from lichen.
Experimental Section
Pharmacophore-based virtual screening
The three-dimensional Chinese herbal medicine (CHM) database, containing approximately 10000 unique compounds, was generated as previously described.[11] Two recently reported mPGES-1 inhibitor pharmacophore models[10b] were employed to virtually screen the CHM database using the “search 3D database” protocol of Discovery Studio 2.0 in FAST mode (Accelrys Inc., San Diego, CA, USA).
Isolation of depsides and depsidones
Perlatolic acid (8) was obtained from an Et2O extract of Cladonia portentosa subjected to medium-pressure liquid chromatography (pre-packed Macherey Nagel C18 column, 40–63 μm) using an Armen Spot Flash Chromatography system. A gradient of MeOH/H2O (60:40→100:0) over 1 h at a flow rate of 20 mL min−1 was used, with depside 8 eluting at tR=40 min (approximate solvent system at time of elution: MeOH 80–90%).
Olivetoric acid (9) and physodic acid (2) were both obtained from an acetone extract of Pseudevernia furfuracea purified by vacuum liquid chromatography (silica gel, 70–230 mesh) using a cyclohexane/EtOAc gradient (100:0→30:70, 10% cyclohexane increasing steps). Depside 9 and depsidone 2 eluted at 40% and 60% EtOAc, respectively, and both compounds were further purified by crystallization from cyclohexane/EtOAc mixtures (30% and 40% cyclohexane for compounds 9 and 2, respectively).
Atranorin (10), methyl β-orcinol carboxylate (6) and fumarprotocetraric acid (1) were isolated from Usnea articulata as previously reported.[29] Salazinic acid (3), variolaric acid (4) and scensidin (5) were obtained from Parmotrema tinctorum,[30] Ochrolechia parella,[31] and Diploicia canescens,[32] respectively. Evernic acid (7) was purchased from Extrasynthese (no. 6274, Genay, France).
All compound samples were checked for >95% purity, and spectroscopic data were in agreement with literature data.[33] Lichen compounds were dissolved in dimethyl sulfoxide (DMSO) and kept in the dark at −20°C, and freezing/thawing cycles were kept to a minimum.
Determination of PGE2 synthase and COX-1/2 activities
Materials
High-glucose Dulbecco’s modified Eagle’s medium (DMEM), penicillin, streptomycin, and trypsin/EDTA solution were purchased from PAA Laboratories (Linz, Austria). PGH2 was obtained from Larodan (Malmö, Sweden), while 11β-PGE2 and MK-886 (11) were both purchased from Cayman Chemical (Ann Arbor, MI, USA). All other chemicals were obtained from Sigma–Aldrich (Deisenhofen, Germany) unless stated otherwise.
Human A549 cells were cultured in high-glucose DMEM (4.5 gL−1) supplemented with heat-inactivated fetal calf serum (FCS, 10% v/v), penicillin (100 UmL−1), and streptomycin (100 μgmL−1) at 37°C and 5% CO2. After 20 h, cells were first transferred into fresh medium (high-glucose DMEM with FCS (2% v/v), penicillin (100 UmL−1), and streptomycin (100 μgmL−1)), and then stimulated with 2 ngmL−1 interleukin-1β (IL-1β) and cultured for a further 24 h at 37°C. After this time, confluent cells were detached using 1×trypsin/EDTA solution and resuspended in fresh medium for subsequent experiments.
Preparation of microsomes of A549 cells and determination of mPGES-1 activity was performed as described previously.[14] In brief, cells were treated with 1 ngmL−1 IL-1β for 48 h. After sonification, the homogenate was subjected to differential centrifugation at 10000 g for 10 min and 174000 g for 1 h at 4°C. The pellet (microsomal fraction) was resuspended in 1 mL homogenization buffer (100 mm potassium phosphate buffer (pH 7.4), 1 mm phenylmethanesulphonyl fluoride, 60 μgmL−1 soybean trypsin inhibitor, 1 μgmL−1 leupeptin, 2.5 mm glutathione, and 250 mm sucrose), and the total protein concentration was determined. Microsomal membranes were diluted in potassium phosphate buffer (0.1 m, pH 7.4) containing 2.5 mm glutathione. Test compound or vehicle (DMSO, 0.3%) were added, and after 15 min at 4°C, the reaction was initiated by addition of PGH2 (20 μm, final concentration). After 1 min at 4°C, the reaction was terminated using stop solution (100 μL; 40 mm FeCl2, 80 mm citric acid, and 10 μm of 11β-PGE2 as an internal standard). PGE2 was separated by solid-phase extraction and analyzed by RP-HPLC as previously described.[14]
Assays to determine the inhibitory activities of test compounds against isolated ovine COX-1 and human recombinant COX-2 were performed as previously described.[14] Briefly, purified COX-1 (ovine, 50 U) or COX-2 (human recombinant, 20 U) were diluted in 1 mL reaction mixture containing 100 mm Tris buffer (pH 8), 5 mm glutathione, 5 μm hemoglobin, and 100 μm EDTA at 4°C and pre-incubated with test compound for 5 min. Samples were pre-warmed for 60 s at 37°C, and arachidonic acid (5 μm for COX-1; 2 μm for COX-2) was added to start the reaction. After 5 min at 37°C, COX product 12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid (12-HHT) was extracted and analyzed by HPLC.[34]
Statistics
Data are expressed as the mean ± standard error (SE). IC50 values are approximations determined by graphical analysis (linear interpolation between the points at 50% activity). The program Graphpad InStat 3.05 (Graphpad Software Inc., San Diego, CA, USA) was used for statistical comparisons. Statistical evaluation of the data was performed by one-way analysis of variance (ANOVA) for independent or correlated samples followed by Tukey’s honestly significant difference (HSD) post-hoc tests. A P value of <0.05 (*) was considered significant.
Acknowledgements
Bianca Jazzar and Daniela Müller (both University of Tuebingen, Germany) are acknowledged for the expert technical assistance. The authors thank AureliaSan GmbH (Tuebingen, Germany), the Standortagentur Tirol (TWF), and the Austrian Science Fund (FWF) [NFN S10711, S10703, S10701] for financial support.
References
- [1].Rainsford KD. Subcell. Biochem. 2007;42:3–27. doi: 10.1007/1-4020-5688-5_1. [DOI] [PubMed] [Google Scholar]
- [2] a).Funk CD. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]; b) Smith WL. Biochem. J. 1989;259:315–324. doi: 10.1042/bj2590315. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang D, Dubois RN. Nat. Rev. Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Abdel-Tawab M, Zettl H, Schubert-Zsilavecz M. Curr. Med. Chem. 2009;16:2042–2063. doi: 10.2174/092986709788682209. [DOI] [PubMed] [Google Scholar]
- [4].Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Proc. Natl. Acad. Sci. USA. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Samuelsson B, Morgenstern R, Jakobsson PJ. Pharmacol. Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- [6].Koeberle A, Werz O. Curr. Med. Chem. 2009;16:4274–4296. doi: 10.2174/092986709789578178. [DOI] [PubMed] [Google Scholar]
- [7] a).Rådmark O, Samuelsson B. J. Intern. Med. 2010;268:5–14. doi: 10.1111/j.1365-2796.2010.02246.x. [DOI] [PubMed] [Google Scholar]; b) Chang HH, Meuillet EJ. Future Med. Chem. 2011;3:1909–1934. doi: 10.4155/fmc.11.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jegerschold C, Pawelzik SC, Purhonen P, Bhakat P, Gheorghe KR, Gyobu N, Mitsuoka K, Morgenstern R, Jakobsson PJ, Hebert H. Proc. Natl. Acad. Sci. USA. 2008;105:11110–11115. doi: 10.1073/pnas.0802894105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pawelzik SC, Uda NR, Spahiu L, Jegerschold C, Stenberg P, Hebert H, Morgenstern R, Jakobsson PJ. J. Biol. Chem. 2010;285:29254–29261. doi: 10.1074/jbc.M110.114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10] a).Rörsch F, Wobst I, Zettl H, Schubert-Zsilavecz M, Grosch S, Geisslinger G, Schneider G, Proschak E. J. Med. Chem. 2010;53:911–915. doi: 10.1021/jm9012505. [DOI] [PubMed] [Google Scholar]; b) Waltenberger B, Wiechmann K, Bauer J, Markt P, Noha SM, Wolber G, Rollinger JM, Werz O, Schuster D, Stuppner H. J. Med. Chem. 2011;54:3163–3174. doi: 10.1021/jm101309g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fakhrudin N, Ladurner A, Atanasov AG, Heiss EH, Baumgartner L, Markt P, Schuster D, Ellmerer EP, Wolber G, Rollinger JM, Stuppner H, Dirsch VM. Mol. Pharmacol. 2010;77:559–566. doi: 10.1124/mol.109.062141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Müller K. Appl. Microbiol. Biotechnol. 2001;56:9–16. doi: 10.1007/s002530100684. [DOI] [PubMed] [Google Scholar]
- [13].Sankawa U, Shibuya M, Ebizuka Y, Noguchi H, Kinoshita T, Iitaka Y, Endo A, Kitahara N. Prostaglandins. 1982;24:21–34. doi: 10.1016/0090-6980(82)90174-5. [DOI] [PubMed] [Google Scholar]
- [14].Koeberle A, Siemoneit U, Buhring U, Northoff H, Laufer S, Albrecht W, Werz O. J. Pharmacol. Exp. Ther. 2008;326:975–982. doi: 10.1124/jpet.108.139444. [DOI] [PubMed] [Google Scholar]
- [15].Riendeau D, Aspiotis R, Ethier D, Gareau Y, Grimm EL, Guay J, Guiral S, Juteau H, Mancini JA, Methot N, Rubin J, Friesen RW. Bioorg. Med. Chem. Lett. 2005;15:3352–3355. doi: 10.1016/j.bmcl.2005.05.027. [DOI] [PubMed] [Google Scholar]
- [16].Koeberle A, Northoff H, Werz O. Mol. Cancer Ther. 2009;8:2348–2355. doi: 10.1158/1535-7163.MCT-09-0290. [DOI] [PubMed] [Google Scholar]
- [17].Koeberle A, Rossi A, Bauer J, Dehm F, Verotta L, Northoff H, Sautebin L, Werz O. Front. Inflammation Pharmacol. 2011;2:7. doi: 10.3389/fphar.2011.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bauer J, Koeberle A, Dehm F, Pollastro F, Appendino G, Northoff H, Rossi A, Sautebin L, Werz O. Biochem. Pharmacol. 2011;81:259–268. doi: 10.1016/j.bcp.2010.09.025. [DOI] [PubMed] [Google Scholar]
- [19].Koeberle A, Pollastro F, Northoff H, Werz O. Br. J. Pharmacol. 2009;156:952–961. doi: 10.1111/j.1476-5381.2009.00070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Siemoneit U, Koeberle A, Rossi A, Dehm F, Verhoff M, Reckel S, Maier TJ, Jauch J, Northoff H, Bernhard F, Doetsch V, Sautebin L, Werz O. Br. J. Pharmacol. 2011;162:147–162. doi: 10.1111/j.1476-5381.2010.01020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Koeberle A, Bauer J, Verhoff M, Hoffmann M, Northoff H, Werz O. Biochem. Biophys. Res. Commun. 2009;388:350–354. doi: 10.1016/j.bbrc.2009.08.005. [DOI] [PubMed] [Google Scholar]
- [22].Koeberle A, Northoff H, Werz O. Biochem. Pharmacol. 2009;77:1513–1521. doi: 10.1016/j.bcp.2009.02.005. [DOI] [PubMed] [Google Scholar]
- [23] a).da Costa Silva JA, Bomfim RR, Cdos SE, Antoniolli AR, Araujo AA, Thomazzi SM. Pharm. Biol. 2010;48:745–752. doi: 10.3109/13880200903273914. [DOI] [PubMed] [Google Scholar]; b) Freysdottir J, Omarsdottir S, Ingolfsdottir K, Vikingsson A, Olafsdottir ES. Int. Immunopharmacol. 2008;8:423–430. doi: 10.1016/j.intimp.2007.11.007. [DOI] [PubMed] [Google Scholar]; c) Choudhary MI, Azizuddin, Jalil S, Atta-ur-Rahman Phytochemistry. 2005;66:2346–2350. doi: 10.1016/j.phytochem.2005.06.023. [DOI] [PubMed] [Google Scholar]; d) Bugni TS, Andjelic CD, Pole AR, Rai P, Ireland CM, Barrows LR. Fitoterapia. 2009;80:270–273. doi: 10.1016/j.fitote.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hieke M, Greiner C, Dittrich M, Reisen F, Schneider G, Schubert-Zsilavecz M, Werz O. J. Med. Chem. 2011;54:4490–4507. doi: 10.1021/jm200092b. [DOI] [PubMed] [Google Scholar]
- [25].Hara S, Kamei D, Sasaki Y, Tanemoto A, Nakatani Y, Murakami M. Biochimie. 2010;92:651–659. doi: 10.1016/j.biochi.2010.02.007. [DOI] [PubMed] [Google Scholar]
- [26].Gerrard JM, Peterson DA. Prostaglandins Leukotrienes Med. 1984;13:139–142. doi: 10.1016/0262-1746(84)90003-9. [DOI] [PubMed] [Google Scholar]
- [27] a).Kumar K. C. S, Müller K. J. Nat. Prod. 1999;62:817–820. doi: 10.1021/np9803777. [DOI] [PubMed] [Google Scholar]; b) Ingólfsdóttir K, Gudmundsdóttir GF, Ogmundsdóttir HM, Paulus K, Haraldsdóttir S, Kristinsson H, Bauer R. Phytomedicine. 2002;9:654–658. doi: 10.1078/094471102321616481. [DOI] [PubMed] [Google Scholar]
- [28].Bucar F, Schneider I, Ogmundsdottir H, Ingolfsdottir K. Phytomedicine. 2004;11:602–606. doi: 10.1016/j.phymed.2004.03.004. [DOI] [PubMed] [Google Scholar]
- [29].Lohézic-Le Dévéhat F, Tomasi S, Elix JA, Bernard A, Rouaud I, Uriac P, Boustie J. J. Nat. Prod. 2007;70:1218–1220. doi: 10.1021/np070145k. [DOI] [PubMed] [Google Scholar]
- [30].Eifler-Lima VL, Sperry A, Sinbandhit S, Boustie J, Tomasi S, Schenkel E. Magn. Reson. Chem. 2000;38:472–474. [Google Scholar]
- [31].Millot M, Tomasi S, Articus K, Rouaud I, Bernard A, Boustie J. J. Nat. Prod. 2007;70:316–318. doi: 10.1021/np060561p. [DOI] [PubMed] [Google Scholar]
- [32].Millot M, Tomasi S, Studzinska E, Rouaud I, Boustie J. J. Nat. Prod. 2009;72:2177–2180. doi: 10.1021/np9003728. [DOI] [PubMed] [Google Scholar]
- [33].Huneck S, Yoshimura I. Identification of Lichen Substances. Springer; New York: 1996. [Google Scholar]
- [34].Albert D, Zündorf I, Dingermann T, Müller WE, Steinhilber D, Werz O. Biochem. Pharmacol. 2002;64:1767–1775. doi: 10.1016/s0006-2952(02)01387-4. [DOI] [PubMed] [Google Scholar]