

Abstract
Study Objectives:
Narcolepsy is a sleep disorder characterized by loss of orexin neurons. Previously, our group demonstrated that transfer of the orexin gene into surrogate neurons in the lateral hypothalamus and the zona incerta significantly reduced cataplexy bouts in the orexin-ataxin-3 mice model of narcolepsy. The current study determined the effects of orexin gene transfer into the dorsolateral pontine neurons in the orexin knockout (KO) mice model of narcolepsy. The dorsolateral pons was chosen because it plays a critical role in regulating muscle tone and thus it is conceivable to be involved in cataplexy as well. Cataplexy is the pathognomonic symptom in narcolepsy.
Design:
Independent groups of orexin KO mice were given bilateral microinjections (0.75 μL each side) of either recombinant adenoassociated virus-orexin (rAAV-orexin; n = 7), or rAAV-green fluorescent protein (rAAV-GFP; n = 7) into the dorsolateral pons. A group of orexin KO mice that did not receive rAAV (n = 7) and a group of wild-type mice (C57BL/J6; n = 5) were used as controls. Three weeks after rAAV-mediated gene transfer narcolepsy symptoms were examined using sleep and behavioral recordings. Number, location of the orexin-immunoreactive neurons, and relative density of orexin immunoreactive fibers were determined.
Measurements and Results:
Orexin gene transfer into the dorsolateral pons significantly decreased cataplexy and modestly improved wake maintenance compared to the orexin KO mice that did not receive rAAV. In contrast, GFP gene transfer worsened narcoleptic symptoms compared to the no-rAAV orexin KO group.
Conclusion:
Orexin gene transfer into the dorsolateral pontine neurons can control cataplexy attacks and modestly improve wake maintenance.
Citation:
Blanco-Centurion C; Liu M; Konadhode R; Pelluru D; Shiromani PJ. Effects of orexin gene transfer in the dorsolateral pons in orexin knockout mice. SLEEP 2013;36(1):31–40.
Keywords: Narcolepsy, hypocretin, orexin; recombinant adenoassociated virus; gene transfer, cataplexy; sleep, REM sleep
INTRODUCTION
Narcolepsy is a complex sleep disorder characterized by sudden episodes of flaccid muscle paralysis (known as cataplexy), excessive daytime sleepiness, sleep paralysis, hypnagogic hallucinations, and sleep fragmentation.1,2 The motor deficits observed in patients with narcolepsy range from localized muscle weakness to total collapse of the body. Interestingly, cataplexy attacks are usually triggered by strong emotions such as laughter or anger3,4 but the underlying mechanisms are unknown. Prevalence of narcolepsy with cataplexy in the United States ranges from 25–50 per 100,000.5
Narcolepsy is now considered a neurodegenerative disorder resulting from massive loss of neurons synthesizing the neuropeptide orexin, also named hypocretin.6–8 The orexin neurons are present in the perifornical region extending into the posterior aspect of the lateral hypothalamus. From there orexin neurons project to the entire brain and spinal cord,9–11 particularly to regions involved in sleep/wake control. Consistent with the hypothesis of orexin neuronal loss, patients with narcolepsy have low to negligible levels of orexin-A in their cerebrospinal fluid.12 Two other peptides, dynorphin and neuronal activity-related pentraxin, that colocalize with orexin are also absent in postmortem brain tissue from humans with narcolepsy.13,14 Because most of the orexin neurons have already died in patients with narcolepsy, it is important to identify surrogate neurons where the ectopic expression of the orexin gene could block some or all of the symptoms of narcolepsy.
Over the past few years, our group has been investigating whether orexin gene transfer can be used to block narcoleptic symptoms. In the first study in orexin KO mice cataplexy was blocked by transferring the orexin gene into neurons in the lateral hypothalamus using the herpes simplex-1 viral vector.15 In the second study a recombinant adenoassociated virus (rAAV) vector was used to induce orexin gene expression for a longer time period (3 wk) in the brains of orexin-ataxin-3 mice.16 rAAV-orexin gene transfer in the lateral hypothalamus and zona incerta significantly reduced cataplexy whereas orexin gene transfer into the striatum was ineffective, indicating site-specific effects of the orexin gene transfer. The surrogate neurons that were effective in decreasing cataplexy were active, as measured by c-Fos, during wake and received inputs from areas controlling strong emotional states such as the amygdala and the cingulate cortex. These neurons projected to the dorsolateral pons, which has been implicated in muscle control and in the various tonic and phasic aspects of rapid eye movement (REM) sleep.17 We also previously established that rAAV-orexin gene transfer into several types of neurons produced release of the gene product into the cerebrospinal fluid, suggesting the transduced neurons released orexin onto postsynaptic neurons as well.16 Moreover, it has been determined that orexin receptors are still functional in narcoleptic mice.18 Thus, the amelioration of narcoleptic symptoms observed in our previous studies was likely the result of stimulation of orexin receptors. To identify whether the pons represents the next link in the control of cataplexy, in the current study, rAAV vector was used to deliver the mouse prepro-orexin gene (rAAV-orexin) into the dorsolateral pons of the orexin KO mice.
METHODS
Construction of rAAV Vector
Mouse prepro-orexin complementary deoxyribonucleic acid (cDNA) (393 bp from Dr. Yanagisawa's laboratory; University of Texas Southwestern Medical Center, Dallas, TX, USA.) was used as a template to get the orexin fragment by polymerase chain reaction (PCR) with the following primers:
5'-AAATATGCGGCCGCGCCACCATGGATACGTATCGCTACATATC-3' and
5'-GACCGG CTCGAGTCAGACTCCGGACCCTC-3'.
After cutting with the restriction endonucleases xhoI and NotI, this PCR fragment was inserted into the plasmid pAAV-MCS (MCS = multiclonal site) from Harvard Gene Therapy Initiative Laboratory, (77 Avenue Louis Pasteur, Room HIM-412, Boston, MA) to form pAAV-MCS-orexin (plasmid adenoasociated virus-multiclonal site-orexin). A similar method was used to create pAAV-MCS-GFP by inserting gene fluorescent protein (GFP) complementary DNA into pAAV-MCS. Vector packaging and titration were done by Harvard Gene Therapy Initiative Laboratory. Briefly, the tripartite transfection (AAV-rep/cap expression plasmid, adenovirus miniplasmid, and our vector plasmid) was done in 293A cells and virus was purified by iodixanol gradient followed by Q sepharose column chromatography. Purified virus was dialyzed against phosphate buffered saline, concentrated by Amicon spin column (Millipore, Billerica, MA), and titrated by dot blot hybridization. The titers of all vectors were between 2.7–3.0 × 1013 genomic copies/mL.
Delivery of the Vector and Implant of Sleep Recording Electrodes
Under deep anesthesia (isofluorane 0.5–2.0%) viral particles were microinjected into the mice brains using a stereotaxic instrument (Kopf, Tujunga, CA). The stereotaxic coordinates for delivering the vectors into the dorsolateral pons were A = −5.0, L = ± 0.8, V = −2.5. Bregma and the dura mater were used as landmark references for the calculations of the anterior-posterior and vertical coordinates, respectively.19 The experimental (rAAV-orexin) and control (rAAV-GFP) viral vectors were both delivered in a volume of 0.75 μL on each side of the brain using a Hamilton syringe (2.0 μL; Hamilton Company, Reno, NV) coupled to a stainless steel injector microcannula (33G, Plastics One, Roanoke, VA). Injections were done gradually over 15 min. After the microinjection, the injector needle was left in place for 10 min and then withdrawn slowly. At the same time the mice were implanted under continuous inhaled anesthesia (isofluorane 0.5–2.0%) with electrodes to record the electroencephalogram (EEG) and electromyogram (EMG) as described elsewhere.20 Briefly, to record the EEG four stainless steel miniature screw electrodes were positioned in the skull to sit on the surface of the cerebral cortex. Two electrodes were positioned to sit on both sides of the motor cortex while the other two sit on the visual cortex. In addition, two flexible multistranded electrodes embedded in the nuchal muscles were implanted to record the EMG activity. All electrode leads were then secured onto the skull using dental cement.
Mice Strain and Groups
Orexin knockout (KO) mice were bred in our facility from founders supplied by Dr. Yanagisawa (University of Texas Southwestern Medical Center, Dallas, TX). These mice have been backcrossed for many generations on a C57BL/6J line. Only homozygous orexin KO mice, identified by PCR from tail snips, were used for the experiment. Twenty-one orexin KO mice with narcolepsy (female and male, 6–12 mo old, 20–35 g) were randomly assigned to the following groups: no rAAV (n = 7), rAAV-GFP (n = 7), and rAAV-orexin (n = 7). Five wild-type (WT) mice of the same age and background strain (C57BL/6J) as the orexin KO mice were used as additional controls (Jackson Laboratory, Bar Harbor, ME).
Experiment Protocol
One wk after the surgery the mice were connected to custom-made lightweight sleep recording cables and allowed 2 wk for adaptation to the cables. Mice were housed singly in shoebox-size colorless Plexiglass cages containing dry corncob as bedding material. Food and water were available ad libitum. Adaptation and sleep recordings were done inside a quiet room with controlled temperature (18–22°C) and a 12:12 h light-dark cycle (07:00-07:00 lights on; 100 lux) was maintained at all times. On day 21, a 48-h sleep recording was made. Thereafter, the mice were anesthetized and the brains perfused for histologic analysis. All manipulations done to the mice adhered to the National Institutes of Health Guide for the humane care and use of laboratory animals and were approved by the local Institutional committee competent on these matters (Institutional Animal Care and Use Committee).
Sleep Recording
The EEG and EMG signals and behavior data were continuously recorded over the 48-h period. The EEG signal was obtained from two contralateral electrodes (motor cortex versus contralateral visual cortex) whereas the EMG signal was acquired from the two contralateral nuchal muscles. Using a polygraph (AstroMed, Model 12; West Warwick, RI) EEG and EMG signals were amplified, filtered (0.3–100 Hz for EEG; 100-1KHz for EMG), and then recorded onto a computer hard disk using an analog-digital board controlled by a sleep data acquisition software (SleepSign; Kissei Comtec Co, Nagano, Japan). EEG data were acquired at a sampling rate of 128 Hz. Behavioral data were also continuously recorded via CCD night vision surveillance cameras located in front of the animal's cage. The sleep data acquisition software synchronized video streams to the EEG signal and stored it onto the hard disk.
Identification of Sleep-Wake States
The 48 h of EEG, EMG, and video recordings were scored semiautomatically in 12-sec epochs on a computer screen using a sleep recording analysis software (SleepSign, Kissei Comtec Co, Nagano, Japan). Staff blind to the type of vector delivered to the animal used waveform (EEG/EMG), EEG power spectral analysis and behavioral data for scoring the recording as wake, non-rapid eye movement (NREM) sleep, rapid eye movement (REM) sleep, sleep attacks, and cataplexy. Wake was identified by the presence of desynchronized EEG and high EMG activity. NREM sleep consisted of high-amplitude, slow-frequency waves together with low EMG tone relative to waking. REM sleep was identified by the presence of EEG theta activity coupled with low EMG relative to NREM sleep. Criteria for identification of cataplexy and sleep attacks are given in the following paragraphs. The amount of time spent in wakefulness, NREM sleep, REM sleep, cataplexy, and sleep attacks was determined for each hour. After the data were scored, the code was broken to reveal the identity of each mouse.
Identification of Cataplexy
The EEG, EMG, and behavioral data were examined for signs of sudden loss of muscle tone when the mice were engaged in behavioral activity.21 In rats and mice with loss of the orexin or of the orexin neurons, such behavioral incidences are evident on the EEG/EMG and on the video recordings. Strict criteria were used to identify cataplexy attacks. Such episodes had to occur while the mouse was awake for at least 36 sec (three epochs since we scored sleep-wake states in 12-sec epochs) and engaged in an active behavior such as walking, running, grooming, eating, or drinking (from video recording); the cataplexy episode had to last at least 12 sec (one epoch); during the cataplexy episode EEG theta activity had to be predominant while EEG delta activity diminished and the EMG signal clearly had to indicate a complete loss of nuchal muscle tone. Number and duration of each of these cataplexy attacks during the 12-h day and night periods were determined.
Identification of Sleep Attacks
During these attacks, the mice entered briefly into NREM sleep during active behaviors such as grooming, eating, or drinking. These episodes were separate and distinct from cataplexy because the mice did not lose muscle tone and there was a mixture of delta and theta activity in the EEG, in contrast with cataplexy, where theta activity dominated.
Immunolabeling, Mapping, and Tally of Transfected Neurons
All mice were deeply anesthetized with sodium pentobarbital and their brains perfused with 0.9% saline solution (15 mL) followed by 10% phosphate buffered formalin (20 mL). The brains were then placed in 30% sucrose/0.1M phosphate buffered saline and allowed to equilibrate at 4°C. After equilibration, the whole brain was sliced in a cryostat (40-μm thickness; coronal angle) and one of four series of sections were processed for visualization of orexin-A immunoreactive (orexin-A-ir) neurons. Briefly, free-floating brain sections were incubated overnight at room temperature in the primary antibody (goat anti-orexin-A, 1:10,000; Santa Cruz Biotechnology, Santa Cruz, CA), then incubated for 1 h with a biotinylated secondary antibody (rabbit antigoat immunoglublin G (IgG); Jackson Immunoresearch Laboratories, West Grove, PA) and finally the label was visualized using the avidin/biotin/peroxidase-diaminobenzidine-nickel staining method (Vector Laboratories Inc, Burlingame, CA). Labeled sections were then mounted onto gelatin-coated glass slides and coverslipped using bright field mounting media (Richard-Allan, Kalamazoo, MI). In the case of the GFP group, coronal sections were directly mounted and coverslipped using fluorescence mounting media (ProLong Gold Antifade Reagent; Invitrogen, NY).
Immunofluorescence was used to detect colocalization of the gene product, orexin, with well-established endogenous markers for cholinergic, noradrenergic, and gamma aminobutyric acid (GABA)ergic neurons in the dorsolateral pontine neurons. To detect orexin in cholinergic neurons, sections were incubated with goat antiorexin A (1:500; Santa Cruz Biotechnology, Santa Cruz, CA) and rabbit anticholine acetyltransferase (1:500; Millipore, Billerica, MA) overnight at room temperature. To detect orexin in noradrenergic neurons sections were incubated with mouse antityrosine hydroxylase (1:200; Millipore) and goat anti-orexin A (1:500) overnight at 4°C in. To detect orexin in GABAergic neurons, the sections were incubated with rabbit antivesicular GABA transporter (1:500) and goat antiorexin A (1:1000) for 72 h at 4°C. Primary antibodies were always coincubated with 1% normal donkey serum (blocker) and 0.25% triton (permeabilizer) in phosphate buffered saline. After primary incubation, donkey antigoat IgG tagged with Alexa Fluor 488 (1:500 Invitrogen, Grand Island, NY) and donkey antirabbit conjugated to Alexa Fluor 568 (1:500; Invitrogen) were incubated with the sections for 1 h and then washed with phosphate buffered saline. Sections were then mounted onto gelatin-coated glass slides and coverslipped with bleaching retardant mounting media (ProLong Gold Antifade Reagent; Invitrogen). Stained tissue sections were visualized with a Fluoview 1000 Olympus confocal laser scanning microscope (Olympus America Inc, Center Valley, PA) with wavelength emission for Alexa Fluor 488 (520 nm; orexin) and Alexa Fluor 568 (603 nm; ChAT = choline acetyltransferase, TH = tyrosine hydroxylase, and vGABAT = vesicular GABA transporter).
Mapping and cell counting of infected neurons were done semiautomatically using high- resolution (20× magnification) tiled images of all sections containing either orexin-A-ir or GFP neurons (mapping only). For mapping, sections containing labeled cells were used to outline the infected area. The infected area was defined as the area where all orexin- positive labeled cells were found. Using Adobe Photoshop tools the semitransparent tiled image containing the infected area was overlapped onto the corresponding brain atlas coronal plate19 and the boundaries of the labeled neurons plotted. Size transformation of the tiled image and common landmarks were considered to achieve the best match between the section image and the corresponding plate. For each mouse a series of maps was created to display the total extent of the labeled area. For cell count, individual orexin-A-ir somata were tagged on a computer screen. Only somata showing strong labeling were tagged. Both incomplete somata (no nucleus) and weakly labeled somata were omitted from tagging. Image J analysis software (National Institutes of Health) was then used to automatically count all tags. The tally was derived from all coronal sections (one well in one in four series) that contained orexin-A labeled cells and represents the average number of labeled neurons per section. The average number of sections per mouse used for counting was 9 ± 2.
Density of orexin-ir fibers was also manually assessed in one in four series of brain coronal sections for individual mice injected with rAAV-orexin. The density in brain regions was scored according to the following numeric scale: 0 = no fibers, 1 = few fibers, 2 = some fibers, 3 = many fibers, 4 = maximum fiber density and 5 = orexin-A-ir somata and fibers. Labeling of orexin fibers and somata was done using the avidin-biotin complex and 3.3'-diaminobenzidine-nickel method (Vector Laboratories).
Statistical Analysis
One-way analysis of variance with appropriate post hoc tests (Holm-Sidak) compared the group means. Statistical significance was evaluated at the P < 0.05 (two-tailed) level.
RESULTS
Effects on Cataplexy
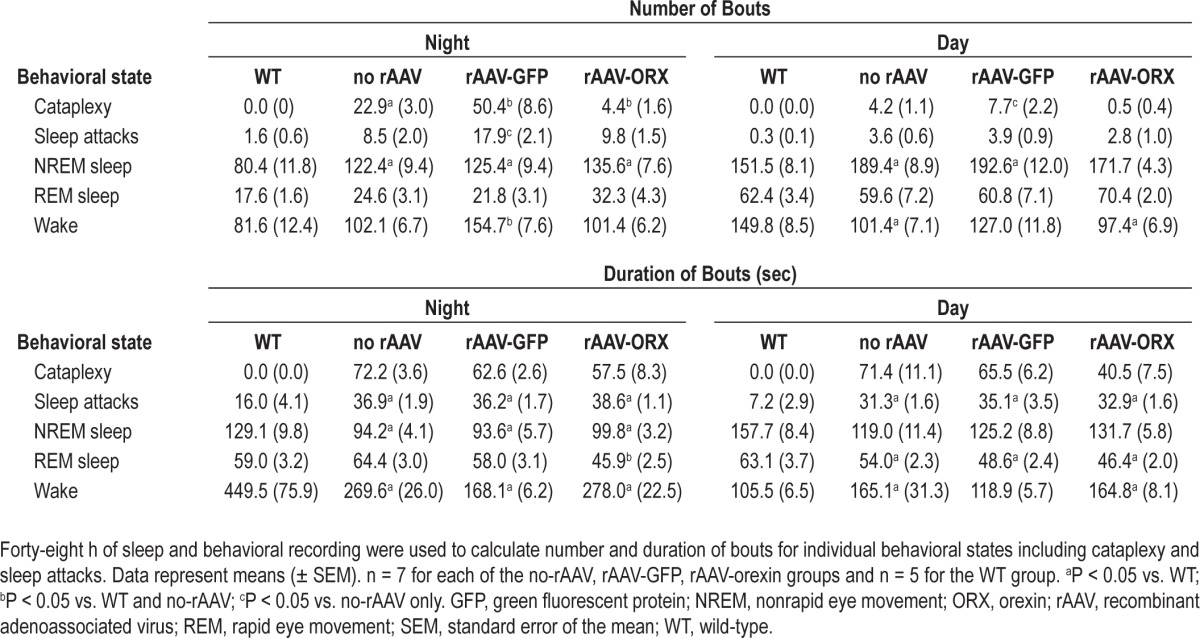
Consistent with previous reports21 orexin KO mice not given orexin gene transfer had numerous episodes of cataplexy (Figure 1 and Table 1). Most (84.5%) of the cataplexy attacks occurred at night, which corresponds with the active period of the animals. Cataplexy attacks were never observed in WT mice. Orexin gene transfer into the dorsolateral pontine neurons significantly decreased cataplexy attacks at night (P < 0.01) and during the second half of the day cycle (P < 0.001; Figure 1). Orexin gene transfer decreased the number of cataplexy episodes by 80.7% (22.9 versus 4.4 per night, P = 0.012; Table 1). There was a decrease in length of the cataplexy bout but this was not significant. Surprisingly, orexin KO mice given the reporter gene, GFP, had more cataplexy compared with the no-rAAV orexin-KO group. Altogether rAAV-GFP mice had a 114% increase in the number of cataplexy attacks compared with the no-rAAV group (Table 1; P = 0.049).
Figure 1.
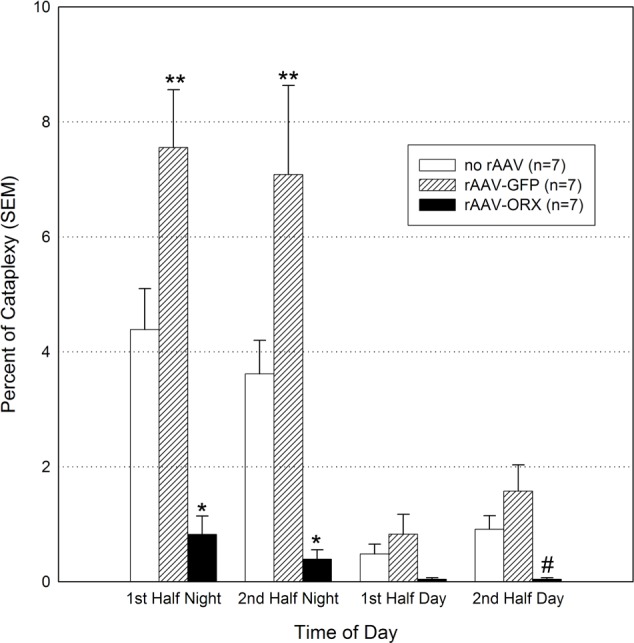
Orexin gene transfer into the dorsolateral pontine neurons significantly decreases cataplexy in orexin (ORX) knockout mice. Twenty-one days after recombinant adenoassociated virus (rAAV)-mediated gene transfer sleep and behavior were recorded for 48 h. The data summarize the percent of time spent in cataplexy in 6-h bins. *P < 0.01 versus no rAAV and rAAV-green fluorescent protein (GFP); #P < 0.001 vs. rAAV-GFP; **P < 0.01 versus no rAAV. SEM, standard error of the mean.
Table 1.
Number and length (in sec) of bouts in wild type and orexin knockout mice
Effects on Sleep Attacks
In contrast with WT mice in whom sleep attacks were rare, the orexin KO mice (no-rAAV) had numerous sleep attacks during the night and day (Table 1). Orexin gene transfer into dorsolateral pons neurons had no effect on sleep attacks. In contrast, mice given the control gene GFP had significantly more sleep attacks at night compared with the no-rAAV group (17.9 ± 2.1 versus 8.5 ± 2.0, P < 0.05; Table 1).
Effects on Wake Maintenance and Overall Wake Levels
Wake maintenance was evaluated by calculating the weighted percent of bouts of different durations of waking at night (Figure 2). During the night WT mice spent more than 74% of their total wake time in episodes lasting more than 32.2 min (Figure 2). In contrast, the orexin KO mice spent only 8% of their wake time in episodes lasting more than 32.2 min (−89% versus WT mice; P < 0.001). Orexin gene transfer into the dorsolateral pons of orexin KO mice improved wake maintenance, increasing the percent of wake bouts longer than 32.2 min to 23% (+180% versus no rAAV; P < 0.001). Deficit in wake maintenance in orexin KO mice was aggravated by rAAV-GFP gene transfer because wake bouts lasting longer than 32.2 min decreased to 1% (P = 0.001; Figure 2). Neither orexin nor GFP changed overall percent of wakefulness compared with no rAAV orexin KO mice (Figure 3).
Figure 2.
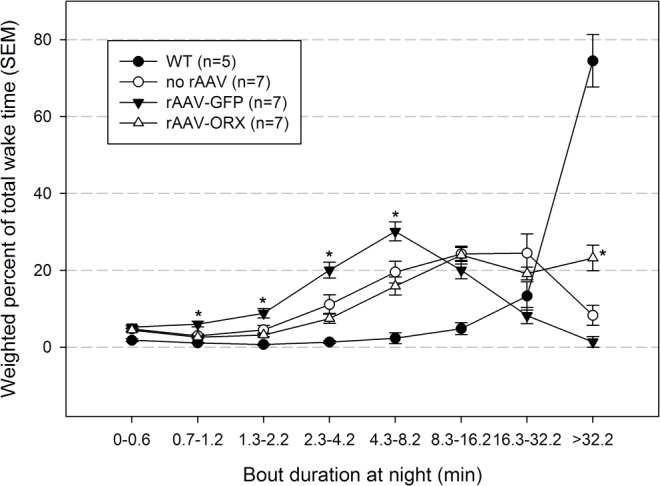
Effects of recombinant adenoassociated virus (rAAV)-mediated gene transfer on wake maintenance during the night. Individual wake bouts were grouped into bins of different durations. Data represent group means ± standard error of the mean (SEM). Orexin (ORX) knockout mice given rAAV-orexin into the dorsolateral pons had a significant increase in the percent of time spent in long bouts. *P < 0.05 versus no rAAV. GFP, green fluorescent protein; WT, wild-type.
Figure 3.
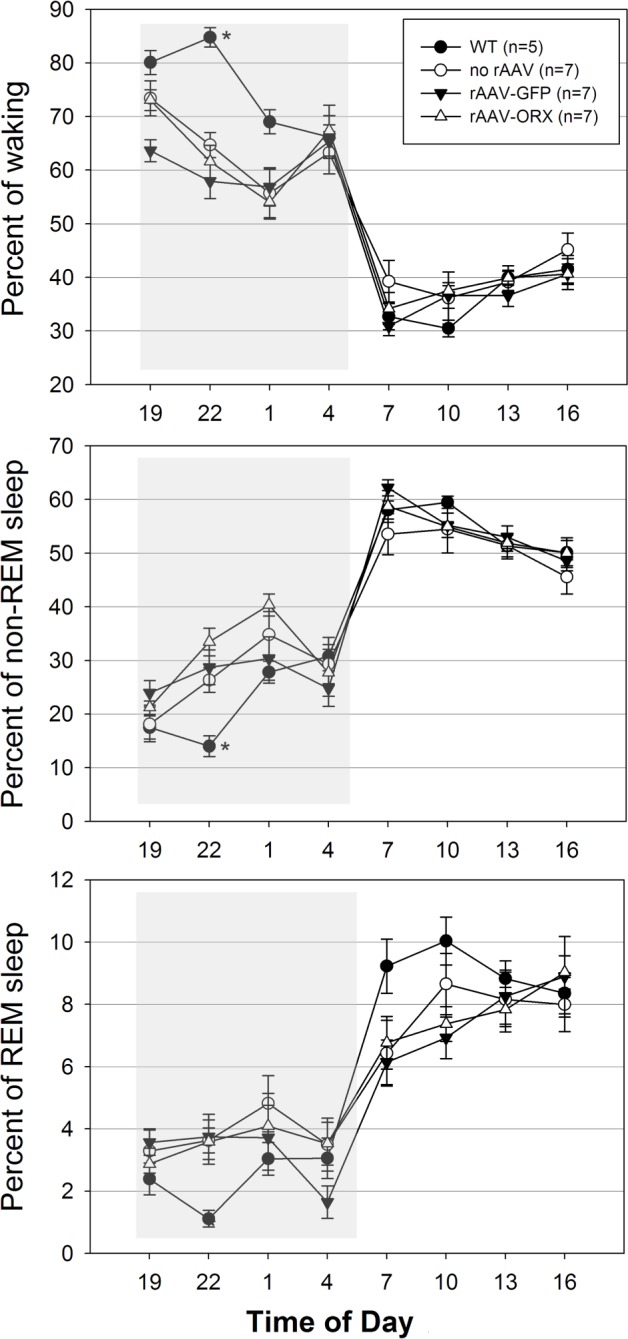
Effects of gene transfer on percent of sleep-wake states across 24 h in orexin (ORX) knockout and wild-type (WT) mice. Percent of time spent in vigilance states was determined 21 days after microinjection of rAAV-GFP, rAAV-ORX or no injection into the dorsolateral pons. WT (C57BL/6J) mice were also recorded and represent additional controls. Shaded boxes represent the lights-off period (19:00-07:00). *P < 0.05 versus all experimental groups. GFP, green fluorescent protein; rAAV, recombinant adenoassociated virus.
Effects on NREM and REM Sleep
Consistent with a previous report,21 the no-rAAV orexin KO mice in the current study had significantly more NREM sleep bouts compared with WT mice (Table 1; P < 0.05). After rAAV-orexin administration into the pons, the number of NREM sleep bouts significantly decreased during the day, suggesting less sleep fragmentation. However, at night the rAAV orexin mice continued to have more NREM sleep bouts. rAAV-orexin mice also showed significantly shorter REM sleep bouts (P < 0.05; Table 1). However, the number of REM sleep bouts in these mice was no different from either WT or orexin KO mice (Table 1). Percentage of NREM or REM sleep was not changed by orexin or GFP gene transfer compared with the no-rAAV orexin KO group (Figure 3).
Anatomic Findings
Orexin-A immunoreactive (ir) somata or fibers were not observed in orexin-KO mice given no-rAAV or rAAV-GFP. On the other hand, 3 wk after delivering rAAV-orexin into the pons of orexin KO mice, orexin-A-ir was clearly visible in the somata and terminals (Figure 4). Similarly, GFP expression was observed in somata and terminals of mice given rAAV-GFP (Figure 4; Panel A). As shown in Table 2, orexin KO mice given rAAV-orexin in the pons had similar numbers of orexin-A-ir neurons compared with WT mice, indicating that the titer and volume used in the current study produced comparable numbers of surrogate neurons as orexin neurons in the WT mice. Figure 5 summarizes the distribution maps of immunolabeled neurons either with the rAAV-orexin or with the rAAV-GFP. In mice given rAAV-orexin, the orexin-A-ir neurons were primarily seen within the ventrolateral periaqueductal gray area (vlPAG) and the laterodorsal tegmental nucleus (LDT) extending into the cuneiform, dorsal raphe, pedunculopontine tegmental nucleus, parabrachial nucleus, and the locus coeruleus (LC). A very similar distribution of the infected neurons was observed in the group of mice injected with rAAV-GFP, although in some mice (K587 and K588) infected neurons were also seen within the pontine reticular formation (Figure 5). Consistent with the distribution of orexin immunoreactivity in these brainstem nuclei, orexin-A-ir was observed within cholinergic neurons in the LDT, noradrenergic neurons in the LC, and GABAergic neurons in the vlPAG (Figure 6).
Figure 4.
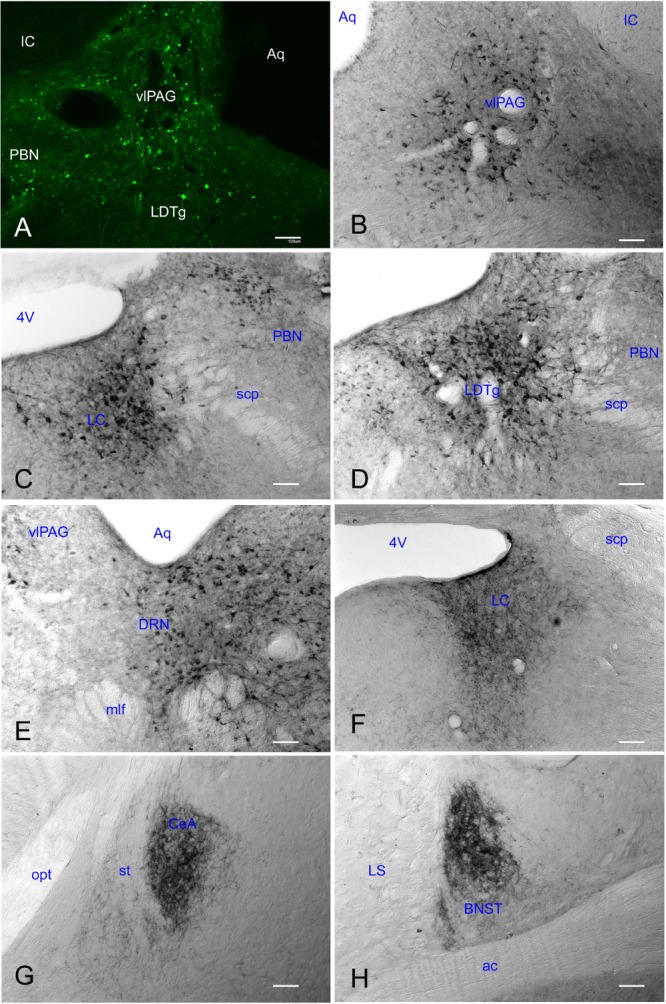
Recombinant adenoassociated viral vector (rAAV) gene transfer produced robust gene product in brains of orexin knockout mice 3 wk after its administration. Reporter gene (GFP) expression was clearly seen in somata and proximal dendrites in the dorsolateral pontine region (panel A). Robust expression of the orexin-A-ir was observed in this area including the ventrolateral periaqueductal gray (vlPAG; panel B), locus coeruleus (LC; panel C), laterodorsal tegmental nucleus (LDTg; panel D), and dorsal raphe nucleus (DRN; panel E). Orexin-A-ir was also observed in distal terminals within the LC area (Panel F), the central nucleus of the amygdala (CeA; panel G) and the bed nucleus of the stria terminalis (BNST; panel H) among others (see Table 3). White scale bars represent 100 μm. 4V, 4th ventricle; ac, anterior commissure; Aq, aquedectus of silvyus; IC, inferior colliculus, mlf, medial longitudinal fasciculus; LS, lateral septal nucleus; opt, optic tract; PBN, parabrachial nucleus; scp, superior cerebellar peduncle; st, stria terminalis.
Table 2.
Tally of orexin-A immunoreactive (ir) neurons in different groups of orexin knockout (KO) and wild-type (WT) mice
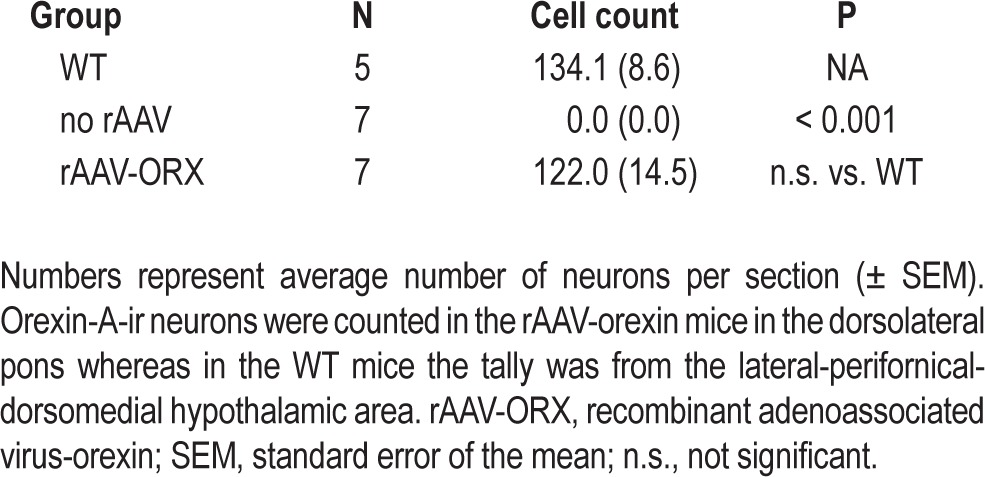
Figure 5.

Schematic distribution of infected neurons. All coronal sections containing cells expressing either GFP (green, top panel) or orexin-A (red, bottom panel) were plotted onto plates from Franklin and Paxinos mouse brain atlas.18 The numbers below each series represent individual mice. GFP, green fluorescent protein; rAAV, recombinant adenoassociated virus.
Figure 6.

rAAV-orexin gene transfer into the dorsolateral pons produced orexin A-ir in cholinergic (top row), noradrenergic (middle row) and gamma aminobutyric acid (GABA)ergic neurons (bottom row). GABAergic neurons were labeled using a vesicular GABA transporter (vGABAT) antibody. Images were acquired using a Fluoview 1000 Olympus confocal laser scanner microscope and two excitation lines (488 and 543 nm) tuned to detect Alexa Fluor 488 (520 nm; ORX, orexin) and Alexa Fluor 568 (603 nm; ChAT, choline acetyltransferase; TH, tyrosine hydroxylase; and vGABAT, vesicular GABA transporter) dyes, respectively.
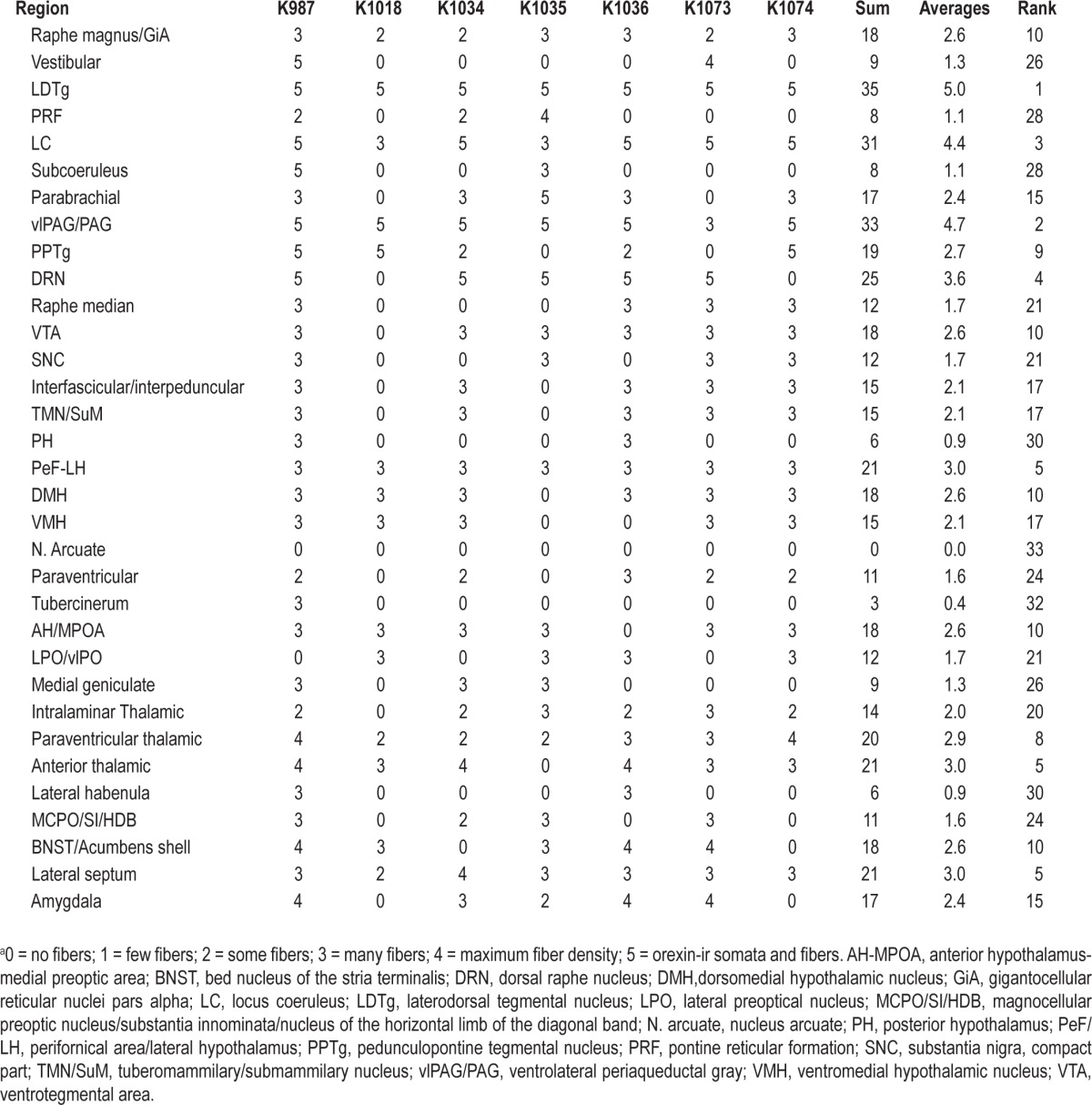
After infection numerous orexin-A-ir fibers were observed in the following regions: raphe magnus-gigantocellular reticular nucleus (pars alpha), LC, LDT, parabrachial nucleus, periaqueductal gray area, dorsal raphe nucleus, pedunculopontine tegmental nucleus, interpeduncular and ventral tegmental area, lateral ventromedial and dorsomedial hypothalamus, anterior and paraventricular thalamic nucleus, lateral septum, and the amygdala. Table 3 shows the relative density of orexin-A-ir fibers.
Table 3.
Relative density of orexin immunoreactive (ir) fibers 21 days after microinjection of rAAV-orexin into the dorsolateral pons of orexin KO micea
DISCUSSION
The primary finding of this study was that the transfer of the orexin gene into neurons of the dorsolateral pons of orexin KO mice significantly decreased the incidence of cataplexy. Unexpectedly, the insertion of the reporter gene, GFP, into the same area worsened cataplexy. In our previous studies, in orexin KO and orexin-ataxin-3 mice models of narcolepsy GFP did not worsen cataplexy when it was delivered to other areas of the brain.15,16 As this is the first study to examine the effects of gene transfer in the dorsolateral pons in narcoleptic mice models, we suggest that the dorsolateral pons is especially sensitive to manipulation. This is consistent with extensive evidence from pharmacology, lesion, and recent optogenetic stimulation studies that changes in muscle tone can be reliably elicited by manipulation of the dorsolateral pons.22–24
GFP might have unknown effects because cell lines stably expressing this reporter gene have been largely unsuccessful even though transgenic GFP-expressing mice have been developed. Likewise, examples of GFP cytotoxicity have been reported for rat liver stem cells,25 fibroblasts (NIH3t3, BHK21), hepatocarcinoma (Huh7,Hep G2), or even for plant cells lines.26 The cytotoxicity of GFP found in these stable cell lines was independent of the viral vector type or the level of GFP gene expression.27 Further electrophysiologic and/or calcium imaging studies will be necessary to determine the effect of GFP in pontine neurons as the biologic functions of the fluorescent proteins currently used, as common gene reporters are not well understood.28
Cataplexy occurs primarily during waking behavior, and proper motor control likely requires a fine coordination in the activity of dorsolateral pontine neurons. Previously, we determined that in orexin KO mice many neurons in the dorsolateral pons fired at their highest rate during wake.29 The gene product for orexin or GFP was present in these neurons, and in other neurons that are wake-active such as the LDT,30 the LC,31 and the dorsal raphe nucleus.32 A disturbance in the activity of these neurons, as produced by optogenetic stimulation of LC neurons, or by localized lesion, pharmacologic imbalance, or lack of proper orexin signal, is able to disturb motor tone. In the current study, insertion of the orexin gene may have stabilized the neuronal activity, whereas insertion of GFP produced further destabilization increasing cataplexy.
In the current study, the dorsolateral pons was selected as the site for orexin gene delivery because of its role in muscle tone control. Moreover, in our previous studies the orexin gene was inserted into neurons upstream of the pons and these surrogate neurons were found to heavily innervate the dorsolateral pons.15,16 In rats, orexin neurons from the lateral hypothalamus project to the pons and also to the medulla and spinal cord.10 In the current study, orexin gene transfer into the pontine neurons may have reestablished the orexin tone onto the downstream medullary and spinal cord neurons. Previously, the widespread and constitutive ectopic expression of orexin peptide has been shown to rescue narcoleptic symptoms in narcoleptic mice.18 Our studies have, however, identified specific neurons and the network that could be recruited to block a specific symptom.
Orexin gene transfer into the dorsolateral pons also improved wake maintenance, albeit modestly. Orexin KO mice given rAAV-orexin spent 23% of their time awake in long episodes. Without orexin, these mice spent only 8% of their time on long wake bouts. Thus, even though rAAV-orexin mice did not reach the level of wake maintenance observed in WT mice, these mice stayed awake significantly longer than orexin KO mice not given orexin gene transfer. We believe that wake maintenance was improved in infected mice because orexin was being released by neurons that fire predominantly during waking and that these wake-on neurons have been implicated in maintaining arousal. The important role of orexin signal in the tuberomammillary nucleus (TMN) for wake maintenance has been recently reported.33 TMN neurons also fire predominantly during attentive waking.34 As noted earlier, wake-on neurons have been recorded in the orexin gene transfer zone such as LC, LDT/PPT, dorsal raphe nucleus, and vlPAG. More importantly, orexin gene transfer into these wake-on neurons significantly reduced the number of NREM sleep bouts suggesting lower sleep drive in the infected mice.
In addition to cataplexy, the orexin KO mice also have more REM sleep at night.21 In the current study, the orexin KO mice not given rAAV-orexin also had more REM sleep at night compared with the WT mice, which is consistent with study results by Chemelli et al.21 We hypothesized that orexin gene transfer should decrease both cataplexy and REM sleep in the orexin KO mice given the role of the pons in triggering both. In the current study, although cataplexy was decreased, REM sleep was not. Previously, we had noted that cataplexy is a unique state different from REM sleep.29 The current data once again underscore the difference between cataplexy and REM sleep because the current study found that cataplexy can be manipulated independently of REM sleep, which is consistent with other data.35 We suggest that cataplexy was decreased because the LC and other neurons in the region that contained orexin are also wake-active and likely releasing orexin to block cataplexy, which occurs during waking. On the other hand, during REM sleep these neurons are silent and unlikely releasing orexin that would decrease REM sleep. This finding further demonstrates that the activity of the surrogate neurons is another important variable to be considered.
We have now established that cataplexy can be effectively blocked with ectopic expression of orexin in neurons at the following regions: the lateral hypothalamus, zona incerta, TMN, and now in the dorsolateral pons. In all these areas, wake-active neurons are known to be present. Likewise, these neurons receive limbic inputs from the amygdala. Cataplexy neurons have been recorded within the amygdala36 and dysfunction at the level of the amygdala has been reported in patients with narcolepsy.37,38 Orexin gene transfer into this region also blocks cataplexy.39 Incidentally, this manipulation may also decrease REM sleep because the cataplexy active cells also discharge at equal rates during REM sleep.36 Likewise, cataplexy-active neurons have also been recorded in the ventromedial medulla.40 For many years it is known that the ventromedial medulla is crucial to control skeletal muscle tone.41 Orexin also plays a role in the control of muscle tone in the ventromedial medulla.42 Therefore, it is important to assess the effects of orexin gene transfer into the ventromedial medulla.
DISCLOSURE STATEMENT
This was not an industry supported study. The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
Supported by Medical Research Service of the Department of Veterans Affairs grant number 1I01BX000798-01 and NIH grants HL091363, MH055772, NS030140, NS052287 (Dr. Shiromani), and J. Christian Gillin research grant from the Sleep Research Foundation (Dr. Blanco-Centurion). Drs. Blanco-Centurion and Liu contributed equally to the manuscript.
REFERENCES
- 1.Aldrich MS. The neurobiology of narcolepsy-cataplexy. Prog Neurobiol. 1993;41:533–41. doi: 10.1016/0301-0082(93)90042-q. [DOI] [PubMed] [Google Scholar]
- 2.Overeem S, Mignot E, van Dijk JG, Lammers GJ. Narcolepsy: clinical features, new pathophysiologic insights, and future perspectives. J Clin Neurophysiol. 2001;18:78–105. doi: 10.1097/00004691-200103000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Anic-Labat S, Guilleminault C, Kraemer HC, Meehan J, Arrigoni J, Mignot E. Validation of a cataplexy questionnaire in 983 sleep-disorders patients. Sleep. 1999;22:77–87. [PubMed] [Google Scholar]
- 4.Krahn LE, Lymp JF, Moore WR, Slocumb N, Silber MH. Characterizing the emotions that trigger cataplexy. J Neuropsychiatry Clin Neurosci. 2005;17:45–50. doi: 10.1176/jnp.17.1.45. [DOI] [PubMed] [Google Scholar]
- 5.Longstreth WT, Jr, Koepsell TD, Ton TG, Hendrickson AF, van Belle G. The epidemiology of narcolepsy. Sleep. 2007;30:13–26. doi: 10.1093/sleep/30.1.13. [DOI] [PubMed] [Google Scholar]
- 6.Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–7. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- 7.Thannickal TC, Moore RY, Nienhuis R, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–74. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thannickal TC, Nienhuis R, Siegel JM. Localized loss of hypocretin (orexin) cells in narcolepsy without cataplexy. Sleep. 2009;32:993–8. doi: 10.1093/sleep/32.8.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Lecea L, Kilduff TS, Peyron C, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–7. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peyron C, Tighe DK, van den Pol AN, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:1. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 12.Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355:39–40. doi: 10.1016/S0140-6736(99)05582-8. [DOI] [PubMed] [Google Scholar]
- 13.Blouin AM, Thannickal TC, Worley PF, Baraban JM, Reti IM, Siegel JM. Narp immunostaining of human hypocretin (orexin) neurons: loss in narcolepsy. Neurology. 2005;65:1189–92. doi: 10.1212/01.wnl.0000175219.01544.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crocker A, Espana RA, Papadopoulou M, et al. Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology. 2005;65:1184–8. doi: 10.1212/01.wnl.0000168173.71940.ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu M, Thankachan S, Kaur S, et al. Orexin (hypocretin) gene transfer diminishes narcoleptic sleep behavior in mice. Eur J Neurosci. 2008;28:1382–93. doi: 10.1111/j.1460-9568.2008.06446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu M, Blanco-Centurion C, Konadhode R, et al. Orexin gene transfer into zona incerta neurons suppresses muscle paralysis in narcoleptic mice. J Neurosci. 2011;31:6028–40. doi: 10.1523/JNEUROSCI.6069-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siegel J. Brain mechanisms that control sleep and waking. Naturwissenschaften. 2004;91:355–65. doi: 10.1007/s00114-004-0541-9. [DOI] [PubMed] [Google Scholar]
- 18.Mieda M, Willie JT, Hara J, Sinton CM, Sakurai T, Yanagisawa M. Orexin peptides prevent cataplexy and improve wakefulness in an orexin neuron-ablated model of narcolepsy in mice. Proc Natl Acad Sci U S A. 2004;101:4649–54. doi: 10.1073/pnas.0400590101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franklin BJK, Paxinos G. ed 2. San Diego, CA USA: Academic Press Inc; 1997. The mouse brain in stereotaxic coordinates, [Google Scholar]
- 20.Shiromani PJ, Xu M, Winston EM, Shiromani SN, Gerashchenko D, Weaver DR. Sleep rhythmicity and homeostasis in mice with targeted disruption of mPeriod genes. Am J Physiol Regul Integr Comp Physiol. 2004;287:R47–R57. doi: 10.1152/ajpregu.00138.2004. [DOI] [PubMed] [Google Scholar]
- 21.Chemelli RM, Willie JT, Sinton CM, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–51. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 22.Shiromani PJ, McGinty DJ. Pontine neuronal response to local cholinergic infusion: relation to REM sleep. Brain Res. 1986;386:20–31. doi: 10.1016/0006-8993(86)90137-x. [DOI] [PubMed] [Google Scholar]
- 23.Henley K, Morrison AR. A re-evaluation of the effects of lesions of the pontine tegmentum and locus coeruleus on phenomena of paradoxical sleep in the cat. Acta Neurobiol Exp (Warsz) 1974;34:215–32. [PubMed] [Google Scholar]
- 24.Carter ME, Yizhar O, Chikahisa S, et al. Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat Neurosci. 2010;13:1526–33. doi: 10.1038/nn.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taghizadeh RR, Sherley JL. CFP and YFP, but not GFP, provide stable fluorescent marking of rat hepatic adult stem cells. J Biomed Biotechnol. 2008;2008:453590. doi: 10.1155/2008/453590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haseloff J, Amos B. GFP in plants. Trends Genet. 1995;11:328–9. doi: 10.1016/0168-9525(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 27.Liu HS, Jan MS, Chou CK, Chen PH, Ke NJ. Is green fluorescent protein toxic to the living cells? Biochem Biophys Res Commun. 1999;260:712–7. doi: 10.1006/bbrc.1999.0954. [DOI] [PubMed] [Google Scholar]
- 28.Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90:1103–63. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- 29.Thankachan S, Kaur S, Shiromani PJ. Activity of pontine neurons during sleep and cataplexy in hypocretin knock-out mice. J Neurosci. 2009;29:1580–5. doi: 10.1523/JNEUROSCI.5151-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shiromani PJ, Armstrong DM, Bruce G, Hersh LB, Groves PM, Gillin JC. Relation of pontine choline acetyltransferase immunoreactive neurons with cells which increase discharge during REM sleep. Brain Res Bull. 1987;18:447–55. doi: 10.1016/0361-9230(87)90019-0. [DOI] [PubMed] [Google Scholar]
- 31.Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–86. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGinty DJ, Harper RM. Dorsal raphe neurons: depression of firing during sleep in cats. Brain Res. 1976;101:569–75. doi: 10.1016/0006-8993(76)90480-7. [DOI] [PubMed] [Google Scholar]
- 33.Mochizuki T, Arrigoni E, Marcus JN, et al. Orexin receptor 2 expression in the posterior hypothalamus rescues sleepiness in narcoleptic mice. Proc Natl Acad Sci U S A. 2011;108:4471–6. doi: 10.1073/pnas.1012456108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi K, Lin JS, Sakai K. Neuronal activity of histaminergic tuberomammillary neurons during wake-sleep states in the mouse. J Neurosci. 2006;26:10292–8. doi: 10.1523/JNEUROSCI.2341-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burgess CR, Tse G, Gillis L, Peever JH. Dopaminergic regulation of sleep and cataplexy in a murine model of narcolepsy. Sleep. 2010;33:1295–1304. doi: 10.1093/sleep/33.10.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gulyani S, Wu MF, Nienhuis R, John J, Siegel JM. Cataplexy-related neurons in the amygdala of the narcoleptic dog. Neuroscience. 2002;112:355–65. doi: 10.1016/s0306-4522(02)00089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poryazova R, Schnepf B, Werth E, et al. Evidence for metabolic hypothalamo-amygdala dysfunction in narcolepsy. Sleep. 2009;32:607–13. doi: 10.1093/sleep/32.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartz S, Ponz A, Poryazova R, et al. Abnormal activity in hypothalamus and amygdala during humor processing in human narcolepsy with cataplexy. Brain. 2008;131:514–22. doi: 10.1093/brain/awm292. [DOI] [PubMed] [Google Scholar]
- 39.Liu M, Blanco-Centurion C, Konadhode R, Pelluru D, Shiromani PJ. New Orleans, LA: Society for Neuroscience; 2012. Orexin gene transfer in amygdala inhibits cataplexy in narcoleptic mice. Program No. 799.17. Neuroscience Meeting Planner. Online. [Google Scholar]
- 40.Siegel JM, Nienhuis R, Fahringer HM, et al. Neuronal activity in narcolepsy: identification of cataplexy-related cells in the medial medulla. Science. 1991;252:1315–8. doi: 10.1126/science.1925546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magoun HW, Rhines R. An inhibitory mechanism in the bulbar reticular formation. J Neurophysiol. 1946;9:165–71. doi: 10.1152/jn.1946.9.3.165. [DOI] [PubMed] [Google Scholar]
- 42.Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Muscle tone facilitation and inhibition after orexin-a (hypocretin-1) microinjections into the medial medulla. J Neurophysiol. 2002;87:2480–9. doi: 10.1152/jn.2002.87.5.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]