

Abstract
NAD(P)H:quinone oxidoreductase type I (NQO1) is a target enzyme for triggered delivery of drugs at inflamed tissue and tumor sites, particularly those that challenge traditional therapies. Prodrugs, macromolecules, and molecular assemblies possessing trigger groups that can be cleaved by environmental stimuli are vehicles with the potential to yield active drug only at prescribed sites. Furthermore, quinone propionic acids (QPAs) covalently attached to prodrugs or liposome surfaces can be removed by application of a reductive trigger stimulus, such as that from NQO1; their rates of reductive activation should be tunable via QPA structure. We explored in detail the recombinant human NAD(P)H:quinone oxidoreductase type I (rhNQO1)-catalyzed NADH reduction of a family of substituted QPAs and obtained high precision kinetic parameters. It is found that small changes in QPA structure—in particular, single atom and function group substitutions on the quinone ring at R1—lead to significant impacts on the Michaelis constant (Km), maximum velocity (Vmax), catalytic constant (kcat), and catalytic efficiency (kcat/Km). Molecular docking simulations demonstrate that alterations in QPA structure result in large changes in QPA alignment and placement with respect to the flavin isoalloxazine ring in the active site of rhNQO1; a qualitative relationship exists between the kinetic parameters and the depth of QPA penetration into the rhNQO1 active site. From a quantitative perspective, a very good correlation is observed between log(kcat/Km) and the molecular-docking-derived distance between flavin hydride donor site and quinone hydride acceptor site in the QPAs, an observation that is in agreement with developing theories. The comprehensive kinetic and molecular modeling knowledge obtained for the interaction of recombinant human NQO1 with the quinone propionic acid analogues provides insight into the design and implementation of the QPA trigger groups for drug delivery applications.
In the past two decades, there has been remarkable interest in the activation of drug delivery systems and cellular processes by enzymes associated with inflammatory diseases and cancer.1 Such avenues have been addressed in two ways: 1) through the use of such endogenous, upregulated proteins to selectively activate antitumor compounds,2 prodrugs,3–6 and very recently liposomes;7–13 and 2) targeting these enzymes with molecules that deactivate them, thereby causing cell death through accumulation of damaging species, such as superoxide.14 Among the enzymes used as stimuli for these potential therapeutic routes, a significant research effort has been directed at a subgroup of reductase enzymes, of which NAD(P)H:quinone oxidoreductase-1 (NQO1, DT-diaphorase, EC 1.6.99.2)15, 16 has become of great interest.17 This homodimeric flavoprotein is unique among reductases, as it catalyzes the direct two-electron reduction of a wide variety quinones using NADH or NADPH as cofactor.15, 18 NQO1 is located mainly in the cytosol of cells, but can be found also in the nucleus, endoplasmic reticulum, cellular membrane, and mitochondria, as well as extracellularly.15, 19–25 The overexpression of human NQO1 in certain cancer tumor tissues (e.g. non-small cell lung, pancreas, and colon)26–30 makes it a valuable target for activating stimuli-responsive drug delivery systems based on quinone derivatives, such as prodrugs31 and liposomes.32 As a result, many groups have been actively studying the molecular structure of NQO1 using crystallographic methods so as to improve knowledge regarding its native structure (Protein Data Bank (PDB) accession code 1D4A),33 as well as its behavior with substrates, such as duroquinone (PDB accession code 1DXO), or inhibitors, including dicoumarol (PDB accession code 2F1O).33–38 Importantly, the ability of NQO1 to reductively activate sophisticated quinone and quinoidal compounds makes it an ideal target for the development of antitumor compounds39, 40 and prodrugs.31, 41 However, the quantitative behavior of human NQO1 with structurally similar, simple quinones, or quinones that can be used as trigger groups for prodrugs and delivery vehicles, has to date not been assessed.
Trigger groups that can be activated by a reductive stimulus, and as a result subsequently undergo cleavage from a targeted group,6 hold much promise in the development of endogenously-triggered prodrugs and drug delivery vehicles, such as liposomes, as well as imaging strategies based on prodyes.42, 43 Our group has shown that reductively-activated liposomes can have their contents delivered upon self-cleavage of the reduced quinone propionic acid headgroup of the fusogenic lipids composing the liposomes; similarly, such cleavage of the reduced quinone from cloaked probes (prodyes) results in fluorescent signaling of human NQO1 activity.32, 42 Variants of the quinone propionic acid (QPA) trigger group have also been investigated for prodrugs, such as those based on mustard44 and oxindoles.45
The key step for these applications is the initial reduction process of the cleavable quinone head group that is attached to the target molecule (lipid, drug, dye), a process that should be controlled by the structure of the trimethyl-locked (Thorpe-Ingold or gem-disubstituent effect)46–48 quinone propionic acid trigger, Scheme 1. The trimethyl-lock effect comes about when R3=R4=CH3, and it leads to a more rapid cyclization of the hydroquinone to yield at higher rates the lactone and the released target molecule, R’H, Scheme 1.32 Tunability in the reductive triggering of QPA groups for either prodrug or liposome delivery applications should be possible as a result of substituent variation at R1, R2, and R3.
Scheme 1.

Reduction and cyclization of quinone propionic acid (QPA) trigger groups.
Although there exists a good number of reports on structure-relationship activity49–56 and docking studies of NQO1 inhibitors and substrates for anticancer therapy,14, 35, 57–62 there is only one published report that provides Michaelis constant (Km) and maximum velocity (Vmax) values for the interaction of human NAD(P)H:quinone oxidoreductase-1 with a simple quinone.44 In addition, the significant differences in reactivity of human NQO1 and rat NQO1 with specific quinone species63–65 suggest limited applicability of structure-reactivity relationship predictions based on rat NQO1.66–68
In this report, we examine a family of substituted quinone propionic acid (QPA) derivatives as potential substrates for recombinant human NQO1. We report on detailed solution-phase rhNQO1 kinetic parameters, namely the Michaelis constant (Km), maximum velocity (Vmax), catalytic constant (kcat), and catalytic efficiency (kcat/Km). Using the known crystal structure of the hNQO1-duroquinone complex (1DXO),33 six different molecular docking receptors were delineated (changing interface radius, oxidation state of cofactor, and active site definition). Quinone derivatives were docked in each receptor to determine if there were any differences in the outcomes and to have a better prediction for the orientation of quinone propionic acids in the active site of rhNQO1. Furthermore, we explored possible correlations between the experimental observables extracted from the kinetic assays and those obtained from the docking studies.
MATERIALS AND METHODS
Materials
Recombinant human enzyme NAD(P)H:quinone oxidoreductase-1 (EC 1.6.99.2) (rhNQO1), nicotinamide adenine dinucleotide reduced (β-NADH), and bovine serum albumin (BSA) were purchased from Sigma (D1315, N8129, A3294). Potassium dihydrogen phosphate (KH2PO4, Fluka 60219), dipotassium hydrogen phosphate (K2HPO4, Fisher P288), potassium chloride (KCl, Sigma P9541) and potassium hydroxide pellets (KOH, Mallinckrodt) were used to make the buffer. Ethyl alcohol (200 Proof) was purchased from Pharmaco-AAPER. High-purity water was obtained from a Nanopure Diamond Barnstead System (18.2 MΩ-cm). The quinone propionic acid substrates were synthesized as previously described69 or in the Supporting Information. The quartz 96-well microplate used in the enzyme assays was purchased from Hellma (730.009-QG). A FLUOstar Optima plate reader from BMG Labtech was used to follow NADH consumption during the enzyme assay.
Enzyme Kinetic Assay
Enzyme kinetic parameters were determined using UV-visible spectroscopy by adapting a previous assay70 to a well plate reader and 96-well quartz plate. The oxidation of NADH at 340 nm was measured using an experimentally determined extinction coefficient of 4390 M−1cm−1 (see Supporting Information). Kinetic experiments were performed on three different days with at least three replicates of each condition performed. The assay solution consisted of 0.1 M phosphate buffer/0.1 M KCl solution at pH 7.10 containing 0.007% of bovine serum albumin, 100 µL of appropriate concentration of quinone substrate, 50 µL of a 400 µM NADH solution, and 50 µL of enzyme solution (0.11–3.00 µg). The total assay volume was 200 µL. All stock quinone solutions were prepared by dissolving each quinone in ethanol (100 µL) with subsequent dilution to 10.00±0.02 mL with 0.007% BSA solution. Quinone solutions of the desired concentrations were made by taking the appropriate volume from the quinone stock solution and diluting it to 10.00±0.02 mL in a volumetric flask. The solutions were kept in the dark at room temperature (22±2 °C), for approximately 2 h before the experiments were started. Once the 96-well plate was filled with the assay solutions, except the NADH solution, it was placed into the instrument and left to sit for 5 min before starting the measurements. The enzyme reaction was initiated by automated dispensing of the NADH solution into the wells, and the absorbance change at 340 nm was measured for 1.6 min at room temperature (22 to 26 °C). The linear portion of the absorbance vs. time graphs (the first 20 seconds to 1 min) were fitted, and the slopes were calculated (velocity). The average velocity (slope) from the replicates was used to calculate the kinetic parameters, with the Q–test being applied so as to reject values outside of the 90% confidence level.71 Plots of average velocity versus quinone concentration were used to obtain apparent Km and Vmax values from non-linear least squares analysis using algorithms developed by Cleveland for Michaelis-Menten kinetics.72 In some cases, the t-test was applied to kinetic parameter values for different substrates so as to determine if they were statistically different at the 95% confidence level.73 Attempts to obtain the kinetic parameters for quinone propionic acids with R1 substituents of N-methylamine (QMe-N-COOH) and N-(n-propyl)amine (Qn-pr-NH-COOH) were met with frustration, because the results obtained are not reliable due to both substrates absorbing light at the wavelength (340 nm) that NADH consumption is measured.
Docking
Docking studies were performed using FlexX from LeadIT (1.3.0 version).74–81 Quinone structures were prepared in Chemdraw3D and subsequently minimized using Sybyl-X (Version 1.1.1) using the Tripos force field for suitable bond distances and angles, and the Gasteiger-Hückel method for charge calculation; the minimized quinone structures were used without modification in the docking studies. Enzyme coordinates used in the FlexX software were obtained from The Protein Data Bank (code: 1DXO). A molecular visualization system called PyMOL was used to prepare the docking frames. Construction of six different receptors was done with the FlexX software by selecting the active site containing amino acids from monomer A and C of the enzyme crystal structure. Two water molecules were present in each of the receptors. Receptor 1 includes FAD (as it is in the X-ray coordinates) as a cofactor with duroquinone as the reference ligand (X-ray coordinates edited by FlexX for correct atom hybridization and bond type), and the binding site is defined within an interface of 6 Å encompassing the reference ligand. Receptor 2 includes FADH2 (FAD X-ray coordinates modified) as a cofactor with duroquinone as the reference ligand (X-ray coordinates edited by FlexX for correct atom hybridization and bond type) and the binding site is defined within an interface of 6 Å encompassing the reference ligand. Receptor 3 is the same as Receptor 1 but has an interface radius of 8 Å. Receptor 4 is the same as Receptor 2 but has an interface radius of 8 Å. Receptor 5 includes FAD as a cofactor with the active site defined by a sphere of 8.5 Å with N5 from FAD as its center. Receptor 6 includes FADH2 as a cofactor with the active site defined by a sphere of 8.5 Å with N5 from FADH2 as its center.
In FlexX flexible docking calculations, the initial position of the substrate is outside the active site. Then, an algorithm for fragmenting the quinone substrate is executed, and a base fragment is automatically selected and placed in the active site on which an incremental construction algorithm is performed. From these calculations, each substrate examined using different receptors has a set of solutions where the closest poses (based on score, position and interactions with the protein) were selected for detailed investigation. On these selected poses, the influence of the cofactor state (oxidized - FAD, or reduced - FADH2 form), receptor radius, and the inclusion/lack of inclusion of a reference ligand was studied. In addition, the total score was used as an estimate of the free energy of binding and was subsequently used in a plot of theoretical free energies of binding versus experimental free energies of binding.75
The docking method is based on the experimental observed range of nonbonded contact geometries revealed by statistical analysis of the Cambridge Structural Database (CSD); the analysis of the CSD is used to define the range of allowed angles and dihedrals describing the nonbonded contact geometry.82 The program discards any solution with electrostatic repulsion (based on a threshold limit distance). The scoring function accounts for hydrogen bonds, ionic interactions, hydrophobic contact surface, and the number of rotatable bonds in the substrate; it is a modification of Böhm’s function.78–80 The limitations of the scoring function are that it does not account for differences in binding strengths between various neutral hydrogen bonds or ionic interactions, and it does not include the conformational energy of the substrate.75, 82 The FlexX algorithm breaks down the substrate into fragments where one is automatically elected as the base fragment,78 and then the fragments are connected flexibly during docking in an incremental way.77 In the triangle algorithm, triangles of interaction centers are formed using the base fragment, and those are mapped onto triangle points lying on the receptor’s surface. As an alternative to the triangle algorithm, a line algorithm is used when only two interactions exist simultaneously between the substrate and the enzyme. The receptor is rigid and the ligand is flexible for both algorithms.
Voltammetry
Cyclic voltammetry (CV) experiments were performed with a computer-controlled EG&G PAR model 273A potentiostat and a three-electrode setup that included a (pretreated) glassy carbon working electrode (A = 0.07 cm2, CH Instruments, Austin, TX), a homemade 99.9% platinum counter electrode (d = 0.05 cm), and Ag/AgCl (3.0 M KCl, CH Instruments) reference electrode. The working electrode was polished, prior to its electrochemical pretreatment, on a Buehler microcloth with 1 micron Alpha alumina slurry micropolish. Pretreatment of the working electrode was achieved by applying +1.5 V for 10 min, a method that essentially serves to eliminate any electrochemically-active species on the surface of the electrode that may interfere with data collection. Scans were conducted at a rate of 0.1 V s−1 at room temperature in 0.1 M phosphate buffer/0.1 M KCl solution at pH 7.10.
RESULTS AND DISCUSSION
Kinetic Studies on Triggerable Quinones
It is known that NQO1 from various mammalian species can reduce—albeit with greatly varying rates—a broad range of small- and large-sized quinone substrates, ranging from the simple 1,4-benzoquinone to the very complex geldanamycin quinone.83 This apparent “universal” ability to catalyze the reduction of such different-sized quinones has been attributed to the quite large quinone/nicotinamide binding site in human and mouse NQO1 (360 Å3).33 However, to the best of our knowledge, there are no studies that quantitatively address the effects of systematic variations in quinone ring substituent size on human NQO1 Vmax and Km values under similar experimental conditions. Having in hand these readily comparable kinetic parameters is crucial to gaining a more thorough understanding of the reactivity of different hNQO1 substrates, particularly those that can be used in the delivery of drugs via prodrug or triggered-release methodologies.
Presented in Table 1 are the recombinant human NQO1 kinetic parameters (Km, Vmax, kcat, and kcat/Km), two-electron aqueous reduction potentials (E1/2) and van der Waals volume for the family of quinone propionic acid (QPA) derivatives studied here. For comparison purposes, the collection of QPA derivatives is subdivided into four groups: a) quinone propionic acids having different substituents at the R1 position of the quinone ring for the trimethyl lock series (R3=R4=CH3), b) a charged and neutral version of a quinone propionic acid having the trimethyl lock (R1=R2=R3=R4=CH3), c) trimethyl-locked quinone propionic acids with substituent variations at R1 and R2, and d) quinone propionic acids that do not possess the trimethyl lock motif.
Table 1. Kinetic parameters for the reduction of quinone propionic acid derivatives by hNQO1.
Values reported are the mean ± one standard deviation for three independent determinations. [NADH] = 1.00 × 10−4 M in all cases. The van der Waals volumes were calculated according to the literature.89
| Quinone |
Vmax (µmol·min−1·mg−1) |
Km (µM) |
kcat (s−1) |
kcat/Km (M−1·s−1) |
E1/2 vs. SHE (V) |
van der Waals volume (Å3) |
|---|---|---|---|---|---|---|
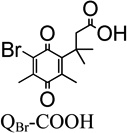 |
88±7 | 41±8 | 45±4 | 1.1±0.2 × 106 | 0.095±0.001 | 251 |
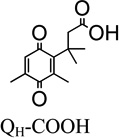 |
83±8 | 50±11 | 43±4 | 8.5±2.0 × 105 | 0.117±0.002 | 232 |
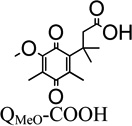 |
42±5 | 447±102 | 22±3 | 4.8±1.2 × 104 | 0.098±0.001 | 258 |
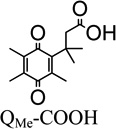 |
38±5 | 158±41 | 20.±3 | 1.2±0.4 × 105 | 0.047±0.002 | 249 |
 |
60±7 | 132±32 | 31±4 | 2.3±0.6 × 105 | 0.041±0.001 | 295 |
 |
14±1 | 376±87 | 7.2±0.5 | 1.9±0.5 × 104 | 0.128±0.001 | 282 |
 |
78±3 | 20±3 | 40.±2 | 2.0±0.3 × 106 | 0.144±0.001 | 232 |
 |
66±4 | 5±1 | 34±2 | 6.8±1.4 × 106 | 0.143±0.002 | 215 |
Substituent Variation at the R1 position on the Quinone Ring of the Trimethyl Lock Series
We obtained the kinetic parameters for the rhNQO1-catalyzed reduction of four trimethyl-locked quinone propionic acids (R2–R4= CH3) that differ in their functional group at the R1 position on the quinone ring (R1 = Br, H, CH3, CH3O). We selected the R1 position for substituent variation due to its minimal influence on the geometry of the trimethyl lock “side” of the quinone ring that dictates the rate of cyclization of the hydroquinone resulting from reduction.32, 69 Upon initial perusal of the data in Table 1, there is no apparent well-defined relationship between Vmax, kcat, or kcat/Km and the two-electron quinone reduction potential E1/2, an interesting outcome and one that indicates steric effects play a dominant role in the enzyme-catalyzed, two-electron reduction of these quinones; we elaborate on these observations later.
It is noteworthy that QBr-COOH (R1=Br) and QH-COOH (R1=H) are the best rhNQO1 substrates in this trimethyl lock series based on their Km, Vmax, kcat, and kcat/Km values. The rhNQO1 is able to reduce these two substrates quickly (about 45 molecules per second) and efficiently (~106 M−1 s−1). The observed Michaelis constant of Km = 41 µM (QBr-COOH) and Km = 50 µM (QH-COOH), as well as the maximum velocity values of Vmax = 88 µmol·min−1·mg−1 (QBr-COOH) and Vmax = 83 µmol·min−1·mg−1 (QH-COOH), were found to be statistically indistinguishable at the 95% confidence level. However, the Km and Vmax of QMe-COOH (158 µM, 38 µmol·min−1·mg−1) and QMeO-COOH (447 µM, 42 µmol·min−1·mg−1) are substantially different from each other and from those for the QBr-COOH and QH-COOH, with their higher Km values reflecting their lower rhNQO1 affinity.
The impact of substituent at the R1 position for the trimethyl-locked quinone propionic acids (R2–R4= CH3) follows the general trend of the larger is the functionality at R1, the smaller is the Vmax and binding capability (larger Km). When comparing the quinone propionic acids (R2–R4= CH3) with increasing size of the substituent at R1 (H < CH3 < CH3O), the Km increases, as expected for steric interactions between the quinone ring and the hNQO1 binding motif that result in an unfavorable alignment and position of the quinone ring in the active site.35 Interestingly, Vmax decreases with increased size of R1 substituent on the quinone propionic acid ring. This is quite evident when comparing the QH-COOH, QMeO-COOH, and QMe-COOH species, as the Vmax of the hydrogen variant (83 µmol·min−1·mg−1) is roughly two times greater than that of the methyl-substituted quinone propionic acid (38 µmol·min−1·mg−1) and the methoxy derivative (42 µmol·min−1·mg−1). Steric hindrance has previously been suggested as the cause of lower reduction rates for indolequinones and benzoquinone mustard compounds.53, 56, 62 Although it could be argued that the differences in Vmax observed here are due solely to differences in the electronic properties of the quinone propionic acids, it is clear this is not the case. Such a conclusion is supported by our observation that the methoxy derivative has a reduction potential (E1/2 = 0.098 V) very close to that of QH-COOH (E1/2 = 0.117 V), but the Vmax of the methoxy derivative (42 µmol·min−1·mg−1) is roughly half that of QH-COOH (83 µmol·min−1·mg−1) and is almost identical to that of QMe-COOH (38 µmol·min−1·mg−1) which has a much more negative E1/2 (0.047 V). Thus, steric factors appear to be dominant in the rhNQO1-catalyzed reduction of these trimethyl-locked quinone propionic acids.
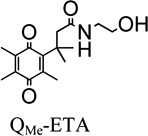
The Effect of Neutral versus Charged Trimethyl-locked Quinone Propionic Acid
To investigate the rhNQO1-catalyzed reduction of a neutral quinone versus a negatively-charged quinone, we studied one trimethyl-locked quinone propionic acid (R1=R2=R3=R4=CH3) derivatized with ethanolamine via an amide bond (QMe-ETA) that is electrically neutral at the pH values used here;84 we compared its rhNQO1 kinetic values to those obtained for QMe-COOH that is anionic at physiological pH due to its deprotonated carboxylic acid group.47 QMe-ETA yields a significantly higher maximum reduction velocity (60 µmol·min−1·mg−1) in comparison to the anionic QMe-COOH derivative (38 µmol·min−1·mg−1); however, the Michaelis constants are statistically indistinguishable. The increase of 22 µmol·min−1·mg−1 in Vmax for the QMe-ETA is possibly due to the lack of charge on this substrate, or the QMe-ETA may adopt a conformation in the active site of the enzyme that promotes more favorable interactions. These hypotheses will be discussed later in the sections addressing outcomes from molecular docking studies.
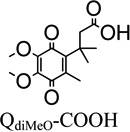
Substituent Variation at the R1 and R2 positions on the Quinone Ring of the Trimethyl Lock Series—QdiMeO-COOH
Although this analog possesses the trimethyl-lock structural motif, QdiMeO-COOH does not fall into the category discussed in the previous section, because it does not have a methyl group at the R2 position of the quinone ring. But it was synthetically facile and possesses the core portion of Coenzyme Q10, and thus it was added to our library of quinone propionic acids for investigation. The Km value of the dimethoxy derivative is statistically indistinguishable from that of the mono-methoxy quinone propionic acid. However, the maximum reduction velocity for QdiMeO-COOH is only one-third that of the mono-methoxy derivative, QMeO-COOH (Vmax = 42 µmol·min−1·mg−1). The significantly lower rhNQO1 reduction velocity most likely results from the exceptional amount of steric interactions between the methoxy groups and the NQO1 binding site that arise from the presence of adjacent methoxy groups in QdiMeO-COOH. It is known for ortho-dimethoxy-1,4-benzoquinones that one of the methoxy groups lie out of the plane of the quinone ring.85, 86 Thus, the out-of-plane methoxy substituent of QdiMeO-COOH will provide for unfavorable steric interactions with the NQO1 active site.
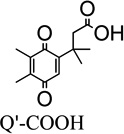
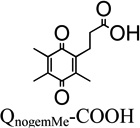
The Presence versus the Absence of the Trimethyl-Lock Motif in the Quinone
In order to probe the effect of trimethyl-lock motif presence/absence on the rhNQO1 kinetic parameters, we examined two quinone propionic acids that do not possess the trimethyl lock. The Q’-COOH derivative does not have a methyl substituent at R3, and QnogemMe-COOH lacks methyl groups at the geminal R4 position. For Q’-COOH that possesses a hydrogen (instead of a methyl) at the R3 position on the quinone ring, its observed Michaelis constant is 20 µM, a value that reflects substantially tighter binding of Q’-COOH by rhNQO1 than QMe-COOH (158 µM) which possesses the trimethyl-lock motif. In addition, QH-COOH and Q’-COOH both have a hydrogen group on the quinone ring, but the difference in hydrogen group position at R1 versus R3 makes QH-COOH a trimethyl-lock derivative and Q’-COOH not; the presence of the trimethyl lock in QH-COOH leads to a ~21° deviation from planarity of the quinone ring when compared to Q’-COOH.69 These two positional isomers have significantly different Km values of 20 µM (Q’-COOH) and 50 µM (QH-COOH), most likely because of the impact of ring distortion on binding of QH-COOH in the active site of rhNQO1. To further probe the effect of the trimethyl-lock motif, we examined the quinone propionic acid with no germinal methyl groups on the propionic side chain (R4 position), QnogemMe-COOH, whose quinone ring is planar.69 It was found that QnogemMe-COOH was even more specific toward the enzyme, as noted by its Km value of roughly 5 µM and Vmax of 66 µmol·min−1·mg−1; in addition, its catalyzed reduction is quite efficient (6.8 × 106 M−1 s−1).
Possible Correlations Between hNQO1-catalyzed Kinetic Parameters and Quinone Propionic Acid Characteristics
Based on the widely accepted two-electron reduction (hydride-transfer) mechanism for the NQO1-catalyzed reduction of various quinones,18 it is anticipated that there should exist a relationship between the physical and chemical properties of the quinone substrates and the kinetic observables associated with their reduction by hNQO1. From previous studies with other mammalian (rat and mouse) NQO1, it has been suggested that the “reactivity” of quinones toward murine NQO1 does not strictly depend on the two-electron reduction potential87 or lipophilicity88 of the quinone substrates; others have noted qualitative trends between rat NQO1 activity and the octanol-water partition coefficient,70 as well as van der Waals volume,66 of certain quinones. Due to the differences in the structure of human NQO1 and its capability to activate chemotherapeutic agents49, 63 and menadione (vitamin K3)64, 65 versus that from murine sources, it is of great importance to learn what if any quantitative relationships exist between human NQO1 reduction kinetic parameters for analogous quinone substrates and the quinone substrate properties. To the best of our knowledge, there are no studies that quantitatively address the effects of systematic variations in quinone ring substituent size on human NQO1 reactivity.
Upon inspection of the reactions governing the mechanism for the ping-pong, bi-bi, hNQO1-catalyzed reduction of quinones (Q) to hydroquinones (H2Q) using NADH as the hydride (electron) source, kcat and kcat/Km are predicted to have a dependency on the characteristics of the quinone substrate:
| Equation 1 |
| Equation 2 |
| Equation 3 |
| Equation 4 |
In particular, kcat is a function of the first-order rate constants for hNQO1 reaction with NADH (k2, Equation 2) and the quinone substrate (k4, Equation 4), with kcat = k2k4/(k2+k4); while kcat/KmQuinone = k3k4/(k−3+k4), where k3 and k−3 are the forward and reverse rate constants for quinone substrate binding with reduced hNQO1 (Equation 3). Under the assumption of the first-order reaction of reduced hNQO1 with quinone (Equation 4) being the rate-limiting step, kcat = k4 and kcat/KmQuinone = k3k4/k−3. It was found that there is no apparent correlation between log kcat and quinone propionic acid two-electron reduction potential E1/2 or log kcat and quinone propionic acid van der Waals volume89 (Figures S2–S3). It is possible that for some of the substrates studied, the first-order reaction of reduced hNQO1 with quinone (Equation 4) is not the rate-limiting step, leading to kcat = k2. In addition, in the case of comparable reaction rates (k2 ≈ k4), kcat may depend on k2 and k4. Both of these scenarios would lead to decreased correlation between log kcat and E1/2 or log kcat and quinone propionic acid van der Waals volume. However, kcat/KmQuinone should not be sensitive to these concerns of k2 versus k4.
Interestingly, a plot of log(kcat/Km) vs. van der Waals volume, Figure 1, gives rise to two identifiable relationships, yielding two sets of quinone propionic acids that we refer to as fast (green points) or slow (red points) with respect to their reduction by rhNQO1. A similar observation has previously been noted in the rat NQO1-catalyzed reduction of a wide variety of quinone substrates, such as anthraquinones and naphthoquinones.66 In addition, the same slow and fast quinone substrates are noted in a plot of log(kcat/Km) vs. E1/2, with the increased size of the slow substrates yielding a negative correlation, Figure S4. This latter observation is similar to what has been found in attempted correlations of glucose oxidase activity with the reduction potentials of substituted ferrocenes, where it was found that oxidase reactivity decreased with more favorable ferrocene reduction potential; this outcome was attributed to steric interactions between the glucose oxidase and the substituted ferrocenes.90 The relationships we observe for the two groups of quinone propionic acids in Figure 1 are quite reasonable (R = 0.9008 for the fast substrates, green line; 0.96661 for the slow quinones, red line), but this may be due to the small range of van der Waals volumes studied here. In total, our observations clearly point to the significant role sterics plays in the hNQO1-catalyzed NADH reduction of the quinone propionic acids. As a result, we undertook computational substrate-enzyme docking studies in order to possibly develop a more comprehensive relationship between the observed kinetic parameters and information about quinone substrate interactions with rhNQO1.
Figure 1. Relationship between substrate specificity and quinone propionic acid van der Waals volume for fast (green points/line) and slow (red points/line) substrates.

Lines are the best fit to the data points as obtained by linear least-squares analysis. Error bars represent one standard deviation (n = 3). The van der Waals volumes were calculated according to the literature.89
Docking Studies on Triggerable Quinones
To elucidate the possible influence of all the factors associated with the computed interactions between the quinone propionic acids (QPAs) and hNQO1, a variety of receptors were defined and evaluated as outlined below. In docking, the way in which a receptor is defined affects in different ways the predictions of substrate interactions. Specifically, it is important to know what amino acids are to be included as defined by the radius of interaction (“interface radius”). Also, for the case at hand, the cofactor oxidation state (FAD vs. FADH2) of the receptor must be selected. The docking methods employing the selected receptors were validated using a known hNQO1-substrate crystal structure. Then, all the QPAs evaluated in the kinetic assays were docked in the active site of hNQO1 using the defined receptors. In addition, the QPA amine derivatives, QN-Me-COOH and Qn-pr-NH-COOH, were also docked in the active site of hNQO1with the aim of predicting their position and binding energy because of the lack of enzyme kinetic data for them.
Which hNQO1 environment should be used for validation of the docking methods?
For method validation purposes, we examined docking outcomes for a quinone whose interactions with hNQO1 are well known from an X-ray crystal structure of the quinone bound in the active site of hNQO1.33 We selected duroquinone (2,3,5,6-tetramethyl-1,4-benzoquinone) due to its structural similarity to the core of the quinone propionic acids studied here. For docking of duroquinone with all of the receptors described below, it was found that excellent results could be achieved, with an observed root-mean-square deviation (RMSD) < 0.5 Å for the best prediction and an RMSD < 1.34 Å for the poorest one. In all of these docking outcomes, it was found that the orientation of the duroquinone ring was very similar, with differences of less than 1.2 kJ·mol−1 being observed between the lowest energy versus 20th-lowest energy outcome for each receptor. In Figure 2 is shown the best pose (RMSD = 0.45 Å) for duroquinone (pink) in comparison to its position in the original crystal structure (yellow).
Figure 2. Best prediction for docked duroquinone in receptor 1 compared with the position of the duroquinone in the original crystal structure.

The position of the docked duroquinone (pink) differs from the original duroquinone (yellow) as noted by the 0.45 Å root-mean-square deviation (RMSD). Representation of amino acids (stick display; color by atom type, carbon atoms colored in purple) and FAD (sticks display; color by atom type, carbon atoms colored in cyan) in receptor 1.
FAD (oxidized) versus. FADH2 (reduced)
In the NQO1-catalyzed reduction of quinones by NADH, there is still much debate as to where the negative charge resides after the first hydride transfer occurs (flavin reduction by NADH). The most prominent proposal in the literature is that in which the negative charge is delocalized in the region of the N(1)-C=O(2) portion of the flavin, with it being stabilized through hydrogen bonding interactions with the phenol functionality of Tyr-155. During NAD+ displacement, it is thought that the O(2) of the flavin is protonated to form FADH2.34, 40, 91, 92 Subsequent reduction of the quinone occurs via a mechanism that is effectively the reverse of that for formation of FADH2. These unproven mechanistic processes are outlined in Scheme 2.40, 93 There are also concerns about the geometry of FAD versus FADH2 and how protonation can affect its conformation. In the literature, it has been reported that the shape of FADH2 in solution is butterfly-like (versus planar for FAD), but in most modeling studies, FADH2 is modeled as being planar in the active site.33, 91
Scheme 2.

Hydride transfer mechanism for possible atom sites of QPAs.
The outcome solutions from docking the quinone propionic acid analogs into the different receptors having an active site possessing FAD or FADH2 as cofactor were examined. For this, we compared the results from receptor 1 versus receptor 2, receptor 3 versus receptor 4, and receptor 5 versus receptor 6. In all cases, the positions of the quinones were effectively the same. For the poorest score in each case, a slight increase of 0.6 kJ·mol−1 was observed for the receptor containing FAD versus the receptor containing FADH2; for the best score, the increase was 0.8 kJ·mol−1.
Interface Radius of 6 Å vs. Interface Radius of 8 Å
The binding conformations of the quinone analogs were examined for the case wherein the only difference was the interface radius of the active site. We compared receptor 1 to receptor 3, and receptor 2 to receptor 4. For the two interface radii, the orientation of the quinone analogs in all the receptors mentioned above were the same, and the outcome solution with the lowest score energy for the duroquinone in each receptor differed by only 1 kJ·mol−1.
Defining the Active Site (Reference Ligand versus Sphere)
The binding conformations of the quinone propionic acids were examined by defining the active site differently. In the case of receptors 3 and 4, the ligand bound to the enzyme was used as reference ligand (duroquinone), and it had an interface radius of 8 Å defined from its position. In the case of receptor 5 and 6, the nitrogen N5 of the FAD or FADH2 cofactor was used as the sphere center, and an interface radius of 8.5 Å was defined from it. We compared receptor 3 versus receptor 5, and receptor 4 versus receptor 6. In these scenarios, the outcomes with the lowest score and the best prediction differ in their orientations only for duroquinone but not for the other quinone analogs. However, the outcome for the lowest energy value and the one associated with the best orientation is different for all of the quinones. The lower energy score is smaller by 1 kJ·mol−1 in the receptors possessing the active site defined by the 8.5 Å radius sphere. The best prediction corresponds to the quinone located deeper in receptors 5 and 6. In this case, the energy is 0.6 kJ·mol−1 lower than in receptors 3 and 4. We conclude in this case for the quinone propionic acids, even though there is a difference in score energy, this difference is minimal and does not affect the outcome. Therefore, there is no apparent influence of the chosen active site definitions on quinone positions or score energies.
After comparing all the receptors, we observed that there is no significant effect of cofactor oxidation state or interface radius, on the docking outcomes. In addition, the definition of the active site has little influence on the docking solutions. In Figures S5 and S6 are represented the lower score frame of QBr-COOH (shown as a representative example of the outcomes observed for all of the quinone propionic acid homologs) in each receptor and the corresponding poseview, so as to help to visually support the conclusions made above. These conclusions are also based on data in Table S1 that contains the lowest score energies for QBr-COOH in all the receptors.
Outcomes from Docking Studies for the Quinone Propionic Acids
The solutions using receptor 1 were utilized in comparisons of the quinone propionic acids, and those outcomes are an accurate representation of what is observed in all the receptors as visualized in Figure 3 and their superimposed images for the trimethyl-lock motif (Figure S7), quinones with the trimethyl-lock motif present and absent (Figure S8), and quinones differing in their charge (Figure S9). For the trimethyl-lock series, it was found that QBr-COOH penetrates most deeply into the active site of hNQO1, whereas QMeO-COOH penetrates the least. In addition, QBr-COOH, QH-COOH and QMe-COOH all lay parallel to the isoalloxazine ring of the FAD cofactor, but the distance between the sites of the quinone ring for possible hydride transfer and the flavin is different due to slight variations in quinone ring orientation with respect to the flavin, vide infra. The methoxy-, dimethoxy-, and amine-substituted quinone derivatives do not bind as tightly as the other quinone analogs. The QPAs lacking the trimethyl-lock motif penetrate into the active site most deeply, with Q’-COOH being closest to the flavin isoalloxazine ring. Interestingly, the neutral QMe-ETA is located deeper in the active site of the enzyme than is the anionic QMe-COOH. Using the criteria of quinone orientation with respect to and distance from the flavin of hNQO1, all of the observations from docking studies are in qualitative agreement with the kcat/Km values. The obvious possible explanation for the differences in quinone propionic acid orientation and distance from the flavin is that of steric interactions resulting from the presence of more bulky substituents on the quinone ring or changes in quinone ring planarity due to the presence of the trimethyl lock.69 Other research groups have reported that a bulky amine at the 2-position of the quinone ring of indolequinones and quinolinequinones result in a tremendous decrease in their rate of hNQO1-catalyzed reduction, thereby making them poor substrates.51, 52, 54
Figure 3. Structural frames of FlexX-docked quinone propionic acids in the active site of hNQO1.

Stick display style in all the frames, FAD (color by atom type; carbon atoms colored in cyan), amino acids (color by atom type, carbon atoms colored in purple) and docked-quinones (color by atom type, carbon atoms in pink).
Quantitative Relationships Between Docking Outcomes and NQO1-catalyzed QPA Reduction Parameters
We investigated a possible correlation between the theoretical binding energies from the docking outcomes75 and those derived from the experimentally determined Km values. Unfortunately, there was no relationship between these experimental and theoretical energies, as noted in Figure S10. However, this is not necessarily surprising, based on the controversy that exists in the literature about whether or not binding energies from docking studies can be correlated with experimental reactivities.38, 59–61, 94
Based on our observations in Figure 1 that indicate QPA substrate specificity or catalytic efficiency (kcat/Km) is strongly influenced by the size of the QPA, we posited that the second-order rate constant is a function of the distance of substrate approach to the reduced flavin site in hNQO1. Thus, we explored the possible application of Marcus-like95 theories regarding hydride-transfer (H tunneling) in enzyme systems. A basic tenet of these theories is the rate constant for hydride-transfer between a donor and an acceptor is exponentially dependent on the distance of separation between the hydride donor and acceptor atoms when the system is in the tunneling ready state.96 This adaptation of Marcus theory to describe the rate of hydride transfer is achieved by inclusion of a Franck-Condon term that relates the efficiency of tunneling through a barrier to the donor-acceptor distance (DAD) and the mass of the tunneling species. Thus, we examined docking outcomes and measured the hydride donor-acceptor distance for each of the quinone propionic acids as a function of the possible paths in Scheme 2.
The values of the distance from N5 of the flavin center in hNQO1 to the possible hydride transfer site of the quinones,40, 93, 97 denoted here as the hydride donor-acceptor distance (DAD), are presented in Table 2 for the best score energy obtained for each quinone propionic acid substrate. We assumed that all paths in Scheme 2 are possible. Upon examining possible relationships between the substrate specificity or catalytic efficiency (kcat/Km) and the DAD for the three paths in Scheme 2, we found that there exists a an acceptable correlation between the log(kcat/Km) and DAD for the scenario wherein the hydride is transferred from the N5 of the flavin of NQO1 to the carbon ortho to the carbonyl (Path A in Scheme 2, CA) of the QPAs; this is reflected by the R value of 0.9447 for this plot in Figure 4 for the set of eight different quinone propionic acids studied here and is most likely due to the similar structural characteristics of the hNQO1 substrates. No apparent correlation exists between the log(kcat/Km) and DAD for either Path B (hydride transfer to the carbonyl carbon, CB) or Path C (transfer to the carbonyl oxygen, OC), Figures S11 (R = 0.4841) and S12 (R = 0.2534). Although the outcomes are preliminary in nature in regard to evaluating application of this data treatment route, the suggested 1,4 addition of hydride from the correlation in Figure 4 makes sense from an energetics point of view.59, 93
Table 2. Energy scores and hydride donor-acceptor distances (DAD, distance from N5 of flavin to the possible hydride transfer atoms) for the quinone propionic acids.
| Donor-Acceptor Distance (Å) |
||||
|---|---|---|---|---|
| Quinone | Score (kJ·mol−1) | [N5-CA] | [N5-CB=O] | [N5-OC] |
| QBr-COOH | −25.2 | 3.25 | 3.47 | 3.58 |
| QH-COOH | −24.0 | 3.40 | 4.11 | 4.50 |
| QMe-COOH | −26.4 | 3.58 | 3.54 | 3.71 |
| QMeO-COOH | −20.3 | 3.83 | 3.90 | 3.50 |
| QdiMeO-COOH | −28.4 | 4.05 | 4.05 | 3.68 |
| Q’-COOH | −27.0 | 3.18 | 3.81 | 3.92 |
| QnogemMe-COOH | −25.8 | 3.22 | 3.49 | 3.68 |
| QMe-ETA | −28.5 | 3.43 | 3.73 | 3.81 |
| Q(Me-N)-COOH | −24.8 | 3.95 | 3.90 | 3.45 |
| Q(n-pr-NH)-COOH | −21.0 | 3.68 | 4.36 | 4.32 |
Figure 4. Correlation between quinone propionic acid specificity and FAD N5-quinone acceptor site (CA) distance.

R = 0.9447 for the best linear-least-squares fit (green line). Error bars represent one standard deviation (n = 3).
Due to the goodness of the correlation for the log(kcat/Km)-DAD relationship above, we calculated the predicted value of kcat/Km for Q(Me-N)-COOH and Q(n-pr-NH)-COOH, two substrates whose kinetic parameters cannot be obtained due to their spectral overlap at 340 nm with the NADH that is used to obtain the rates of reaction. This was achieved using the best fit equation from Figure 4 of log(kcat/Km) = 14.668±0.405 − 2.601±0.118*DAD. The kcat/Km value for Q(n-pr-NH)-COOH was calculated to be 1.2 × 105 M−1 s−1 and that for Q(Me-N)-COOH is 2.5 × 104 M−1 s−1, with the former being comparable to that of QMe-COOH and the latter similar to the kcat/Km value for QdiMeO-COOH. These values clearly place the amino-substituted quinone propionic acids in the slow set of hNQO1 substrates, as one might expect based in the steric bulk of the amine substituent.
We are unaware of any published reports that have investigated application of Marcus-like theory to reactivities of NQO1-catalyzed NADH reduction of quinones and the distance from N5 of the flavin center in hNQO1 to the possible hydride transfer site of the quinone substrates. Our initial positive outcomes with this model using a family of quinone propionic acids opens up opportunities of exploration with other classes of quinones, as well as investigations of NQO1 structural changes. For example, studies are necessary to elucidate the impact of NQO1 mutations on the rate of substrate reduction, wherein positioning of the flavin in the enzyme, and with respect to the bound substrate, can be tailored through site-directed mutagenesis of amino acids behind the flavin site.64 In addition, investigations are warranted regarding the influence of protein motion (reorganization energy)95 on the rate of hydride transfer to substrates; this can be probed via kinetic isotope effect experiments and those using the apo enzyme reconstituted with flavins having different E1/2 values.98
In summary, through the use of a 96-well microplate enzyme assay, we have been able to obtain highly reproducible kinetic parameters for the human recombinant NQO1-catalyzed NADH reduction of quinone propionic acids, including those that can be used as a trigger group for drug delivery systems.32 As a result, we were able to observe significant differences in enzymatic activity resulting from minor structural changes to the quinone ring and the propionic acid side chain of the rhNQO1 substrates. The differences in reactivity for the quinone propionic acids are proposed to be the result of variations in enzyme-substrate steric interactions, an observation similar in nature for benzoquinone mustards.53, 56 Outcomes from molecular docking studies qualitatively confirm this hypothesis, as the more active quinone propionic acids are found to penetrate deeper into the flavin active site of the rhNQO1. A very strong correlation is found between log(kcat/Km) and molecular-docking-derived hydride donor-acceptor distance, DAD, for the case of hydride transfer from the N5 of the flavin of NQO1 to the carbon ortho to the carbonyl (Path A in Scheme 2, CA), in agreement with Marcus-like theories concerning enzymatically facilitated hydride transfer reactions. Overall, our observations enrich the knowledge base for simple quinone-hNQO1 interactions, and provide valuable information for the design of enzyme triggerable systems. For example, the docking and kinetics studies reveal that the presence of a longer side chain attached to the quinone (i.e. QMe-ETA versus QMe-COOH) can lead to the quinone core being able to penetrate deeper into the active site of rhNQO1; this suggests that addition of spacers to the quinone derivatives99 may facilitate quinone-enzyme binding and perhaps their rate of enzymatic conversion.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Grover Waldrop for helpful discussions on enzyme assay experiments. This work was made possible in part by financial support from the US National Science Foundation (CHE-0910845) and the US National Institutes of Health (5R21CA135585).
Abbreviations
- rhNQO1
recombinant human NAD(P)H:quinone oxidoreductase type 1
- QBr-COOH
3-(5-bromo-2,4-dimethyl-3,6-dioxocyclohexa-1,4-dien-1-yl)-3-methylbutanoic acid
- QH-COOH
3-(2,4-dimethyl-3,6-dioxocyclohexa-1,4-dien-1-yl)-3-methylbutanoic acid
- QMe-COOH
3-methyl-3-(2,4,5-trimethyl-3,6-dioxocyclohexa-1,4-dien-1-yl)butanoic acid
- QMeO-COOH
3-(5-methoxy-2,4-dimethyl-3,6 dioxocyclohexa-1,4-dien-1-yl)-3-methylbutanoic acid
- Q’-COOH
3-(4,5-dimethyl-3,6-dioxocyclohexa-1,4-dien-1-yl)-3-methylbutanoic acid
- QnogemMe-COOH
3-(2,4,5-trimethyl-3,6-dioxocyclohexa-1,4-dien-1-yl)propanoic acid
- QdiMeO-COOH
3-(4,5-dimethoxy-2-methyl-3,6-dioxocyclohexa-1,4-dien-1-yl)-3-methylbutanoic acid
- QMe-ETA
N-(2-hydroxyethyl)-3-methyl-3-(2,4,5-trimethyl-3,6-dioxocyclohexa-1,4-dien-1-yl)butanamide
- Q(Me-N)-COOH
3-(2,4-dimethyl-5-(methylamino)-3,6-dioxocyclohexa-1,4-dien-1-yl)-3-methylbutanoic acid
- Q(n-pr-NH)-COOH
3-(2,4-dimethyl-3,6-dioxo-5-(propylamino)cyclohexa-1,4-dien-1-yl)-3-methylbutanoic acid
- QPA
quinone propionic acid
Footnotes
This work was supported by the U.S. National Institutes of Health (5R21CA135585) and the U.S. National Science Foundation (CHE-0910845).
SUPPORTING INFORMATION
Detailed enzyme protocols, synthesis and characterization of QMe-ETA, and structural frames of the quinone analogs from molecular docking simulations. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Andresen TL, Jensen SS, Jorgensen K. Advanced strategies in liposomal cancer therapy: Problems and prospects of active and tumor specific drug release. Prog. Lipid Res. 2005;44:68–97. doi: 10.1016/j.plipres.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Asche C. Antitumour quinones. Mini-Rev. Med. Chem. 2005;5:449–467. doi: 10.2174/1389557053765556. [DOI] [PubMed] [Google Scholar]
- 3.de Groot FMH, Damen EWP, Scheeren HW. Anticancer prodrugs for application in monotherapy: Targeting hypoxia, tumor-associated enzymes, and receptors. Curr. Med. Chem. 2001;8:1093–1122. doi: 10.2174/0929867013372634. [DOI] [PubMed] [Google Scholar]
- 4.Jaffar M, Abou-Zeid N, Bai L, Mrema I, Robinson I, Tanner R, Stratford IJ. Quinone bioreductive prodrugs as delivery agents. Curr. Drug Delivery. 2004;1:345–350. doi: 10.2174/1567201043334560. [DOI] [PubMed] [Google Scholar]
- 5.Rooseboom M, Commandeur JNM, Vermeulen NPE. Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol. Rev. 2004;56:53–102. doi: 10.1124/pr.56.1.3. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Hu LQ. Design of anticancer prodrugs for reductive activation. Med. Res. Rev. 2009;29:29–64. doi: 10.1002/med.20137. [DOI] [PubMed] [Google Scholar]
- 7.Davis SC, Szoka FC. Cholesterol phosphate derivatives: Synthesis and incorporation into a phosphatase and calcium-sensitive triggered release liposome. Bioconjugate Chem. 1998;9:783–792. doi: 10.1021/bc980047y. [DOI] [PubMed] [Google Scholar]
- 8.Davidsen J, Vermehren C, Frokjaer S, Mouritsen OG, Jorgensen K. Drug delivery by phospholipase A(2) degradable liposomes. Int. J. Pharm. 2001;214:67–69. doi: 10.1016/s0378-5173(00)00634-7. [DOI] [PubMed] [Google Scholar]
- 9.Jorgensen K, Davidsen J, Mouritsen OG. Biophysical mechanisms of phospholipase A2 activation and their use in liposome-based drug delivery. FEBS Lett. 2002;531:23–27. doi: 10.1016/s0014-5793(02)03408-7. [DOI] [PubMed] [Google Scholar]
- 10.Davidsen J, Jorgensen K, Andresen TL, Mouritsen OG. Secreted phospholipase A(2) as a new enzymatic trigger mechanism for localised liposomal drug release and absorption in diseased tissue. Biochim. Biophys. Acta. 2003;1609:95–101. doi: 10.1016/s0005-2736(02)00659-4. [DOI] [PubMed] [Google Scholar]
- 11.Meers P. Enzyme-activated targeting of liposomes. Advanced Drug Delivery Reviews. 2001;53:265–272. doi: 10.1016/s0169-409x(01)00205-8. [DOI] [PubMed] [Google Scholar]
- 12.Sarkar N, Banerjee J, Hanson AJ, Elegbede AI, Rosendahl T, Krueger AB, Banerjee AL, Tobwala S, Wang RY, Lu XN, Mallik S, Srivastava DK. Matrix metalloproteinase-assisted triggered release of liposomal contents. Bioconjugate Chem. 2008;19:57–64. doi: 10.1021/bc070081p. [DOI] [PubMed] [Google Scholar]
- 13.Banerjee J, Hanson AJ, Gadam B, Elegbede AI, Tobwala S, Ganguly B, Wagh AV, Muhonen WW, Law B, Shabb JB, Srivastava DK, Mallik S. Release of liposomal contents by cell-secreted matrix metalloproteinase-9. Bioconjugate Chem. 2009;20:1332–1339. doi: 10.1021/bc9000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reigan P, Colucci MA, Siegel D, Chilloux A, Moody CJ, Ross D. Development of indolequinone mechanism-based inhibitors of NAD(P)H : quinone oxidoreductase 1 (NQO1): NQO1 inhibition and growth inhibitory activity in human pancreatic MIA PaCa-2 cancer cells. Biochemistry. 2007;46:5941–5950. doi: 10.1021/bi700008y. [DOI] [PubMed] [Google Scholar]
- 15.Ernster L. DT-Diaphorase - a historical review. Chem. Scripta. 1987;27A:1–13. [Google Scholar]
- 16.Ernster L, Navazio F. Soluble diaphorase in animal tissues. Acta Chem. Scand. 1958;12:595–595. [Google Scholar]
- 17.Siegel D, Reigan P, Ross D. One- and two-electron-mediated reduction of quinones: enzymology and toxicological implications. Biotechnology: Pharmaceutical Aspects. 2008;9:169–197. [Google Scholar]
- 18.Iyanagi T, Yamazaki I. One-electron-transfer reactions in biochemical systems.5. Difference in mechanism of quinone reduction by NADH dehydrogenase and NAD(P)H dehydrogenase (DT-diaphorase) Biochim. Biophys. Acta. 1970;216:282–294. doi: 10.1016/0005-2728(70)90220-3. [DOI] [PubMed] [Google Scholar]
- 19.Winski SL, Koutalos Y, Bentley DL, Ross D. Subcellular localization of NAD(P)H : quinone oxidoreductase 1 in human cancer cells. Cancer Res. 2002;62:1420–1424. [PubMed] [Google Scholar]
- 20.Villalba JM, Navarro F, Arroyo A, Martin SF, Bello RI, de Cabo R, Burgess JR, Navas P. Protective role of ubiquinone in vitamin E and selenium-deficient plasma membranes. Biofactors. 1999;9:163–170. doi: 10.1002/biof.5520090211. [DOI] [PubMed] [Google Scholar]
- 21.Ernster L. DT Diaphorase. Methods Enzymol. 1967;10:309–317. [Google Scholar]
- 22.Nakamura M, Hayashi T. One-electron and 2-electron reduction of quinones by rat-liver subcellular-fractions. J. Biochem.-Tokyo. 1994;115:1141–1147. doi: 10.1093/oxfordjournals.jbchem.a124470. [DOI] [PubMed] [Google Scholar]
- 23.Siegel D, McGuinness SM, Winski SL, Ross D. Genotype-phenotype relationships in studies of a polymorphism in NAD(P)H : quinone oxidoreductase 1. Pharmacogenetics. 1999;9:113–121. doi: 10.1097/00008571-199902000-00015. [DOI] [PubMed] [Google Scholar]
- 24.Sreerama L, Hedge MW, Sladek NE. Identification of a class 3 aldehyde dehydrogenase in human saliva and increased levels of this enzyme, glutathione S-transferases, and DT-diaphorase in the saliva of subjects who continually ingest large quantities of coffee or broccoli. Clin. Cancer Res. 1995;1:1153–1163. [PubMed] [Google Scholar]
- 25.Reicks MM, Appelt LC. Soy induces phase II enzymes but does not inhibit dimethylbenz[a]anthracene-induced carcinogenesis in female rats. J. Nutr. 1999;129:1820–1826. doi: 10.1093/jn/129.10.1820. [DOI] [PubMed] [Google Scholar]
- 26.Cresteil T, Jaiswal AK. High-levels of expression of the NAD(P)H-quinone oxidoreductase (NQO1) gene in tumor-cells compared to normal-cells of the same origin. Biochem. Pharmacol. 1991;42:1021–1027. doi: 10.1016/0006-2952(91)90284-c. [DOI] [PubMed] [Google Scholar]
- 27.Fitzsimmons SA, Workman P, Grever M, Paull K, Camalier R, Lewis AD. Reductase enzyme expression across the national cancer institute tumor cell line panel: Correlation with sensitivity to mitomycin C and E09. J. Natl. Cancer Inst. 1996;88:259–269. doi: 10.1093/jnci/88.5.259. [DOI] [PubMed] [Google Scholar]
- 28.Siegel D, Franklin WA, Ross D. Immunohistochemical detection of NAD(P)H : Quinone oxidoreductase in human lung and lung tumors. Clin. Cancer Res. 1998;4:2065–2070. [PubMed] [Google Scholar]
- 29.Siegel D, Ross D. Immunodetection of NAD(P)H : quinone oxidoreductase 1 (NQO1) in human tissues. Free Radical Biol. Med. 2000;29:246–253. doi: 10.1016/s0891-5849(00)00310-5. [DOI] [PubMed] [Google Scholar]
- 30.Jamieson D, Wilson K, Pridgeon S, Margetts JP, Edmondson RJ, Leung HY, Knox R, Boddy AV. NAD(P)H : quinone oxidoreductase1 and NRH : quinone oxidoreductase 2 activity and expression in bladder and ovarian cancer and lower NRH : quinone oxidoreductase 2 activity associated with an NQO2 exon 3 single-nucleotide polymorphism. Clin. Cancer Res. 2007;13:1584–1590. doi: 10.1158/1078-0432.CCR-06-1416. [DOI] [PubMed] [Google Scholar]
- 31.Volpato M, Abou-Zeid N, Tanner RW, Glassbrook LT, Taylor J, Stratford I, Loadman PM, Jaffar M, Phillips RM. Chemical synthesis and biological evaluation of a NAD(P)H : quinone oxidoreductase-1-targeted tripartite quinone drug delivery system. Mol. Cancer Ther. 2007;6:3122–3130. doi: 10.1158/1535-7163.MCT-07-0519. [DOI] [PubMed] [Google Scholar]
- 32.Ong W, Yang YM, Cruciano AC, McCarley RL. Redox-triggered contents release from liposomes. J. Am. Chem. Soc. 2008;130:14739–14744. doi: 10.1021/ja8050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faig M, Bianchet MA, Talalay P, Chen S, Winski S, Ross D, Amzel LM. Structures of recombinant human and mouse NAD(P)H : quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. U. S. A. 2000;97:3177–3182. doi: 10.1073/pnas.050585797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li R, Bianchet MA, Talalay P, Amzel LM. The three-dimensional structure of NAD(P)H:quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: Mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. U. S. A. 1995;92:8846–8850. doi: 10.1073/pnas.92.19.8846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faig M, Bianchet MA, Winski S, Hargreaves R, Moody CJ, Hudnott AR, Ross D, Amzel LM. Structure-based development of anticancer drugs: Complexes of NAD(P)H : quinone oxidoreductase 1 with chemotherapeutic quinones. Structure. 2001;9:659–667. doi: 10.1016/s0969-2126(01)00636-0. [DOI] [PubMed] [Google Scholar]
- 36.Winski SL, Faig M, Bianchet MA, Siegel D, Swann E, Fung K, Duncan MW, Moody CJ, Amzel LM, Ross D. Characterization of a mechanism-based inhibitor of NAD(P)H: Quinone oxidoreductase 1 by biochemical, X-ray crystallographic, and mass spectrometric approaches. Biochemistry. 2001;40:15135–15142. doi: 10.1021/bi011324i. [DOI] [PubMed] [Google Scholar]
- 37.Asher G, Dym O, Tsvetkov P, Adler J, Shaul Y. The crystal structure of NAD(P)H quinone oxidoreductase 1 in complex with its potent inhibitor dicoumarol. Biochemistry. 2006;45:6372–6378. doi: 10.1021/bi0600087. [DOI] [PubMed] [Google Scholar]
- 38.Nolan KA, Doncaster JR, Dunstan MS, Scott KA, Frenkel AD, Siegel D, Ross D, Barnes J, Levy C, Leys D, Whitehead RC, Stratford IJ, Bryce RA. Synthesis and biological evaluation of coumarin-based inhibitors of NAD(P)H: quinone oxidoreductase-1 (NQO1) J. Med. Chem. 2009;52:7142–7156. doi: 10.1021/jm9011609. [DOI] [PubMed] [Google Scholar]
- 39.Danson S, Ward TH, Butler J, Ranson M. DT-diaphorase: a target for new anticancer drugs. Cancer Treatment Reviews. 2004;30:437–449. doi: 10.1016/j.ctrv.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Colucci MA, Moody CJ, Couch GD. Natural and synthetic quinones and their reduction by the quinone reductase enzyme NQO1: from synthetic organic chemistry to compounds with anticancer potential. Org. Biomol. Chem. 2008;6:637–656. doi: 10.1039/b715270a. [DOI] [PubMed] [Google Scholar]
- 41.Weerapreeyakul N, Anorach R, Khuansawad T, Yenjai C, Isaka M. Synthesis of bioreductive esters from fungal compounds. Chemical & Pharmaceutical Bulletin. 2007;55:930–935. doi: 10.1248/cpb.55.930. [DOI] [PubMed] [Google Scholar]
- 42.Silvers WC, Payne AS, McCarley RL. Shedding light by cancer redox—human NAD(P)H:quinone oxidoreductase 1 activation of a cloaked fluorescent dye. Chem. Commun. 2011 doi: 10.1039/c1cc14578a. [DOI] [PubMed] [Google Scholar]
- 43.Huang ST, Lin YL. New latent fluorophore for DT diaphorase. Org Lett. 2006;8:265–268. doi: 10.1021/ol052635e. [DOI] [PubMed] [Google Scholar]
- 44.Volpato M, Abou-Zeid N, Tanner RW, Glassbrook LT, Taylor J, Stratford I, Loadman P, Jaffar M, Phillips RM. Chemical synthesis and biological evaluation of a NAD(P)H:quinone oxidoreductase-1-targeted tripartite quinone drug delivery system. Mol. Cancer Ther. 2007;6:3122–3130. doi: 10.1158/1535-7163.MCT-07-0519. [DOI] [PubMed] [Google Scholar]
- 45.Moody CJ, Maskell L, Blanche EA, Colucci MA, Whatmore JL. Synthesis and evaluation of prodrugs for anti-angiogenic pyrrolylmethylidenyl oxindoles. Bioorg. Med. Chem. Lett. 2007;17:1575–1578. doi: 10.1016/j.bmcl.2006.12.108. [DOI] [PubMed] [Google Scholar]
- 46.Ingold CK, Sako S, Thorpe JF. Influence of substituents on the formation and stability of heterocyclic compounds. I. Hydantoins. J. Chem. Soc., Trans. 1922;121:1177–1198. [Google Scholar]
- 47.Milstien S, Cohen LA. Stereopopulation control.1. rate enhancement in lactonizations of ortho-hydroxyhydrocinnamic acids. J. Am. Chem. Soc. 1972;94:9158–9165. doi: 10.1021/ja00781a029. [DOI] [PubMed] [Google Scholar]
- 48.Jung ME, Piizzi G. gem-Disubstituent effect: theoretical basis and synthetic applications. Chem. Rev. 2005;105:1735–1766. doi: 10.1021/cr940337h. [DOI] [PubMed] [Google Scholar]
- 49.Beall HD, Murphy AM, Siegel D, Hargreaves RHJ, Butler J, Ross D. Nicotinamide adenine-dinucleotide (phosphate):quinone oxidoreductase (DT-Diaphorase) as a target for bioreductive antitumor quinones - quinone cytotoxicity and selectivity in human lung and breast-cancer cell-lines. Mol. Pharmacol. 1995;48:499–504. [PubMed] [Google Scholar]
- 50.Phillips RM. Bioreductive activation of a series of analogues of 5-aziridinyl-3-hydroxymethyl-1-methyl-2-[1H-indole-4,7-dione] prop-beta-en-alpha-ol (EO9) by human DT-diaphorase. Biochem. Pharmacol. 1996;52:1711–1718. doi: 10.1016/s0006-2952(96)00521-7. [DOI] [PubMed] [Google Scholar]
- 51.Beall HD, Winski S, Swann E, Hudnott AR, Cotterill AS, O'Sullivan N, Green SJ, Bien R, Siegel D, Ross D, Moody CJ. Indolequinone antitumor agents: Correlation between quinone structure, rate of metabolism by recombinant human NAD(P)H : quinone oxidoreductase, and in vitro cytotoxicity. J. Med. Chem. 1998;41:4755–4766. doi: 10.1021/jm980328r. [DOI] [PubMed] [Google Scholar]
- 52.Swann E, Barraja P, Oberlander AM, Gardipee WT, Hudnott AR, Beall HD, Moody CJ. Indolequinone antitumor agents: Correlation between quinone structure and rate of metabolism by recombinant human NAD(P)H : quinone oxidoreductase. Part 2. J. Med. Chem. 2001;44:3311–3319. doi: 10.1021/jm010884c. [DOI] [PubMed] [Google Scholar]
- 53.Fourie J, Oleschuk CJ, Guziec F, Guziec L, Fiterman DJ, Monterrosa C, Begleiter A. The effect of functional groups on reduction and activation of quinone bioreductive agents by DT-diaphorase. Cancer Chemother. Pharmacol. 2002;49:101–110. doi: 10.1007/s00280-001-0395-1. [DOI] [PubMed] [Google Scholar]
- 54.Fryatt T, Pettersson HI, Gardipee WT, Bray KC, Green SJ, Slawin AMZ, Beall HD, Moody CJ. Novel quinolinequinone antitumor agents: structure-metabolism studies with NAD(P)H : quinone oxidoreductase (NQO1) Bioorg. Med. Chem. 2004;12:1667–1687. doi: 10.1016/j.bmc.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 55.Newsome JJ, Swann E, Hassani M, Bray KC, Slawin AMZ, Beall HD, Moody CJ. Indolequinone antitumour agents: correlation between quinone structure and rate of metabolism by recombinant human NAD(P)H : quinone oxidoreductase. Org. Biomol. Chem. 2007;5:1629–1640. doi: 10.1039/b703370b. [DOI] [PubMed] [Google Scholar]
- 56.Begleiter A, El-Gabalawy N, Lange L, Leith MK, Guziec LJ, Guziec FS., Jr A model for NAD(P)H:Quinoneoxidoreductase 1 (NQO1) targeted individualized cancer chemotherapy. Drug Target Insights. 2009;4:1–8. doi: 10.4137/dti.s1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fagan V, Bonham S, Carty MP, Saenz-Médez P, Eriksson LA, Aldabbagh F. COMPARE analysis of the toxicity of an iminoquinone derivative of the imidazo[5,4-f]benzimidazoles with NAD(P)H:quinone oxidoreductase 1 (NQO1) activity and computational docking of quinones as NQO1 substrates. Bioorg. Med. Chem. 2012;20:3223–3232. doi: 10.1016/j.bmc.2012.03.063. [DOI] [PubMed] [Google Scholar]
- 58.Skelly JV, Sanderson MR, Suter DA, Baumann U, Read MA, Gregory DSJ, Bennett M, Hobbs SM, Neidle S. Crystal structure of human DT-diaphorase: A model for interaction with the cytotoxic prodrug 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB1954) J. Med. Chem. 1999;42:4325–4330. doi: 10.1021/jm991060m. [DOI] [PubMed] [Google Scholar]
- 59.Suleman A, Skibo EB. A comprehensive study of the active site residues of DT-diaphorase: Rational design of benzimidazolediones as DT-diaphorase substrates. J. Med. Chem. 2002;45:1211–1220. doi: 10.1021/jm0104365. [DOI] [PubMed] [Google Scholar]
- 60.Hassani M, Cai W, Koelsch KH, Holley DC, Rose AS, Olang F, Lineswala JP, Holloway WG, Gerdes JM, Behforouz M, Beall HD. Lavendamycin antitumor agents: Structure-based design, synthesis, and NAD(P)H : Quinone oxidoreductase 1 (NQO1) model validation with molecular docking and biological studies. J. Med. Chem. 2008;51:3104–3115. doi: 10.1021/jm701066a. [DOI] [PubMed] [Google Scholar]
- 61.Nolan KA, Humphries MP, Barnes J, Doncaster JR, Caraher MC, Tirelli N, Bryce RA, Whitehead RC, Stratford IJ. Triazoloacridin-6-ones as novel inhibitors of the quinone oxidoreductases NQO1 and NQO2. Bioorg. Med. Chem. 2010;18:696–706. doi: 10.1016/j.bmc.2009.11.059. [DOI] [PubMed] [Google Scholar]
- 62.Phillips RM, Naylor MA, Jaffar M, Doughty SW, Everett SA, Breen AG, Choudry GA, Stratford IJ. Bioreductive activation of a series of indolequinones by human DT-diaphorase: Structure-activity relationships. J. Med. Chem. 1999;42:4071–4080. doi: 10.1021/jm991063z. [DOI] [PubMed] [Google Scholar]
- 63.Chen S, Knox R, Lewis AD, Friedlos F, Workman P, Deng PS, Fung M, Ebenstein D, Wu K, Tsai TM. Catalytic properties of NAD(P)H:quinone acceptor oxidoreductase: study involving mouse, rat, human, and mouse-rat chimeric enzymes. Mol. Pharmacol. 1995;47:934–939. [PubMed] [Google Scholar]
- 64.Chen S, Knox R, Wu K, Deng PSK, Zhou DJ, Bianchet MA, Amzel LM. Molecular basis of the catalytic differences among DT-diaphorase of human, rat, and mouse. J. Biol. Chem. 1997;272:1437–1439. doi: 10.1074/jbc.272.3.1437. [DOI] [PubMed] [Google Scholar]
- 65.Chen S, Wu K, Zhang D, Sherman M, Knox R, Yang CS. Molecular characterization of binding of substrates and inhibitors to DT-Diaphorase: combined approach involving site-directed mutagenesis, inhibitor-binding analysis, and computer modeling. Mol. Pharmacol. 1999;56:272–278. doi: 10.1124/mol.56.2.272. [DOI] [PubMed] [Google Scholar]
- 66.Anusevicius Z, Sarlauskas J, Cenas N. Two-electron reduction of quinones by rat liver NAD(P)H : quinone oxidoreductase: quantitative structure-activity relationships. Arch. Biochem. Biophys. 2002;404:254–262. doi: 10.1016/s0003-9861(02)00273-4. [DOI] [PubMed] [Google Scholar]
- 67.Čėas N, Anusevicius Z, Nivinskas H, Miseviciene L, Sarlauskas J. Structure-activity relationship in two-electron reduction of quinones. Methods Enzymol. 2004;382:258–277. doi: 10.1016/S0076-6879(04)82015-9. [DOI] [PubMed] [Google Scholar]
- 68.Zhou ZG, Fisher D, Spidel J, Greenfield J, Patson B, Fazal A, Wigal C, Moe OA, Madura JD. Kinetic and docking studies of the interaction of quinones with the quinone reductase active site. Biochemistry. 2003;42:1985–1994. doi: 10.1021/bi026518s. [DOI] [PubMed] [Google Scholar]
- 69.Hollabaugh Carrier NM. Ph.D. Dissertation. Baton Rouge, LA: Department of Chemistry, Louisiana State University; 2011. Redox-active liposome delivery agents with highly controllable stimuli-responsive behavior. [Google Scholar]
- 70.Osman AM, Boeren S. Studies on the DT-diaphorase-catalysed reaction employing quinones as substrates: evidence for a covalent modification of DT-diaphorase by tetrachloro-p-benzoquinone. Chem.-Biol. Interact. 2004;147:99–108. doi: 10.1016/j.cbi.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 71.Harris DC. Statistics. In: Byrd ML, editor. Quantitative Chemical Analysis. 7th ed. New York: W. H. Freeman and Company; 2007. p. 65. [Google Scholar]
- 72.Cleland WW. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 73.Harris DC. Statistics. In: Byrd ML, editor. Quantitative Chemical Analysis. 7th ed. New York: W. H. Freeman and Company; 2007. p. 59. [Google Scholar]
- 74.Bohm HJ. The computer-program Ludi - a new method for the denovo design of enzyme-inhibitors. J. Comput. Aid. Mol. Des. 1992;6:61–78. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- 75.Bohm HJ. The development of a simple empirical scoring function to estimate the binding constant for a protein ligand complex of known 3-dimensional structure. J. Comput. Aid. Mol. Des. 1994;8:243–256. doi: 10.1007/BF00126743. [DOI] [PubMed] [Google Scholar]
- 76.Rarey M, Wefing S, Lengauer T. Placement of medium-sized molecular fragments into active sites of proteins. J. Comput. Aid. Mol. Des. 1996;10:41–54. doi: 10.1007/BF00124464. [DOI] [PubMed] [Google Scholar]
- 77.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 78.Rarey M, Kramer B, Lengauer T. Multiple automatic base selection: Protein-ligand docking based on incremental construction without manual intervention. J. Comput. Aid. Mol. Des. 1997;11:369–384. doi: 10.1023/a:1007913026166. [DOI] [PubMed] [Google Scholar]
- 79.Kramer B, Rarey M, Lengauer T. CASP2 experiences with docking flexible ligands using FLEXX. Proteins. 1997:221–225. doi: 10.1002/(sici)1097-0134(1997)1+<221::aid-prot30>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 80.Kramer B, Rarey M, Lengauer T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins. 1999;37:228–241. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 81.Rarey M, Kramer B, Lengauer T. Docking of hydrophobic ligands with interaction-based matching algorithms. Bioinformatics. 1999;15:243–250. doi: 10.1093/bioinformatics/15.3.243. [DOI] [PubMed] [Google Scholar]
- 82.Bohm HJ. Ludi - rule-based automatic design of new substituents for enzyme-inhibitor leads. J. Comput. Aid. Mol. Des. 1992;6:593–606. doi: 10.1007/BF00126217. [DOI] [PubMed] [Google Scholar]
- 83.Ross D. Quinone reductases multitasking in the metabolic world. Drug Metab. Rev. 2004;36:639–654. doi: 10.1081/dmr-200033465. [DOI] [PubMed] [Google Scholar]
- 84.Robitaille PML, Robitaille PA, Brown GG, Brown GG. An analysis of the pH-dependent chemical-shift behavior of phosphorus-containing metabolites. J. Magn. Reson. 1991;92:73–84. [Google Scholar]
- 85.Silverman J, Stam-Thole I, Stam CH. Crystal and molecular structure of 2-methyl-4,5-dimethoxy para quinone (fumigatin methyl ether), C9H10O4. Acta Cryst. B-Struc. B. 1971;27:1846–1851. [Google Scholar]
- 86.Burie JR, Boullais C, Nonella M, Mioskowski C, Nabedryk E, Breton J. Importance of the conformation of methoxy groups on the vibrational and electrochemical properties of ubiquinones. J. Phys. Chem. B. 1997;101:6607–6617. [Google Scholar]
- 87.Lind C, Cadenas E, Hochstein P, Ernster L. DT-Diaphorase: purification, properties, and function. Methods Enzymol. 1990;186:287–301. doi: 10.1016/0076-6879(90)86122-c. [DOI] [PubMed] [Google Scholar]
- 88.Buffinton GD, Ollinger K, Brunmark A, Cadenas E. DT-diaphorase-catalyzed reduction of 1,4-naphthoquinone derivatives and glutathionyl-quinone conjugates. Biochem. J. 1989;257:561–571. doi: 10.1042/bj2570561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhao YH, Abraham MH, Zissimos AM. Fast calculation of van der Waals volume as a sum of atomic and bond contributions and its application to drug compounds. J. Org. Chem. 2003;68:7368–7373. doi: 10.1021/jo034808o. [DOI] [PubMed] [Google Scholar]
- 90.Ryabov AD, Ryabova ES, Reshetova MD. Enzymatic chemistry of ferrocenes: micellar tuning of the glucose oxidase reactivity toward solubilized electrochemically generated n-alkylferricenium cations. J. Organomet. Chem. 2001;637–639:469–475. [Google Scholar]
- 91.Cavelier G, Amzel LM. Mechanism of NAD(P)H : quinone reductase: Ab initio studies of reduced flavin. Proteins. 2001;43:420–432. [PubMed] [Google Scholar]
- 92.Deller S, Macheroux P, Sollner S. Flavin-dependent quinone reductases. Cell. Mol. Life Sci. 2008;65:141–160. doi: 10.1007/s00018-007-7300-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bianchet MA, Faig M, Amzel LM. Structure and mechanism of NAD[P]H:quinone acceptor oxidoreductases (NQO) In: Helmut S, Lester P, editors. Quinones and quinone enzymes, Part B. Elsevier Academic Press; 2004. pp. 144–174. [DOI] [PubMed] [Google Scholar]
- 94.Nolan KA, Timson DJ, Stratford IJ, Bryce RA. In silico identification and biochemical characterization of novel inhibitors of NQO1. Bioorg. Med. Chem. Lett. 2006;16:6246–6254. doi: 10.1016/j.bmcl.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 95.Marcus RA. H and other transfers in enzymes and in solution: theory and computations, a unified view. 2. applications to experiment and computations. J. Phys. Chem. B. 2007;111:6643–6654. doi: 10.1021/jp071589s. [DOI] [PubMed] [Google Scholar]
- 96.Roston D, Amnon K. Elusive transition state of alcohol dehydrogenase unveiled. Proc. Natl. Acad. Sci. U. S. A. 2010;107:9572–9577. doi: 10.1073/pnas.1000931107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carlson BW, Miller LL. Mechanism of the oxidation of NADH by quinones -energetics of one-electron and hydride routes. J. Am. Chem. Soc. 1985;107:479–485. [Google Scholar]
- 98.Tedeschi G, Chen S, Massey V. Active site studies of DT-diaphorase employing artificial flavins. J. Biol. Chem. 1995;270:2512–2516. doi: 10.1074/jbc.270.6.2512. [DOI] [PubMed] [Google Scholar]
- 99.Tranoy-Opalinski I, Fernandes A, Thomas M, Gesson JP, Papot S. Design of self-immolative linkers for tumour-activated prodrug therapy. Anti-Cancer Agent. Med. Chem. 2008;8:618–637. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.