

Abstract
Linkage maps have been invaluable for the positional cloning of many genes involved in severe human diseases. Standard genetic linkage maps have been constructed for this purpose from the Centre d'Etude du Polymorphisme Humain and other panels, and have been widely used. Now that attention has shifted towards identifying genes predisposing to common disorders using linkage disequilibrium (LD) and maps of single nucleotide polymorphisms (SNPs), it is of interest to consider a standard LD map which is somewhat analogous to the corresponding map for linkage. We have constructed and evaluated a cosmopolitan LD map by combining samples from a small number of populations using published data from a 10-megabase region on chromosome 20. In support of a pilot study, which examined a number of small genomic regions with a lower density of markers, we have found that a cosmopolitan map, which serves all populations when appropriately scaled, recovers 91 to 95 per cent of the information within population-specific maps. Recombination hot spots appear to have a dominant role in shaping patterns of LD. The success of the cosmopolitan map might be attributed to the co-localisation of hot spots in all populations. Although there must be finer scale differences between populations due to other processes (mutation, drift, selection), the results suggest that a whole-genome standard LD map would indeed be a useful resource for disease gene mapping.
Keywords: linkage disequilibrium, single nucleotide polymorphism, genetic map, map integration
Introduction
Linkage disequilibrium (LD) is the non-random association of alleles at linked loci reflecting the presence of ancestral haplotype segments. The extent of LD varies across the genome, and its pattern is dominated by recombination and the number of generations over which this has taken place. Chromosome segments with high LD and low haplotype diversity are punctuated by narrow regions with variable rates of recombination, some of which are best described as 'hot spots' [1,2]. Patterns of LD are also influenced by other processes, such as genetic drift, selection and mutation, but recombination and time have the greatest impact on long-range LD structure. With the advent of panels of single nucleotide polymorphisms (SNPs) typed at high density in a number of chromosome regions, [3,4] it has become possible to determine the LD structure for whole chromosomes.
Maniatis et al. [5] describe an approach for determining patterns of LD which computes distances between adjacent SNPs in the map, measured in LD units (LDUs). Locations on the LDU map are somewhat analogous to the centimorgan (cM) locations of linkage maps. Unlike linkage maps, however, LDU map patterns also reflect duration since the last major population bottleneck, or, more correctly, the partly cumulative effects of multiple historical bottlenecks. Population differences in duration account for some substantial variations in LD patterns between populations.
To the extent that LD patterns reflect recombination, it is observed that recombination hot spots correspond to regions where LD breaks down rapidly within a unit physical distance (a high LDU/kilobase [kb] ratio). By contrast, regions with a low LDU/kb ratio have been shown to correspond to recombination cold areas and regions of relatively low haplotype diversity [6,7]. When the LDU locations for markers are plotted against the corresponding kb locations, a series of plateaux and steps is revealed, the latter typically spanning very few kb in high density SNP maps. This supports the finding of Jeffreys et al., that much recombination in the genome is 'intensely punctate' [1]. In a study by McVean et al., LD data were used to estimate local recombination rates [8]. Their approach, based on an approximation to the coalescent, identifies recombination hot spots and assigns a uniform recombination rate across intervening regions. An LD map differs, in that it accommodates variation in the extent of LD among different genomic regions and between populations, reflecting differences in population history and duration.
The discovery that a proportion of the genome comprises blocks of low haplotype diversity prompted the International HapMap project [9] to examine a range of populations in the hope of determining panels of haplotype tag SNPs that will be useful for disease gene mapping. There is a high degree of divergence in haplotype composition and diversity between different populations, [10] however, complicating the use of tag SNPs for multiple populations. By contrast, and despite the underlying diversity in haplotype structure, long-range patterns of LD tend to be conserved and differences due to duration can be modelled. For this purpose, Lonjou et al. proposed the development of a standard 'cosmopolitan' LD map [11]. The data of Gabriel et al. [12] and other samples for small chromosome regions were used to construct a cosmopolitan LD map for each region, and this was compared with population-specific maps. The authors concluded that a cosmopolitan map, when linearly scaled to reflect population duration, recovers 95 per cent of the information contained in the individual population maps. Differences in duration to a major bottleneck, or to cumulative effects of bottlenecks, are accommodated using 'scaling factors' which are population specific. The observed difference in scaling was particularly striking when contrasting African-derived populations with the non-African groups. This presumably reflects the dominant effect of the bottleneck imposed by the 'out of Africa' migrations which were undertaken by the ancestors of modern non-African populations.
LD maps are constructed in such a way that one LDU corresponds to one 'swept radius', [13] which is the extent of LD on the kb scale that is useful for disease gene localisation. Wide variations in the extent of LD in different genomic regions make uniform spacing of SNPs on the kb or cM scale inadequate for coverage of a disease candidate region. By contrast, even spacing of SNPs on the LDU scale incorporates relatively more markers in regions where LD is declining rapidly within a unit physical distance, ensuring more complete coverage.
If a standard LDU map is constructed for a whole chromosome, a direct assessment of the number of LDUs requiring coverage in a chromosome region for any population might be made. With this in mind, we constructed and examined the properties and utility of a standard cosmopolitan LDU map in a sample of SNPs typed in a large genomic region and four populations. If the utility of such a map is shown to be supported, the development of standard LD maps for every chromosome and a database of population-specific scaling factors would be a useful resource for disease gene mapping. Applications would include the determination of adequate screening densities and spacing of SNPs, and the provision of maps for multilocus disease gene mapping by association -- for which LD maps have been shown to have high power [14].
Materials and methods
Data sample
We used the dataset described by Ke et al., [4] which comprises a sample spanning 10,098 kb of chromosome 20q12-13.2 typed at high density (5,954 SNPs in total) in four populations. These data are available from: http://www.sanger.ac.uk/HGP/Chr20/ld-hmg/. The sample has an average density of one SNP per 1.7 kb. Markers were typed in four populations, comprising 97 African-Americans (AF), 96 UK Caucasians (CA), 47 Utah Centre d'Etude du Polymorphisme Humain (CEPH) founders (CE) and 42 East Asians (Japanese and Chinese; AS). We tested all SNPs in the data sample provided at the above website address and found no departures from Hardy-Weinberg equilibrium [15].
In order to construct a cosmopolitan LD map, we reduced each population sample of diplotypes to the corresponding haplotype counts (alleles were coded 1 or 2 and the haplotype designations were therefore 11, 12, 21 and 22). Pairwise SNP haplotype frequencies, converted into counts, were computed for each population sample following the algorithm described by Hill [16]. The haplotype counts were combined across populations by summation among matching locus pairs. To achieve this, we ensured that all markers shared among more than one population were coded consistently. We therefore ensured that, for example, an allele coded as '1' in one population was coded the same in all others.
From the haplotype counts, pairwise association probabilities (ρ) and the corresponding information (Kρ) were computed following Collins et al. [17] For a given pair of SNPs, the ρ metric takes values between zero (no association) and one (complete association). For the special case of association between SNP pairs, ρ is equal to the absolute value of D'. The computation of ρ in the more general case of disease and marker association, where disease haplotypes are identified, has also been described [18].
We constructed cosmopolitan LD maps from the SNP pairwise association data at different marker densities to examine whether map distances are additive. We then selected a single mean density of 6 kb for evaluating the cosmopolitan map against population-specific alternatives. The 6 kb spacing was selected because, to cover the human genome at this density, approximately 500,000 SNPs would be required, corresponding to the initial target of the International HapMap Consortium [9]. Unlike Ke et al., [4] we selectively removed SNPs to achieve uniform spacing on the kb map, thereby avoiding large gaps. The following algorithm was applied to achieve this.
Starting from the end of the map closest to the p telomere, the first typed SNP was chosen as the 'starting SNP', and two other SNPs were identified on either side of a position a selected number of kb away. The SNP closest to that position was chosen. The chosen SNP then became the new 'starting SNP' and the process was continued along the whole map. The length of the region (10,098 kb) was then divided by the number of SNPs selected to calculate the average density over the region. The process was repeated using a range of positions at various kb distances until the desired mean density was created.
LD map construction
LD maps [5] are constructed using the 'Malecot equation', ρ = (1 - L)Me -εd + L, which describes the decline in association ρ as a function of distance d. This has the same form, with different parameters, as the equation derived by Malecot for isolation by distance [18]. The population genetics theory underlying the derivation and application of this model for LD is described by Morton et al. [13]. The parameters of the model include M -- the maximum association at zero distance, reflecting association at the last major bottleneck. L reflects both the residual association at large distance together with the bias in ρ, which is related to sample size, and ε is the exponential decline of ρ with distance in kb. The parameters ε and M are estimated by fitting multiple pairwise values of ρ and corresponding information, Kρ, using composite likelihood. We used the predicted L (Lp), [13] which depends on sample size, rather than estimating the L parameter. Lonjou et al. [11] found that the local effect of block structure can inflate the estimate of L, leading to distortions in the LD map through the creation of 'holes' between adjacent SNPs. The LDMAP program (http://cedar.genetics.soton.ac.uk/pub/PROGRAMS/LDMAP/, with the online manual at: http://cedar.genetics.soton.ac.uk/public_html/helpld.html, [5] computes ε for each interval between pairs of SNPs. The parameters are estimated so that the composite likelihood is maximised and the length of the ith interval, in LDU, is given by εidi. The treatment of map holes, where εidi is assigned an upper limit value of 3 LDUs, has been described [6]. The authors established this upper limit by maximum likelihood and found a strong correlation between the location of holes and the recombination rate. The identification of these intervals is clearly important for determining regions which require further SNP typing. LD units have been shown to be additive in high-density SNP maps for a wide range of marker densities [4]. There is, however, likely to be some loss of additivity in regions of the map where holes are present. One of the additional advantages of combining samples to create a cosmopolitan map is that the increased sample size and addition of markers in poorly covered regions reduces the number of holes.
Tests on marker density
As the full data sample contained markers typed at a very high density, we were able to construct cosmopolitan maps using sub-samples at reduced densities by means of the method described. This allowed us to examine the stability of map lengths and the effect of the number of holes. Maps with mean SNP densities 6 kb, 8 kb, 10 kb, 12 kb and 15 kb were created for this purpose. The average minor allele frequency remained stable across the different density samples at ~0.23.
Tests on cosmopolitan maps
LD maps were created for each population and a cosmopolitan map was constructed from combined data, as described. The fit of the multiple pairwise data to the cosmopolitan map was examined and the corresponding error variances were computed for the population-specific LDU maps. The relative efficiency of the cosmopolitan map can be judged by comparing error variances. The composite log likelihood has the form ; where is the observed association between the ith pair of SNPs and ρi is estimated from the Malecot model replacing distance d in kb with the corresponding distance in LD units [5,18]. Therefore, the fit of the population-specific pairwise data to any map (kb, population-specific LDU or cosmopolitan LDU) can be evaluated. The error variances for a given population for the kb map (Vkb), the population-specific LDU map (VLDU) and the cosmopolitan map (VCOS) follow Lonjou et al. [11]. The degrees of freedom are computed as N - (m - 1) - r, [6] where N is the number of pairs, m is the number of loci (therefore, there are m-1 intervals in which ε may be estimated) and r is the number of additional parameters estimated.
We define Ni and Nc as the number of pairs in the ith population sample and cosmopolitan data sample, respectively. The number of SNP markers in the ith population sample and cosmopolitan sample is, respectively, mi and mc.
The error variances for kb, LDU and cosmopolitan maps are computed as:
where ε and M are estimated;
where M is estimated and ε is estimated in each map interval;
where mc - 1 intervals in the cosmopolitan map have been previously computed using the proportion of data represented by the ith population sample, as Ni/Nc, and ε and M are estimated.
The relative efficiency (RE) of the cosmopolitan maps was calculated to determine how much of the information was recovered; RE = VLDU/VCOS. Values of ε obtained when the population-specific data are fitted to the cosmopolitan map are each divided by the 1 for the cosmopolitan map to obtain the scaling factors.
Results
Table 1 describes the characteristics of cosmopolitan maps constructed at different marker densities. The 6 kb cosmopolitan map contains 1,691 markers, of which 405 are represented in a single population, 74 in two populations, 181 in three populations and 1,031 are found in all four. The LDU map lengths vary over the range 187 - 204 LDUs and the number of intervals where the limit of 3 LDUs has been assigned is between 2 and 7. Given the large number of loci in the maps (670 -1,691), the relatively small number of intervals in this category suggests that there is good marker coverage at all densities and that the LD map is well characterised. Figure 1 illustrates the conservation of the LD map contours under the different density selections.
Table 1.
Cosmopolitan linkage disequilibrium maps at different kilobase marker densities
| Density | N | m | ε | M | Lp | -2lnL | df | VLDU | No. LDUs | No. holes |
|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 132,171 | 1,691 | 1.1521 | 0.894 | 0.091 | 179,822 | 130,480 | 1.378 | 187.15 | 2 |
| 8 | 76,236 | 1,289 | 1.1609 | 0.877 | 0.092 | 105,937 | 74,947 | 1.413 | 198.02 | 5 |
| 10 | 45,221 | 992 | 1.1399 | 0.895 | 0.090 | 61,091 | 44,229 | 1.381 | 204.41 | 5 |
| 12 | 31,497 | 833 | 1.1331 | 0.897 | 0.091 | 40,581 | 30,664 | 1.323 | 204.56 | 6 |
| 15 | 20,483 | 670 | 1.1381 | 0.870 | 0.090 | 28,439 | 19,813 | 1.435 | 196.28 | 7 |
N: Number of pairs; m: number of loci; ε, M; Lp: Malecot parameters; -2lnL: composite -2 log likelihood; df: degrees of freedom; VLDU: residual error variance for the linkage disequilibrium unit map.
Figure 1.
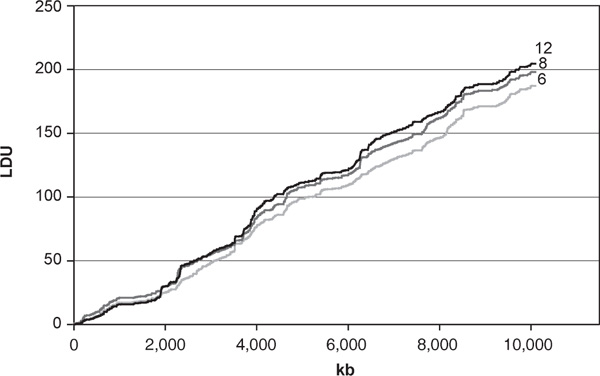
Contours of the linkage disequilibrium maps at a range of marker densities (6, 8 and 12 kilobase mean spacing).
From Table 1, it is evident that maps with more holes tend to be somewhat longer, although this is not always the case, for example in the comparison of the 12 kb and 15 kb mean spacing maps. The effect of holes on map length is difficult to predict, since addition of markers within a recombination-intense segment creates more intervals and may therefore generate additional holes indicating LDU = 3 as an underestimate. Presumably, however, in most cases the limit is an overestimate and addition of further markers in the interval will reduce map length. Given that the 6 kb cosmopolitan map corresponds to approximately 500,000 SNPs in the genome and has only two holes, it seems the most suitable to use as a basis for evaluating cosmopolitan maps.
The fit of the multiple pairwise data to the kb map is described in Table 2. The mean swept radii for the four populations are in the range 80 - 105 kb, with the AF sample having the least extensive LD. This is consistent with a previous study [11] and reflects the longer duration since the last major population bottleneck. The intercept M has an evolutionary interpretation by reflecting association at the bottleneck and is in the range 0.66 - 0.88 with, again, the lowest value found in the AF population. Higher values in the non-African populations are consistent with the bottleneck from the 'out of Africa' migration. This had the effect of creating LD by restricting the founding haplotype set and is consistent with the elevated M parameter. The cosmopolitan sample (COS) shows intermediate values, with a value of 1/ε of 98 kb and an M parameter of 0.74.
Table 2.
Fitting the pairwise data to the physical (kilobase) map
| Population | N | m | ε | M | Lp | -2lnL | df | Vkb | Swept radius(kb) |
|---|---|---|---|---|---|---|---|---|---|
| COS | 132,171 | 1,691 | 0.01024 | 0.738 | 0.091 | 339,109 | 132,169 | 2.566 | 97.6 |
| AF | 87,135 | 1,338 | 0.01243 | 0.661 | 0.136 | 114,123 | 87,133 | 1.310 | 80.4 |
| CA | 71,097 | 1,211 | 0.01043 | 0.877 | 0.135 | 109,046 | 71,095 | 1.534 | 95.9 |
| CE | 111,067 | 1,518 | 0.00953 | 0.805 | 0.197 | 102,478 | 111,065 | 0.923 | 104.9 |
| AS | 64,586 | 1,153 | 0.01117 | 0.861 | 0.204 | 52,781 | 64,584 | 0.817 | 89.6 |
N: Number of pairs; m: number of loci; ε M, Lp: Malecot parameters; -2lnL: composite -2 log likelihood; df: degrees of freedom; swept radius: 1/ε; Vkb: residual error variance on fitting pairwise data to the kilobase map; COS: Cosmopolitan sample; AF: African - American; CA: UK Caucasians; CE: Utah CEPH founders; AS: Japanese and Chinese.
The population-specific LD maps (Table 3, Figure 2) give LDU maps of length 204 - 272 LDUs for the individual populations with the longest LD map for the AF population, again reflecting the greater time for recombination to erode LD. The cosmopolitan map is somewhat shorter (187 LDUs) but the number of holes in the population-specific maps is considerably higher (in the range 9 - 17 compared with only 2 in the cosmopolitan map). The discrepancy suggests that increased SNP typing in the intervals with holes may reduce the overall length of the population-specific maps. To test this, we rebuilt LD maps for the four populations but included all of the SNPs typed in each population sample within each interval containing a hole. We were then able to examine the effect on map length under conditions where some additional typing is possible. There were 51 holes for the four populations (Table 3), 37 of which had additional SNPs available in the full dataset. A total of 184 additional SNPs were then added into these 37 intervals. The corresponding revised map lengths and residual number of holes are shown in brackets in Table 3. Following this procedure, the CE population had the fewest number of SNPs added; six of the 12 holes had no additional SNPs. Overall, only 11 of the total 51 holes were resolved and the corresponding map lengths were only reduced by between 1 and 4 LDU at best, with a minimal increase in map length for the CE population. Tapper et al. [6]. found a strong relationship between number of holes and high recombination intensity on chromosome 22, so that particularly high marker densities may be required within some narrow intervals. Preliminary LD maps are therefore useful to target regions requiring additional typing.
Table 3.
Linkage disequilibrium unit maps constructed for each population
| Population | E | M | -2lnL | df | VLDU | No. LDUs* | No. holes* |
|---|---|---|---|---|---|---|---|
| COS | 1.1521 | 0.894 | 179,822 | 130,480 | 1.378 | 187.15 | 2 |
| AF | 1.1661 | 0.842 | 77,916 | 85,797 | 0.908 | 272.49(268.22) | 13 (10) |
| CA | 1.0754 | 0.957 | 54,057 | 69,880 | 0.774 | 209.62(208.06) | 9 (8) |
| CE | 1.1290 | 0.924 | 67,225 | 109,549 | 0.614 | 204.19(204.66) | 12 (9) |
| AS | 1.0811 | 0.923 | 33,777 | 63,434 | 0.532 | 223.20(222.29) | 17 (13) |
ε, M: Malecot parameters; -2lnL: composite -2 log likelihood; df: degrees of freedom; VLDU: residual error variance for the linkage disequilibrium unit map; COS: Cosmopolitan sample; AF: African - American; CA: UK Caucasians; CE: Utah CEPH founders; AS: Japanese and Chinese.
* Figures in brackets are corresponding values when the additional single nucleotide polymorphisms are included, whenever the linkage disequilibrium unit distance in an interval is bounded at 3, and is therefore a hole.
Figure 2.
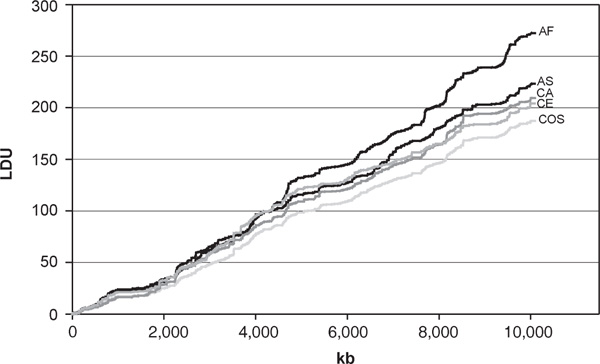
Population-specific linkage disequilibrium map contours for African-American (AF), CEPH (CE), Caucasian (CA) and Asian (AS) populations, together with the cosmopolitan map (COS), which combines all data.
Table 4 examines the fit of the population-specific data to the cosmopolitan map. The ε parameters for each population, divided by that for the cosmopolitan map (ε = 1.152, Table 3), give the appropriate scaling factors (Table 5) to apply to the cosmopolitan map so that it can represent any population. For the AF population, the scaling factor is 1.33, suggesting that the cosmopolitan map should be lengthened by 33 per cent. Relative to the Caucasian population, the scaling factor would be 1.43 (Table 4). This figure compares with previous scaling factor estimates (11) in the range 1.63 - 1.84, obtained in much smaller genomic regions. There is considerably greater similarity in the ε parameter estimates for the other populations which do not have recent African ancestry.
Table 4.
Fitting the data for each population to the COS map
| Population | ε | M | -2lnL | df | VCOS |
|---|---|---|---|---|---|
| AF | 1.5323 | 0.811 | 82,334 | 86,019 | 0.957 |
| CA | 1.0659 | 0.968 | 59,172 | 70,186 | 0.843 |
| CE | 1.0231 | 0.927 | 70,952 | 109,645 | 0.647 |
| AS | 1.1859 | 0.931 | 37,198 | 63,758 | 0.583 |
ε, M: Malecot parameters; -2lnL: composite -2 log likelihood; df: degrees of freedom; VCOS: residual error variance for the individual population data fitted to the cosmopolitan sample map; COS: Cosmopolitan sample; AF: African-American; CA: UK Caucasians; CE: Utah CEPH founders; AS: Japanese and Chinese.
Table 5.
Relative efficiency of different maps and scaling factors for different populations
| Population | Vkb | VLDU | Vcos | Relative effciency of kb map (VLDU/Vkb) | Relative effciency of COS map (VLDU/VCOS) | Scaling factor relative to COS map |
|---|---|---|---|---|---|---|
| AF | 1.310 | 0.908 | 0.957 | 0.693 | 0.949 | 1.330 |
| CA | 1.534 | 0.774 | 0.843 | 0.504 | 0.918 | 0.925 |
| CE | 0.923 | 0.614 | 0.647 | 0.665 | 0.949 | 0.888 |
| AS | 0.817 | 0.532 | 0.583 | 0.651 | 0.913 | 1.029 |
Vkb: Residual error variance on fitting pairwise data to the kilobase map; VLDU: residual error variance for the linkage disequilibrium unit map; VCOS: residual error variance for the individual population data fitted to the cosmopolitan sample map; AF: African-American; CA: UK Caucasians; CE: Utah CEPH founders; AS: Japanese and Chinese.
Table 5 gives the REs of the kb and cosmopolitan maps as ratios of the corresponding error variances. The RE of the kb map, compared with the individual population LD maps, varies between 50 and 69 per cent, the relatively low figure being expected, because kb maps do not reflect patterns of LD. By contrast, there is good support for the use of the cosmopolitan map to represent LD patterns for all populations, when ε and M are estimated. The relative efficiency is between 91 and 95 per cent, suggesting a small loss of information. Figure 3 illustrates the utility of a cosmopolitan map by comparing the AF LD map with a cosmopolitan map transformed by the appropriate scaling factor. The rescaled map is somewhat shorter, but the likely effect of the relatively large number of holes for the AF population map suggests that the map may be somewhat inflated in overall length.
Figure 3.
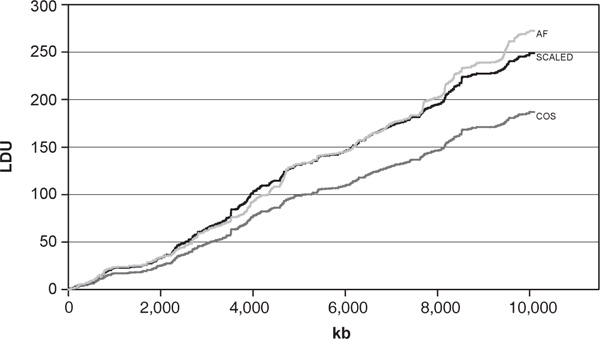
The contour of the cosmopolitan map (COS) after applying the scaling factor for the African-American population (SCALED). The African - American linkage disequilibrium map is also shown (AF).
Discussion
This study supports and extends previous findings [11] which demonstrated that careful modelling of LD patterns in human populations reveals both similarities and differences that have biological interpretation and reflect differences in population history. Recent studies have shown the importance of narrow, intense, recombination hot spots in determining LD patterns and the tendency for these to be co-localised in all human populations, which accounts for the remarkable convergence in the overall LD 'plateau' and 'step' structure. We have shown that the predominant source of differences between the maps arises from population duration, and that appropriate linear scaling of a 'standard' map recovers most of the information in population-specific maps. The loss of information, after appropriate scaling for a given population, is between 5 and 9 per cent. A cosmopolitan LD map, which combines data over a range of populations, has advantages for map integration, through being derived from a larger data sample and therefore having fewer regions of uncertainty (holes). Such a map may also have a larger number of loci and is, therefore, of potentially higher resolution. The integration of standard LD maps with genetic and physical chromosome maps will be useful for disease gene mapping by association. Ke et al. [19] described the integrated location database, LDB2000 http://cedar.genetics.soton.ac.uk/public_html/, which gives locations on alternative scales for genes and polymorphisms. The integrated maps specify kb locations -- obtained from the current human genome sequence -- together with male and female genetic maps in cM and cytogenetic band assignments. Once LD maps are integrated, it will be possible to infer locations for all markers on all scales, by interpolation. The LD map might therefore be useful for local improvement of the resolution of the genetic linkage map, which is currently unreliable below ~1 cM, reflecting the relatively small numbers of meioses in the linkage families.
Genetic linkage maps have greatly facilitated the multipoint linkage mapping of many major disease genes. The importance of exploiting linkage map locations for this purpose, rather than using the physical (kb) map, is maximal for candidate regions where there is a substantial deviation from the genome-wide average of 1 cM ≃ 1 megabase. As an illustration, in the hemochromatosis gene region, [20] a low recombination rate was initially misinterpreted as a small physical distance and this may have delayed the positional cloning of the gene. It is likely that the correct characterisation of LD patterns will be of equal, or perhaps greater, importance for association mapping. A study by Maniatis et al. [14] has shown substantially increased power and precision for multilocus modelling of disease-marker association when using an LD rather than a kb map. Localisation within an LD map was shown to increase power by ~38 per cent on average, compared with a kb map, in extensive simulation studies. To effectively apply association mapping for complex traits, it is not sufficient to construct a high-resolution recombination map, [8] since association mapping requires the characterisation of LD patterns. Although recombination hot spots are colocalised among populations, differences in duration means that the steps have different heights, and this is particularly apparent when comparing African-derived populations with other populations. Furthermore, a recombination map does not accommodate the effects of other processes that shape LD patterns, such as drift, selection and mutation.
Methods to construct high-resolution LD maps are likely to improve. It is fortunate that a substantial body of data is now available for a number of populations http://www.hapmap.org/; this will allow whole-chromosome LD map construction to be undertaken. The samples from HapMap offer high marker density and, therefore, LD maps with well-defined LD structure can be constructed, permitting further evaluation of the utility of LD maps and integrated maps for disease gene mapping. Maps constructed at relatively low densities [6] remain useful, in that they provide credible starting estimates for LDU distances in a candidate region which may be revised through the addition of more markers and larger sample sizes.
Recent evidence points to a substantial loss of power through SNP selection to retain haplotype diversity [21]. A proposed multistage design, in which tested models and marker density change adaptively, uses an LD map to guide the addition of SNPs into a candidate region, rather than selective removal. In the multistage design, stage 1 is a genome scan by linkage or association at relatively low-resolution, stage 2 refines an identified candidate region at moderate marker densities and the final stage requires functional tests on SNPs at high resolution, with the goal of identifying one or more causal polymorphisms. Appropriately scaled LD maps will be valuable throughout, but particularly in the early stages, to identify poorly covered regions which particularly require additional SNPs, thereby reducing the probability of failing to screen a critical region.
Acknowledgements
This work was supported by research grants from the Biotechnology and Biological Sciences Research Council, the Medical Research Council and Applied Biosystems. We are grateful to Panos Deloukas and colleagues at the Wellcome Trust Sanger Institute for making these data available.
References
- Jeffreys AJ, Kauppi L, Neumann R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet. 2001;29:217–222. doi: 10.1038/ng1001-217. [DOI] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF. et al. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- Dawson E, Abecasis GR, Bumpstead S. et al. A first-generation linkage disequilibrium map of human chromosome 22. Nature. 2002;418:544–548. doi: 10.1038/nature00864. [DOI] [PubMed] [Google Scholar]
- Ke X, Hunt S, Tapper W. et al. The impact of SNP density on fine-scale patterns of linkage disequilibrium. Hum Mol Genet. 2004;13:577–588. doi: 10.1093/hmg/ddh060. [DOI] [PubMed] [Google Scholar]
- Maniatis N, Collins A, Ku X-F. et al. The first linkage disequilibrium (LD) maps: Delineation of hot, cold blocks by diplotype analysis. Proc Natl Acad Sci USA. 2002;99:2228–2233. doi: 10.1073/pnas.042680999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper WJ, Morton NE, Dunham I. et al. A sequence-based map of chromosome 22. Genome Res. 2001;11:1290–1295. doi: 10.1101/gr.161301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Collins A, Maniatis N. et al. Properties of linkage disequilibrium (LD) maps. Proc Natl Acad Sci USA. 2002;99:17004–17007. doi: 10.1073/pnas.012672899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GAT, Myers SR, Hunt S. et al. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. The International HapMap project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Kauppi L, Sajantila A, Jeffreys AJ. Recombination hotspots rather than population history dominate linkage disequilibrium in the MHC class II region. Hum Mol Genet. 2003;12:33–40. doi: 10.1093/hmg/ddg008. [DOI] [PubMed] [Google Scholar]
- Lonjou C, Zhang W, Collins A. et al. Linkage disequilibrium in human populations. Proc Natl Acad Sci USA. 2003;100:6069–6074. doi: 10.1073/pnas.1031521100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H. et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Morton NE, Zhang W, Taillon-Miller P. et al. The optimal measure of allelic association. Proc Natl Acad Sci USA. 2001;98:5217–5222. doi: 10.1073/pnas.091062198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis N, Collins A, Gibson J. et al. Positional cloning by linkage disequilibrium. Am J Hum Genet. 2004;74:846–855. doi: 10.1086/383589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez I, Collins A, Lonjou C. et al. Hardy-Weinberg quality control. Ann Hum Genet. 1999;63:535–538. doi: 10.1046/j.1469-1809.1999.6360535.x. [DOI] [PubMed] [Google Scholar]
- Hill WG. Estimation of linkage disequilibrium in randomly mating populations. Heredity. 1974;33:229–239. doi: 10.1038/hdy.1974.89. [DOI] [PubMed] [Google Scholar]
- Collins A, Lonjou C, Morton NE. Genetic epidemiology of single nucleotide polymorphisms. Proc Natl Acad Sci USA. 1999;96:15173–15177. doi: 10.1073/pnas.96.26.15173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins A, Morton NE. Mapping a disease locus by allelic association. Proc Natl Acad Sci USA. 1998;95:1741–1745. doi: 10.1073/pnas.95.4.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke X, Tapper W, Collins A. LDB2000: Sequence-based integrated maps of the human genome. Bioinformatics. 2001;17:581–586. doi: 10.1093/bioinformatics/17.7.581. [DOI] [PubMed] [Google Scholar]
- Lonjou C, Collins A, Ajioka RS. et al. Allelic association under map error, recombinational heterogeneity: A tale of two sites. Proc Natl Acad Sci USA. 1998;95:11366–11370. doi: 10.1073/pnas.95.19.11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Collins A, Morton NE. Does haplotype diversity predict power for association mapping of disease susceptibility? Hum Genet. 2004;115:157–164. doi: 10.1007/s00439-004-1122-x. [DOI] [PubMed] [Google Scholar]