

Abstract
Substantial progress has been made in human genetics and genomics research over the past ten years since the publication of the draft sequence of the human genome in 2001. Findings emanating directly from the Human Genome Project, together with those from follow-on studies, have had an enormous impact on our understanding of the architecture and function of the human genome. Major developments have been made in cataloguing genetic variation, the International HapMap Project, and with respect to advances in genotyping technologies. These developments are vital for the emergence of genome-wide association studies in the investigation of complex diseases and traits. In parallel, the advent of high-throughput sequencing technologies has ushered in the 'personal genome sequencing' era for both normal and cancer genomes, and made possible large-scale genome sequencing studies such as the 1000 Genomes Project and the International Cancer Genome Consortium. The high-throughput sequencing and sequence-capture technologies are also providing new opportunities to study Mendelian disorders through exome sequencing and whole-genome sequencing. This paper reviews these major developments in human genetics and genomics over the past decade.
Keywords: Human Genome Project, International HapMap Project, 1000 Genomes Project, genome-wide association studies, single nucleotide polymorphisms, copy number variations, next-generation sequencing technologies, cancer genome sequencing, exome sequencing, complex disease, Mendelian disorders, personalised genomic medicine
Introduction
Substantial progress has been made in human genetics and genomics research over the past 10 years since the publication of the draft sequence of the human genome [1,2]. The Human Genome Project (HGP) provided the basic raw DNA sequence that spawned a plethora of secondary studies which together greatly improved our knowledge of the architecture and function of the genome, yielding new insights with respect to (i) gene number and density, (ii) non-protein-coding RNA genes (or RNA genes), (iii) pervasive transcription, (iv) high copy number repeat sequences and (v) evolutionary conservation. These developments also have challenged the classical definition of the gene (see below).
In parallel, the design of studies investigating complex diseases and traits has gradually shifted from candidate-gene association and linkage studies to genome-wide association studies (GWASs). The first proper GWAS study was published in 2005. This succeeded in identifying a common risk variant with a large effect size in the complement factor H (CFH) gene, which was associated with age-related macular degeneration [3]. By 2007, approximately 100 new GWASs had been published, relating to various complex diseases and traits [4]. There has, however, been some criticism of the inability of GWASs to identify many of the presumed disease-associated variants. Indeed, the validity of the common-disease common-variant (CD/CV) model has recently been challenged by virtue of the perceived 'missing heritability' [5-7]. This notwithstanding, the GWAS approach has dramatically changed the field of human disease genetics, from identifying mostly irreproducible disease associations in the pre-GWAS era to revealing thousands of statistically robust single nucleotide polymorphism (SNP) associations today [8-11]. The focus has also gradually shifted back to Mendelian disorders, with the advent of high-throughput sequence capture and sequencing technologies which have potentiated exome and whole-genome (re)sequencing (WGS) [12-16].
The rapid advances made in genotyping technologies over the past decade, from the arrival of the first 'whole-genome' SNP genotyping array (the Affymetrix GeneChip 10K [Affymetrix; Santa Clara, CA] in 2003) to current capacity able to genotype five million SNPs per array (Illumina Omni5.0 Beadchip [Illumina; San Diego, CA]),[17,18] have contributed substantially to GWASs (http://www.genome.gov/gwastudies). A total of 874 publications and 4,327 SNP associations with p-values < 1.0 × 10-5 for approximately 500 complex diseases and traits had been included in the catalogue as of 13th May 2011.
The genotyping arrays have also contributed significantly to population genetics studies [19-21]. These arrays have been used to identify and characterise copy number variations (CNVs)[22,23] and regions of homozygosity (ROHs) [24,25]. Research on CNVs and ROHs has also progressed rapidly since CNVs were first reported to be widespread in the human genome,[26,27] and ROHs have been found to be common in outbred populations [28]. In recognition of the progress achieved in the context of both GWASs and CNVs, 'human genetic variation' was considered the 'Breakthrough of The Year' in 2007 by Science [4].
Advances have also been made in sequencing technologies, with the advent of the first next-generation sequencer in 2004 (Roche GS 20 System [Roche 454; Branford, CT]) and later, third-generation sequencing (TGS) technologies such as true single molecule sequencing (Helicos Biosciences, Cambridge, MA) and single molecule real-time sequencing (SMRT) (Pacific Biosciences Menlo Park, CA) [29-33]. Developments of other more promising TGS or single-molecule sequencing technologies are on the horizon, such as nanopore sequencing and sequencing using transmission electron microscopy [32,34-37]. These developments have also marked the end of the era of the Sanger dideoxynucleotide or chain termination sequencing method, which has dominated the field since its introduction in 1977 [38].
The arrival of next-generation sequencing (NGS) technologies has also significantly changed the approaches applied in structural and functional genomics studies. Several microarray-based methods have been swiftly supplanted by sequencing-based approaches such as ChIP-Seq, RNA-Seq, Methyl-Seq and CNV-Seq (paired-end mapping [PEM] and depth-of-coverage approaches). Studies using these sequencing approaches have contributed significantly to both fields [39-41]. In addition to a variety of different applications in functional genomic studies, these sequencing technologies have also made it feasible, both technically and in terms of cost, to sequence a whole human genome within weeks, for tens rather than hundreds of thousands of US dollars [42,43]. Currently, the cost of WGS at several tenfold depth of sequencing coverage has been reduced to less than 5,000 US dollars [44]. The number of WGS studies for both normal and cancer genomes has grown rapidly over the past three years [45]. These studies have led to important discoveries in the context of both heritable genetic variation [42,43,46] and somatic mutations in cancer genomes [47-49].
Such progress would not have been possible without the reference genome generated by the HGP. Also made possible by the high-throughput genotyping and sequencing technologies, several large-scale international projects have been launched, such as the International HapMap Project; the Encyclopedia of DNA Elements (ENCODE) Project, the 1000 Genomes Project, the International Cancer Genome Consortium, the National Institute of Health (NIH) Roadmap Epigenomics Program and the Human Microbiome Project. These projects have contributed substantially to our understanding and knowledge of human genetics and genomics.
This paper aims to review these major developments in human genetics and genomics over the past decade. Major developments and landmarks in human genetics and genomics are summarised in Table 1.
Table 1.
Major developments and landmarks in human genetics and genomics, 1977 to date
| Year | Development | References |
|---|---|---|
| 1977 | Sanger dideoxynucleotide/chain termination sequencing method developed | [38] |
| Mammalian genes shown to contain introns | [50] | |
| 1978 | First report of characterisation of gross gene deletions responsible for human inherited disease (α- and β-thalassaemia) by Southern blotting | [51] |
| 1979 | First single base-pair substitution causing a human inherited disease (β-thalassaemia) characterized by DNA sequencing | [52] |
| 1980 | Construction of a genetic linkage map in humans using restriction fragment length polymorphisms | [53] |
| 1990 | Initiation of the Human Genome Project (HGP) | [54] |
| 1992 | Second-generation linkage map of the human genome | [55] |
| 1996 | The Human Gene Mutation Database (HGMD), an attempt to collate known (published) gene lesions responsible for human inherited disease, established and made available at http://www.hgmd.org | [56] |
| Genome-wide association studies (GWAS) approach for genetic studies of complex diseases first proposed | [57] | |
| 2001 | Completion of draft DNA sequences of the human genome by the International Human Genome Sequencing Consortium (IHGSC) and Celera Genomics | [1,2] |
| International SNP Map Working Group identifies 1.42 million SNPs in the human genome | [58] | |
| Genetic architecture of complex diseases subjected to intense debate | [59,60] | |
| Linkage disequilibrium (LD) patterns documented between SNPs in regions of the human genome | [61,62] | |
| 2003 | Initiation of the International HapMap Project | [63] |
| First whole-genome SNP genotyping array - Affymetrix GeneChip 10K | [17] | |
| 2004 | IHGSC publishes the 'finished version' of the DNA sequence of the human genome | [64] |
| Initiation of the ENCODE project | [65] | |
| Discovery of hundreds of copy number variations (CNVs) in the human genome | [26,27] | |
| Database of Genomic Variants (DGV) established to catalogue CNVs | [27] | |
| First new-generation sequencing (NGS) technology - Roche 454 GS 20 System | [29,30] | |
| 2005 | Completion of the International HapMap Phase I Project | [66] |
| First proper GWAS using a commercial whole-genome SNP genotyping array | [3] | |
| 2005-present | Rapid developments of whole-genome and custom SNP genotyping arrays and technologies | [18] |
| Rapid developments of sequencing technologies | [31,33] | |
| 2006 | Discovery of more than 1,000 regions of homozygosity > 1 megabase (Mb) in the genomes of outbred populations | [28] |
| First comprehensive map of CNVs in the HapMap populations | [22] | |
| An initial map of insertion and deletion variants in the human genome | [67] | |
| Illumina sequencing platform commercially marketed | [29,30] | |
| 2007 | The first human diploid genome (Craig Venter's genome) sequenced by the Sanger sequencing method | [68] |
| Completion of the International HapMap Phase II Project and extension to Phase III | [69] | |
| Genome-wide detection and characterisation of positive selection in human populations | [70] | |
| Completion of the ENCODE project | [71] | |
| Explosion of GWAS publications ('Year of GWAS'), approximately 100 new GWASs | [4] | |
| 'Human Genetic Variation' considered to be the 'Breakthrough of The Year' in 2007 by Science | [4] | |
| Sequence capture or enrichment methods and technologies developed | [72-74] | |
| Pervasive transcription documented | [75] | |
| Demonstration of paired-end mapping (PEM) to detect structural variation using NGS technologies | [76] | |
| Demonstration of ChIP-Seq to map transcription factor binding sites | [77] | |
| Demonstration of ChIP-Seq to interrogate histone modifications | [78] | |
| Life Technologies SOLiD sequencing platform commercially marketed | [29,30] | |
| A community resource project launched to sequence large-insert clones from many individuals, systematically discovering and resolving these complex variants at the DNA sequence level (The Human Genome Structural Variation Working Group) | [79] | |
| 2007-Present | Microarray-based methods increasingly supplanted by sequencing-based approaches such as ChIP-Seq, RNA-Seq, Methyl-Seq and CNV-Seq | [39,41,80,81] |
| 2008 | First human diploid genome (James Watson's genome) sequenced by NGS technologies | [46] |
| First whole cancer genome (acute myeloid leukaemia [AML]) sequenced | [82] | |
| Initiation of the 1000 Genomes Project | [83] | |
| Vast majority of human genes shown to undergo alternative splicing (RNA-Seq) | [84,85] | |
| Large scale mapping and sequencing of structural variation using a clone-based method | [86] | |
| Demonstration of depth-of-coverage approach to detect CNVs using NGS technologies | [87] | |
| First GWAS meta-analysis using imputation methods | [88] | |
| The issue of 'missing heritability' in GWASs recognised | [89] | |
| 2009 | Feasibility of exome sequencing approach to identify a causal mutation for a Mendelian disorder first demonstrated | [12] |
| Exome sequencing as a useful tool for diagnostic application demonstrated | [90] | |
| Third generation sequencing (TGS; single molecule sequencing) technology introduced --Heliscope Single Molecule Sequencer (Helicos Biosciences) commercially marketed | [91] | |
| First human diploid genome sequenced by TGS technology | [92] | |
| Latest assembly of the human genome (Genome Reference Consortium, release GRCh37, February 2009), Genebuild published by Ensembl (database version 56.37a) includes 23,616 protein-coding genes, 6,407 putative RNA genes and 12,346 pseudogenes | http://www.ensembl.org/Homo_sapiens/Info/StatsTable | |
| Large intergenic non-coding RNAs (lincRNAs) found to represent a novel category of evolutionarily conserved RNAs | [93,94] | |
| Direct single molecule RNA sequencing without prior conversion of RNA to cDNA | [95] | |
| First human DNA methylomes at base resolution | [96] | |
| Comprehensive mapping of long-range chromatin interactions | [97,98] | |
| 2010 | Number of disease-causing/disease-associated germline mutations collated in the Human Gene Mutation Database exceeds 100,000 in > 3,700 different nuclear genes | [99,100] |
| More than 17 million SNPs in the human genome catalogued in the SNP Database (dbSNP; http://www.ncbi.nlm.nih.gov/projects/SNP/) | [101] | |
| As of 2nd November 2010, DGV catalogued 66,741 CNVs, 953 inversions and 34,229 insertions and deletions (indels) (100 base pairs (bp) -- 1 kilobase (kb) from 42 published studies | http://projects.tcag.ca/variation/ | |
| 1,048 microRNAs found in the human genome | miRBase, Release 16.0: September 2010, http://www.mirbase.org/ | |
| Completion of the International HapMap Phase III Project | [21] | |
| Completion of pilot phase of the 1000 Genomes Project | [102] | |
| Second generation whole-genome SNP genotyping array (with SNP selection from the 1000 Genomes Project) launched | http://www.illumina.com/applications/gwas.ilmn | |
| Cost of whole-genome sequencing (at several tenfold of sequencing coverage depth) reduced to less than $5,000 | [44] | |
| Metagenomic sequencing of human gut microbes accomplished using NGS technologies | [103] | |
| Exome sequencing study identifies causal mutations and genes for previously unexplained Mendelian disorders | [13,14] | |
| GWAS meta-analysis involving total sample size of > 249,000 | [104] | |
| Comprehensive mapping of CNVs using high-resolution tiling oligonucleotide microarrays (42 million probes) | [105] | |
| Characterisation of 20 sequenced human genomes to evaluate the prospects for identifying rare functional variants | [106] | |
| Neanderthal genome sequenced | [107] | |
| The genome of an extinct Palaeo-Eskimo sequenced | [108] | |
| Exome sequencing of 200 individuals identifies an excess of low-frequency non-synonymous coding variants | [109] | |
| International Cancer Genome Consortium (ICGC) launched | [110] | |
| Largest GWAS of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls performed | [111] | |
| 2011 | As of 13th May 2011, 874 publications and 4,327 SNPs documented in the National Human Genome Research Institute (NHGRI) 'A Catalog of Published Genome-wide Association Studies' | http://www.genome.gov/gwastudies/ |
| Comprehensive mapping of copy number variations based on whole-genome DNA sequencing data | [112] | |
| Developments of other TGS technologies, such as single-molecule real-time sequencing and nanopore sequencing, are on the horizon | [32] | |
| New addition to the NGS market -- the Ion Torrent Personal Genome Machine (PGM), produced by Life Technologies (Carlsbad, CA) | http://www.iontorrent.com/ | |
| Single-cell sequencing to infer tumour evolution | [113] |
The HGP
Rapid progress has been made since the completion of the HGP, with the provision of a 'finished' reference DNA sequence for the human genome [64]. The project was initiated in 1990 and, upon its completion in 2003 it yielded important new insights into the architecture and function of the human genome. The sequencing of the HGP relied almost entirely upon the Sanger sequencing method.
The draft sequences of the HGP were imperfect because of the incomplete coverage of the euchromatic regions (euchromatin) -- approximately 10 per cent of these regions were missing. In reality, the coverage was even less complete when the whole genome was considered (ie when the heterochromatic regions were included). Thus, in all, some 30 per cent of the genome was not initially covered. Furthermore, there was an extensive number of gaps between contigs, which rendered the genome sequence discontinuous [1,2]. The IHGSC subsequently published an improved version of the human genome sequence in 2004 and the HGP was then deemed to be 'complete'. This 'finished' version of the genome had achieved an almost complete coverage of all the euchromatic regions (ie approximately 99 per cent) and also significantly reduced the number of gaps between contigs to 341 from the initial hundreds of thousands [64].
Significant further progress toward the total completion of the human genome sequence continued until 2006; the complete euchromatic sequences of all individual human chromosomes, including the annotation of genes and other features, have now been published (summarised in Table S1 (Table 2)). Since November 2005, the National Center for Biotechnology Information (NCBI) Build 36 assembly of the human genome sequence has been available in public databases. The data comprise a reference assembly of the complete genome sequence plus the Celera WGS (Celera; Alameda, CA) and a number of alternative assemblies of individual haplotypic chromosomes or regions. The full list of assemblies in NCBI 36, as well as the genome sequences, is available through the following genome browsers:
Table S1.
Special features of human autosomes 1-22 and the sex chromosomes, including respective lengths, gene number and density
| Chromosome | Chromosome length (bp)a | Number of known protein-coding genes per chromosomea | Gene density (genes/Mb) | Special features | Reference |
|---|---|---|---|---|---|
| 1 | 247,249,719 | 2,189 | 8.85 | Largest human chromosome. Rich in disease genes. Huge (~30 Mb) pericentromeric heterochromatic region at 1q12 spans ~5% of the length of the chromosome. Contains clusters of amylase genes (1p21), U1 snRNA genes (1q12-q22) and 5S RNA genes (1q) as well as multiple (~250) tRNA genes | 1 |
| 2 | 242,951,149 | 1,328 | 5.47 | Chromosome 2 (along with chromosome 4) exhibits the lowest recombination rate of all the autosomes. Contains at 2q13 an ancient telomere-telomere fusion junction at the position where two ape chromosomes once fused to give rise to this human chromosome | 2 |
| 3 | 199,501,827 | 1,112 | 5.57 | Lowest rate of segmental duplication of all human chromosomes. Contains several olfactory receptor gene clusters | 3 |
| 4 | 191,273,063 | 797 | 4.17 | Chromosome 4 (along with chromosome 2) exhibits the lowest recombination rate of all the autosomes. Highest percentage of LINE elements among all chromosomes | 2 |
| 5 | 180,857,866 | 903 | 4.99 | Rich in intra-chromosomal duplications. Contains interleukin and protocadherin gene clusters on 5q31 | 4 |
| 6 | 170,899,992 | 1,133 | 6.62 | Harbours the major histocompatibility complex and the largest tRNA gene cluster in the human genome. Contains at least three imprinted genes | 5 |
| 7 | 158,821,424 | 1,023 | 6.44 | Contains the highest number of intra-chromosomal duplications among all human chromosomes. Contains at least six imprinted genes | 6, 7 |
| 8 | 146,274,826 | 747 | 5.11 | Contains a fast-evolving 15 Mb region on distal 8p with genes related to the innate immunity and nervous systems that appear to have evolved under positive selection | 8 |
| 9 | 140,273,252 | 929 | 6.62 | Structurally highly polymorphic. Contains the large (~14 Mb) block of pericentromeric heterochromatin. Contains large numbers of intra- and inter-chromosomal segmental duplications, as well as the largest interferon gene cluster in the human genome (9p22) | 9 |
| 10 | 135,374,737 | 834 | 6.16 | Region of extensive segmental duplication located on 10q11 | 10 |
| 11 | 134,452,384 | 1,385 | 10.30 | Rich in both genes and disease genes. Contains 40% of all olfactory receptor gene clusters. Contains at least nine imprinted genes | 11 |
| 12 | 132,349,534 | 1,080 | 8.16 | Chromosome 12 has a unique history of evolutionary rearrangements that occurred in the rodent and primate lineages. Contains clusters of proline-rich protein and type II keratin genes at 12q13 | 12 |
| 13 | 114,142,980 | 361 | 3.16 | Low gene density in general; contains a central 38 Mb segment where the gene density drops to only 3.1 genes per Mb. This acrocentric chromosome contains ribosomal RNA genes at 13p12 and at least one imprinted gene | 13 |
| 14 | 106,368,585 | 669 | 6.29 | This acrocentric chromosome contains ribosomal RNA genes at 14p12. Contains two 1 Mb regions of crucial importance to the immune system (T cell receptor and immunoglobulin heavy chain genes). Contains serpin gene cluster at 14q32.1 and several regions with imprinted genes | 14 |
| 15 | 100,338,915 | 641 | 6.39 | This acrocentric chromosome contains ribosomal RNA genes at 15p12. Two large clusters of clinically important segmental duplications are located in the proximal and distal regions of 15q. Contains a number of imprinted genes | 15 |
| 16 | 88,827,254 | 925 | 10.41 | Relatively high gene density. Contains a large number of segmental duplications | 16 |
| 17 | 78,774,742 | 1,236 | 15.69 | High gene density. Has undergone extensive intra-chromosomal rearrangement, many of which were probably mediated by segmental duplications. High G + C content of 45% (genome average: 41%) | 17 |
| 18 | 76,117,153 | 295 | 3.88 | Low gene density overall. Contains serpin gene cluster at 18q21.3 | 18 |
| 19 | 63,811,651 | 1,443 | 22.61 | Highest gene density of all human chromosomes. One quarter of the genes on chromosome 19 belong to tandemly arranged gene families, encompassing 25% of the length of the chromosome. High G + C content of 48-49% (genome average: 41%). Repetitive sequences constitute 53-57% of the chromosome, as compared with a genome average of 40-44%. Contains clusters of olfactory receptor genes and cytochrome P450 genes, and multiple clusters of zinc finger genes, and at least two imprinted genes | 19 |
| 20 | 62,435,964 | 617 | 9.88 | Smallest metacentric autosome. Rich in both genes and disease genes. Contains type 2 cystatin gene cluster and at least two imprinted genes | 20 |
| 21 | 46,944,323 | 284 | 6.05 | Smallest human chromosome with fewer genes than any other autosome. This acrocentric chromosome contains ribosomal RNA genes at 21p12 | 21 |
| 22 | 49,691,432 | 519 | 10.44 | This acrocentric chromosome contains ribosomal RNA genes at 22p12. Relatively high gene density. Clusters of segmental duplications at 22q11.2 are associated with several genomic disorders | 22 |
| X | 154,913,754 | 891 | 5.75 | Contains the pseudoautosomal regions, PAR1 and PAR2, at the tips of the short and long arms, respectively. These regions are essential for normal male meiosis and recombination. PAR1 undergoes an obligate crossover with the Y chromosome, thereby giving this region the highest recombination rate in the human genome, at least in males. One X chromosome is subject to inactivation in females. Highly enriched in interspersed repeats and has a low G + C content of 39% (genome average: 41%) | 23 |
| Y | 57,772,954 | 80 | 1.38 | Lowest gene density of all human chromosomes (contains only 82 known genes). Contains the male-specific region which is a mosaic of heterochromatin and euchromatic X-transposed, X-degenerate and ampliconic sequences that make up 30% of the euchromatin. PAR1 undergoes an obligate crossover with the X chromosome. The virtual absence of homologous recombination between the X and the Y chromosomes has led to a gradual degeneration of Y chromosomal genes over evolutionary time. However, the absence of recombination, at least within the extensive non-recombining region of the Y chromosome, has also favoured the evolutionary accumulation of transposable elements on the Y chromosome | 24 |
aChromosome lengths and the numbers of genes per chromosome are according to the Ensembl database, version 47.36. The chromosome length corresponds to the length of each chromosome that has been sequenced so far. The number of known protein-coding genes represents a conservative estimate of the likely total number, comprising genes which have been fully annotated. An earlier version of this table was published by Kehrer-Sawatzki and Cooper.25
1Gregory, S.G., Barlow, K.F., McLay, K.E., Kaul, R. et al. (2006), 'The DNA sequence and biological annotation of human chromosome 1', Nature Vol. 441, pp. 315-321.
2Hillier, L.W., Graves, T.A., Fulton, R.S., Fulton, L.A. et al. (2005), 'Generation and annotation of the DNA sequences of human chromosomes 2 and 4', Nature Vol. 434, pp. 724-731.
3Muzny, D.M., Scherer, S.E., Kaul, R., Wang, J. et al. (2006), 'The DNA sequence, annotation and analysis of human chromosome 3', Nature Vol. 440, pp. 1194-1198.
4Schmutz, J., Martin, J., Terry, A., Couronne, O. et al. (2004), 'The DNA sequence and comparative analysis of human chromosome 5', Nature Vol. 431, pp. 268-274.
5Mungall, A.J., Palmer, S.A., Sims, S.K., Edwards, C.A. et al. (2003), 'The DNA sequence and analysis of human chromosome 6', Nature Vol. 425, pp. 805-811.
6Hillier, L.W., Fulton, R.S., Fulton, L.A., Graves, T.A. et al. (2003), 'The DNA sequence of human chromosome 7', Nature Vol. 424, pp. 157-164.
7Scherer, S.W., Cheung, J., MacDonald, J.R., Osborne, L.R. et al. (2003), 'Human chromosome 7: DNA sequence and biology', Science Vol. 300, pp. 767-772.
8Nusbaum, C., Mikkelsen, T.S., Zody, M.C., Asakawa, S. et al. (2006), 'DNA sequence and analysis of human chromosome 8', Nature Vol. 439, pp. 331-335.
9Humphray, S.J., Oliver, K., Hunt, A.R., Plumb, R.W. et al. (2004), 'DNA sequence and analysis of human chromosome 9', Nature Vol. 429, pp. 369-374.
10Deloukas, P., Earthrowl, M.E., Grafham, D.V., Rubenfield, M. et al. (2004), 'The DNA sequence and comparative analysis of human chromosome 10', Nature Vol. 429, pp. 375-381.
11Taylor, T.D., Noguchi, H., Totoki, Y., Toyoda, A. et al. (2006), 'Human chromosome 11 DNA sequence and analysis including novel gene identification', Nature Vol. 440, pp. 497-500.
12Scherer, S.E., Muzny, D.M., Buhay, C.J., Chen, R. et al. (2006), 'The finished DNA sequence of human chromosome 12', Nature Vol. 440, pp. 346-351.
13Dunham, A., Matthews, L.H., Burton, J., Ashurst, J.L. et al. (2004), 'The DNA sequence and analysis of human chromosome 13', Nature Vol. 428, pp. 522-528.
14Heilig, R., Eckenberg, R., Petit, J.L., Fonknechten, N. et al. (2003), 'The DNA sequence and analysis of human chromosome 14', Nature Vol. 421, pp. 601-607.
15Zody, M.C., Garber, M., Sharpe, T., Young, S.K. et al. (2006), 'Analysis of the DNA sequence and duplication history of human chromosome 15', Nature Vol. 440, pp. 671-675.
16Martin, J., Han, C., Gordon, L.A., Terry, A. et al. (2004), 'The sequence and analysis of duplication-rich human chromosome 16', Nature Vol. 432, pp. 988-994.
17Zody, M.C., Garber, M., Adams, D.J., Sharpe, T. et al. (2006), 'DNA sequence of human chromosome 17 and analysis of rearrangement in the human lineage', Nature Vol. 440, pp. 1045-1049.
18Nusbaum, C., Zody, M.C., Borowsky, M.L., Kamal, M. et al. (2005), 'DNA sequence and analysis of human chromosome 18', Nature Vol. 437, pp. 551-555.
19Grimwood, J., Gordon, L.A., Olsen, A., Terry, A. et al. (2004), 'The DNA sequence and biology of human chromosome 19', Nature Vol. 428, pp. 529-535.
20Deloukas, P., Matthews, L.H., Ashurst, J., Burton, J. et al. (2001), 'The DNA sequence and comparative analysis of human chromosome 20', Nature Vol. 414, pp. 865-871.
21Hattori, M., Fujiyama, A., Taylor, T.D., Watanabe, H. et al. (2000), 'The DNA sequence of human chromosome 21', Nature Vol. 405, pp. 311-319.
22Dunham, I., Shimizu, N., Roe, B.A., Chissoe, S. et al. (1999), 'The DNA sequence of human chromosome 22', Nature Vol. 402, pp. 489-495.
23Ross, M.T., Grafham, D.V., Coffey, A.J., Scherer, S. et al. (2005), 'The DNA sequence of the human X chromosome', Nature Vol. 434, pp. 325-337.
24Skaletsky, H., Kuroda-Kawaguchi, T., Minx, P.J., Cordum, H.S. et al. (2003), 'The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes', Nature Vol. 423, pp. 825-837.
25Kehrer-Sawatzki, H. and Cooper, D.N. (2008), 'Sequencing the human genome: novel insights into its structure and function', in: Encyclopedia of Life Sciences (ELS), John Wiley & Sons Ltd, Chichester.
• Ensembl (http://www.ensembl.org/)[114]
• UCSC (http://genome.ucsc.edu/)[115]
• NCBI (http://www.ncbi.nlm.nih.gov/mapview/map_search.cgi?taxid = 9606) [116].
Although both HGP and Celera Genomics had only sequenced the human haploid genome, the availability of the reference DNA sequence initiated a new era in the study of genetic variation and the functional characterisation of the human genome. The two global projects that subsequently ensued were the International HapMap Project and the ENCODE project [63,65]. The aim of the HapMap initiative was to validate several million SNPs that were identified during and after the completion of the HGP, and then to characterise the extent of their linkage disequilibrium (LD) patterns in populations of European, Asian and African ancestry. The ENCODE project was conceived to identify all the functional and regulatory elements in the human genome.
Architecture and function of the human genome
To coincide with the tenth anniversary of the release of the draft human genome sequences, the key findings from the HGP and their importance for the results of subsequent studies will now be recalled briefly. The findings emanating from the HGP and follow-on studies have had an enormous impact on the understanding of the architecture and function of the human genome.
Gene number and density
Initial annotation data indicated that the human genome encodes at least 20,000-25,000 protein-coding genes, with an indeterminate number of additional 'computationally derived genes' supported by somewhat weaker in silico evidence [2,64]. Many genes are now known to encode RNAs rather than proteins as their final products [117] but many still remain unannotated [75]. In the latest assembly of the human genome (Genome Reference Consortium, release GRCh37, February 2009), the Genebuild published by Ensembl (database version 56.37a) includes 23,616 protein-coding genes, 6,407 putative RNA genes and 12,346 pseudogenes (http://www.ensembl.org/Homo_sapiens/Info/StatsTable). The HUGO Human Gene Nomenclature Committee (http://www.genenames.org/index.html) has so far approved more than 28,000 human gene symbols, although some of these may yet turn out to correspond to functionally meaningless open reading frames [118]. It is nevertheless encouraging that at least 17,052 human genes have been shown to have orthologous counterparts in the mouse genome, suggesting that they do indeed correspond to real proteins [119]. The definition of what constitutes a gene is still fairly fluid, and hence, depending upon the precise definition adopted, it may be that many additional human 'genes' still remain to be described and annotated.
To appreciate why definition is an issue here, one need only be aware of the many exceptions to genes being contiguous (as well as functionally and spatially distinct) entities, as classically envisaged. Thus, some genes are known to occur within the introns of other genes [120-122]. Some genes can overlap with each other either on the same or on different DNA strands,[123] resulting in the sharing of some of their coding and/or regulatory elements [124,125]. In addition, the vast majority of human genes are now known to undergo alternative splicing,[84] leading in some cases to quite different proteins being encoded by the same gene. For example, the human cyclin-dependent kinase inhibitor 2A gene (CDKN2A) (MIM# 600160) encodes an alternatively spliced variant (p14ARF) which, through the inclusion of an alternative first exon, acquires an altered reading frame so as to specify a protein product that is structurally unrelated to the other p16 isoforms encoded by this gene.
Gene density varies between the human chromosomes and the gene distribution within chromosomes is also rather uneven. Strikingly gene-poor regions have been identified and are known as 'gene deserts' [126]. These are regions that are devoid of protein-coding genes over distances of several Mb but which may nevertheless contain regulatory sequences (Box 1).
RNA genes or non-protein-coding RNAs
A large proportion of the human transcriptome still remains to be annotated [136]. Although some of the overall transcriptional activity may simply be 'transcriptional noise',[137,138] at least a portion of it is likely to be associated with functional non-coding RNA genes, many of which are located in regions previously regarded as intergenic and/or non-coding [71]. Non-coding RNA genes are as widespread as they are diverse,[139] are transcribed from both strands of the genome and may well exceed protein-coding genes in terms of their number [140,141].
Non-coding RNAs of known function include structural RNAs such as transfer RNAs, ribosomal RNAs and small nuclear RNAs, but also putative regulatory RNAs (microRNAs, small interfering RNAs [siRNAs], piwi-interacting RNAs, transcription initiation RNAs [tiRNAs], transcription start site-associated RNAs [TSSa-RNAs], promoter upstream transcripts [PROMPTs], promoter-associated sRNAs [PASRs and PALRs] and longer non-coding RNAs such as XIST), which are involved in sequence-specific transcriptional and post-transcriptional modulation of gene expression [142-148]. Thus, more than 1,000 microRNA genes already have been identified in the human genome, with many more probably awaiting discovery (Box 2). In total, at least 1,500 non-coding RNA genes already have been annotated in the human genome reference sequence, with up to 5,000 more predicted by homology-based methods [117] (see Ensembl, database version 56.37a).
Indeed, large intergenic non-coding RNAs (lincRNAs) recently have been found to represent a novel category of evolutionarily conserved RNAs, with a diverse array of functions ranging from stem cell pluripotency to cellular proliferation;[93,94] lincRNAs appear to number at least 3,000 in the human genome [155-158]. Some lincRNAs guide chromatin-modifying complexes to specific genomic loci, to regulate gene expression [94]. LincRNAs also play an important role in the derivation of human-induced pluripotent stem cells [156]. Collectively, non-coding RNAs have been intensively studied over the past several years [159,160].
Pervasive transcription: Transcripts of unknown function and unannotated transcripts
The ENCODE project, designed to analyse 30 Mb of DNA from 44 genomic regions to characterise the functional elements present, has identified complex patterns of regulation and 'pervasive transcription' of the human genome [71]. Although > 90 per cent of the human genome appears to be represented in nuclear primary transcripts, it has become clear that only 35-50 per cent of processed transcripts have so far been annotated as genes, implying that many genes may not yet have been recognised as such [71,85,161,162]. Thus, large numbers of hitherto unannotated transcripts may well yet turn out to be of functional significance [161]. Such transcripts have been collectively classified as transcripts of unknown function (TUFs) and are thought to include (i) antisense transcripts of protein-coding genes, (ii) isoforms of protein-coding genes and (iii) transcripts that either overlap introns of annotated gene transcripts (on the same strand) or which are derived entirely from inter-genic regions. Although both the complexity and abundance of TUFs are remarkable, it should be realised that there is often no firm evidence for these transcripts being of functional significance. Indeed, unannotated non-polyadenylated transcripts originating from intergenic regions have been found to represent the bulk of the > 90 per cent of the human genome that now appears to be transcribed [161,163,164]. Although the functional significance of pervasive transcription remains unclear, it is much more extensive than had previously been realised [165].
In both humans and mice, up to 70 per cent of genomic loci exhibit evidence of transcription from the antisense strand, as well as the sense strand [166-168]. These naturally occurring antisense transcripts may modulate the level of expression of their associated sense transcripts (or otherwise influence their processing), thereby adding another level of complexity to the regulation of gene expression [169,170]. Although there is, as yet, no suggestion that the genomic sources of such antisense transcripts should be regarded as genes in their own right, their prevalence clearly renders our task of defining the gene that much more difficult.
High copy number repeat sequences
The HGP revealed that repeat sequences account for at least 50 per cent of the human genome sequence. These repeats may be classified as (i) transposon-derived repeats, (ii) partially retroposed copies of genes (referred to as processed pseudo-genes), (iii) simple sequence repeats, (iv) blocks of tandemly repeated sequences at centromeres, telomeres and the short arms of acrocentric chromosomes and (v) segmental duplications (SDs) or low copy number repeats.
Segmental duplications
Both the number and the breadth of the distribution of SDs in the human genome (5 per cent) were surprising. SDs represent extensive inter- and intra-chromosomal duplications of genomic regions that contain genes as well as intergenic sequences [1,2]. She et al. extended the initial analyses of these low copy number repeats or SDs and initiated the characterisation of the duplicational landscape of the human genome [171]. SDs may be viewed as mutational hotspots, since they are prone to aberrant recombination events occurring between highly homologous paralogous SDs, and give rise to large deletions or duplications of the intervening sequences resulting in human genomic disorders [172]. Indeed, SDs have been shown to represent frequent sites of CNV between individuals, thereby contributing considerably to human genomic diversity [173]. The mechanism that generates CNVs in SDs is known as non-allelic homologous recombination [174]. These interspersed SDs confer susceptibility to recurrent microdeletions and microduplications upon approximately 10 per cent of the human genome through unequal crossing over. Furthermore, data have accumulated showing that specific recurrent rearrangements within these genomic hotspots are associated with both syndromic and non-syndromic diseases. Studies of common complex diseases have shown that these recurrent events play an important role in autism, schizophrenia and epilepsy [175-177].
The above notwithstanding, the duplicated genomic regions have remained largely intractable, owing to difficulties in accurately resolving their structure, copy number and sequence content. New algorithms have been developed to map comprehensively next-generation sequence reads, allowing the prediction of absolute CNVs of duplicated segments and genes. On average, 73-87 genes vary in copy number between any two individuals and these differences overwhelmingly correspond to segmental duplications [178].
Pseudogenes
Whether processed or non-processed (duplicational), it has become clear that pseudogenes are almost as abundant as genes ('classical' or otherwise) in the human genome, with ~20 per cent of known pseudogenes being transcribed [179-181]. By means of a comparison of cytochrome P450 genes (CYP) from the mouse and human genomes, Nelson et al. (2004) demonstrated that the complete identification of all human pseudogene sequences is likely to be clinically important and proposed a naming procedure for CYP pseudogenes [182].
It should, however, be appreciated that, although some pseudogenes may well be readily identifiable as lacking protein-coding potential by virtue of the interruption of their open-reading frames by premature stop codons or frameshift mutations, others will be less easily recognisable, especially if they are transcribed. The recent identification of short (≤ 300 bp) human pseudogenes generated via the retrotransposi-tion of mRNAs,[183] however, suggests that pseudo-genes may be even more common in the human genome than previously appreciated. Intriguingly, some of these pseudogenes are polymorphic, in that they have functional as well as non-functional alleles segregating in the extant human population [184].
With the realisation that pseudogene-derived RNA transcripts may harbour functional elements,[181,185] the distinction between genes and pseudogenes has become somewhat blurred [186]. Indeed, some 'pseudogenes' appear to have a regulatory role,[187,188] providing additional examples of the potential functional significance of non-coding RNAs. At present it is unclear what proportion of the pseudogenes identified to date have either retained or acquired a function via their non-coding RNAs.
Transposable elements
Transposable elements, including Long INterspersed Elements (LINE-1), Alu and SINE-VNTR-Alu (SVA) elements (SVA is an unusual composite element derived from three other repeats: Short INterspersed Elements [SINE]-R, variable number tandem repeats [VNTR] and Alu), make up ~40 per cent of the human genome [189] and constitute a major source of inter-individual structural variability [190]. Some of these transposable elements have contributed gene-coding sequences to the human genome via 'exonisation' [191]. Other transposable elements have contributed functional non-coding sequence -- for example, as regulatory elements,[192,193] microRNAs [194] or naturally occurring antisense transcripts [195]. Many more are likely to have functional significance, as suggested by their evolutionary conservation [196,197].
Evolutionary conservation
Extensive evolutionary conservation of non-coding DNA sequences is evident in the human genome because only ~40 per cent of the evolutionarily constrained sequence occurs within protein-coding exons or their associated untranslated regions [71]. Studies of evolutionarily conserved non-coding sequences [198-201] have suggested that 5-20 per cent of the genome may be of functional importance, rather than just the ~2 per cent associated with the protein-coding portion [202,203]. Some non-coding regions (the genomic 'dark matter') contain 'ultra-conserved elements' which not only exhibit enhancer function, but are also transcribed and often appear to have been subject to selection to the same extent as protein-coding regions [204-206]. Some non-coding regions contain CpG islands, which, although located far from the transcriptional initiation sites of genes, may nevertheless have some regulatory significance [207]. It should be appreciated, however, that the absence of evolutionary conservation does not necessarily denote lack of function. Indeed, human specific functional elements have been shown to be present within rapidly evolving non-coding sequences [208,209].
Towards a new definition of the gene
It is clear from the above that precisely what constitutes a gene has become somewhat contentious. The unanticipated scale of the extent of transcription in the genome, coupled with the widespread occurrence of overlapping genes and shared functional elements, hampers attempts to demarcate precisely and unambiguously where one gene ends and another one begins. As a consequence, the notion of the gene has become diffuse [161,210]. Indeed, as Kapranov et al.[211] opined, 'it is not unusual that a single base-pair can be part of an intricate network of multiple isoforms of overlapping sense and antisense transcripts, the majority of which are unannotated'. Gene regulatory elements that are often distant from the genes they regulate,[212] the existence of trans- as well as cis-regulatory elements [213] and the formation of non-co-linear transcripts through trans-splicing,[214] taken together with the abundance of non-coding RNA genes [215] and evolutionarily conserved non-coding regions,[199,201] have combined to challenge the classical notion of the gene.
On the basis of the findings of the ENCODE project, Gerstein et al.[210] proposed an updated definition of the gene as 'a union of genomic sequences encoding a coherent set of potentially overlapping functional products'. An alternative definition of the gene as: 'A discrete genomic region whose transcription is regulated by one or more promoters and distal regulatory elements and which contains the information for the synthesis of functional proteins or non-coding RNAs, related by the sharing of a portion of genetic information at the level of the ultimate products (proteins or RNAs)' has been proposed by Pesole [216]. Irrespective of its precise definition, it is clear that the concept of the gene is inadequate to the task of building a lexicon of those functional genomic sequences that could harbour mutations causing human inherited disease. It is likely in the context of mutation detection, that we shall eventually have to consider the universe of functional genetic elements in the human genome as our hunting ground, rather than simply genes per se.
Development of the GWAS approach to complex diseases and traits
In this section, developments in cataloguing genetic variation (SNP and CNV), initiation and completion of the International HapMap Project, and advances in genotyping technologies are discussed. These developments are important prerequisites for the use of GWASs in the investigation of complex diseases and traits.
SNP discovery after the HGP
While the HGP was being completed, genetic variants, in particular SNPs, were also being discovered. By 2001, the International SNP Map Working Group had identified 1.42 million SNPs in the human genome [58]. Currently, more than 17 million SNPs in the human genome have been catalogued in the SNP Database (dbSNP; http://www.ncbi.nlm.nih.gov/projects/SNP/). It is, however, likely that at least some of the entries in the database are errors or artefacts rather than 'genuine' variants. A false-positive rate for the dbSNP of 15-17 per cent has been estimated [101]. Therefore, large-scale validation in population-based studies is necessary. The HapMap Project was conceived in 2003 with the aim of validating several million SNPs in order to obtain SNP and genotype frequency information, as well as to study their LD patterns in different populations.
SNPs are the most abundant type of genetic variation in the human genome. They occur at intervals of approximately one SNP to every kb of DNA sequence throughout the genome when the DNA sequences of any two unrelated individuals are compared. This is approximately equivalent to three million SNPs being carried by each individual genome. Therefore, the DNA sequences of any two unrelated genomes are estimated to be about 99.9 per cent identical; the 0.1 per cent comprises mainly SNPs, and these are believed to be responsible for many of the phenotypic differences noted among individuals in populations -- for example, disease susceptibility, drug responses and physical traits such as height [217].
The discovery of thousands of CNVs that collectively encompass hundreds of Mb of the genome [22,23,105] and the several hundred thousand short indels identified by WGS studies,[42,43] however, have cast doubt upon the initial estimate of '99.9 per cent similarity' between any two genomes. Indeed, the DNA sequences of individuals within and between populations are genetically rather more diverse and varied than previously thought. This has been corroborated by a recent study demonstrating that the Craig Venter genome differs from the consensus reference sequence by approximately 1.2 per cent when indels and CNVs are considered, a further 0.1 per cent when SNPs are considered and ~0.3 per cent when inversions are considered -- a grand total of ~1.6 per cent [218].
Linkage disequilibrium and the International HapMap Project
Most SNPs are predicted to be neutral, without any functional effects. Owing to their abundance in the human genome, they may serve as useful genetic markers in GWASs, by comparison with other genetic variations, such as microsatellites, which in any case exhibit a mutation rate that is too high to be useful in this context. Early reports documented LD patterns between SNPs in parts of the human genome;[61,62,219] however, no large-scale effort had been undertaken to study the LD patterns in the whole genome until the initiation of the International HapMap Project. A total of more than three million SNPs were genotyped and validated in Phase I and Phase II of the project in four populations [66,69]. These populations were the US Utah population of Northern and Western European ancestry (CEU), Han Chinese from Beijing (CHB), Japanese from Tokyo (JPT) and the Yoruba from Ibadan, Nigeria (YRI).
One novel finding has been that 10-30 per cent of pairs of individuals within a population share at least one region of extended genetic identity arising from recent common ancestry. An additional discovery was that up to 1 per cent of all common variants are not tagged by SNPs, primarily because they are located within recombination hotspots [69]. Importantly, increased population differentiation with respect to non-synonymous SNPs was noted, by comparison with synonymous SNPs. These observations have also indicated systematic differences in the strength or efficacy of natural selection between populations from different geographical areas involving genes linked to the Lassa virus in West Africa, skin pigmentation in Europe and hair follicle development in Asia [70].
The discovery of millions of SNPs has created a significant challenge in genotyping. It is neither technically feasible nor cost-effective to genotype all the SNPs in a GWAS, even with the latest genotyping technologies; however, the existence of LD significantly reduces the number of SNPs that need to be genotyped. The indirect association approach of GWASs is dependent on surrogate markers ('tag' SNPs) to locate disease variants through LD. As shown by the HapMap Project [69] and other published work,[220-222] approximately half a million SNPs are adequate to capture most of the SNPs that have been genotyped in the HapMap Phase I and II projects. However, the genome coverage of commercial genotyping arrays is population dependent (Box 3).
The HapMap project has created a useful and valuable resource for GWASs. In parallel, the public availability of the HapMap resource has driven the rapid development of genotyping arrays, in which the data are used to guide the selection of tag SNPs. Once the HapMap Phase I and II projects were completed, a number of genotyping arrays were designed and introduced onto the market [223,224]. The newer arrays (eg the Illumina Human 1M Beadchip and Affymetrix SNP Array 6.0) have significantly improved genome coverage and are also designed for CNV detection [225]. The HapMap Phase I and II projects led to the development of higher resolution genotyping arrays, which in turn were used in the HapMap Phase III project to investigate genetic variations (both SNPs and CNVs) in additional populations of diverse ancestry [21].
The Phase III project, building on the success of the HapMap Phase I and II projects, included an additional seven populations and has recently been completed [21]. These additional populations involved people of African ancestry in the south-western USA (ASW), the Chinese community in Metropolitan Denver, CO (CHD), Gujarati Indians in Houston, TX (GIH), the Luhya in Webuye, Kenya (LWK), people of Mexican ancestry in Los Angeles, CA (MEX), the Maasai in Kinyawa, Kenya (MKK) and Tuscans in Italy (TSI). The ethos behind the HapMap Phase III project was that, in order to obtain a more complete understanding of human genetic variation, populations with a wider geographical/ancestral range needed to be studied. In total, the HapMap Phase III project genotyped approximately 1.6 million SNPs (using both the Illumina Human 1M Beadchip and Affymetrix SNP Array 6.0) in 1,184 individuals from 11 populations (four original and seven additional populations). The population-specific differences among low-frequency variants were characterised in addition to SNPs and common CNVs or copy number polymorphisms (CNPs). More importantly, it also demonstrated the feasibility of imputing newly discovered CNPs and SNPs, which are important for future GWASs and meta-analyses [21].
Whole-genome SNP genotyping technologies
The paradigm shift from candidate-gene association and family linkage studies to GWASs has been attributed to several important developments, most notably the rapid advances in high-throughput SNP genotyping technologies, which have enabled researchers to interrogate up to one million SNPs simultaneously in a microarray [18]. GWASs employ an 'agnostic' approach in the search for unknown disease variants, and hence the ability to interrogate a large number of SNPs covering the entire human genome is a prerequisite for this study design. In parallel with the decreasing cost of genotyping, it has recently become technically feasible to genotype thousands of samples in GWASs. As a result, more than 800 GWASs have been published since 2005 (http://www.genome.gov/gwastudies/), of which almost all have used the commercially available whole-genome SNP genotyping arrays from Illumina or Affymetrix.
A series of whole-genome genotyping arrays have been introduced since 2005, such as the Affymetrix Human Mapping 100K 500K sets, and the Illumina HumanHap300 and HumanHap550 BeadChips [223,224]. These genotyping arrays provide different degrees of genome coverage in different populations; lower coverage was achieved in African populations because of the greater genetic diversity in these populations. For example, the Illumina HumanHap550 Beadchip, which contains approximately 550,000 tag SNPs selected from the HapMap Phase I and II projects, achieved genome coverage of 87 per cent and 83 per cent in CEU and CHB + JTP populations, respectively, but only 50 per cent in YRI [220-222]. Whole-genome genotyping arrays such as the Illumina Human 1M Beadchip and Affymetrix SNP Array 6.0 offer almost complete genome coverage (.90 per cent) for HapMap CEU and CHB + JPT populations (Box 3).
The more recent genotyping arrays, such as the Illumina Human 1M BeadChip and Affymetrix SNP Array 6.0, have enabled genotyping of up to one million SNPs and increased the sensitivity to detect CNVs because of higher marker density and more uniform marker distribution [225]. For example, the Affymetrix SNP Array 6.0 contains more than 1.8 million markers, half of which are SNPs, the remainder being non-polymorphic or copy number probes to enhance the power of detection of CNVs. Copy-number probes were deliberately selected so as to cover regions lacking SNPs or regions where SNPs are difficult to assay, such as repetitive sequences within segmental duplications [226]. In addition, markers were also chosen to target known copy number variable regions as reported in the Database of Genomic Variants (http://projects.tcag.ca/variation/). Employing such a design, these genotyping arrays have enabled researchers to discover novel CNVs, as well as to validate previously known CNVs. These more recent arrays were designed for the application of GWASs and CNV detection.
The first wave of GWASs utilised first-generation SNP genotyping arrays and focused mainly on common SNPs with MAF > 5 per cent [132]. Thus, expanding the coverage to include less common or rarer SNPs (MAF 1-5 per cent) is essential for new discoveries to be made in future GWASs. This step is now technically feasible and practically achievable with the arrival of second-generation SNP genotyping arrays (Illumina HumanOmni2.5 and Omni5.0) in 2010; these are capable of genotyping 2.5 to 5.0 million SNPs (Illumina Whole-Genome Genotyping Product Roadmap; http://www.illumina.com/applications/gwas.ilmn). These arrays were designed to increase the coverage of SNPs down to a MAF of 1 per cent. In contrast to the first-generation arrays, the SNP selection in these latest genotyping arrays leverages the data from the 1000 Genomes Project [102]. However, the promise of second-generation genotyping arrays for new discoveries in GWASs is conditional upon the adequacy of the statistical power of the studies to identify the associations of rarer SNPs with complex traits. This suggests that larger sample sizes will be needed in future GWASs.
The era of GWASs
More than 4,000 SNPs have been reported to be associated with various human complex diseases and traits with varying degrees of replication and success (http://www.genome.gov/gwastudies/).
Despite some notable successes in revealing numerous novel SNPs and loci associated with complex phenotypes, the results from GWASs have been disappointing, in that all the GWAS-SNPs collectively account for only a small proportion of the heritability of complex phenotypes. This is due mainly to the small effect sizes of most GWAS-SNPs (odds ratio < 1.5) [5,10,89]. The small effect sizes of the GWAS-SNPs have also limited their applications in disease risk prediction [227].
Although several diseases have been claimed to be investigated by GWASs and meta-analyses of sufficiently large sample sizes, most of their heritability still remains unaccounted for. This missing heritability has stimulated much discussion on future strategies for detecting the remaining genetic variants associated with complex phenotypes. The proposed strategies range from increasing the sample sizes by combining several GWASs through meta-analysis in order to attain a higher statistical power, to more complicated experiments such as epigenetic studies [5,228]. The methodologies for meta-analysis and for the merging of SNP genotype data from multiple GWASs employing different genotyping arrays are now well developed and rely upon newly developed genotype imputation methods [229-231]. By contrast, there are still many experimental and analytical uncertainties and challenges to be faced in the context of epigenetic studies of complex phenotypes [232,233]. Other approaches are summarised in Figure 1.
Figure 1.
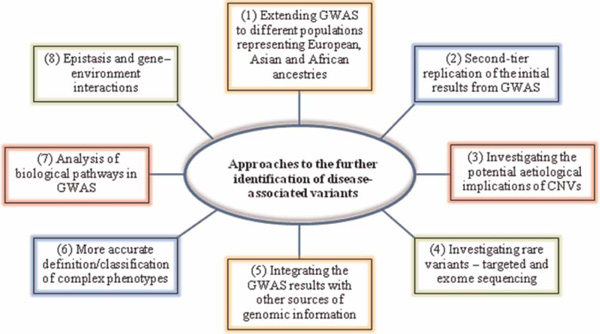
Summary of the approaches identifying disease-associated variants.
Figure 1 summarises a variety of approaches to the further identification of disease-associated variants: (1) GWASs of various complex diseases and traits ideally should be performed in different populations representing European, Asian and African ancestries, as most published studies have focused primarily on populations of European ancestry [234,235]. (2) Most GWASs have done fast-track replication by selecting the top few or top tens of SNPs with the most significant p-values in stage 1 and then proceeded to replicate them in stage 2 or stage 3 with larger sample sizes. Therefore, the next step should be to conduct a second tier of replication, where more SNPs from stage 1 are tested to assess their associations [236]. (3) The role of CNVs is increasingly recognised as being associated with complex diseases and traits; thus, it is important to investigate their associations with these complex phenotypes [111]. (4) Resequencing of the GWAS loci will be needed to uncover additional rarer variants. The success of this approach has been demonstrated in the discoveries of multiple rare variants for type 1 diabetes and hypertriglyceridaemia [237,238]. (5) Integrating GWAS results with other sources of genomic data, such as expression quantitative trait loci (eQTL) and ChIP-Seq, has led to the discovery of novel SNP associations [239,240]. (6) Subgroup analysis of disease phenotypes is a powerful approach to identifying genetic variants that are specific to certain subtypes. For instance, differences in SNP associations for oestrogen receptor-positive and -negative breast cancer have been shown [241]. (7) Pathway-based approaches have been developed using prior biological knowledge of gene function to facilitate more powerful analysis of GWAS datasets [242]. (8) Most studies have not taken epistasis and gene-environment interactions into account, which could account for a proportion of the missing heritability of complex phenotypes; however, challenges associated with studying these interactions should also be noted [243,244].
Genetic architecture of complex diseases
The genetic architecture of complex diseases has been the subject of intense debate over the past decade [59,60] and has been polarised by the emergence of two opposing models: the CD/CV hypothesis and the multiple rare variant or common-disease rare-variant (CD/RV) hypothesis [245]. The CD/CV model formed the basis of the HapMap Project and largely influenced the development of commercial genotyping arrays with respect to SNP selection. Therefore, the published GWAS using the HapMap data mainly involved the interrogation of the association of common SNPs (MAF > 5 per cent) with complex diseases and traits.
One of the reasons that the CD/CV model became favoured was because of the sequencing technologies available at that time. Sanger sequencing did not allow the survey of rare variants in the whole genome. By contrast, the convenient high-throughput genotyping platforms have enabled efficient interrogation of up to one million SNPs throughout the genome, which eventually indirectly leads to the capture of almost all the SNPs in the HapMap Project. Furthermore, it is more affordable to genotype (rather than to sequence) the entire genomes of several thousand cases and controls as part of an adequately powered association study.
Currently, the results from the GWASs focus on common SNPs and explain only a small fraction of the heritability of complex phenotypes [5]. The missing heritability has challenged the validity of the CD/CV hypothesis, and has also diverted research endeavours toward rare variants;[109,237,238,246,247] however, published data have revealed the contributions of both common and rare variants to complex phenotypes. The results from GWASs have strongly supported the involvement of common variants, especially common SNPs, in complex phenotypes [132]. Moreover, recent studies have shown that common SNPs can explain a greater proportion of the heritability than has been accounted for by recent GWASs. These SNPs, however, are often 'hidden' within the GWAS data, and will require larger sample sizes to be uncovered [248,249].
The data supporting the roles of rare variants have also been accumulating from an increasing number of studies of less-common SNPs [109,237,238,246] and rare CNVs [250-253]. This suggests that the genetic architecture of complex phenotypes is likely to comprise both common and rare variants. The relative proportions of these variants remain to be determined and will remain unclear until all the genetic variants for most complex phenotypes are found; furthermore, the relative proportions are likely to vary between different complex phenotypes, with some phenotypes having a greater influence on the genetic susceptibility risk by common variants, whereas other phenotypes may be more affected by rare variants. Being able to predict the genetic architecture of complex phenotypes is critical, however, as it will determine the future strategies to be adopted in seeking disease variants.
Homozygosity mapping
Homozygosity mapping has been shown to be useful in the identification of disease susceptibility genes in complex diseases [254,255]. An ROH defines an uninterrupted stretch of a DNA sequence lacking heterozygosity in the diploid state (ie in the presence of both copies of the homologous DNA segment). Thus, all the genetic variants within the homologous DNA segments are represented by two identical alleles that contribute to the homozygosity [28]. Currently, there are no standardised criteria to define an ROH. Previous studies have focused on regions ≥ 1 Mb, however, and hence the true extent of homozygosity in the human genome could have been underestimated because shorter regions were not considered [28,256,257]. More recent studies have defined ROHs as having a minimum length of 500 kb,[258] the intention being to avoid underestimation of the number of such regions in the human genome.
Although long continuous ROHs were first documented a decade ago, until recently no large-scale population-based studies had been performed to assess the extent of ROHs in the human genome [259]. The recent advances in the genome-wide detection and characterisation of ROHs have been driven mainly by the availability of highly accurate SNP databases such as the HapMap project [28] and advanced genotyping technologies [24,25]. Genotyping a large number of SNPs on a microarray platform presents a powerful tool for detecting ROHs comprehensively across the whole genome, thereby enabling investigation of the number, length, location and distribution of the ROHs in the human genome in a more unbiased manner, as compared with microsatellite markers. It was not previously expected that the genomes of outbred populations would contain ROHs of several Mb in length until the early reports appeared in 2006/2007 [28,256,257].
Many novel causal genes or mutations underlying autosomal recessive disorders have been identified through homozygosity mapping. This approach is particularly useful for investigating these disorders in populations with a high prevalence of consanguinity, as is evident from the many recent studies that have identified causal mutations [260-265].
The effects of consanguinity and recessive variants or heterozygosity levels on the risk of complex phenotypes (diseases and quantitative traits) are well established [266-268]. Higher levels of relative heterozygosity have been shown to be associated with lower blood pressure and total and low-density lipoprotein (LDL) cholesterol by measuring genome-wide heterozygosity [268]. In addition to quantitative traits, inbreeding has also been found to be a significant positive predictor for a number of late-onset complex diseases, such as coronary heart disease, stroke, cancer and asthma [266]. These studies have strongly supported the hypothesis that the genetics of complex phenotypes include a component which corresponds to recessively acting variants. The importance of ROHs to complex phenotypes remains largely unexplored; however, several studies have shown significant differences in ROHs between cases and controls in genome-wide investigations for schizophrenia [269] and late-onset Alzheimer's disease [270]. Success was also achieved for complex quantitative traits such as height, where strong statistical evidence for an association of a particular ROH with height was obtained in a total sample size of > 10,000. The height of individuals with this ROH was significantly higher (increased by 3.5 cm) than the individuals lacking the region [258]. Cataloguing ROHs in human genomes and investigating their associations with complex phenotypes by building on existing GWAS data should be fruitful areas for future research.
Beyond SNPs: CNVs
A new era of CNV discovery began when two separate studies, published concurrently in 2004, identified several hundred deletions and duplications in the human genome [26,27]. Such genetic abnormalities had actually been documented decades before, however, in clinical cytogenetics studies that found them to be a cause of various genomic or cytoge-netic disorders [271]. The distinguishing feature of the recent studies was that these CNVs were found to be much more prevalent in the human genome than previously expected. These changes in copy number did not result in any clinical disorder or pathological phenotype and were found in the genomes of phenotypically normal individuals. As these submicroscopic (< 5 Mb) deletions and duplications were below the detection limit of traditional cytogenetics tools such as fluorescence in situ hybridisation (FISH), these recent discoveries were credited to the use of whole-genome microarray technologies [272].
Although these early whole-genome microarray studies discovered several hundred new CNVs, it was clear from the outset that that this would be a gross underestimate of the true total. These studies used 'low-resolution' microarrays such as representational oligonucleotide microarray analysis (ROMA) containing 85,000 probes with a resolution of approximately one probe per 35 kb [26] or the bacterial artificial chromosome-comparative genomic hybridisation (BAC-CGH) array with a resolution of approximately one probe per 1 Mb [27]. Further, these studies investigated a small sample size, which limited the efficiency of detection of less common CNVs. CNVs smaller than 50-100 kb would not have been detected because their size was below the resolution limit for these microarrays. Thus, both the sample size and the resolution of the microarray are critical factors that contribute to the discovery of less common and/or smaller CNVs.
The contribution of CNVs as a major source of genetic variation in human populations has become appreciated despite the limitations of the microarrays. The first comprehensive mapping of CNVs in 270 samples from the HapMap Phase I project identified a total of 1,447 copy number variable regions, covering 360 Mb. These regions contained hundreds of genes, disease loci, functional elements and segmental duplications [22]. The limitations of ROMA and the BAC-CGH arrays have been overcome in later studies by the use of higher-resolution microarrays and larger sample sizes comprising several hundred samples [23,105,273-276]. High-resolution tiling oligonu-cleotide microarrays, comprising 42 million probes, were used to generate a comprehensive map of 11,700 CNVs [105]. Yim et al.[275] screened CNVs in 3,578 healthy, unrelated Korean individuals, using the Affymetrix SNP Array 5.0.
Other types of chromosomal rearrangement, particularly inversions and balanced translocations, have received considerably less attention [277-279]. Inversions and translocations are also known as 'copy-neutral variations' or 'balanced chromosomal rearrangements', since they do not involve changes in copy number. These copy-neutral variations have also been found to be associated with disease [279]. Collectively, these copy number and copy-neutral variations are broadly classified as 'structural variations'. As discussed, the genome-wide mapping and detection of CNVs in different populations has advanced considerably since 2004, being driven mainly by microarray technologies such as oligonucleotide-CGH and SNP microar-rays. By contrast, the pace in identifying inversions and translocations in the human genome has been slower because more powerful and effective methods were not available until the advent of NGS technologies [76] (Boxes 4 and 5).
The discovery of a 20 kb deletion located immediately upstream of the immunity-related GTPase family M gene (IRGM) underlying Crohn's disease, and the identification of a 45 kb deletion that is in perfect LD with body mass index-associated SNPs near the neuronal growth regulator 1 gene (NEGR1),[287,288] together with other studies reporting evidence for LD of CNVs with GWAS-SNPs at r2 > 0.5, suggest possible associations of CNVs with a variety of different human complex diseases and traits [105]. The genome-wide study performed by the Wellcome Trust Case Control Consortium (WTCCC) investigating the association between ~3,400 common CNVs and eight complex diseases in 19,000 samples did not yield any novel discoveries;[111] however, rare CNVs associated with various complex phenotypes have been identified in studies of schizophrenia,[250,289,290] epilepsy [251] and severe early-onset obesity [252,253]. The studies on schizophrenia found that rare structural variations that disrupt multiple genes in neurodevelopmental pathways are over-represented in cases, as compared with controls [250,289].
High-throughput sequencing technologies and their impact on genomic studies
The advent of high-throughput sequencing technologies has initiated the 'personal genome sequencing' era for both normal and cancer genomes, and large-scale genome sequencing studies such as the 1000 Genomes Project and the International Cancer Genome Consortium. The high-throughput sequencing technologies also provide new opportunities to study Mendelian disorders through exome sequencing and WGS. Several international projects have also been launched to explore functional genomics.
High-throughput sequencing technologies
NGS technologies have only been on the market since 2004, but have now largely replaced Sanger sequencing technologies (owing to the ultra-high-throughput production capacity of NGS technologies, which is a thousand times greater than that of traditional sequencing). One of the major differences is the ability of next-generation sequencers to simultaneously sequence millions of DNA fragments; hence, they are also referred to as massively parallel sequencing technologies. This feature has considerably increased the number of nucleotides that can be sequenced per instrument run when compared with Sanger sequencing. The sequencing chemistry of NGS technologies, together with their ultra-high-throughput production capacity, has also reduced sequencing costs significantly, making large-scale or WGS studies much more affordable [29-31]. The sequencing technologies currently available can be broadly grouped into NGS technologies such as the Roche 454 Genome Sequencer FLX (GS FLX) System, Illumina Genome Analyzer (GA) and HiSeq and Life Technologies Supported Oligonucleotide Ligation Detection System (SOLiD), and TGS (or single-molecule sequencing) technologies such as the HeliScope Single Molecule Sequencer (Helicos Biosciences) [32].
One of the more laborious steps in WGS using the Sanger method was the in vivo amplification step using bacterial cloning. This has now been substituted by the in vitro amplification of millions of DNA fragments by NGS technologies using emulsion polymerase chain reaction (PCR) (Roche GS FLX and Life Technologies SOLiD) or bridge amplification on a solid surface (Illumina GA and HiSeq). The sequencing approach for NGS technologies broadly can be divided into: (1) sequencing-by-synthesis mediated by DNA poly-merase (ie pyrosequencing for Roche GS FLX and sequencing by reversible terminator chemistry for the Illumina sequencing platform); and (2) sequencing-by-synthesis mediated by DNA ligase for Life Technologies SOLiD [29-31].
Whole-genome resequencing can now be accomplished relatively rapidly because of the availability of the HGP template for alignment of the billions of short sequence reads produced by next-generation sequencers. This is necessary because the NGS technologies are characterised by short sequence read lengths of approximately 50-125 bp for both Illumina and Life Technologies sequencing platforms [29-31]. This feature makes de novo sequencing, or the assembly of billions of short sequence reads into large contigs challenging -- especially for large and complex genomes like the human genome [291]. A longer read length is key to obtaining larger contigs with fewer gaps between them during the assembly steps. Although the latest improvements in sequencing chemistry and systems allow the Roche GS FLX to achieve a sequence read length of 500 bp on average, this is still markedly lower than the 800 bp to 1 kb length achieved by Sanger sequencing (http://www.454.com/) [292]. In addition to a short read length, NGS technologies have higher sequence error rates, although this gradually has been improving [293].
A relatively new addition in the NGS market is the Ion Torrent Personal Genome Machine (PGM) produced by Life Technologies (http://www.iontorrent.com/). The earlier NGS technologies relied on emission of either fluor-escent (Illumina and Life Technologies SOLiD sequencing platforms) or chemiluminescent (Roche GS FLX) light to detect and distinguish the nucleotides incorporated during sequencing. However, the Ion Torrent PGM uses proprietary semiconductor sensors to perform direct real-time measurement of the hydrogen ions released upon incorporation of nucleotides during sequencing. Several ion semiconductor sequencing chips will be available, with throughputs ranging from > 10 Mb to > 1 gigabase (Gb) per instrument run, but these are many-fold lower than the several hundred Gb of sequencing data generated by the latest Illumina HiSeq and Life Technologies SOLiD machines. The Ion Torrent PGM is therefore more suitable for smaller-scale targeted sequencing.
The first TGS instrument -- the Heliscope Single Molecule Sequencer -- is now commercially marketed by Helicos Biosciences. The Heliscope Single Molecule Sequencer or true single-molecule sequencing (tSMS) is vaguely classified as a TGS technology because it has features of both NGS and TGS technologies. It is considered to be a TGS platform because of its ability to perform single DNA molecule sequencing without the need for whole-genome amplification but the sequencing is still based on 'cyclic sequencing' (repeated cycles of sequencing) comprising several steps, such as flow of fluorescent-labelled nucleotides and reagents, nucleotides incorporation, washing and imaging steps, in each cycle [32]. Therefore, one of the major distinctions between NGS and TGS is that TGS does not require whole-genome amplification steps.
Numerous other TGS technologies, such as SMRT sequencing, are on the horizon and will soon be marketed commercially,[294] whereas others -- such as nanopore sequencing -- may take several years to become a mature technology [34,35]. SMRT sequencing is performed by synthesising complementary strands of the single DNA molecules by DNA polymerase through incorporation of four different fluorescent colourlabelled nucleotides. The incorporation of each nucleotide into the synthesising DNA strands is monitored in real time by visualisation of 'pulses' of coloured light emitted from each zero-mode waveguide. Each waveguide corresponds to a single molecule of DNA fragment and the incorporation of nucleotides is distinguished by emission of four different colours of light. Similarly, nanopore sequencing requires no cyclic sequencing steps [32]. By comparison, companies such as Complete Genomics (Mountain View, CA) provide a sequencing service, rather than selling their sequencing machines to end-users. The sequencing platform achieves efficient imaging and low reagent consumption with combinatorial probe anchor ligation chemistry independently to assay each base from patterned nanoarrays of self-assembling DNA nanoballs [44]. As TGS is characterised by single DNA molecule sequencing, it has the potential further to increase the number of sequence reads or throughput per instrument run above their current capacity.
Whole-genome (re)sequencing
NGS and TGS technologies have now made possible the sequencing of the entire human genome within a few days. The first human WGS study using a next-generation sequencer was completed in 2008;[46] this marked the beginning of a new era in personalised genome sequencing. To date, more than 20 WGS studies have been completed using NGS and TGS technologies [45]. The number of genomes being sequenced is expected to increase in the coming years, as sequencing technologies and analytical and bioinformatics tools become more advanced and affordable [295]. The reference genome sequence from the HGP is needed for alignments of the large amount of sequence reads produced by the high-throughput sequencers. Clearly, these studies do not involve the de novo assembly of human genome sequences, but rather constitute genome resequencing studies.
The first human diploid genome sequence -- Craig Venter's genome -- appeared in 2007 and was sequenced using the Sanger sequencing method [68]. A year later, the genome of James Watson, who discovered the double-helical structure of the DNA molecule half a century ago, was also sequenced [46]. In contrast to Venter's genome, Watson's genome was sequenced using NGS technologies. A number of additional human genomes have now also been fully sequenced. For example, a single Caucasian/European;[92] a single African (ie NA18507 from the HapMap project, sequenced using two different NGS technologies);[42,296] two Koreans;[297,298] a single Han Chinese;[43] a single Japanese;[299] a single Irish individual [300] and a single Gujarati Indian [301] have been sequenced.
Two whole genomes of the indigenous hunter-gatherer peoples of southern Africa (Khoisan and Bantu) have also been sequenced, together with the protein-coding regions from an additional three hunter-gatherers from the Kalahari. This study has been important for understanding human diversity, as these genomes represent the oldest known lineage of modern humans. A better understanding of genomic differences between the hunter-gatherers and others may help to pinpoint genetic adaptations to an agricultural lifestyle [302]. In addition, the genome of an extinct Palaeo-Eskimo (~4,000-years old)[108] and a Neanderthal genome [107] have been sequenced. The sequencing work of most of these individual genomes was accomplished using NGS technologies.
These WGS studies have identified several hundred thousand new SNPs that had not been previously catalogued in the dbSNP database. For example, Bentley et al. (2008)[42] found about one million new SNPs in the African genome (NA18507), and several hundred thousand new SNPs for other genomes. Most of the common SNPs in human populations have already been captured; thus, the new SNPs identified in these studies are probably representative of those from the lower-frequency spectrum. Data on population frequencies of the new SNPs are not available, since they were derived from individual genome-sequencing studies; however, these data should be available upon completion of the 1000 Genomes Project. In addition to SNPs, several hundred thousand short indels and several thousand structural variants have also been identified.
Schuster et al. characterised the extent of whole-genome and exome diversity among five individuals (two whole genomes and three exomes were sequenced) and identified 1.3 million novel DNA differences genome-wide [302]. Interestingly, in terms of nucleotide substitutions, the Bushmen would appear to be genetically more different from each other than Europeans and Asians are to each other. This is consistent with the view that the genetic diversity between African individuals is greater than between individuals from other ethnogeographic origins [302]. A total of 353,151 high-confidence SNPs were identified in the genome of the extinct Palaeo-Eskimo [108]. By comparing the high-confidence SNPs in this extinct human genome with contemporary populations to identify the populations most closely related to this individual, this study provided evidence for a migration from Siberia into the New World some 5,500 years ago. Comparisons of the Neanderthal genome with the genomes of five extant humans from different parts of the world identified a number of genomic regions that may have been affected by positive selection in ancestral modern humans, regions that include genes involved in metabolism and in cognitive and skeletal development [107].
The WGS studies also identified a portion of the sequence reads that could not be mapped to the NCBI human reference genome, indicating that some sequences are 'missing' from the reference genome. For example, Wheeler et al. found that 1.5 million reads (approximately 1.4 per cent of the total sequence data) did not map to the reference genome [46]. These 'unmappable' sequence reads were then assembled into ~170,000 contigs spanning 48 Mb. Even after the removal of contigs that were < 100 bp in size, there were still ~110,000 contigs spanning 29 Mb. This concurs with the estimated 25 Mb of euchromatic sequence that is absent from the reference genome. More recent studies using sequencing data have also identified new sequences that are absent in the human reference genome [303,304].
1000 Genomes Project
The 1000 Genomes Project was initiated in 2008 with the aim of sequencing the genomes of at least 1,000 individuals from different populations around the world (http://www.1000genomes.org/). The main aim of this international collaborative project has been to provide a comprehensive map of human genetic variation for future disease association studies and population genetics. As with the HapMap project, the data from this project also will be made available publicly.
Owing to the ease of high-throughput genotyping technologies, SNPs have been widely used as genetic markers in GWASs to search for disease variants. Evidence has been accumulating to suggest that (common) SNPs alone are unlikely to account for all the heritable risk of complex disease, however [5]. Concurrently, the amount of data supporting associations of CNVs with complex diseases has been growing [305]. Similarly, the importance of rare variants in complex diseases is also increasingly being recognised [306,307]. This indicates that future disease association studies need to interrogate non-SNP and rare genetic variants, requiring a comprehensive catalogue of human genetic variants. Common SNPs have been well documented in the dbSNP, but rarer (or lower frequency) SNPs are still under-represented in the database and information on indels and structural variations is still incomplete.
The completion of the pilot phase of the 1000 Genomes Project identified approximately 15 million SNPs, one million short indels and 20,000 structural variations, most of which were previously unreported [102]. In addition, the location, allele frequency and local haplotype structure of these genetic variants were described. The sequencing data also enabled characterisation of CNVs within heavily duplicated and near-identical regions [308]. Recently, a map of CNVs was constructed based on WGS data from 185 human genomes in the pilot phase of the project; this encompasses 22,025 deletions and 6,000 additional structural variations, including insertions and tandem duplications. More importantly, approximately half of the structural variations were mapped to single nucleotide resolution, thereby facilitating analysis of their origin and functional impact [112]. Precision in terms of the breakpoint delineation of structural variations is a prerequisite to obtain insights into their underlying mutational mechanisms [286]. The nucleotide resolution analysis of the breakpoints was hampered by the low resolution of the microarrays used in previous studies.
A recent study also identified approximately two million small indels, ranging from 1 bp to 10,000 bp in length, in the genomes of 79 humans. Interestingly, approximately half of these variants (ie 819,363 small indels) mapped to human genes. These small indels were frequently found in the coding exons of these genes, and several lines of evidence indicate that such variation is a major determinant of human biological diversity [309]. This study also found that many of the small indels had high levels of LD with both HapMap-SNPs and GWAS-SNPs, suggesting that a proportion of these indels have already been interrogated indirectly for their associations with complex phenotypes in GWASs through LD with the SNPs as surrogate markers. This also indicates that, in addition to SNPs and larger CNVs, small indel variation is likely to be a key factor underlying the genetics of human complex diseases and traits.
By comparison with WGS, which relies on a reference genome for aligning the sequence reads, de novo genome assembly will enable the more thorough and comprehensive detection of various genetic variations in the human genome ranging from single nucleotide variants and small indels, to large structural variations. Currently, de novo genome assembly is challenging and less practical because of the short sequence reads generated by NGS technologies, especially the Illumina and Life Technologies sequencing platforms. Recent studies have attempted to perform de novo human genome assembly using short sequence reads, with limited success [291,310,311]. One such study showed that de novo assemblies were 16.2 per cent shorter than the reference genome, with thousands of coding exons being completely absent [312]. De novo genome assembly and haplotype phasing will eventually become more feasible with longer sequence read lengths of up to tens of kb being generated by future sequencing technologies [33].
Cancer genome sequencing and somatic mutations
Cancers differ from other complex diseases in several aspects. The involvement of somatic mutations in cancer initiation and progression, in addition to germline variations, is well recognised. Sporadic cancer is considered to be an 'acquired disease' caused by the accumulation of somatic mutations in the genome of the original cancer cell type over the lifespan of a patient. Direct sequencing of the cancer genome, and comparison with the genome sequence from constitutional DNA from the same individual as a reference, is required for the proper assessment of somatic mutations [313,314].
Recent advances in the understanding of the somatic mutational profile of cancer genomes have been driven by NGS technologies, which have enabled numerous whole cancer genomes to be sequenced for the first time [47-49]. Nevertheless, many large-scale targeted resequencing studies of collections of cancer-relevant candidate genes, gene families or the RefSeq genes also have been performed previously using traditional PCR isolation and Sanger sequencing methods. The scale of these targeted studies previously has been limited by the lack of high-throughput sequence capture and sequencing methods [315,316]. By contrast, sequencing of the entire collection of exons in acute monocytic leukaemia was completed without PCR isolation and Sanger sequencing methods [317].
Although somatic mutations have been found in many genes, only a few genes have been found to be frequently mutated across the tumour samples screened (ie mutated in a significant proportion of cancer samples). These genes have been referred to as 'mountains' -- as opposed to the 'hills', which correspond to genes that are infrequently mutated or mutated at low frequency [316,318-320]. For example, the gene encoding V-erb-a erythroblastic leukaemia viral oncogene homolog 4 (avian) (ERBB4) was found to be the most highly mutated gene in melanoma and hence may be considered to be a 'mountain'; a considerable proportion of samples (19 per cent) were found to have somatic mutations in this gene, with some samples containing more than one mutation. The role of ERBB4 was also supported by extensive functional studies showing that various missense mutations increased kinase activity and transformation ability, and the demonstration of reduced cell growth after knockdown of the gene in melanoma cells expressing mutant ERBB4 [320]. Targeted cancer genome sequencing has demonstrated the potential to identify potential therapeutic targets for melanoma.
Despite cost constraints, the number of WGS studies performed on different cancers has been increasing [47-49] since the milestone first study that sequenced the cancer genome of an AML patient;[82] however, these WGS studies have generally sequenced only a few samples [321-323]. The ability of WGS to detect somatic mutations in abundance requires us to be able to identify the 'driver' mutations from among the myriad 'passenger' mutations. It has been predicted that approximately ten functional driver mutations are required to cause most cancers, yet up to tens of thousands of mutations may be identified in an analysis of a cancer genome [313,314]. Effective methods for identifying driver mutations in cancer genomes are not well developed, and the criteria for distinguishing driver mutations are not well defined. In addition, the set of driver mutations can be very different for different cancer types.
Although frequently mutated genes and recurrent mutations are of particular interest,[324] all of the current studies have interrogated only one or a few cancer genomes. Thus, these studies are unable to distinguish 'mountains' from 'hills', and recurrent mutations from other mutations that occur only once in the samples. Therefore, testing a subset of somatic mutations identified in the cancer genome in a larger number of cancer samples is required to identify this subset of genes or mutations [325] before the application of WGS in larger samples can be regarded as not only technically feasible, but also affordable (Box 6).
At present, somatic mutations in non-coding regions have received relatively scant attention and should be given more importance, since pervasive transcription beyond the protein-coding regions has now been demonstrated,[165,333,334] suggesting a regulatory role for the non-coding regions. These somatic mutations, and possibly driver mutations in the non-coding regions, can only be revealed by sequencing the whole cancer genome, as opposed to a targeted approach.
Revisiting Mendelian disorders
Mendelian or monogenic disorders make up approximately 7,000 known or suspected disorders and contribute significantly to the disease burden in society [335-338]. Over the past two decades, much progress has been made in identifying the causal mutations and candidate genes for Mendelian disorders through mainly traditional linkage studies [339]. Currently, causal mutations for > 4,000 Mendelian disorders have been identified [99]. Indeed, a total of 112,864 different disease-causing and disease-associated mutations in 4,078 human genes are currently (as of May 2011) catalogued in the HGMD (http://www.hgmd.org/) (Box 7).
Although classical linkage studies have been the main tool for elucidating the genetics of Mendelian disorders, not all of these disorders are amenable to this study design. Homozygosity mapping is a more powerful and effective approach to studying recessive disorders in consanguineous families. For those disorders that are not amenable to these two conventional approaches, their causal mutations remain elusive. These disorders include: (a) extremely rare Mendelian disorders where only a small number of cases are available; (b) unrelated cases from different families; and (c) sporadic cases due to de novo mutations. Exome sequencing now offers new opportunities to study extremely rare disorders and sporadic cases caused by de novo mutations, such as Kabuki syndrome and Schinzel-Giedion syndrome [14,340].
High-throughput sequence capture methods are able to isolate the universe of exons (the 'exome') in a more efficient and cost-effective way than traditional PCR-based methods. These methods are commercially marketed -- for example, the NimbleGen Sequence Capture technology (NimbleGen, Madison, WI: http://www.nimblegen. com/) and the Agilent SureSelect Target Enrichment technology (Agilent; Santa Clara, CA: http://www. home.agilent.com). They allow researchers to target custom genomic regions of interest in the human genome of up to tens of Mb in length, and also enable isolation of the exome in a single experiment. This development, coupled with the high-throughput sequencing data produced by NGS technologies, ensures an adequate depth of sequencing coverage accurately to detect the genetic variations in the exome or targeted regions [295,341,342].
Causal mutations have been identified for a number of previously unexplained rare disorders, such as Miller syndrome,[13] Sensenbrenner syndrome,[343] Perrault syndrome [344] and Fowler syndrome [345]. Exome sequencing is also a useful tool for diagnostic application and is anticipated to be used increasingly in molecular diagnosis [90,346-348]. The genetic diagnosis of congenital chloride diarrhoea in a patient with suspected Bartter syndrome was made through exome sequencing, which revealed a homozygous missense variant in the solute carrier family 26, member 3 gene (SLC26A3) [90]. The position of this variant is completely conserved from invertebrates to humans. The diagnostic application was further illustrated by Lupski et al. through WGS of a proband with Charcot-Marie-Tooth disease [16]. One missense variant and one nonsense variant were detected in SH3TC2, and all affected individuals in the family of the proband were found to be compound heterozygotes for these variants.
Studying Mendelian disorders can, paradoxically, reveal genes for complex diseases and traits. For example, numerous GWAS-identified common SNPs which are associated with triglyceride, high-density lipoprotein (HDL) cholesterol and LDL cholesterol levels were also found in the candidate genes causing the monogenic form of these lipid metabolism disorders [349,350]. The discovery of causal mutations in the disease genes responsible for Mendelian disorders should help in acquiring an understanding of the underlying pathophysiology. For example, the identification of causal mutations in the gene encoding dihydroorotate dehydrogenase (DHODH) for Miller syndrome has provided new insights into the role of pyrimidine metabolism in craniofacial and limb development [13]. The potential discovery of new drug targets through study of the genetics of Mendelian disorders should also be emphasised. Thus, statins, the most commonly used drugs to lower cholesterol levels by inhibiting the enzyme 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase, were discovered by studying familial hypercholesterolaemia [351].
Currently, the return to Mendelian disorder research has been mainly due to the 'attraction' of the exome sequencing approach, coupled with the disappointment engendered by GWAS results that have served to explain only a small fraction of the heritability of complex diseases and traits. Nevertheless, studying complex diseases should not be abandoned, as GWASs have also revealed new biological insights, such as unravelling the autophagy and interleukin (IL)-23 receptor pathways for Crohn's disease [352-354]. The knowledge gained from studying Mendelian disorders and complex diseases will eventually complement each other and come together synergistically to enhance our understanding of genotype-phenotype relationships.
New efforts to identify further causal mutations underlying Mendelian disorders include a recent initiative by the National Human Genome Research Institute (USA) to establish 'A Center for Mendelian Disorders' whose mission will be to take on the sequencing of Mendelian disorders. This centre will be expected to explain the molecular basis of 40-50 disorders per year (NHGRI Large-Scale Sequencing Program May 2010, http://www.genome.gov/).
Sequencing-based approaches to the study of functional genomics
The NGS technologies, since their introduction in 2004, have been increasingly applied in studies of protein-DNA interactions and histone modifications (ChIP-Seq), transcriptomic profiling of mRNAs and non-coding RNAs (RNA-Seq), and bisulphite sequencing of DNA methylation (Methyl-Seq) [39-41].
ChIP-Seq
Previous studies of protein-DNA interactions --such as the identification of transcription factor binding sites -- have relied on several low-throughput methods and have been focused on a few specific genomic regions. In the era of micro-arrays, the genome-wide studies of protein-DNA interactions and histone modifications were performed using a method known as ChIP-chip [355]. Undeniably, microarray development has enabled interrogation on a genome-wide scale but the detection of the immunoprecipitated DNA sequences is still dependent upon the availability of probes to capture them. Although the development of high-density tiling arrays,[356] where oligonucleotide probes are placed in high density throughout the whole genome, has improved the sensitivity of the ChIP-chip, the cost for such tiling arrays is expensive, especially for large genomes like the human genome [357]. By contrast, for ChIP-Seq, the immunoprecipitated DNA sequences are not hybridised on microarrays (thereby avoiding the problems inherent in probe hybridisation experiments) but instead are directly sequenced to detect their presence and measure their abundance. This allows detection of all the DNA fragments or sequences that are immunoprecipitated without any bias in relation to probe selection [357]. This is a key advantage of ChIP-Seq over microarrays.
ChIP-Seq or chromatin immunoprecipitation with the paired-end ditag sequencing (ChIP-PET) methods have led to major advances in the genome-wide mapping of binding sites for transcription factors (eg p53 transcription factor binding sites),[358] and for DNA binding proteins such as neurone restrictive silencer factor (NRSF) and signal transducer and activator of transcription (STAT1) [77,359]. Studies of histone modifications have also been revolutionised by means of ChIP-Seq methodology;[78] this has expanded our knowledge of how this epigenetic mechanism regulates gene expression in the human genome. ChIP-Seq has made an important contribution to the studies of protein-DNA interactions and histone modifications [360,361].
RNA-Seq
Studies of gene expression are important because they constitute immediate molecular traits that are directly affected by variation in DNA sequences and epigenetics. The term 'gene expression' usually refers to the expression of protein-coding genes. Previous studies were focused on mRNA expression, as mRNAs serve as the templates for protein synthesis; however, this perception changed after the completion of the pilot phase of the ENCODE project. This project and other studies revealed 'pervasive transcription' in the human genome [165,333,334]. Previously it had been thought that only the protein-coding regions or sequences (ie genes) would undergo transcription followed by translation; however, accumulating data are compatible with the view that transcription also occurs in non-protein-coding regions, indicating the importance of studying non-coding RNAs.
The advent of NGS technologies has spawned new approaches to exploring the transcriptome (eg RNA-Seq) [362,363]. This method allows the study of the expression of mRNAs and non-coding RNAs, and is also able to detect and identify new transcripts (coding and non-coding) that have not been formally annotated. The applications of sequencing-based approaches in transcriptomic studies have included genome annotation and the discovery of new transcripts,[364] the investigation of the alternative splicing patterns,[84,365] detection of gene fusions in cancer [366] and allele-specific expression analysis,[367] as well as the discovery and measurement of non-coding RNA expression.
Methyl-Seq
Substantial progress has also been achieved in the context of DNA methylation analysis with the advent of NGS technologies allowing the determination of the DNA methylome at a single-base resolution [96,368-371]. The 'gold standard' for the detection of DNA methylation (or cytosine methylation) is sodium bisulphite conversion of DNA followed by sequencing. The sodium bisulphite treatment will convert the unmethylated cytosine to uracil (subsequently read as thymine during sequencing), whereas methylated cytosine remains unchanged. One of the limitations of this method, however, is that it cannot distinguish between 5-methycytosine and 5-hydroxy-methylcytosine. The importance of studying 5-hydroxymethylcytosine for its biological roles will become clearer when more powerful methods to distinguish them become available [372]. The SMRT sequencer produced by Pacific Biosciences holds out great promise directly to sequence (and distinguish) 5-methycytosine and 5-hydroxymethylcytosine [373]. Nanopore sequencing technologies have also demonstrated the ability to directly detect methylated cytosines [35]. The revolution in sequencing approaches to exploring functional genomics in the human genome has also led to the initiation of several international projects (Box 8).
Personalised genomic medicine
The translation of genomic information to the clinical setting has shown great promise. In the field of pharmacogenetics, the US Food and Drug Administration (FDA) has approved genotyping tests for the screening of genetic variants in candidate genes that influence the responses and adverse effects of several commonly used anticancer drugs (eg the genes encoding thiopurine S-methyltransferase [TPMT] and UDP-glucuronosyltransferase 1A1 [UGT1A1] for thiopurine drugs and irrinotecan, respectively). Pharmacogenetic information is important to guide the optimal dose prescription [384]. Similarly, the FDA has also approved genotyping tests for two genes (CYP2C9 and the vitamin K epoxide reductase complex, subunit 1 gene [VKORC1]) in the prescription of warfarin, a drug of low therapeutic index [385].
The over-expression status of human epidermal growth factor receptor 2 (HER-2) receptors in breast cancer patients is clinically informative in deciding whether a given patient would benefit from trastuzumab treatment. Similarly, the deletion of CYP2D6 predicts whether a patient would benefit from tamoxifen treatment, as this prodrug requires bioactivation into its active metabolite, 4-hydroxytamoxifen, which is catalysed by the CYP2D6 enzyme. Thus, breast cancer patients who would not benefit from trastuzumab and tamoxifen treatments should be prescribed alternative drugs, such as aromatase inhibitors. In terms of prognosis, breast cancer prognostic gene expression arrays such as MammaPrint and Oncotype DX are informative and relevant to clinical management, as they help to determine which patients should receive adjuvant therapy after surgery [386-388]. These examples highlight the potential clinical utility of genomic information in prescribing and optimising treatments.
Genomics information has also been used to develop molecular-targeted cancer therapies. The discovery of the breakpoint cluster region-c-abl oncogene 1 nav-receptor tyrosine kinase (BCR-ABL) genomic translocation ultimately led to the development of a molecular-targeted drug as a treatment for chronic myeloid leukaemia (CML), namely imatinib -- a tyrosine kinase inhibitor targeting the tyrosine kinase domain of the fusion protein [389]. The identification of somatic mutations in the epidermal growth factor receptor (EGFR) in non-small-cell lung cancer led to the development of gefitinib. Further, somatic mutations in EGFR have also been found to be informative in predicting sensitivity to gefitinib and in explaining inter-ethnic variability in drug responses [390]. Advances in epigenetics have led to drug developments such as inhibitors of DNA methylation (DNMTs); indeed, 5-azacytidine and 5-aza-2'-deoxycytidine have been approved in the treatment of AMLs and mye-lodysplastic syndromes by the US FDA [391,392]. These show that genomic discoveries can be directly translated into clinical applications.
Given the advances in the field, more discoveries will eventually translate into clinical applications and management of patients. For example, GWASs have led to several promising discoveries, such as the identification of genetic variants in IL28B that influence the spontaneous clearance of hepatitis C virus and affect the individual response to chronic hepatitis C of interferon-α plus ribavirin therapy [393,394]. Similarly, cancer genome sequencing has identified promising somatic mutations in candidate genes (eg the isocitrate dehydrogenase 1 gene [IDH1]) as potential targets for drug interventions. Recurrent mutations in IDH1 have been found in 12 per cent of glioblastoma multiforme patients [318]. The importance of this gene is not confined to glio-blastoma multiforme, as mutations in IDH1 were also found in 16 per cent of AML patients [325].
In the era of GWASs and WGS, the great challenge lies in data interpretation and how genomic information can be used to discover new drugs or molecular biomarkers for clinical applications that will eventually translate into patient benefit. The ultimate goal of these studies is to improve the clinical management of patients and to bring about personalised medicine [395,396] through the development of new therapeutic agents tailored to the individual, based upon their genetic information. Although progress made towards achieving these goals has been promising, many challenges in the translational phase remain. Hence, it is still unclear how long it will take for personalised genomic medicine to become an everyday reality.
Summary
The analysis of the sequence of the human genome has had a major impact on biomedical research over the past few years. The HGP has made possible a multitude of genome-wide scale analyses and has thus provided a wealth of information about the architecture of the human genome. In many ways, the HGP has paved the way for what is coming to be called individualised or personalised genome medicine. The development of new (genotyping and sequencing) technologies for improved, less cost-intensive and more precise genome sequencing and assembly has been driven by the overwhelming success of the HGP.
In summary, the advances discussed in this review would not have been possible without the reference genome sequence produced a decade ago by the HGP. These advances have greatly improved our understanding of human genetic diversity, disease genetics and functional genomics. The development of powerful analytical and bioinformatics tools is crucially important in the era of genome sequencing (Box 9). The ongoing large-scale international projects will further contribute to the fields of human genetics, as well as human genomics, transcriptomics, epigenomics and metagenomics upon their completion. These projects will provide vital resources for future studies. Continued progress over the next ten years will bring us closer to the final goal of personalised genomic medicine.
Box 1. Gene deserts and their potential relevance to human inherited disease
A functional role(s) for gene deserts [127] has been supported by results from GWASs. Thus, multiple SNPs on chromosome 5p13.1 have been shown to be strongly associated with Crohn's disease, even though the region is located within a 1.2 Mb gene desert and the nearest annotated gene, that encoding prostaglandin E receptor EP4 (PTGER4), is about 270 kb away from the association signals [128-131]. Although the SNPs were consistently associated with the disease, their functional effect is not easy to infer because these SNPs could exert an effect either on the nearest gene or on other genes that are located further away. However, Libioulle et al. (2007)[128] integrated the GWAS results with gene expression data and found that the associated SNPs influenced the level of expression of PTGER4.
The majority of GWAS-SNPs are located in either intronic, intergenic or gene desert regions rather than within gene-coding or promoter sequences. These SNPs could nevertheless be of direct functional significance if their locations coincide with regulatory elements, either already known or yet to be characterised, such as enhancers, transcription factor binding sites and sequences encoding for microRNAs [132].
The association of the SNP rs6983267 at 8q24 with colorectal and prostate cancer has been a mystery since its discovery because the risk allele is located in a gene desert > 300 kb away from the nearest annotated gene, MYC. Recent studies have, however, found that the region containing the risk allele is a transcriptional enhancer that interacts with the MYC proto-oncogene [133,134]. In a similar vein, GWAS-SNPs in a 9p21-located gene desert (associated with coronary artery disease) have been found to impair the interferon-γ signalling response [135].
Box 2. MicroRNAs
MicroRNA has been the most intensively studied non-coding RNA in the human genome. MicroRNA gene loci may be fairly numerous: already more than 15,000 microRNA gene loci have been identified in various species (miRBase, Release 16.0: September 2010; http://www.mirbase.org/), with 1,048 microRNAs being found in the human genome.
Biogenesis and function
The synthesis of microRNAs starts with the transcription of primary microRNAs by RNA polymerase II. The primary microRNAs will be processed further to become precursor microRNAs and then mature microRNAs. The mature microRNAs are short sequences of 18-25 nucleotides; they are incorporated into RNA-induced silencing complex (RISC) to exert their post-transcriptional regulatory roles through binding to the 3' untranslated region (UTR) of target mRNAs. The binding of microRNA to target mRNAs can lead to two possible outcomes; either degradation or cleavage of the mRNAs or suppression of the translation of mRNAs into protein [149].
Relevance to diseases
The importance of microRNAs as functional regulators increasingly has been interrogated by microarray and sequencing studies. Deregulation in the expression patterns of microRNAs was commonly associated with various cancers [150-152]. SNPs in the (i) sequences encoding microRNAs and (ii) 3' UTR of mRNAs also have been found to be associated with various cancers [153,154].
Box 3. Genome coverage
High genome coverage is important, since the underlying principle of this approach is the use of LD to detect disease variants. In SNP-scarce regions, bona fide disease variants could be missed because they are not in strong LD with any of the SNPs genotyped on the array. Genome coverage is an estimate of the proportion of SNPs (using the International HapMap data as a reference) that can be captured by the SNPs which are directly genotyped in an array with a preset r2 threshold. Usually, a threshold of 0.8 is used to estimate genome coverage. These first-generation genotyping arrays used the International HapMap database for SNP selection and have poor coverage for SNPs with minor allele frequency (MAF) < 5 per cent [220-222].
Box 4. Characterising structural variation by means of sequencing
The discovery of copy-neutral variations has been attributed to the development of the PEM method and concurrent advances in NGS technologies. The PEM method has also contributed greatly to the discovery of CNVs in the human genome [76,81,280]. Further studies have also taken advantage of an important feature of sequencing data generated by NGS technologies, where several hundred million short sequence reads are produced per instrument run to detect CNVs based on the abundance or density of the sequence reads aligned to the reference genome. This approach is known as depth-of-coverage (DOC) and is similar to microarray-based methods, in that it is also unable to detect copy-neutral variations [281].
PEM
In the PEM method, a library of DNA fragments with a fixed insert size is prepared and both ends of the DNA fragments are sequenced to generate 'paired-end sequences' (the sequences at both ends of the DNA fragments). This sequence information is then aligned against the reference genome. The underlying principle of PEM in detecting structural variations is reliance upon the discordance in insert size and orientation of the paired-end sequences being aligned to the reference genome to infer 'simple' deletion, insertion and inversion. Thus, when paired-end sequences aligned to the reference sequence display discordance from the expected insert size or distance, this is indicative of either a deletion or an insertion, whereas discordance in orientation suggests the presence of an inversion (ie paired-end sequences are incorrectly oriented by comparison with the reference genome). Hence, the paired-end sequences are usually classified as 'concordant pairs' or 'discordant pairs'; only the discordant pairs are informative for inferring structural variants. Other, more complex, rearrangements -- such as 'everted duplications', 'linked insertions' and 'hanging insertions' -- can also be detected [282].
DOC
The DOC method utilises NGS data for CNV detection. This method is based on the DOC of the sequence reads to infer deletions and duplications. The DOC method is made possible by the production of several hundred million short sequence reads per instrument-run by NGS technologies. The principle underlying the DOC approach is based on the assumption that the sequencing process is uniform, so that the number of sequence reads mapping to a region follows a Poisson distribution. As such, the number of sequence reads should be proportional to the number of times that a particular region appears in the genome. Therefore, it is expected that a duplicated region will have more reads aligned with it, with the converse being true for deletions [281,282]. The assumption that the sequencing process is uniform may not be valid, however, because of the sequencing bias of the NGS technologies, which leads to certain regions of the genome being over- or under-sampled, resulting in spurious signals [283]. Despite their shortcomings, the PEM and DOC methods will continue to play a role in the discovery of structural variations until de novo genome assembly becomes more feasible.
Application in cancer studies
Both PEM and DOC have also proven useful in dissecting somatically acquired rearrangements in cancer genomes [87,284]. Sequencing of both ends of the DNA fragments derived from the genomes of two individuals with lung cancer was performed and 306 germline structural variations and 103 somatic rearrangements were identified to the single nucleotide level of resolution [87].
Box 5. International effort to characterise structural variants using PEM
Proof-of-concept studies
The PEM method for detecting structural variants was first demonstrated by Tuzun et al. by mapping paired-end sequences data from a human fosmid DNA genomic library [285]. The average insert size of a fosmid library is approximately 40 kb. This study identified 297 structural variants (139 insertions, 102 deletions and 56 inversions); however, sequencing of fosmid clones by means of Sanger sequencing is laborious and costly [285]. These limitations have been overcome by NGS technologies which directly sequence the paired-end or mate-pair libraries without the need for cloning steps [76]. Both of these studies applied the PEM approach to investigate structural variants in the same sample (NA15510) from the International HapMap Project. Their library insert sizes differed, however, and this has enabled a comparison of the sensitivity between these studies. Korbel et al.[76] were able to confirm 41 per cent of all deletion and inversion events detected by fosmid paired-end sequencing. Moreover, they identified an additional 407 structural variants in NA15510 that previously had not been detected by fosmid paired-end sequencing. This further suggests that several libraries with different insert sizes are needed to increase the sensitivity of PEM.
Human Genome Structural Variation Working Group
In addition to individual studies, a large-scale effort is currently being undertaken by the Human Genome Structural Variation Working Group comprehensively to map structural variants in phenotypically normal individuals using the PEM approach [79]. More specifically, the objective is to characterise the pattern of human structural variants at the nucleotide sequence level from a collection of 48 individuals of European, Asian and African ancestry. This project plans to make fosmid clone libraries of approximately 40 kb insert size from the genomic DNA of 48 unrelated females. These samples were already genotyped by the HapMap Project. A larger insert size of approximately 150 kb prepared from BAC clone libraries will also be constructed from 14 unrelated HapMap males. This will aim to provide sequence information on structural variants that are too large to be included in the fosmid libraries, such as those associated with segmental duplications. As such, both the fosmid and BAC libraries will ensure the comprehensive capture of structural variants of varying sizes across the human genome.
Structural variation is biased toward complex duplicated and repetitive regions. Hence, developing clone libraries for a modest number of human genomes should serve as a valuable resource for characterising complex and difficult-to-assay regions of genome structural variation. Since the underlying clones can be retrieved, the complete sequence context of the discovered structural variant can also be obtained [79]. This is crucial for precise breakpoint delineation of structural variation, which is then important for understanding the mutational mechanisms responsible for human genome structural variation. A total of 1,695 structural variants were discovered with fosmid libraries derived from nine individuals. The study also showed that 50 per cent were seen in more than one individual and that nearly half lay outside regions of the genome previously described as structurally variant, indicating novel discoveries. More importantly, 525 new insertion sequences (that are not present in the human reference genome) were discovered and many of these were found to be variable in copy number between individuals [86]. This is important because it suggests that structural variants or CNVs could have gone undetected as part of the 'missing sequences' in the human reference genome. Complete sequencing of 261 structural variants provided insights into the different mutational processes that have shaped the human genome. This study therefore provided the first high-resolution sequence map of human structural variation [86]. A subsequent study then expanded the Human Genome Structural Variation clone resource by including capillary end sequencing of 4.1 million additional fosmid clones from eight additional human genomes. The combined set includes 13.8 million clones derived from the genomes of six YRI, five Centre d'Etude du Polymorphisme Humain (CEPH) Europeans, three JPT, two CHB and one individual of unknown ancestry [286]. This study characterised the complete sequence of 1,054 large structural variants and analysed their breakpoint junctions to infer their potential mechanisms of origin. Three mechanisms were found to account for the bulk of germline structural variation: microhomology-mediated processes involving short (2-20 bp) stretches of sequence (28 per cent), non-allelic homologous recombination (22 per cent) and L1 retrotransposition (19 per cent).
Box 6. Challenges in cancer genome sequencing
Several major challenges at the forefront of cancer genome sequencing studies are outlined and discussed. The first relates to the collection of 'high-quality' samples of cancer cells or tissues for DNA extraction for sequencing [48,49]. Primary cancer tissues are usually contaminated by other normal cells that hamper our ability to detect somatic mutations in cancer genomes. The contamination with (or mixture of) DNA from non-cancerous cells is particularly problematic, and a higher depth of sequencing coverage will be required to detect somatic mutations in 'mixed DNA', increasing the cost of sequencing. For example, Ding et al. studied 188 primary lung adenocarcinoma samples, each containing a minimum of 70 per cent tumour cells independently determined by pathologists [326]. Single-cell sequencing is now emerging as a promising approach to resolving cancer tissue heterogeneity or mixed populations of cells, however, because it is potentially able to resolve genetic and/or cellular heterogeneity among the cancer cells. This single-cell sequencing approach was applied to investigate tumour population structure and evolution in two cases of human breast cancer. Analysis of 100 single cells from a polygenomic tumour revealed three distinct clonal subpopulations that probably represent sequential clonal expansions. Analysis of 100 single cells from a monogenomic primary tumour and its liver metastasis indicated that a single clonal expansion formed the primary tumour and seeded the metastasis [113].
The second most important challenge is accurately to identify different types of somatic mutations in the cancer genome. NGS technologies are characterised by shorter sequence read lengths and higher sequencing error rates, by comparison with Sanger sequencing [295]. Data quality could be adversely affected if these sequencing errors are not properly filtered out.
Thirdly, the cost of whole-genome resequencing is still prohibitively expensive when it is to be applied to hundreds of samples. Furthermore, there are also significant bioinformatics and analytical challenges to processing and analysing huge amounts of sequencing data. These two constraints currently restrict whole-genome resequencing studies to studies of only a few cancer genomes. This in itself becomes a major barrier to identifying recurrent mutations (which are more likely to be functionally important) and driver mutations. Although the current approach to identifying recurrent mutations is to select a subset of somatic mutations detected in cancer genomes and then to test them in a larger study,[325] this approach cannot be used to screen for all mutations, resulting in many recurrent mutations remaining undetected. For example, a total of 64 mutations were detected in protein-coding genes, regulatory RNAs and highly conserved non-coding regions in the AML genome, but only four of these mutations were subsequently found in additional samples when tested for in more than 180 AML patients. By contrast, targeted resequencing in large sample sizes is able to identify recurrent mutations. This targeted approach focuses only on certain genes, however, and, as a consequence, those recurrent mutations located outside the targeted regions remain undetected. In addition to identifying recurrent mutations, a large sample size is also needed to distinguish 'mountains' from 'hills'.
Although the findings from targeted,[315,316,320,326,327] exome [317,328,329] and whole-genome resequen-cing [82,321-323,330-332] studies have increasingly provided new insights into cancer genomes, the greatest challenge for cancer genome sequencing lies in discerning driver mutations from the multitude of other (passenger) mutations. Effective methods for identifying driver mutations in cancer genomes are not yet well developed. In addition, driver mutations may differ between cancer types.
Box 7. Human Gene Mutation Database and the 'human mutome'
As the number of disease-causing or disease-associated germline mutations or variants increases, proper cataloguing is critically important. In this regard, the HGMD represents an attempt to collate all known (published) gene lesions responsible for human inherited disease.
Disease-causing or disease-associated germline mutations/variants collated in the HGMD now exceed 110,000 in > 4,000 different nuclear genes. Newly described human gene mutations are currently being reported at a rate of ~10,000 per annum, with ~300 new 'inherited disease genes' being recognised every year. The HGMD has provided useful insights into the 'human mutome' (ie disease-causing or disease-associated germline mutations/variants in the entire human genome) [99,100]. For a variety of reasons, however, this figure is likely to represent only a small proportion of the clinically relevant genetic variants present in the human genome. Those disease-causing or disease-associated variants that are located outside the gene-coding regions are likely to have been overlooked often as a direct consequence either of focusing exclusively on screening the protein-coding sequence or of the inherent limitations of the mutation detection techniques used. Such considerations are important for improving mutation screening strategies, as well as for facilitating the interpretation of findings from GWASs, exome sequencing and WGS.
Box 8. International projects that are exploring functional genomics
The advent of NGS and TGS will facilitate the undertaking of several international projects (http://commonfund.nih.gov/). These large-scale projects would not have been technically feasible without NGS and TGS technologies, which have potentiated sequencing-based approaches in studying functional genomics. These projects will contribute significantly to functional genomics.
The NIH Roadmap Epigenomics Program
The NIH Roadmap Epigenomics Program aims to generate new research tools, technologies, datasets and infrastructure to accelerate our understanding of the role of epigenetics [374]. This will improve our understanding of instances of transcriptional regulation that are not dependent on the DNA sequence. This will be important in understanding diseases attributed to epigenetic aberrations involving DNA methylation or histone modifications [375]. For example, many cancers are commonly associated with epigenetic aberrations [376].
The Genotype-Tissue Expression (GTeX) Project
Transcriptional regulation is modulated not only by epigenetics, but also by genetic variation in the DNA sequence. Therefore, the GTeX Project aims to study human gene expression and regulation in multiple tissues, providing valuable insights into the mechanisms of gene regulation and, in the future, its disease-relevant aberrations. Genetic variation between individuals will be examined for a correlation with differences in gene expression level. Major advances have been made in studies of eQTL through the use of high-throughput genotyping and sequencing technologies [377-381]. For example, Montgomery et al. sequenced the mRNA fraction of the transcriptome in 60 HapMap individuals of European descent and integrated the data with SNP information from the HapMap Phase III project, an undertaking which led to discoveries of novel eQTLs and sequence variants responsible for alternative splicing [380].
The Human Microbiome Project
The Human Microbiome Project aims to characterise the microbial communities found at several different sites in the human body, such as oral cavities, skin, gastrointestinal tract and the urogenital tract. This project is important in providing insights into the roles of these microbes in human health and disease [382]. The first metagenomic sequencing of gut microbes was accomplished using NGS technologies [103]. A human gut microbial gene catalogue was established by characterisation of 3.3 million non-redundant microbial genes derived from faecal samples from 124 European individuals. This research is important in gaining better understanding of the influence of gut microbes on human health and disease [103].
The International Cancer Genome Consortium
New developments have also occurred in cancer genomics, where the International Cancer Genome Consortium aims to obtain a comprehensive description of genomic, transcriptomic and epigenomic changes in 50 different tumour types and subtypes [110]. This is in accordance with the notion of integrative analyses incorporating multiple sources of genomics data [383]. This project will be important in dissecting the somatic genetic heterogeneity, a general hallmark of cancer, through studying various tumour types and subtypes.
Box 9. Bioinformatics -- computational and analytical tools -- in the NGS era
Bioinformatics -- and computational and analytical tools -- play a key role in the NGS era, an era in which huge amounts of sequencing data are being generated. Parallel developments in bioinformatics tools have contributed greatly to recent advances in the field of human structural and functional genomics where NGS technologies have been applied. A detailed discussion of the development of these analytical tools and methodological pipelines is beyond the scope of this paper. However, bioinformatics, computational and analytical tools have been developed for a variety of applications at different stages of the analysis of data generated by both structural and functional genomics studies. Exemplars are given below.
Base calling, alignment, mapping and assembly
1. Base-calling for NGS platforms [397].
2. Survey of sequence alignment algorithms for NGS [398].
3. Evaluation of NGS software in mapping and assembly [399].
4. De novo assembly of short sequence reads [291].
5. Assembly algorithms for NGS data [400].
Structural genomics (discovery of genetic variations)
6. Computational methods for discovering structural variation with NGS [282].
7. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes [401].
8. A framework for variation discovery and genotyping using NGS DNA data [402].
Functional genomics
9. Introduction to the analysis of high-throughput-sequencing based epigenome data [403].
10. Computation for ChIP-seq and RNA-seq studies [404].
11. Bioinformatics approaches for genomics and post-genomics applications of NGS [405].
Association studies
12. Association studies for NGS [406].
References
- Lander ES, Linton LM, Birren B, Nusbaum C. et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW. et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY. et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi E. Breakthrough of the year. Human genetic variation. Science. 2007;318:1842–1843. doi: 10.1126/science.318.5858.1842. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB. et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AJ, Cooper DN. GWAS: Heritability missing in action? Eur J Hum Genet. 2010;18:859–861. doi: 10.1038/ejhg.2010.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly P. Progress and challenges in genome-wide association studies in humans. Nature. 2008;456:728–731. doi: 10.1038/nature07631. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS. The HapMap and genome-wide association studies in diagnosis and therapy. Annu Rev Med. 2009;60:443–456. doi: 10.1146/annurev.med.60.061907.093117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feero WG, Guttmacher AE, Collins FS. Genomic medicine -- An updated primer. N Engl J Med. 2010;362:2001–2011. doi: 10.1056/NEJMra0907175. [DOI] [PubMed] [Google Scholar]
- Ng SB, Turner EH, Robertson PD, Flygare SD. et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW. et al. Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC. et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios J, Stein E, Shendure J, Hobbs HH. et al. Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia. Hum Mol Genet. 2010;19:4313–4318. doi: 10.1093/hmg/ddq352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D. et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy GC, Matsuzaki H, Dong S, Liu WM. et al. Large-scale genotyping of complex DNA. Nat Biotechnol. 2003;21:1233–1237. doi: 10.1038/nbt869. [DOI] [PubMed] [Google Scholar]
- Ragoussis J. Genotyping technologies for genetic research. Annu Rev Genomics Hum Genet. 2009;10:117–133. doi: 10.1146/annurev-genom-082908-150116. [DOI] [PubMed] [Google Scholar]
- Jakobsson M, Scholz SW, Scheet P, Gibbs JR. et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- Abdulla MA, Ahmed I, Assawamakin A, Bhak J. et al. Mapping human genetic diversity in Asia. Science. 2009;326:1541–1545. doi: 10.1126/science.1177074. [DOI] [PubMed] [Google Scholar]
- Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E. et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L. et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S. et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS. et al. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothnagel M, Lu TT, Kayser M, Krawczak M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Hum Mol Genet. 2010;19:2927–2935. doi: 10.1093/hmg/ddq198. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J. et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML. et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Gibson J, Morton NE, Collins A. Extended tracts of homozygosity in outbred human populations. Hum Mol Genet. 2006;15:789–795. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies -- The next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Schadt EE, Turner S, Kasarskis A. A window into third-generation sequencing. Hum Mol Genet. 2010;19:R227–R240. doi: 10.1093/hmg/ddq416. [DOI] [PubMed] [Google Scholar]
- Mardis ER. A decade's perspective on DNA sequencing technology. Nature. 2011;470:198–203. doi: 10.1038/nature09796. [DOI] [PubMed] [Google Scholar]
- Branton D, Deamer DW, Marziali A, Bayley H. et al. The potential and challenges of nanopore sequencing. Nat Biotechnol. 2008;26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J, Wu HC, Jayasinghe L, Patel A. et al. Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol. 2009;4:265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- Derrington IM, Butler TZ, Collins MD, Manrao E. et al. Nanopore DNA sequencing with MspA. Proc Natl Acad Sci USA. 2010;107:16060–16065. doi: 10.1073/pnas.1001831107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treffer R, Deckert V. Recent advances in single-molecule sequencing. Curr Opin Biotechnol. 2010;21:4–11. doi: 10.1016/j.copbio.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255–264. doi: 10.1016/j.ygeno.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Werner T. Next generation sequencing in functional genomics. Brief Bioinform. 2010;11:499–511. doi: 10.1093/bib/bbq018. [DOI] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang W, Li R, Li Y. et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL. et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- Venter JC. Multiple personal genomes await. Nature. 2010;464:676–677. doi: 10.1038/464676a. [DOI] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y. et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- Mardis ER, Wilson RK. Cancer genome sequencing: A review. Hum Mol Genet. 2009;18:R163–R168. doi: 10.1093/hmg/ddp396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- Robison K. Application of second-generation sequencing to cancer genomics. Brief Bioinform. 2010;11:524–534. doi: 10.1093/bib/bbq013. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Flavell RA. The rabbit beta-globin gene contains a large large insert in the coding sequence. Cell. 1977;12:1097–1108. doi: 10.1016/0092-8674(77)90172-6. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Alter BP, Altay C, Mahoney MJ. et al. Application of endonuclease mapping to the analysis and prenatal diagnosis of thalassemias caused by globin-gene deletion. N Engl J Med. 1978;299:166–172. doi: 10.1056/NEJM197807272990403. [DOI] [PubMed] [Google Scholar]
- Chang JC, Kan YW. Beta 0 thalassemia, a nonsense mutation in man. Proc Natl Acad Sci USA. 1979;76:2886–2889. doi: 10.1073/pnas.76.6.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D, White RL, Skolnick M, Davis RW. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet. 1980;32:314–331. [PMC free article] [PubMed] [Google Scholar]
- Watson JD. The human genome project: Past, present, and future. Science. 1990;248:44–49. doi: 10.1126/science.2181665. [DOI] [PubMed] [Google Scholar]
- Weissenbach J, Gyapay G, Dib C, Vignal A. et al. A second-generation linkage map of the human genome. Nature. 1992;359:794–801. doi: 10.1038/359794a0. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Ball EV, Krawczak M. The human gene mutation database. Nucleic Acids Res. 1998;26:285–287. doi: 10.1093/nar/26.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM. et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ. et al. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S, Ireland J. et al. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Luttig CT, Larkins CE, Beauchamp A. et al. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L. et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR, Hinds DA. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Varilly P, Fry B, Lohmueller J. et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R. et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges E, Xuan Z, Balija V, Kramer M. et al. Genome-wide in situ exon capture for selective resequencing. Nat Genet. 2007;39:1522–1527. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- Okou DT, Steinberg KM, Middle C, Cutler DJ. et al. Microarray-based genomic selection for high-throughput resequencing. Nat Methods. 2007;4:907–909. doi: 10.1038/nmeth1109. [DOI] [PubMed] [Google Scholar]
- Albert TJ, Molla MN, Muzny DM, Nazareth L. et al. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4:903–905. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- Kapranov P, Cheng J, Dike S, Nix DA. et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B. et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY. et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Eichler EE, Nickerson DA, Altshuler D, Bowcock AM. et al. Completing the map of human genetic variation. Nature. 2007;447:161–165. doi: 10.1038/447161a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst M, Marra MA. Next generation sequencing based approaches to epigenomics. Brief Funct Genomics. 2010;9:455–465. doi: 10.1093/bfgp/elq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi R, Kim TM, Park PJ. Detecting structural variations in the human genome using next generation sequencing. Brief Funct Genomics. 2010;9:405–415. doi: 10.1093/bfgp/elq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Mardis ER, Ding L, Fulton B. et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Hayden EC. Genomics sizes up. Nature. 2008;451:234. doi: 10.1038/451234a. [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ. et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Sultan M, Schulz MH, Richard H, Magen A. et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS. et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S. et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, Scott LJ, Saxena R, Voight BF. et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Ji W, Liu T. et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers J, Mitchell J, Beer E, Buzby PR. et al. Virtual terminator nucleotides for next-generation DNA sequencing. Nat Methods. 2009;6:593–595. doi: 10.1038/nmeth.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushkarev D, Neff NF, Quake SR. Single-molecule sequencing of an individual human genome. Nat Biotechnol. 2009;27:847–850. doi: 10.1038/nbt.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M. et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Platt AR, Jones DR, Reifenberger JG. et al. Direct RNA sequencing. Nature. 2009;461:814–818. doi: 10.1038/nature08390. [DOI] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH, Pan YF, Liu J. et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DN, Chen JM, Ball EV, Howells K. et al. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Hum Mutat. 2010;31:631–655. doi: 10.1002/humu.21260. [DOI] [PubMed] [Google Scholar]
- Chen JM, Ferec C, Cooper DN. Revealing the human mutome. Clin Genet. 2010;78:310–320. doi: 10.1111/j.1399-0004.2010.01474.x. [DOI] [PubMed] [Google Scholar]
- Day IN. dbSNP in the detail and copy number complexities. Hum Mutat. 2010;31:2–4. doi: 10.1002/humu.21149. [DOI] [PubMed] [Google Scholar]
- Durbin RM, Abecasis GR, Altshuler DL, Auton A. et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes EK, Willer CJ, Berndt SI, Monda KL. et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L. et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelak K, Shianna KV, Ge D, Maia JM. et al. The characterization of twenty sequenced human genomes. PLoS Genet. 2010;6:e1001111. doi: 10.1371/journal.pgen.1001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T. et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M, Li Y, Lindgreen S, Pedersen JS. et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature. 2010;463:757–762. doi: 10.1038/nature08835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E. et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- Hudson TJ, Anderson W, Artez A, Barker AD. et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, Hurles ME, Cardin N, Pearson RD. et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE. et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P. et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard TJ, Aken BL, Beal K, Ballester B. et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Hsu F, Karolchik D, Kuhn RM. et al. Exploring relationships and mining data with the UCSC Gene Sorter. Genome Res. 2005;15:737–741. doi: 10.1101/gr.3694705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Barrett T, Benson DA, Bryant SH. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–D21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S. Annotating noncoding RNA genes. Annu Rev Genomics Hum Genet. 2007;8:279–298. doi: 10.1146/annurev.genom.8.080706.092419. [DOI] [PubMed] [Google Scholar]
- Clamp M, Fry B, Kamal M, Xie X. et al. Distinguishing protein-coding and noncoding genes in the human genome. Proc Natl Acad Sci USA. 2007;104:19428–19433. doi: 10.1073/pnas.0709013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Harrow J, Harte RA, Wallin C. et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog H, Darby K, Hort YJ, Shine J. Intron 17 of the human retinoblastoma susceptibility gene encodes an actively transcribed G protein-coupled receptor gene. Genome Res. 1996;6:858–861. doi: 10.1101/gr.6.9.858. [DOI] [PubMed] [Google Scholar]
- Vuoristo JT, Berrettini WH, Ala-Kokko L. C18orf2, a novel, highly conserved intronless gene within intron 5 of the GNAL gene on chromosome 18p11. Cytogenet Cell Genet. 2001;93:19–22. doi: 10.1159/000056940. [DOI] [PubMed] [Google Scholar]
- Yu P, Ma D, Xu M. Nested genes in the human genome. Genomics. 2005;86:414–422. doi: 10.1016/j.ygeno.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Denoeud F, Kapranov P, Ucla C, Frankish A. et al. Prominent use of distal 5' transcription start sites and discovery of a large number of additional exons in ENCODE regions. Genome Res. 2007;17:746–759. doi: 10.1101/gr.5660607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven H, Rawson RB, Merkx GF, Cremers FP. et al. cDNA cloning and chromosomal localization of the genes encoding the alpha- and beta-subunits of human Rab geranylgeranyl transferase: The 3' end of the alpha-subunit gene overlaps with the transglutaminase 1 gene promoter. Genomics. 1996;38:133–140. doi: 10.1006/geno.1996.0608. [DOI] [PubMed] [Google Scholar]
- Yang MQ, Elnitski LL. Diversity of core promoter elements comprising human bidirectional promoters. BMC Genomics. 2008;9(Suppl 2):S3. doi: 10.1186/1471-2164-9-S2-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovcharenko I, Loots GG, Nobrega MA, Hardison RC. et al. Evolution and functional classification of vertebrate gene deserts. Genome Res. 2005;15:137–145. doi: 10.1101/gr.3015505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J. Clues to function in gene deserts. Trends Biotechnol. 2005;23:269–271. doi: 10.1016/j.tibtech.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Libioulle C, Louis E, Hansoul S, Sandor C. et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes M, Barrett JC, Prescott NJ, Tremelling M. et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Hansoul S, Nicolae DL, Cho JH. et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A, McGovern DP, Barrett JC, Wang K. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM. et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Ahmadiyeh N, Jia L, Herman P. et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. 2009;41:882–884. doi: 10.1038/ng.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuupanen S, Turunen M, Lehtonen R, Hallikas O. et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat Genet. 2009;41:885–890. doi: 10.1038/ng.406. [DOI] [PubMed] [Google Scholar]
- Harismendy O, Notani D, Song X, Rahim NG. et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters BA, St Croix B, Sjoblom T, Cummins JM. et al. Large-scale identification of novel transcripts in the human genome. Genome Res. 2007;17:287–292. doi: 10.1101/gr.5486607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louro R, Smirnova AS, Verjovski-Almeida S. Long intronic noncoding RNA transcription: Expression noise or expression choice? Genomics. 2009;93:291–298. doi: 10.1016/j.ygeno.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Ponjavic J, Ponting CP, Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007;17:556–565. doi: 10.1101/gr.6036807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel C, Gagnebin M, Gehrig C, Kriventseva EV. et al. Mapping of small RNAs in the human ENCODE regions. Am J Hum Genet. 2008;82:971–981. doi: 10.1016/j.ajhg.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affymetrix ENCODE Transcriptome Project; Cold Spring Harbor Laboratory ENCODE Transcriptome Project. Post-transcriptional processing generates a diversity of 5'-modified long and short RNAs. Nature. 2009;457:1028–1032. doi: 10.1038/nature07759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Hofacker IL, Lukasser M, Huttenhofer A. et al. Mapping of conserved RNA secondary structures predicts thousands of functional noncoding RNAs in the human genome. Nat Biotechnol. 2005;23:1383–1390. doi: 10.1038/nbt1144. [DOI] [PubMed] [Google Scholar]
- Collins LJ, Penny D. The RNA infrastructure: dark matter of the eukaryotic cell? Trends Genet. 2009;25:120–128. doi: 10.1016/j.tig.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Kawaji H, Hayashizaki Y. Exploration of small RNAs. PLoS Genet. 2008;4:e22. doi: 10.1371/journal.pgen.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS. The genetic signatures of noncoding RNAs. PLoS Genet. 2009;5:e1000459. doi: 10.1371/journal.pgen.1000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- Preker P, Nielsen J, Kammler S, Lykke-Andersen S. et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–1854. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- Seila AC, Calabrese JM, Levine SS, Yeo GW. et al. Divergent transcription from active promoters. Science. 2008;322:1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft RJ, Glazov EA, Cloonan N, Simons C. et al. Tiny RNAs associated with transcription start sites in animals. Nat Genet. 2009;41:572–578. doi: 10.1038/ng.312. [DOI] [PubMed] [Google Scholar]
- He L, Hannon GJ. MicroRNAs: Small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- Nevins JR, Potti A. Mining gene expression profiles: Expression signatures as cancer phenotypes. Nat Rev Genet. 2007;8:601–609. doi: 10.1038/nrg2137. [DOI] [PubMed] [Google Scholar]
- Farazi TA, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. J Pathol. 2011;223:102–115. doi: 10.1002/path.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoloso MS, Sun H, Spizzo R, Kim H. et al. Single-nucleotide polymorphisms inside microRNA target sites influence tumor susceptibility. Cancer Res. 2010;70:2789–2798. doi: 10.1158/0008-5472.CAN-09-3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: The implications for cancer research. Nat Rev Cancer. 2010;10:389–402. doi: 10.1038/nrc2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M, Guttman M, Feldser D, Garber M. et al. A large intergenic noncoding RNA induced by pp53 mediates global gene repression in the pp53 response. Cell. 2010;142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewer S, Cabili MN, Guttman M, Loh YH. et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet. 2010;42:1113–1117. doi: 10.1038/ng.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M, Rinn JL. Large non-coding RNAs: Missing links in cancer? Hum Mol Genet. 2010;19:R152–R161. doi: 10.1093/hmg/ddq353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orom UA, Shiekhattar R. Long non-coding RNAs and enhancers. Curr Opin Genet Dev. 2011;21:194–198. doi: 10.1016/j.gde.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15(Spec. No. 1):R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- Taft RJ, Pang KC, Mercer TR, Dinger M. et al. Non-coding RNAs: regulators of disease. J Pathol. 2010;220:126–139. doi: 10.1002/path.2638. [DOI] [PubMed] [Google Scholar]
- Gingeras TR. Origin of phenotypes: Genes and transcripts. Genome Res. 2007;17:682–690. doi: 10.1101/gr.6525007. [DOI] [PubMed] [Google Scholar]
- Rozowsky JS, Newburger D, Sayward F, Wu J. et al. The DART classification of unannotated transcription within the ENCODE regions: Associating transcription with known and novel loci. Genome Res. 2007;17:732–745. doi: 10.1101/gr.5696007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapranov P, Cawley SE, Drenkow J, Bekiranov S. et al. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002;296:916–919. doi: 10.1126/science.1068597. [DOI] [PubMed] [Google Scholar]
- Kapranov P, Willingham AT, Gingeras TR. Genome-wide transcription and the implications for genomic organization. Nat Rev Genet. 2007;8:413–423. doi: 10.1038/nrg2083. [DOI] [PubMed] [Google Scholar]
- Dinger ME, Amaral PP, Mercer TR, Mattick JS. Pervasive transcription of the eukaryotic genome: Functional indices and conceptual implications. Brief Funct Genomic Proteomic. 2009;8:407–423. doi: 10.1093/bfgp/elp038. [DOI] [PubMed] [Google Scholar]
- Grinchuk OV, Jenjaroenpun P, Orlov YL, Zhou J. et al. Integrative analysis of the human cis-antisense gene pairs, miRNAs and their transcription regulation patterns. Nucleic Acids Res. 2010;38:534–547. doi: 10.1093/nar/gkp954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama S, Tomaru Y, Kasukawa T, Waki K. et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- Werner A, Carlile M, Swan D. What do natural antisense transcripts regulate? RNA Biol. 2009;6:43–48. doi: 10.4161/rna.6.1.7568. [DOI] [PubMed] [Google Scholar]
- Faghihi MA, Wahlestedt C. Regulatory roles of natural antisense transcripts. Nat Rev Mol Cell Biol. 2009;10:637–643. doi: 10.1038/nrm2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Vogelstein B, Velculescu VE, Papadopoulos N. et al. The antisense transcriptomes of human cells. Science. 2008;322:1855–1857. doi: 10.1126/science.1163853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She X, Jiang Z, Clark RA, Liu G. et al. Shotgun sequence assembly and recent segmental duplications within the human genome. Nature. 2004;431:927–930. doi: 10.1038/nature03062. [DOI] [PubMed] [Google Scholar]
- Shaw CJ, Lupski JR. Implications of human genome architecture for rearrangement-based disorders: The genomic basis of disease. Hum Mol Genet. 2004;13(Spec. No. 1):R57–R64. doi: 10.1093/hmg/ddh073. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z. et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Hansen S, Selzer RR, Cheng Z. et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev. 2009;19:196–204. doi: 10.1016/j.gde.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Eichler EE. Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet. 2010;19:R176–R187. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Kidd JM, Marques-Bonet T, Aksay G. et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PM, Zheng D, Zhang Z, Carriero N. et al. Transcribed processed pseudogenes in the human genome: An intermediate form of expressed retrosequence lacking protein-coding ability. Nucleic Acids Res. 2005;33:2374–2383. doi: 10.1093/nar/gki531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai H, Koyanagi KO, Imanishi T, Itoh T. et al. Frequent emergence and functional resurrection of processed pseudogenes in the human and mouse genomes. Gene. 2007;389:196–203. doi: 10.1016/j.gene.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Zheng D, Frankish A, Baertsch R, Kapranov P. et al. Pseudogenes in the ENCODE regions: Consensus annotation, analysis of transcription, and evolution. Genome Res. 2007;17:839–851. doi: 10.1101/gr.5586307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ. et al. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 2004;14:1–18. doi: 10.1097/00008571-200401000-00001. [DOI] [PubMed] [Google Scholar]
- Terai G, Yoshizawa A, Okida H, Asai K. et al. Discovery of short pseudogenes derived from messenger RNAs. Nucleic Acids Res. 2010;38:1163–1171. doi: 10.1093/nar/gkp1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZD, Frankish A, Hunt T, Harrow J. et al. Identification and analysis of unitary pseudogenes: Historic and contemporary gene losses in humans and other primates. Genome Biol. 2010;11:R26. doi: 10.1186/gb-2010-11-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachane AN, Harrison PM. Assessing the genomic evidence for conserved transcribed pseudogenes under selection. BMC Genomics. 2009;10:435. doi: 10.1186/1471-2164-10-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Gerstein MB. The ambiguous boundary between genes and pseudogenes: The dead rise up, or do they? Trends Genet. 2007;23:219–224. doi: 10.1016/j.tig.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Hirotsune S, Yoshida N, Chen A, Garrett L. et al. An expressed pseudogene regulates the messenger-RNA stability of its homologous coding gene. Nature. 2003;423:91–96. doi: 10.1038/nature01535. [DOI] [PubMed] [Google Scholar]
- Svensson O, Arvestad L, Lagergren J. Genome-wide survey for biologically functional pseudogenes. PLoS Comput Biol. 2006;2:e46. doi: 10.1371/journal.pcbi.0020046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Bennett EA, Iskow RC, Devine SE. Which transposable elements are active in the human genome? Trends Genet. 2007;23:183–191. doi: 10.1016/j.tig.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Xing J, Zhang Y, Han K, Salem AH. et al. Mobile elements create structural variation: Analysis of a complete human genome. Genome Res. 2009;19:1516–1526. doi: 10.1101/gr.091827.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Jiang P, Shen S, Sato S. et al. Large-scale analysis of exonized mammalian-wide interspersed repeats in primate genomes. Hum Mol Genet. 2009;18:2204–2214. doi: 10.1093/hmg/ddp152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan IK, Rogozin IB, Glazko GV, Koonin EV. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 2003;19:68–72. doi: 10.1016/s0168-9525(02)00006-9. [DOI] [PubMed] [Google Scholar]
- Thornburg BG, Gotea V, Makalowski W. Transposable elements as a significant source of transcription regulating signals. Gene. 2006;365:104–110. doi: 10.1016/j.gene.2005.09.036. [DOI] [PubMed] [Google Scholar]
- Piriyapongsa J, Marino-Ramirez L, Jordan IK. Origin and evolution of human microRNAs from transposable elements. Genetics. 2007;176:1323–1337. doi: 10.1534/genetics.107.072553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley AB, Miller WJ, Jordan IK. Human cis natural antisense transcripts initiated by transposable elements. Trends Genet. 2008;24:53–56. doi: 10.1016/j.tig.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Lowe CB, Bejerano G, Haussler D. Thousands of human mobile element fragments undergo strong purifying selection near developmental genes. Proc Natl Acad Sci USA. 2007;104:8005–8010. doi: 10.1073/pnas.0611223104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara H, Smit AF, Okada N. Functional noncoding sequences derived from SINEs in the mammalian genome. Genome Res. 2006;16:864–874. doi: 10.1101/gr.5255506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthana S, Noble WS, Kryukov G, Grant CE. et al. Widely distributed noncoding purifying selection in the human genome. Proc Natl Acad Sci USA. 2007;104:12410–12415. doi: 10.1073/pnas.0705140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JA, Bird C, Nemesh J, Thomas DJ. et al. Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat Genet. 2006;38:223–227. doi: 10.1038/ng1710. [DOI] [PubMed] [Google Scholar]
- Parker SC, Hansen L, Abaan HO, Tullius TD. et al. Local DNA topography correlates with functional noncoding regions of the human genome. Science. 2009;324:389–392. doi: 10.1126/science.1169050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP, Lunter G. Signatures of adaptive evolution within human non-coding sequence. Hum Mol Genet. 2006;15(Spec. No. 2):R170–R175. doi: 10.1093/hmg/ddl182. [DOI] [PubMed] [Google Scholar]
- Eory L, Halligan DL, Keightley PD. Distributions of selectively constrained sites and deleterious mutation rates in the hominid and murid genomes. Mol Biol Evol. 2010;27:177–192. doi: 10.1093/molbev/msp219. [DOI] [PubMed] [Google Scholar]
- Pheasant M, Mattick JS. Raising the estimate of functional human sequences. Genome Res. 2007;17:1245–1253. doi: 10.1101/gr.6406307. [DOI] [PubMed] [Google Scholar]
- Katzman S, Kern AD, Bejerano G, Fewell G. et al. Human genome ultraconserved elements are ultraselected. Science. 2007;317:915. doi: 10.1126/science.1142430. [DOI] [PubMed] [Google Scholar]
- Licastro D, Gennarino VA, Petrera F, Sanges R. et al. Promiscuity of enhancer, coding and non-coding transcription functions in ultraconserved elements. BMC Genomics. 2010;11:151. doi: 10.1186/1471-2164-11-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean C, Bejerano G. Dispensability of mammalian DNA. Genome Res. 2008;18:1743–1751. doi: 10.1101/gr.080184.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedeva YA, Fridman MV, Oparina NJ, Malko DB. et al. Intergenic, gene terminal, and intragenic CpG islands in the human genome. BMC Genomics. 2010;11:48. doi: 10.1186/1471-2164-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird CP, Stranger BE, Liu M, Thomas DJ. et al. Fast-evolving noncoding sequences in the human genome. Genome Biol. 2007;8:R118. doi: 10.1186/gb-2007-8-6-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S, Noonan JP, Paabo S, Rubin EM. Accelerated evolution of conserved noncoding sequences in humans. Science. 2006;314:786. doi: 10.1126/science.1130738. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Bruce C, Rozowsky JS, Zheng D. et al. What is a gene, post-ENCODE? History and updated definition. Genome Res. 2007;17:669–681. doi: 10.1101/gr.6339607. [DOI] [PubMed] [Google Scholar]
- Kapranov P, Drenkow J, Cheng J, Long J. et al. Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. 2005;15:987–997. doi: 10.1101/gr.3455305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan DA, Lettice LA. Long-range gene control and genetic disease. Adv Genet. 2008;61:339–388. doi: 10.1016/S0065-2660(07)00013-2. [DOI] [PubMed] [Google Scholar]
- Morley M, Molony CM, Weber TM, Devlin JL. et al. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–747. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingeras TR. Implications of chimaeric non-co-linear transcripts. Nature. 2009;461:206–211. doi: 10.1038/nature08452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. MicroRNomics: A newly emerging approach for disease biology. Physiol Genomics. 2008;33:139–147. doi: 10.1152/physiolgenomics.00034.2008. [DOI] [PubMed] [Google Scholar]
- Pesole G. What is a gene? An updated operational definition. Gene. 2008;417:1–4. doi: 10.1016/j.gene.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10:241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- Pang AW, MacDonald JR, Pinto D, Wei J. et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 2010;11:R52. doi: 10.1186/gb-2010-11-5-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM. et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Cardon LR. Evaluating coverage of genome-wide association studies. Nat Genet. 2006;38:659–662. doi: 10.1038/ng1801. [DOI] [PubMed] [Google Scholar]
- Eberle MA, Ng PC, Kuhn K, Zhou L. et al. Power to detect risk alleles using genome-wide tag SNP panels. PLoS Genet. 2007;3:1827–1837. doi: 10.1371/journal.pgen.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Li C, Guan W. Evaluation of coverage variation of SNP chips for genome-wide association studies. Eur J Hum Genet. 2008;16:635–643. doi: 10.1038/sj.ejhg.5202007. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Loi H, Dong S, Tsai YY. et al. Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res. 2004;14:414–425. doi: 10.1101/gr.2014904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson KL, Steemers FJ, Lee G, Mendoza LG. et al. A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet. 2005;37:549–554. doi: 10.1038/ng1547. [DOI] [PubMed] [Google Scholar]
- Cooper GM, Zerr T, Kidd JM, Eichler EE. et al. Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nat Genet. 2008;40:1199–1203. doi: 10.1038/ng.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen F, Huang J, Fitch KR, Truong VB. et al. Improved detection of global copy number variation using high density, non-polymorphic oligonucleotide probes. BMC Genet. 2008;9:27. doi: 10.1186/1471-2156-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft P, Hunter DJ. Genetic risk prediction -- Are we there yet? N Engl J Med. 2009;360:1701–1703. doi: 10.1056/NEJMp0810107. [DOI] [PubMed] [Google Scholar]
- Eichler EE, Flint J, Gibson G, Kong A. et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP. et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC. et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- Maunakea AK, Chepelev I, Zhao K. Epigenome mapping in normal and disease states. Circ Res. 2010;107:327–339. doi: 10.1161/CIRCRESAHA.110.222463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA, Huang L, Jewett EM, Szpiech ZA. et al. Genome-wide association studies in diverse populations. Nat Rev Genet. 2010;11:356–366. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Need AC, Goldstein DB. Next generation disparities in human genomics: Concerns and remedies. Trends Genet. 2009;25:489–494. doi: 10.1016/j.tig.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Eeles RA, Kote-Jarai Z, Al Olama AA, Giles GG. et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet. 2009;41:1116–1121. doi: 10.1038/ng.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M. et al. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen CT, Wang J, Lanktree MB, Cao H. et al. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet. 2010;42:684–687. doi: 10.1038/ng.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen K, Visschedijk MC, van Sommeren S, Fu JY. et al. Analysis of SNPs with an effect on gene expression identifies UBE2L3 and BCL3 as potential new risk genes for Crohn's disease. Hum Mol Genet. 2010;19:3482–3488. doi: 10.1093/hmg/ddq264. [DOI] [PubMed] [Google Scholar]
- Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ. et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: Associations with disease and evolution. Genome Res. 2010;20:1352–1360. doi: 10.1101/gr.107920.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Closas M, Hall P, Nevanlinna H, Pooley K. et al. Heterogeneity of breast cancer associations with five susceptibility loci by clinical and pathological characteristics. PLoS Genet. 2008;4:e1000054. doi: 10.1371/journal.pgen.1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat Rev Genet. 2010;11:843–854. doi: 10.1038/nrg2884. [DOI] [PubMed] [Google Scholar]
- Cordell HJ. Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet. 2009;10:392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. Gene-environment-wide association studies: emerging approaches. Nat Rev Genet. 2010;11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schork NJ, Murray SS, Frazer KA, Topol EJ. Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes J, Lawrence R, Eyre S, Panoutsopoulou K. et al. Rare variation at the TNFAIP3 locus and susceptibility to rheumatoid arthritis. Hum Genet. 2010;128:627–633. doi: 10.1007/s00439-010-0889-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal V, Libiger O, Torkamani A, Schork NJ. Statistical analysis strategies for association studies involving rare variants. Nat Rev Genet. 2010;11:773–785. doi: 10.1038/nrg2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S. et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Wacholder S, Gail MH, Peters U. et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42:570–575. doi: 10.1038/ng.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM. et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- Mefford HC, Muhle H, Ostertag P, von Spiczak S. et al. Genome-wide copy number variation in epilepsy: Novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6:e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RG, Jacquemont S, Valsesia A, de Smith AJ. et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671–675. doi: 10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochukova EG, Huang N, Keogh J, Henning E. et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010;463:666–670. doi: 10.1038/nature08689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa H, Kato M, Awata T, Kohda M. et al. Homozygosity haplotype allows a genomewide search for the autosomal segments shared among patients. Am J Hum Genet. 2007;80:1090–1102. doi: 10.1086/518176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Orr A, Guernsey DL, Robitaille J. et al. Application of homozygosity haplotype analysis to genetic mapping with high-density SNP genotype data. PLoS One. 2009;4:e5280. doi: 10.1371/journal.pone.0005280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LH, Ho SF, Chen CH, Wei CY. et al. Long contiguous stretches of homozygosity in the human genome. Hum Mutat. 2006;27:1115–1121. doi: 10.1002/humu.20399. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J, Scholz S, Fung HC, Matarin M. et al. Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum Mol Genet. 2007;16:1–14. doi: 10.1093/hmg/ddl436. [DOI] [PubMed] [Google Scholar]
- Yang TL, Guo Y, Zhang LS, Tian Q. et al. Runs of homozygosity identify a recessive locus 12q21.31 for human adult height. J Clin Endocrinol Metab. 2010;95:3777–3782. doi: 10.1210/jc.2009-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Weber JL. Long homozygous chromosomal segments in reference families from the Centre d'Etude du Polymorphisme Humain. Am J Hum Genet. 1999;65:1493–1500. doi: 10.1086/302661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harville HM, Held S, Diaz-Font A, Davis EE. et al. Identification of 11 novel mutations in eight BBS genes by high-resolution homozygosity mapping. J Med Genet. 2010;47:262–267. doi: 10.1136/jmg.2009.071365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, Shahin H, Elkan-Miller T, Lee MK. et al. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am J Hum Genet. 2010;87:90–94. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang J, Zhang S, Yang P, Hawkins-Lee B. et al. Loss-of-function mutations in HPSE2 cause the autosomal recessive urofacial syndrome. Am J Hum Genet. 2010;86:957–962. doi: 10.1016/j.ajhg.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapunzina P, Aglan M, Temtamy S, Caparros-Martin JA. et al. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet. 2010;87:110–114. doi: 10.1016/j.ajhg.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas E, Poitelon Y, Chouery E, Salem N. et al. CAMOS, a nonprogressive, autosomal recessive, congenital cerebellar ataxia, is caused by a mutant zinc-finger protein, ZNF592. Eur J Hum Genet. 2010;18:1107–1113. doi: 10.1038/ejhg.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin RW, Safieh C, Littink KW, Shalev SA. et al. Mutations in C2ORF71 cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;86:783–788. doi: 10.1016/j.ajhg.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudan I, Rudan D, Campbell H, Carothers A. et al. Inbreeding and risk of late onset complex disease. J Med Genet. 2003;40:925–932. doi: 10.1136/jmg.40.12.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudan I, Campbell H, Carothers AD, Hastie ND. et al. Contribution of consanguinuity to polygenic and multifactorial diseases. Nat Genet. 2006;38:1224–1225. doi: 10.1038/ng1106-1224. [DOI] [PubMed] [Google Scholar]
- Campbell H, Carothers AD, Rudan I, Hayward C. et al. Effects of genome-wide heterozygosity on a range of biomedically relevant human quantitative traits. Hum Mol Genet. 2007;16:233–241. doi: 10.1093/hmg/ddl473. [DOI] [PubMed] [Google Scholar]
- Lencz T, Lambert C, DeRosse P, Burdick KE. et al. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc Natl Acad Sci USA. 2007;104:19942–19947. doi: 10.1073/pnas.0710021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Guerreiro RJ, Simon-Sanchez J, Bras JT. et al. Extended tracts of homozygosity identify novel candidate genes associated with late-onset Alzheimer's disease. Neurogenetics. 2009;10:183–190. doi: 10.1007/s10048-009-0182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007;39:S48–S54. doi: 10.1038/ng2092. [DOI] [PubMed] [Google Scholar]
- Carter NP. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat Genet. 2007;39:S16–S21. doi: 10.1038/ng2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki H, Wang PH, Hu J, Rava R. et al. High resolution discovery and confirmation of copy number variants in 90 Yoruba Nigerians. Genome Biol. 2009;10:R125. doi: 10.1186/gb-2009-10-11-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Kim JI, Ju YS, Gokcumen O. et al. Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat Genet. 2010;42:400–405. doi: 10.1038/ng.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim SH, Kim TM, Hu HJ, Kim JH. et al. Copy number variations in East-Asian population and their evolutionary and functional implications. Hum Mol Genet. 2010;19:1001–1008. doi: 10.1093/hmg/ddp564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku CS, Pawitan Y, Sim X, Ong RT. et al. Genomic copy number variations in three Southeast Asian populations. Hum Mutat. 2010;31:851–857. doi: 10.1002/humu.21287. [DOI] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- Feuk L. Inversion variants in the human genome: Role in disease and genome architecture. Genome Med. 2010;2:11. doi: 10.1186/gm132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Xuan Z, Makarov V, Ye K. et al. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods. 2009;6:S13–S20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- Harismendy O, Ng PC, Strausberg RL, Wang X. et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, McBride DJ, Lin ML, Varela I. et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R. et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- Kidd JM, Graves T, Newman TL, Fulton R. et al. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell. 2010;143:837–847. doi: 10.1016/j.cell.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Huett A, Kuballa P, Chilewski SD. et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Speliotes EK, Loos RJ, Li S. et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulle JG, Dodd AF, McGrath JA, Wolyniec PS. et al. Microdeletions of 3q29 confer high risk for schizophrenia. Am J Hum Genet. 2010;87:229–236. doi: 10.1016/j.ajhg.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszkiewicz K, Studholme DJ. De novo assembly of short sequence reads. Brief Bioinform. 2010;11:457–472. doi: 10.1093/bib/bbq020. [DOI] [PubMed] [Google Scholar]
- Rothberg JM, Leamon JH. The development and impact of 454 sequencing. Nat Biotechnol. 2008;26:1117–1124. doi: 10.1038/nbt1485. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang J. Faster human genome sequencing. Nat Biotechnol. 2009;27:820–821. doi: 10.1038/nbt0909-820. [DOI] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K. et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- Koboldt DC, Ding L, Mardis ER, Wilson RK. Challenges of sequencing human genomes. Brief Bioinform. 2010;11:484–498. doi: 10.1093/bib/bbq016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan KJ, Peckham HE, Costa GL, McLaughlin SF. et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn SM, Kim TH, Lee S, Kim D. et al. The first Korean genome sequence and analysis: Full genome sequencing for a socioethnic group. Genome Res. 2009;19:1622–1629. doi: 10.1101/gr.092197.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Ju YS, Park H, Kim S. et al. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–1015. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto A, Nakagawa H, Hosono N, Nakano K. et al. Whole-genome sequencing and comprehensive variant analysis of a Japanese individual using massively parallel sequencing. Nat Genet. 2010;42:931–936. doi: 10.1038/ng.691. [DOI] [PubMed] [Google Scholar]
- Tong P, Prendergast JG, Lohan AJ, Farrington SM. et al. Sequencing and analysis of an Irish human genome. Genome Biol. 2010;11:R91. doi: 10.1186/gb-2010-11-9-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzman JO, Mackenzie AP, Adey A, Hiatt JB. et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol. 2011;29:59–63. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SC, Miller W, Ratan A, Tomsho LP. et al. Complete Khoisan and Bantu genomes from southern Africa. Nature. 2010;463:943–947. doi: 10.1038/nature08795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Sampas N, Antonacci F, Graves T. et al. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat Methods. 2010;7:365–371. doi: 10.1038/nmeth.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Zheng H, Luo R. et al. Building the sequence map of the human pan-genome. Nat Biotechnol. 2010;28:57–63. doi: 10.1038/nbt.1596. [DOI] [PubMed] [Google Scholar]
- Wain LV, Armour JA, Tobin MD. Genomic copy number variation, human health, and disease. Lancet. 2009;374:340–350. doi: 10.1016/S0140-6736(09)60249-X. [DOI] [PubMed] [Google Scholar]
- Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer W, Tomlinson I. Rare genetic variants and the risk of cancer. Curr Opin Genet Dev. 2010;20:262–267. doi: 10.1016/j.gde.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C. et al. Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Pittard WS, Mullaney JM, Farooq U. et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 2011;21:830–839. doi: 10.1101/gr.115907.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhu H, Ruan J, Qian W. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hu Y, Bolund L, Wang J. State of the art de novo assembly of human genomes from massively parallel sequencing data. Hum Genomics. 2010;4:271–277. doi: 10.1186/1479-7364-4-4-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Sajjadian S, Eichler EE. Limitations of next-generation genome sequence assembly. Nat Methods. 2011;8:61–65. doi: 10.1038/nmeth.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton MR. Exploring the genomes of cancer cells: Progress and promise. Science. 2011;331:1553–1558. doi: 10.1126/science.1204040. [DOI] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL. et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J. et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Yan XJ, Xu J, Gu ZH, Pan CM. et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–315. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC. et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prickett TD, Agrawal NS, Wei X, Yates KE. et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009;41:1127–1132. doi: 10.1038/ng.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ. et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Stephens PJ, O'Meara S, McBride DJ. et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Jiang Z, Liu J, Haverty PM. et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;465:473–477. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- Ivanov D, Hamby SE, Stenson PD, Phillips AD. et al. Comparative analysis of germline and somatic microlesion mutational spectra in 17 human tumor suppressor genes. Hum Mutat. 2011;32:620–632. doi: 10.1002/humu.21483. [DOI] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE. et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgliesh GL, Furge K, Greenman C, Chen L. et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Walia V, Lin JC, Teer JK. et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43:442–446. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela I, Tarpey P, Raine K, Huang D. et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totoki Y, Tatsuno K, Yamamoto S, Arai Y. et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43:464–469. doi: 10.1038/ng.804. [DOI] [PubMed] [Google Scholar]
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K. et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link DC, Schuettpelz LG, Shen D, Wang J. et al. Identification of a novel TP53 cancer susceptibility mutation through whole-genome sequencing of a patient with therapy-related AML. JAMA. 2011;305:1568–1576. doi: 10.1001/jama.2011.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquier A. The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat Rev Genet. 2009;10:833–844. doi: 10.1038/nrg2683. [DOI] [PubMed] [Google Scholar]
- Alexander RP, Fang G, Rozowsky J, Snyder M. et al. Annotating non-coding regions of the genome. Nat Rev Genet. 2010;11:559–571. doi: 10.1038/nrg2814. [DOI] [PubMed] [Google Scholar]
- Ropers HH. New perspectives for the elucidation of genetic disorders. Am J Hum Genet. 2007;81:199–207. doi: 10.1086/520679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropers HH. Single gene disorders come into focus --Again. Dialogues Clin Neurosci. 2010;12:95–102. [PMC free article] [PubMed] [Google Scholar]
- Antonarakis SE, Beckmann JS. Mendelian disorders deserve more attention. Nat Rev Genet. 2006;7:277–282. doi: 10.1038/nrg1826. [DOI] [PubMed] [Google Scholar]
- Antonarakis SE, Chakravarti A, Cohen JC, Hardy J. Mendelian disorders and multifactorial traits: The big divide or one for all? Nat Rev Genet. 2010;11:380–384. doi: 10.1038/nrg2793. [DOI] [PubMed] [Google Scholar]
- Botstein D, Risch N. Discovering genotypes underlying human phenotypes: Past successes for Mendelian disease, future approaches for complex disease. Nat Genet. 2003;33(Suppl):228–237. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- Hoischen A, van Bon BW, Gilissen C, Arts P. et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- Mamanova L, Coffey AJ, Scott CE, Kozarewa I. et al. Target-enrichment strategies for next-generation sequencing. Nat Methods. 2010;7:111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- Turner EH, Ng SB, Nickerson DA, Shendure J. Methods for genomic partitioning. Annu Rev Genomics Hum Genet. 2009;10:263–284. doi: 10.1146/annurev-genom-082908-150112. [DOI] [PubMed] [Google Scholar]
- Gilissen C, Arts HH, Hoischen A, Spruijt L. et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce SB, Walsh T, Chisholm KM, Lee MK. et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am J Hum Genet. 2010;87:282–288. doi: 10.1016/j.ajhg.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde E, Albrecht S, Ha KC, Jacob K. et al. Unexpected allelic heterogeneity and spectrum of mutations in Fowler syndrome revealed by next-generation exome sequencing. Hum Mutat. 2010;31:918–923. doi: 10.1002/humu.21293. [DOI] [PubMed] [Google Scholar]
- Bonnefond A, Durand E, Sand O, De Graeve F. et al. Molecular diagnosis of neonatal diabetes mellitus using next-generation sequencing of the whole exome. PLoS One. 2010;5:e13630. doi: 10.1371/journal.pone.0013630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthey EA, Mayer AN, Syverson GD, Helbling D. et al. Making a definitive diagnosis: Successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–262. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- Montenegro G, Powell E, Huang J, Speziani F. et al. Exome sequencing allows for rapid gene identification in a Charcot-Marie-Tooth family. Ann Neurol. 2011;69:464–470. doi: 10.1002/ana.22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathiresan S, Musunuru K, Orho-Melander M. Defining the spectrum of alleles that contribute to blood lipid concentrations in humans. Curr Opin Lipidol. 2008;19:122–127. doi: 10.1097/MOL.0b013e3282f70296. [DOI] [PubMed] [Google Scholar]
- Hegele RA. Plasma lipoproteins: Genetic influences and clinical implications. Nat Rev Genet. 2009;10:109–121. doi: 10.1038/nrg2481. [DOI] [PubMed] [Google Scholar]
- Brinkman RR, Dube MP, Rouleau GA, Orr AC. et al. Human monogenic disorders -- A source of novel drug targets. Nat Rev Genet. 2006;7:249–260. doi: 10.1038/nrg1828. [DOI] [PubMed] [Google Scholar]
- Mathew CG. New links to the pathogenesis of Crohn's disease provided by genome-wide association scans. Nat Rev Genet. 2008;9:9–14. doi: 10.1038/nrg2203. [DOI] [PubMed] [Google Scholar]
- Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008;8:458–466. doi: 10.1038/nri2340. [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN. Genomewide association studies --Illuminating biologic pathways. N Engl J Med. 2009;360:1699–1701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- Hanlon SE, Lieb JD. Progress and challenges in profiling the dynamics of chromatin and transcription factor binding with DNA microarrays. Curr Opin Genet Dev. 2004;14:697–705. doi: 10.1016/j.gde.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Mockler TC, Chan S, Sundaresan A, Chen H. et al. Applications of DNA tiling arrays for whole-genome analysis. Genomics. 2005;85:1–15. doi: 10.1016/j.ygeno.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Park PJ. ChIP-seq: Advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP. et al. A global map of pp53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M. et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- Farnham PJ. Insights from genomic profiling of transcription factors. Nat Rev Genet. 2009;10:605–616. doi: 10.1038/nrg2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denoeud F, Aury JM, Da Silva C, Noel B. et al. Annotating genomes with massive-scale RNA sequencing. Genome Biol. 2008;9:R175. doi: 10.1186/gb-2008-9-12-r175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Pai AA, Gilad Y, Pritchard JK. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet. 2010;6:e1001236. doi: 10.1371/journal.pgen.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S. et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heap GA, Yang JH, Downes K, Healy BC. et al. Genome-wide analysis of allelic expression imbalance in human primary cells by high-throughput transcriptome resequencing. Hum Mol Genet. 2010;19:122–134. doi: 10.1093/hmg/ddp473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhu J, Tian G, Li N. et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010;8:e1000533. doi: 10.1371/journal.pbio.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Wang T, Coarfa C, Nagarajan RP. et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 2010;28:1097–1105. doi: 10.1038/nbt.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Ecker JR. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res. 2009;19:959–966. doi: 10.1101/gr.083451.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl C, Gronbaek K, Guldberg P. Advances in DNA methylation: 5-hydroxymethylcytosine revisited. Clin Chim Acta. 2011;412:831–836. doi: 10.1016/j.cca.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Flusberg BA, Webster DR, Lee JH, Travers KJ. et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B. et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- Gilad Y, Rifkin SA, Pritchard JK. Revealing the architecture of gene regulation: The promise of eQTL studies. Trends Genet. 2008;24:408–415. doi: 10.1016/j.tig.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyrieras JB, Kudaravalli S, Kim SY, Dermitzakis ET. et al. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF. et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP. et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Dermitzakis ET. From expression QTLs to personalized transcriptomics. Nat Rev Genet. 2011;12:277–282. doi: 10.1038/nrg2969. [DOI] [PubMed] [Google Scholar]
- Peterson J, Garges S, Giovanni M, McInnes P. et al. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Ren B. Next-generation genomics: An integrative approach. Nat Rev Genet. 2010;11:476–486. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate L, Cuffe S, Horgan A, Hung RJ. et al. Germline genetic variation, cancer outcome, and pharmacogenetics. J Clin Oncol. 2010;28:4029–4037. doi: 10.1200/JCO.2009.27.2336. [DOI] [PubMed] [Google Scholar]
- Tan GM, Wu E, Lam YY, Yan BP. Role of warfarin pharmacogenetic testing in clinical practice. Pharmacogenomics. 2010;11:439–448. doi: 10.2217/pgs.10.8. [DOI] [PubMed] [Google Scholar]
- Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer. 2009;9:576–586. doi: 10.1038/nrc2683. [DOI] [PubMed] [Google Scholar]
- Kim C, Paik S. Gene-expression-based prognostic assays for breast cancer. Nat Rev Clin Oncol. 2010;7:340–347. doi: 10.1038/nrclinonc.2010.61. [DOI] [PubMed] [Google Scholar]
- Hartman M, Loy EY, Ku CS, Chia KS. Molecular epidemiology and its current clinical use in cancer management. Lancet Oncol. 2010;11:383–390. doi: 10.1016/S1470-2045(10)70005-X. [DOI] [PubMed] [Google Scholar]
- Mauro MJ, O'Dwyer M, Heinrich MC, Druker BJ. STI571: A paradigm of new agents for cancer therapeutics. J Clin Oncol. 2002;20:325–334. doi: 10.1200/JCO.2002.20.1.325. [DOI] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S. et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Pinto A, Zagonel V. 5-aza-2'-deoxycytidine (decitabine) and 5-azacytidine in the treatment of acute myeloid leukemias and mye-lodysplastic syndromes: Past, present and future trends. Leukemia. 1993;7(Suppl 1):51–60. [PubMed] [Google Scholar]
- Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DL, Thio CL, Martin MP, Qi Y. et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suppiah V, Moldovan M, Ahlenstiel G, Berg T. et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- Guttmacher AE, McGuire AL, Ponder B, Stefansson K. Personalized genomic information: preparing for the future of genetic medicine. Nat Rev Genet. 2010;11:161–165. doi: 10.1038/nrg2735. [DOI] [PubMed] [Google Scholar]
- Altman RB, Kroemer HK, McCarty CA, Ratain MJ. et al. Pharmacogenomics: Will the promise be fulfilled? Nat Rev Genet. 2011;12:69–73. doi: 10.1038/nrg2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledergerber C, Dessimoz C. Base-calling for next-generation sequencing platforms. Brief Bioinform. 2011. in press . [DOI] [PMC free article] [PubMed]
- Li H, Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Brief Bioinform. 2010;11:473–483. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Jiang R, Kwan W, Wang B, Evaluation of next-generation sequencing software in mapping and assembly. J Hum Genet. 2011. in press . [DOI] [PubMed]
- Miller JR, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010;95:315–327. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Alkan C, Eichler EE, Sahinalp SC. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19:1270–1278. doi: 10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depristo MA, Banks E, Poplin R, Garimella KV. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss M. Introduction into the analysis of high-throughput-sequencing based epigenome data. Brief Bioinform. 2010;11:512–523. doi: 10.1093/bib/bbq014. [DOI] [PubMed] [Google Scholar]
- Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat Methods. 2009;6:S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner DS, Pavesi G, Castrignano T, De Meo PD. et al. Bioinformatics approaches for genomics and post genomics applications of next-generation sequencing. Brief Bioinform. 2010;11:181–197. doi: 10.1093/bib/bbp046. [DOI] [PubMed] [Google Scholar]
- Luo L, Boerwinkle E, Xiong M. Association studies for next-generation sequencing. Genome Res. 2011;21:1099–1108. doi: 10.1101/gr.115998.110. [DOI] [PMC free article] [PubMed] [Google Scholar]