

Abstract
To dissect the haplotype structure of candidate genes for disease association studies, it is important to understand the nature of genetic variation at these loci in different populations. We present a survey of haplotype structure and linkage disequilibrium of chemokine and chemokine receptor genes in 11 geographically-distinct population samples (n = 728). Chemokine proteins are involved in intercellular signalling and the immune response. These molecules are important modulators of human immunodeficiency virus (HIV)-1 infection and the progression of the acquired immune deficiency syndrome, tumour development and the metastatic process of cancer. To study the extent of genetic variation in this gene family, single nucleotide polymorphisms (SNPs) from 13 chemokine and chemokine receptor genes were genotyped using the 5' nuclease assay (TaqMan).
SNP haplotypes, estimated from unphased genotypes using the Expectation-Maximization-algorithm, are described in a cluster of four CC-chemokine receptor genes (CCR3, CCR2, CCR5 and CCRL2) on chromosome 3p21, and a cluster of three CC-chemokine genes [MPIF-1 (CCL23) PARC (CCL18) and MIP- 1α (CCL3)] on chromosome 17q11-12. The 32 base pair (bp) deletion in exon 4 of CCR5 was also included in the haplotype analysis of 3p21. A total of 87.5 per cent of the variation of 14 biallelic loci scattered over 150 kilobases of 3p21 is explained by 11 haplotypes which have a frequency of at least 1 per cent in the total sample. An analysis of haplotype blocks in this region indicates recombination between CCR2 and CCR5, although long-range pairwise linkage disequilibrium across the region appears to remain intact on two common haplotypes. A reduced-median network demonstrates a clear relationship between 3p21 haplotypes, rooted by the putative ancestral haplotype determined by direct sequencing of four primate species. Analysis of six SNPs on 17q11-12 indicates that 97.5 per cent of the variation is explained by 15 haplotypes, representing at least 1 per cent of the total sample. Additionally, a possible signature of selection at a non-synonymous coding SNP (M106V) in the MPIF-1 (CCL23) gene warrants further study. We anticipate that the results of this study of chemokine and chemokine receptor variation will be applicable to more extensive surveys of long-range haplotype structure in these gene regions and to association studies of HIV-1 disease and cancer.
Keywords: chemokine, SNP, population genetics, variation, haplotype estimation, linkage disequilibrium
Introduction
Chemokine signals play a key role in immune and inflammatory responses, wound healing and angiogenesis. Chemokines are 'chemoattractants', directing the migration of cells along a concentration gradient [1] by binding receptors expressed on the surface of leukocytes, natural killer cells, epithelial cells, endothelial cells and smooth muscle cells [2]. The two major subfamilies of chemokine proteins are defined as CC, with two adjacent cysteine residues, or as CXC, with an intervening non-conserved amino acid. Other chemokines have cysteine residues separated by more than one intervening amino acid (eg CX3CL1 or fractalkine),[3,4] or have only one cysteine (eg XCL1 or lymphotactin) [5,6]. Chemokine receptors are defined by the subfamily of chemokine ligand that they bind. The chemokine and the chemokine receptor genes are generally clustered in four distinct chromosomal regions: CC on 17q11-21, CXC on 4q12-21, both CCR and CXCR on 3p21-24 and CXCR on 2q21-35.
The most studied chemokine receptor, CCR5 on 3p21, has been proven to bind the human immunodeficiency (HIV)-1 virus during infection of the host cell [7-9]. Mutations in this gene have been implicated in resistance to HIV-1 infection,[10-12] accelerated progression to the acquired immune deficiency syndrome (AIDS)[10,13,14] and AIDS-related conditions [15,16]. Other chemokine receptors, also co-receptors of the HIV-1 virus, are implicated in affecting disease status [17,18]. Chemokines also have an impact on this infectious disease, presumably by direct interaction with the receptors that serve as entry mechanisms of HIV-1 into the host cell [19-22]. In addition, chemokines and their receptors are a vital part of the cancer process, specifically in tumour development [23-25] and metastasis of cancer cells to a secondary site [26]. Because of this wealth of functional and genetic evidence, chemokine and chemokine receptor genes are ideal candidates for further disease association analyses.
Analyses of genetic polymorphisms and haplotypes in 3p21 have focused primarily on the CCR5 and CCR2 genes and their impact on HIV-1 disease [10,13,18,27-29]. A recent resequencing survey of variation in the CCR5 promoter in four continental population samples has yielded evidence of past balancing selection in this region [30]. Previous work has demonstrated that a high degree of linkage disequilibrium between single nucleotide polymorphisms (SNPs) extends out from CCR5, possibly by several hundred kilobases (kb) [31]. Assessments of haplotype structure in chemokine genes on chromosome 17q11-12 are limited to RANTES (CCL5) [22] and MCP-1 (CCL2) and Eotaxin (CCL11) [32].
An understanding of the nature of genetic variation in a diverse population sample is important for a complete assessment of these disease candidate loci. Comparing haplotype frequencies and patterns of linkage disequilibrium between populations can determine the detailed structure of genetic variation and fine-map haplotype 'blocks' [33]. Also, assaying a diverse sample to find common variants can determine haplotype-tagging SNPs that capture most of the variation in a 'block' to reduce the genotyping for an association analysis [34]. In order to rigorously describe haplotype structure, it is necessary to have either resequencing data readily available (such as the analysis of the LPL [35,36] and APOE[37] genes), or to have a dense array of SNPs, perhaps at minimum intervals of 5 kb [38].
To further describe the global distribution of genetic variation in chemokine and chemokine receptor genes, we have analysed the haplotype structure of bi-allelic loci in three CC-chemokine genes on chromosome 17q11-12 and four CC-chemokine receptor genes on chromosome 3p21 in 11 population samples. Genotype data were also collected from SNP loci in the fractalkine receptor gene (CX3CR1) on 3p21.3 and CXC-chemokine genes on chromosome 4q12-21. The analysis presented here is not intended to be an exhaustive study of variation, but part of an ongoing effort in our laboratory to catalogue SNPs and characterise linkage disequilibrium in chemokine and chemokine receptor genes to aid in association studies of HIV-1 disease and cancer.
Samples and methods
DNA samples
Forty Centre d'Etude Polymorphisme Humain (CEPH) families, comprising 489 individuals, were genotyped for 28 SNPs, and these data were used to validate each genotyping assay and to determine haplotype phase by pedigree analysis. These 28 SNPs were also genotyped in a panel of 625 individuals from ten populations: 96 Mende and 95 Temne from Sierra Leone in West Africa, 49 Papua New Guineans (PNGs) from highland and lowland groups, 50 Indonesians, 37 Mixe Indians, 57 Mixteca and 45 Zapotecs from Oaxaca, Mexico, 31 Chinese, 107 African-Americans, 24 Hispanic-Americans and 34 European-Americans. Two common chimpanzees, two pygmy chimpanzees, two gorillas and two orangutans were genotyped by direct sequencing to determine the ancestral haplotype for each gene region. All human DNA samples were collected with informed consent under the guidelines of the institutions involved in sample collection. The use of all anonymous DNA samples was either reviewed by the National Institutes of Health Internal Review Board or determined as 'exempt' from review.
Genomic regions and SNPs
Table 1 describes the 28 chemokine and chemokine receptor SNPs on chromosomes 3p21, 17q11-12 and 4q12-21. The relative position of each SNP included in the haplotype analysis of 3p21 and 17q11-12 is indicated under 'haplotype position' and 'contig position' in Table 1. The allelic composition of each SNP is indicated as 'allele 1/allele 2' under 'nucleotide position' in the Table.
Table 1.
Biallelic loci typed in total population sample
| Chromosome location | Haplotype position | Gene | NCBI Locus Link | Nuclotide position | NCBI Genbank | NCBI contig | Contig position | Ancestral allele | NCBI dbSNP# |
|---|---|---|---|---|---|---|---|---|---|
| 3p21 | |||||||||
| 1 | CCR3 | 1232 | Y17YA/G | NM_001837 | NT_05827 | 3997337 | A | 4987053 | |
| 2 | CCR2 | 1231 | -5983 G/A | U95626 | NT_05827 | 4083672 | G | ||
| 3 | CCR2 | 1231 | -5048 G/T | U95626 | NT_05827 | 4084607 | G | 3918357 | |
| 4 | CCR2 | 1231 | -4866 G/C | U95626 | NT_05827 | 4084789 | G | 3918370 | |
| 5 | CCR2 | 1231 | -3433 T/C | U95626 | NT_05827 | 4086222 | T | 3092964 | |
| 6 | CCR2 | 1231 | V64I C/T | NM_000647 | NT_05827 | 4089845 | C | 1799864 | |
| 7 | CCR2 | 1231 | N260N A/G | NM_000647 | NT_05827 | 4090435 | G | 1799865 | |
| 8 | CCR5 | 1234 | 208 C/A | NM_000579 | NT_05827 | 4102477 | C | 2734648 | |
| 9 | CCR5 | 1234 | 303 C/T | NM_000579 | NT_05827 | 4102572 | C | 1799987 | |
| 10 | CCR5 | 1234 | 676 T/C | NM_000579 | NT_05827 | 4102945 | T | 1800023 | |
| 11 | CCR5 | 1234 | L55Q T/A | NM_000579 | NT_05827 | 4015194 | T | 1799863 | |
| 12 | CCR5 | 1234 | D32 | NM_000579 | NT_05827 | WT | |||
| 13 | CCRL2 | 9034 | 1243V C/T | NM_003965 | NT_05827 | 4140934 | T | 3204850 | |
| 14 | CCRL2 | 9034 | 1137 C/G | NM_003965 | NT_05827 | 4141344 | C | ||
| 3p21.3 | |||||||||
| CX3CRI | 1524 | V2491 G/A | NM_001337 | NT_037565 | 1246118 | G | 3732379 | ||
| CX3CRI | 1524 | T280M C/T | NM_001337 | NT_037565 | 1246212 | C | 3732378 | ||
| 4q 12-13 | |||||||||
| ENA-78 (CXCI.5) | 6374 | Q56Q G/A | NM_002997 | NT_006216 | 3371007 | G | 425535 | ||
| 4q21 | |||||||||
| GRO1 (CXCL1) | 2919 | 1086 G/A | X54489 | NT_006216 | 3243154 | A | 4074 | ||
| GRO1 (CXCL1) | 2919 | 1176 C/T | X54489 | NT_006216 | 3243245 | C | 1814092 | ||
| IPIO(CXCLIO) | 3627 | 503 C/G | NM_001565 | NT_016354 | 1437648 | C | 3921 | ||
| 17q21.1-21.2 | |||||||||
| MCPI (CCL2) | 6347 | -362 C/G | M37719 | NT_010799 | 7315787 | n/d | 2857656 | ||
| EOTAXIN (CCL11) | 6356 | -1382 C/T | Z92709 | NT_010799 | 7345226 | n/d | 4795895 | ||
| 17q12 | |||||||||
| 1 | MPIF-1 (CCL23) | 6368 | MI06VG/A | U85767 | NT_010799 | 9074064 | A | 1003645 | |
| 2 | PARC (CCL18) | 6362 | -1 16 C/T | AB0121 13 | NT_010799 | 9125397 | C | 2015086 | |
| 3 | PARC (CCL18) | 6362 | 81 G/A | AB0121 13 | NT_010799 | 9125563 | G | 2015070 | |
| 4 | PARC (CCL18) | 6362 | 311 C/A | AB0121 13 | NT_010799 | 9125793 | A | 2015052 | |
| 5 | PARC (CCL18) | 6362 | 6793 A/G | AB0121 13 | NT_010799 | 9132275 | G | 14304 | |
| 6 | MIP-IA(CCL3) | 6348 | - 1541 T/C | M23178 | NT_010799 | 9152727 | C | 1634497 |
Abbreviations: NCBI = National Center for Biotechnology Information; dbSNP = Database for single nucleotide polymorphisms.
SNP detection strategy
The SNPs typed in this study were derived from three sources: SNPs previously characterised in our laboratory; SNPs validated from public database information;[39] and SNPs determined by resequencing and subsequent analysis with the POLYPHRED software package [40]. SNPs in chemokine and chemokine receptor genes discovered by searching public databases were validated in a panel of 16 individuals from four ethnic groups (European-Americans, African-Americans, Asians and self-identified Hispanics) by direct sequencing. Four (CCR2, -3433 T/C, -4866 G/C, -5048 G/T and -5983 G/A) of the 28 SNPs have not previously been reported and were discovered by direct resequencing of 3 kb of the CCR2 promoter region in the aforementioned panel of 16 individuals. Details of this sequencing survey are discussed elsewhere [41] The putative ancestral allele at each SNP (see Table 1) was determined by direct sequencing of the eight primate samples.
The TaqMan (5' nuclease assay) method
All SNPs were genotyped using the 5' nuclease assay under a set of universal assay conditions [41-43]. The polymerase chain reaction (PCR) was performed in 96-well plates that included positive genotypic controls (for both homozygote states and the heterozygote state for each SNP) and reactions with no DNA as a negative control. All 5' nuclease assay plates were read on the ABI 7700 Sequence Detector, and analysed under dye components with the SDS v1.6.3 or v1.7 software package (Applied Biosystems). Genotype determinations for each reaction were made manually by visual inspection of a scatter-plot of the data, with reference to the results of the genotype control samples.
Description of haplotype data
After compliance with Mendelian inheritance had been established for all of the CEPH family data using PedCheck,[44] the haplotype phase of the multi-site genotypes on chromosomes 3p21 and 17q11-12 was determined by visual inspection of the genotype data for all 40 families (a total of 489 individuals) [41]. Haplotype states and frequencies were estimated in sets of unphased genotype data from the population samples with MLOCUS,[45,46] which uses the Expectation-Maximization (EM) algorithm [47] -- a maximum likelihood-based method. The most likely set of haplotypes for each population sample was determined by a previously described three-step procedure [48]. Average haplotype diversity was calculated for haplotypes in each population sample using Nei's heterozygosity statistic [49].
Haplotypes estimated using MLOCUS were used to calculate FST statistics in ARLEQUIN [50]. These data were analysed by a non-parametric approach, where haplotypes are permuted both within and among populations to create a null distribution of FST values [51]. An exact test of population differentiation provided in ARLEQUIN is also used here to describe differences in haplotype frequencies between populations [52]. Haplotype blocks on 3p21 were assessed using HaploBlockFinder [53] which performs the four-gamete test (FGT)[54] between each pairwise combination of SNPs to identify past recombination events [55]. The minimal-D' metho [56,57] (with minimum D' = 80) was also used to assess haplotype block structure in the 150 kb region of 3p21. Haplotype tag SNPs (htSNPs) were chosen using the htSNP utility, which analyses haplotypes generated by SNPHap [34,58]. Reduced Median (RM) network [59,60] were calculated using Network 3.1.1.1 [61] to visualise the mutational relationships between the haplotypes inferred by MLOCUS in population genotype data.
Estimating linkage disequilibrium in population data
Linkage disequilibrium between pairs of SNPs on chromosomes 3p21 and 17q11-12 was estimated in unphased genotype data from each population sample. The D' statistic was calculated and graphical results were generated using the linkage disequilibrium utility on the Innate Immunity website [62].
Results
SNP allele frequencies and Hardy-Weinberg equilibrium
Of the potential 20,384 genotypes generated for this analysis (728 individuals typed for 28 loci), 517 (or 2.53 per cent) were unable to be resolved by the Applied Biosystems SDSv1.6.3 genotyping software. The remaining 19,867 genotypes were included in the estimation of SNP allele and genotype frequencies for each population sample. For each SNP, the most frequent allele in a panel of 88 individuals (from different ethnic groups) is designated as 'allele 1'. The frequency of 'allele 1' for each SNP is reported in Table 2, and those SNP genotype distributions that deviate from the Hardy-Weinberg equilibrium (HWE) are indicated with an underline. Seventeen out of the 28 SNPs typed were polymorphic in all 11 populations. Of the 308 tests performed to check for conformity to HWE, 21 had significant (p < 0.05) deviations. No one SNP showed deviation from HWE in all populations. Most deviations appear to be due to small numbers of rare homozygotes in some sample sets. Some deviations, primarily in the Indonesian and PNG samples, could be due, in part, to substructure in the sample.
Table 2.
Allele frequencies of all loci typed in I I population samples
| Mende (n = 96) | Temne (n = 95) | Afr-Amer-(n = 107) | Euro-Amer-(n = 133) | Hispanic (n = 24) | Mixteca (n = 57) | Mixe (n = 37) | Zapotec (n = 45) | Chinese (n = 31) | Indonesians (n = 50) | PNG (n = 49) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP allele 1 | |||||||||||
| CCR3(YI7Y_A) | 0.865 | 0.872 | 0.924 | 0.904 | 0.938 | 0.91 1 | 1.000 | 0.953 | 0.967 | 0.978 | 1.000 |
| CCR2(-5983 G) | 0.716 | 0.683 | 0.807 | 1.000 | 0.957 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| CCR2(-5048 G) | 0.847 | 0.858 | 0.819 | 0.901 | 0.792 | 0.746 | 0.649 | 0.71 1 | 0.783 | 0.932 | 0.847 |
| CCR2(-4866 C) | 0.699 | 0.621 | 0.745 | 0.996 | 1.000 | 0.974 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| CCR2(-3433 T) | 0.927 | 0.963 | 0.916 | 0.785 | 0.708 | 0.728 | 0.778 | 0.800 | 0.733 | 0.939 | 0.781 |
| CCR2(64V_C) | 0.941 | 0.922 | 0.840 | 0.904 | 0.826 | 0.750 | 0.649 | 0.71 1 | 0.808 | 0.955 | 0.826 |
| CCR2(N260N_T) | 0.637 | 0.580 | 0.646 | 0.682 | 0.667 | 0.536 | 0.757 | 0.716 | 0.710 | 0.755 | 0.910 |
| CCR5(208 G) | 0.620 | 0.632 | 0.676 | 0.639 | 0.625 | 0.640 | 0.797 | 0.778 | 0.484 | 0.538 | 0.850 |
| CCR5(303 G) | 0.321 | 0.245 | 0.412 | 0.533 | 0.583 | 0.500 | 0.797 | 0.733 | 0.446 | 0.525 | 0.847 |
| CCR5(676 A) | 0.941 | 0.962 | 0.883 | 0.643 | 0.667 | 0.643 | 0.797 | 0.767 | 0.516 | 0.700 | 0.850 |
| CCR5(55L_T) | 1.000 | 1.000 | 0.995 | 0.978 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| CCR5(+32) | 1.000 | 0.995 | 0.962 | 0.891 | 0.979 | 1.000 | 1.000 | 0.978 | 1.000 | 1.000 | 1.000 |
| CCRL2(243I_C) | 1.000 | 1.000 | 0.986 | 0.907 | 0.957 | 0.904 | 1.000 | 0.933 | 0.952 | 0.979 | 1.000 |
| CCRL2(II37C) | 0.700 | 0.658 | 0.794 | 0.996 | 0.979 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| MCP-I(-362C) | 0.521 | 0.532 | 0.570 | 0.713 | 0.708 | 0.300 | 0.108 | 0.227 | 0.433 | 0.510 | 0.710 |
| EOTAXIN(-l382C) | 0.995 | 0.994 | 0.949 | 0.776 | 0.848 | 0.991 | 0.986 | 0.988 | 0.919 | 0.904 | 0.860 |
| MIP-la(--1541 A) | 0.816 | 0.830 | 0.801 | 0.728 | 0.771 | 0.792 | 0.757 | 0.739 | 0.638 | 0.633 | 0.826 |
| PARC(6793 G) | 0.984 | 0.989 | 0.953 | 0.699 | 0.761 | 0.789 | 0.608 | 0.716 | 0.650 | 0.670 | 0.820 |
| PARC(3 1 1 A) | 0.696 | 0.676 | 0.745 | 0.970 | 0.938 | 1.000 | 1.000 | 1.000 | 0.950 | 0.938 | 0.833 |
| PARC(81 G) | 0.977 | 0.968 | 0.947 | 0.922 | 0.792 | 0.825 | 0.736 | 0.644 | 0.919 | 0.867 | 0.419 |
| PARC(-116C) | 0.615 | 0.606 | 0.607 | 0.906 | 0.729 | 0.825 | 0.730 | 0.644 | 0.897 | 0.798 | 0.274 |
| MPIF-1(106M_A) | 0.047 | 0.064 | 0.260 | 0.800 | 0.739 | 0.769 | 0.770 | 0.889 | 0.633 | 0.567 | 0.276 |
| CX3CR1(249V_G) | 0.891 | 0.921 | 0.868 | 0.724 | 0.761 | 0.750 | 0.750 | 0.800 | 0.984 | 0.919 | 0.784 |
| CX3CR1(280T_G) | 0.990 | 1.000 | 0.958 | 0.842 | 0.875 | 0.804 | 0.757 | 0.81 1 | 0.983 | 0.889 | 0.796 |
| ENA(Q56Q_A) | 0.521 | 0.500 | 0.605 | 0.890 | 0.938 | 0.873 | 0.905 | 0.898 | 0.962 | 0.830 | 0.631 |
| GROI(I086G) | 0.995 | 1.000 | 0.881 | 0.313 | 0.521 | 0.632 | 0.527 | 0.636 | 0.532 | 0.378 | 0.510 |
| GRO1(1176 C) | 0.737 | 0.720 | 0.778 | 0.996 | 1.000 | 1.000 | 1.000 | 0.989 | 1.000 | 1.000 | 0.990 |
| IP 10 (503 C) | 0.734 | 0.814 | 0.678 | 0.553 | 0.771 | 0.945 | 0.971 | 0.942 | 0.967 | 0.875 | 0.792 |
Those genotype distributions that demonstrate a distortion from the Hardy-Weinberg equilibrium are indicated with an underline.
Abbreviations: Afr-Amer = African-Americans; Euro-Amer = European-Americans; PNG = Papua New Guineans.
Six SNPs (CCR2 (N260N) CCR5 (303G/A), MCP-1 (-362 C/G) MPIF-1 (M106V) ENA-78 (Q56Q) and GRO1 (1086 G/A) show large differences in allele frequencies (high-deltas) between at least two populations. Two SNPs have at least a 60 per cent difference in allele frequencies between Native Africans and European-American populations: a coding variant in the MPIF-1 chemokine on chromosome 17q11-12, M106V, has a frequency difference of 75.3 per cent. Position 1086G/A, in an intron in GRO1, a CXC chemokine on chromosome 4, has a frequency difference of 68.2 per cent. A promoter SNP (-362) in the MCP-1 CC-chemokine gene on chromosome 17q11-12 differs in frequency between the Mixtecs in Mexico (0.108) and European-Americans (0.713) by 65 per cent. The promoter SNP at position 303 in CCR5 has a frequency difference of 60.2 per cent, where allele 1 (303 G) is 0.847 in the PNG sample and 0.245 in the Temne in West Africa. A synonymous change in ENA-78 (Q56Q) on chromosome 4 has a frequency difference of 46.2 per cent between the Chinese sample (0.962) and the Temne in Sierra Leone (0.500).
Haplotypes and heterozygosity on chromosome 3p21
The EM algorithm generated 48 distinct haplotypes from the 14 SNPs typed in the four genes on chromosome 3p21 (only those 25 haplotypes above 3 per cent in any one population are listed in Table 3). The haplotype order of these 14 SNPs is indicated in Table 1. Nine of these haplotypes, listed in Table 3, have previously been resolved unambiguously by pedigree analysis of 40 CEPH families [41]. Eight of the nine pedigree-phased haplotypes were among the most frequent in the global set of haplotypes in the population samples, and the remaining haplotype (11111111112111) is defined by the CCR5(L55Q) allele, which is apparently specific to European-derived populations.
Table 3.
3p21 haplotype frequencies in 11 population samples
| No. | Haplotype | Mende | Temne | Afr-Amer | Euro-Amer | Hispanic | Mixteca | Mixe | Zapotec | Chinese | Indonesian | PNG | Global |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 11111111111111 | 0.143 | 0.100 | 0.134 | 0.294 | 0.308 | 0.245 | 0.391 | 0.422 | 0.062 | 0.415 | 0.546 | 0.279 |
| 5 | 11211211111111 | 0.029 | 0.056 | 0.085 | 0.089 | 0.187 | 0.255 | 0.351 | 0.254 | 0.158 | 0.053 | 0.100 | 0.147 |
| 3 | 11112122221111 | 0.059 | 0.033 | 0.062 | 0.175 | 0.204 | 0.282 | 0.162 | 0.189 | 0.021 | 0.108 | ||
| 9 | 11121121211111 | 0.273 | 0.283 | 0.174 | 0.027 | 0.069 | |||||||
| 2 | 11111112221111 | 0.029 | 0.173 | 0.109 | 0.027 | 0.014 | 0.01 1 | 0.165 | 0.158 | 0.035 | 0.066 | ||
| 10 | 12111112211112 | 0.274 | 0.293 | 0.127 | 0.063 | ||||||||
| 11111122221111 | 0.006 | 0.064 | 0.027 | 0.022 | 0.288 | 0.163 | 0.094 | 0.061 | |||||
| 11112111111111 | 0.169 | 0.033 | 0.133 | 0.031 | |||||||||
| 4 | 21111121211121 | 0.007 | 0.078 | 0.091 | 0.044 | 0.038 | 0.013 | 0.025 | |||||
| 6 | 11111111111211 | 0.006 | 0.024 | 0.105 | 0.028 | 0.015 | |||||||
| 7 | 11112121111111 | 0.023 | 0.046 | 0.055 | 0.011 | ||||||||
| 21121121211111 | 0.028 | 0.075 | 0.009 | ||||||||||
| 11212211111111 | 0.034 | 0.065 | 0.009 | ||||||||||
| 11112112221111 | 0.007 | 0.066 | 0.026 | 0.009 | |||||||||
| 21211211111111 | 0.030 | 0.027 | 0.022 | 0.012 | 0.008 | ||||||||
| 11111112211111 | 0.032 | 0.011 | 0.041 | 0.008 | |||||||||
| 11211111111111 | 0.040 | 0.012 | 0.026 | 0.007 | |||||||||
| 11111121111111 | 0.065 | 0.006 | |||||||||||
| 11111121211121 | 0.042 | 0.009 | 0.013 | 0.006 | |||||||||
| 11111122211111 | 0.005 | 0.053 | 0.005 | ||||||||||
| 11121111211111 | 0.055 | 0.005 | |||||||||||
| 21111111111111 | 0.034 | 0.008 | 0.004 | ||||||||||
| 12111122211112 | 0.036 | 0.003 | |||||||||||
| 8 | 11111111112111 | 0.022 | 0.002 | ||||||||||
| A | 11111121211111 | 0.004 | 0.000 | ||||||||||
| Heterozygosity | 0.82 ±0.016 | 0.81 ± 0.018 | 0.92 ± 0.008 | 0.83 ± 0.012 | 0.82 ± 0.034 | 0.79 ±0.017 | 0.70 ± 0.028 | 0.73 ±0.03 | 0.84 ± 0.026 | 0.76 ± 0.04 | 0.66 ± 0.047 |
Haplotype diversity values are indicated at the bottom of the table. Haplotype numbers describe haplotypes defined in Clark et al. [43].
Only estimated haplotypes above 3 per cent frequency in any population are listed. The ancestral haplotype is denoted by the box.
Abbreviations: Afr-Amer = African-Americans; Euro-Amer = European-Americans; PNG = Papua New Guineans.
MLOCUS was unable to resolve haplotypes for 37 of the 728 individuals in the analysis of all population samples because of missing genotype data. MLOCUS will drop individuals with two or more missing genotypes from the haplotype estimation. This yields an analysis dropout rate of 5.1 per cent for the 3p21 data. Of the 14 SNPs typed for this analysis, only seven were polymorphic in all 11 population samples. All polymorphic SNPs, even those with low frequency (1 per cent), were included in the haplotype estimation for each population sample. Of the 25 haplotypes presented in Table 3, two are present in every population: those designated as haplotype 1 (11111111111111) and haplotype 5 (11211211111111). The 11 haplotypes that have a frequency of at least 1 per cent in the total sample (2n = 1, 382) comprise 87.5 per cent of the total variation of all haplotypes. The remaining 12.5 per cent of the variation is explained by 37 haplotypes, all of which are found in four or fewer population samples.
Almost half (22) of the total number of haplotypes (48) are unique to one population sample. One unique haplotype (11111121211111), which was confirmed empirically by a previous pedigree analysis of the CEPH grandparent sample [41] (included here in the European-American group), was determined by direct sequencing of primates to be the ancestral or 'root' haplotype. Nearly half of the unique haplotypes (nine out of 22) are found in the African-American sample. The African-American sample is the most diverse, with a total of 27 haplotypes. The heterozygosity values calculated from the haplotype frequencies confirm this, as the African-American sample has the highest heterozygosity at 0.92 ± 0.008. The Mixe sample from Mexico is the least diverse, with only six haplotypes. The hetero-zygosity for the Mixtec-E and the PNG samples were the lowest of the 11 population samples, with values of 0.70 ± 0.028 and 0.6 ± 0.047, respectively
Population-specific patterns of linkage disequilibrium in 3p21
Pairwise comparisons between all polymorphic SNPs using unphased genotypes yields a generally high degree of linkage disequilibrium across the entire 150 kb region in most of the populations (Figure 1). Notable patterns include the relatively intact strong linkage disequilibrium between SNPs in CCR2 and CCR5 in the Native African populations [Mende (1a) and Temne (1b)]. In these populations, there is a distinct lack of linkage disequilibrium between CCR3 (Y17Y) and other SNPs in the region. By comparison, the African-Americans (1c) appear to have the least linkage disequilibrium of any of the populations, with CCR3(Y17Y) (SNP 1) and CCR2 (N260N) (SNP 7) showing the most evident breakdown in pairwise linkage disequilibrium with other SNPs. Compared with the Native African populations and the African-Americans, linkage disequilibrium (D' = 1) is maintained between SNPs CCR3(Y17Y) and CCRL2 (I243V) at opposite ends of the region in the Mixteca (1f) and the Chinese (1i), and somewhat less so in the European-Americans (D' = 0.8) (1d). The less frequent alleles at these SNPs occur almost exclusively on haplotype 4 (21111121211121) in these populations. Although the intact haplotype 4 is absent in African-Americans and Africans [as CCRL2(I243V) is not polymorphic in these populations], some long-range linkage disequilibrium is evident in these samples. The other SNP typed in CCRL2, 1137 C/G, is polymorphic in Africans, and shows significant linkage disequilibrium (D' = 1.0) with CCR3 (Y17Y) in the Temne (1b) specifically.
Figure 1.

(a-k) The D' statistic for pairwise comparisons of the 14 SNPs genotyped in 3p21 in each population sample. The axes of each plot indicate those polymorphic SNPs included in each analysis. SNP labels refer to numbering system described in Table 1. Abbreviations: Euro-Americans = European-Americans; PNG = Papua New Guineans.
Long-range linkage disequilibrium (D' = 1.0) exists in the Mende (1a) between an SNP in the CCR2 promoter (-5983 G/A) and CCRL2 (1137 C/G). The less frequent alleles at these SNPs (-5983A and 1137G) are both carried on the same high frequency haplotype 10 (12111112211112) almost exclusively in the Native Africans populations. Of the Native American populations, the Zapotecs (1h) show a distinct lack of long-range linkage disequilibrium, particularly in pairwise comparisons with CCR3(Y17Y), when compared with the Mixteca (1f) and the Mixe (1g) populations. Finally, the Chinese (1i), Indonesians (1j) and PNGs (1k) all exhibit a relative breakdown in linkage disequilibrium between CCR2 (-3455) (SNP 5) and SNPs in CCR5.
Haplotype blocks and 'tag' SNPs on 3p21
Haplotype blocks and htSNPs were accessed in all 11 population samples; these results are catalogued in ftp://ftp.ncifcrf.gov/pub/users/goldb/. For seven of these population samples, the two tests, the FGT and the minimal-D method (set to D' = 80), indicate the same haplotype block structure for 3p21. For the remaining four populations (African-Americans, Mixteca, Mixe and Zapotecs), the FGT detects more blocks than the minimal-D' test. For example, the minimal-D' analysis finds four blocks in the African-American data, while the FGT generates seven blocks by splittin CCR2 (V64I) CCR5 (208) and CCRL2(1137) each into separate blocks. With the FGT, ten of the 11 populations (all except the PNG) demonstrate a break between CCR2(N260N) and CCR5 (208). This result indicates a past recombination event somewhere in the 20 kb between CCR2 and CCR5. The CEPH pedigree haplotypes support this, as, although there is no direct observation of a recombination event in the pedigree data, one haplotype (11112121211121) appears to be a recombinant of haplotypes 4 (211111121211121) and 7 (11112121111111) [41]. Only three analyses (Mixteca, Chinese and Indonesians) indicate that CCR3 and CCR2 are in the same block, while the data from the other eight populations all indicate a break in block structure in the 80 kb between these two genes. Additionally, the FGT detects an intact block that includes SNPs in the CCR5 and CCRL2 genes (30 kb apart) in five populations (African-Americans, European-Americans, Hispanics, Mix-teca and Indonesians). Conversely, the FGT analysis of the Temne (African) and the Zapotec (Native Mexican) samples shows breaks in both of these intergenic regions (ie between CCR3-CCR2 and CCR5-CCRL2), which supports the linkage disequilibrium patterns illustrated in Figure 1.
Analysis of the total sample (n = 728) indicates that nine htSNPs are sufficient to explain all of the haplotypes above 1 per cent (of haplotypes listed in Table 3). The htSNP utility [53] generated eight equivalent sets of nine SNPs (see supplementary data on page 272). This analysis demonstrated that CCR2(-5983) and CCRL2 (1137) are binary equivalents, which corroborates the linkage disequilibrium results for the Native African populations (the Mende and Temne samples). The 32 bp deletion in CCR5 (del32) was chosen as an htSNP, as it describes a distinct haplotype in all populations in which it is polymorphic. CCR5(L55Q) -- a relatively rare SNP specific to European-Americans -- was not chosen as an htSNP, although it would need to be genotyped to capture that particular haplotype (number 8 in Table 3) in European-derived populations.
Ancestral haplotypes and networks on 3p21
The 'root' haplotype, determined by sequencing four primate species for each SNPs, was found very infrequently in the total set of estimated haplotypes of SNPs in 3p21 and 17q11-12. The root haplotype is indicated by an 'A' in the RM haplotype network of the 3p21 SNPs described in Figure 2. The 3p21 'root' haplotype (11111121211111) differs from the most common haplotype (11111111111111), which is found at nearly 30 per cent in the total sample, by two mutations. The 'allele 2' of these two SNPs, CCR2(N260N_C) and CCR5(303A), are at high frequencies in the African populations (at 0.42 and 0.75, respectively, in the Temne population sample). These two SNPs are polymorphic in all 11 populations surveyed.
Figure 2.
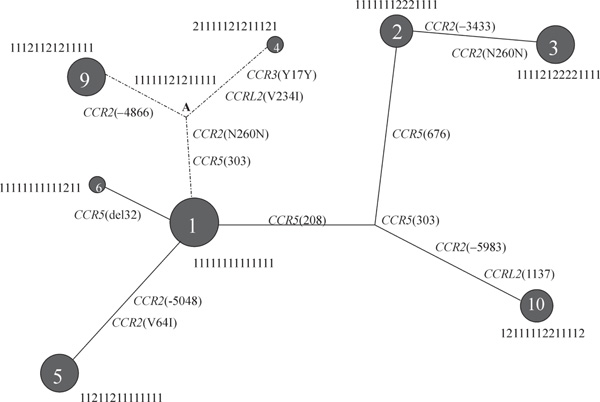
Reduced median (R-M) Network of haplotypes of 14 SNPs in 3p21. Haplotypes indicated comprise nearly 80 per cent of the variation in the total population sample (N = 690). The size of the circle for each haplotype indicates the frequency of the haplotype in the pooled sample.
The 'root' haplotype differs from the most common haplotype in the African samples (111121121211111), by the change from a C to a G at CCR2(-4866), a SNP that is virtually African-specific. The only other populations that are polymorphic at this locus are the Mixtec and the Zapotecs from southern Mexico, and this is probably due to a recurrent mutational event. The 'root' haplotype also differs from hap-lotype 4 (21111121211121) by two mutational events at the extremes of the 150 kb region.
Haplotypes and heterozygosity on chromosome 17q11-12
The EM algorithm generated 30 distinct haplotypes (22 hap-lotypes above 2 per cent in any one population are listed in Table 4) from the six SNPs typed in three chemokine genes in the 79 kb region of chromosome 17q11-12. Twelve of these haplotypes were found in the CEPH grandparent sample and were phased unambiguously by pedigree analysis of 40 CEPH families [41]. MLOCUS estimation was unable to determine haplotypes for 22 of the 724 individuals (in 11 population samples) because of missing genotype data, which yielded a dropout rate of 3.1 per cent for the 17q11-12 haplotype analysis. The two most frequent haplotypes (111111) and (211111) out of the 30 haplotypes in the total set are found in all 11 population samples. These two haplotypes differ by just one mutation, a change from A to G at MPIF1(M106V). 'Allele 2' (106V) at this SNP is at very high frequency in the African populations surveyed, and the haplotype defined by this SNP (211111) is the most frequent in these samples (Mende at 0.49 and Temne at 0.51).
Table 4.
Haplotype frequencies of six single nucleotide polymorphisms in the 17q12 chemokine gene cluster in 11 population samples
| No. | Haplotype | Mende | Temne | Afr-Amer | Euro-Amer | Hispanic | Mixteca | Mixe | Zapotec | Chinese | Indonesian | PNG | Global freq |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 111111 | 0.02 | 0.03 | 0.12 | 0.46 | 0.41 | 0.47 | 0.36 | 0.29 | 0.26 | 0.30 | 0.11 | 0.2570 |
| 2 | 211111 | 0.49 | 0.51 | 0.35 | 0.11 | 0.07 | 0.14 | 0.03 | 0.09 | 0.22 | 0.18 | 0.05 | 0.2034 |
| 3 | 111122 | 0.03 | 0.21 | 0.12 | 0.21 | 0.20 | 0.23 | 0.11 | 0.18 | 0.04 | 0.1217 | ||
| 4 | 222111 | 0.01 | 0.01 | 0.02 | 0.14 | 0.09 | 0.16 | 0.02 | 0.09 | 0.54 | 0.0976 | ||
| 5 | 122111 | 0.04 | 0.05 | 0.08 | 0.09 | 0.06 | 0.32 | 0.08 | 0.05 | 0.03 | 0.0709 | ||
| 6 | 221211 | 0.21 | 0.23 | 0.16 | 0.03 | 0.02 | 0.02 | 0.0607 | |||||
| 7 | 211122 | 0.03 | 0.05 | 0.15 | 0.09 | 0.03 | 0.0323 | ||||||
| 8 | 111121 | 0.06 | 0.05 | 0.14 | 0.02 | 0.08 | 0.0318 | ||||||
| 9 | 211112 | 0.07 | 0.07 | 0.06 | 0.01 | 0.0191 | |||||||
| 10 | 221212 | 0.07 | 0.05 | 0.07 | 0.01 | 0.01 | 0.0199 | ||||||
| 11 | 121211 | 0.02 | 0.02 | 0.03 | 0.04 | 0.07 | 0.0163 | ||||||
| 12 | 111112 | 0.03 | 0.02 | 0.02 | 0.01 | 0.06 | 0.01 | 0.0143 | |||||
| 13 | 221111 | 0.06 | 0.03 | 0.08 | 0.0154 | ||||||||
| 14 | 221222 | 0.01 | 0.03 | 0.06 | 0.0085 | ||||||||
| 15 | 121212 | 0.01 | 0.02 | 0.03 | 0.0056 | ||||||||
| 222112 | 0.02 | 0.01 | 0.0034 | ||||||||||
| 211121 | 0.02 | 0.01 | 0.01 | 0.01 | 0.0045 | ||||||||
| 221112 | 0.02 | 0.0017 | |||||||||||
| 222122 | 0.03 | 0.0029 | |||||||||||
| A | 121112 | 0.02 | 0.0015 | ||||||||||
| 122122 | 0.02 | 0.01 | 0.0025 | ||||||||||
| 122112 | 0.02 | 0.0020 | |||||||||||
| Hap. Diversity | 0.70 ± 0.029 | 0.68 ± 0.029 | 0.81 ± 0.019 | 0.71 ± 0.023 | 0.80 ± 0.042 | 0.71 ± 0.031 | 0.78 ± 0.029 | 0.76 ± 0.021 | 0.84 ± 0.023 | 0.84 ± 0.022 | 0.67 ± 0.054 |
Only those estimated haplotypes above 2 per cent in any one population are listed. The inferred ancestral haplotype is denoted by the box.
Abbreviations: Afr-Amer = African-Americans; Euro-Amer = European-Americans; PNG = Papua New Guineans.
The 15 most frequent haplotypes (at least 1 per cent in the total sample) comprise 97.5 per cent of the total variation of all haplotypes. All 15 of these haplotypes are present in more than one population sample. Of the 15 haplotypes that comprise the remaining 2.5 per cent, 11 are unique to just one population. The 'root' haplotype (121112) -- determined by direct sequencing of four primate species -- is only found in the Temne, a population in West Africa, at a frequency of 0.02. The most haplotypes were found in the African-American (14), Temne (14) and PNG (13) samples. While the African-American sample exhibited one of the highest heterozygosity values (0.81 ± 0.019), however, the Temne (0.68 ± 0.029) and the PNG (0.67 ± 0.054) had the lowest of the 11 population samples. This is because the haplotype distributions of both the Temne and PNG samples were dominated by one haplotype with a frequency of at least 0.50, while the distributions of the haplotypes is more dispersed in the African-American sample. Two Asian populations, the Chinese (0.84 ± 0.023) and the Indonesians (0.84 ± 0.022), also had relatively high heterozygosity values compared with the rest of the samples.
Population-specific patterns of linkage disequilibrium in 17q11-12
Figure 3 describes the results of pairwise comparisons (D') for the six SNPs typed in MPIF-1, PARC an MIP-1a in each of the 11 population samples. Overall, there appears to be more heterogeneity in patterns of linkage disequilibrium when compared with the 3p21 region, although it must be noted that this analysis of 17q11-12 has a somewhat lower resolution. Pairwise comparisons between SNPs indicate strong linkage disequilibrium (D' > 0.8) in seven of the populations between PARC(6793) and MIP-1a(-1541), which are 20.5 kb apart. The notable exception is a Native African population sample (Temne) which has very little linkage disequilibrium between these two SNPs (D' = 0.1). It is interesting to note that the two African populations (the Mende and Temne) have distinctly different patterns of linkage disequilibrium across the 79 kb region, by contrast with the similarity of the analyses of the 3p21 SNPs.
Figure 3.

(a-k). The D' statistic for pairwise comparisons of the 6 SNPs genotyped in MPIF-1, PARC, and MIP-1a on 17q11-12 in each population sample. The axes of each plot indicate those polymorphic SNPs included in each analysis. SNP labels refer to numbering system described in Table 1. Abbreviations: Euro-Americans = European-Americans; PNG = Papua New Guineans.
Strong linkage disequilibrium is observed in the Chinese, and less so in the Mixe, between SNPs in the PARC gene (311 and 6793) and MPIF1(M106V), which is 50 kb away. This result is distinctly different from the pattern observed in the other nine populations as well as the pedigree-phased CEPH grandparents (n = 103), in which there is no significant linkage disequilibrium between MPIF1(M106V) and any of the other five SNPs included in the 79 kb region [41]. Whereas the other populations exhibit more moderate long-range linkage disequilibrium across this region, pairwise comparisons in Mixteca and Zapotec population samples indicate that strong linkage disequilibrium is maintained at the extremes of the region, as every SNP is in linkage disequilibrium with MIP-1a(-1541), even MPIF1(M106V).
Ancestral haplotypes on 17q11-12
The ancestral haplotype (121112) is infrequent in the total population sample (less than 1 per cent). The EM algorithm results indicate that the Temne population sample was the ancestral haplotype, at a frequency of 2 per cent. The minor alleles as the distinguishing loci for this haplotype [PARC(-116 T) and MIP-1a(-1541 C)] are at relatively high frequencies in all population samples, particularly in the African populations, where they range between 40 and 50 per cent. Only two haplotypes in the set of the 15 most frequent are just one mutation away from the ancestral haplotype: haplotype 12 (111112) and haplotype 15 (121212) -- both of which are found at around 1 per cent in the total population sample and in populations other than the Africans.
Recombination, in particular, seems to have generated the diversity of haplotypes observed in the combinations of six SNPs on 17q11-12. This recombination hampers efforts to distinguish the structure of relationships between haplotypes. Attempts to find a definitive haplotype network was impossible due to the parallel pathways that exist in this dataset. To define the overall pattern of variation, and to determine the relationship between haplotypes in each 'block', it will be necessary to type more SNPs within these genes and in intergenic regions.
Patterns of population differentiation
The distributions of estimated haplotype frequencies for both the 3p21 data and the 17q11-12 data were compared between the 11 population samples to calculate FST statistics, or the proportion of haplotype diversity that is attributable to differences between populations. Significance of the FST statistic was determined by randomly permuting haplotypes among populations, and those values that were not significant are highlighted in italics in Tables 5 and 6. For the 3p21 region (Table 5), several populations exhibited limited differentiation, apparently due to geography. The Mende and Temne -- two populations in Sierra Leone, in West Africa -- did not show significant differences in the distribution of haplotypes. Likewise, the Hispanic sample did not have a significantly different haplotype composition from the populations sampled in southern Mexico: the Mixteca, Mixe and the Zapotecs. This could be a result of Native American admixture in the Hispanic individuals sampled. The only two populations not to have significant results for the exact test of population differentiation for the 3p21 haplotypes [52] were the Mixe sample and the Zapotecs. The most diverse populations were the Temne and the PNGs, with an FST of 0.210.
Table 5.
FST values calculated from 3p21 haplotype frequencies
| Mende | Temne | Afr-Amer | Euro-Amer | Hispanics | Mixteca | Mixe | Zapotecs | Chinese | Indonesians | PNG | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mende | 0.000 | ||||||||||
| Temne | 0.001 | 0.000 | |||||||||
| Afr-Amer | 0.023 | 0.029 | 0.000 | ||||||||
| Euro-Amer | 0.126 | 0.144 | 0.067 | 0.000 | |||||||
| Hispanics | 0.122 | 0.141 | 0.059 | 0.006 | 0.000 | ||||||
| Mixteca | 0.137 | 0.151 | 0.073 | 0.041 | 0.013 | 0.000 | |||||
| Mixe | 0.169 | 0.185 | 0.101 | 0.063 | 0.018 | 0.027 | 0.000 | ||||
| Zapotecs | 0.156 | 0.176 | 0.092 | 0.042 | 0.006 | 0.020 | -0.001 | 0.000 | |||
| Chinese | 0.159 | 0.163 | 0.095 | 0.111 | 0.115 | 0.117 | 0.158 | 0.158 | 0.000 | ||
| Indonesians | 0.146 | 0.171 | 0.095 | 0.044 | 0.046 | 0.100 | 0.094 | 0.068 | 0.101 | 0.000 | |
| PNG | 0.183 | 0.210 | 0.133 | 0.088 | 0.076 | 0.130 | 0.089 | 0.064 | 0.167 | 0.028 | 0.000 |
Those FST values that did achieve significance with the permutation test [50] are indicated in italics. Those comparisons between populations that did not yield significant differences with the exact test of population differentiation [52] are indicated with a box.
Abbreviations: Afr-Amer = African-Americans; Euro-Amer = European-Americans; PNG = Papua New Guineans.
The 17q12 haplotypes showed a similar pattern of population clustering (Table 6), although for this dataset the distribution of haplotypes in the Indonesian sample showed similarity to the Chinese (FST = 002) and, surprisingly, the Hispanic sample (FST = 0.011). The overall level of higher population differentiation is apparent with the 17q11-12 data, as the FST values were generally greater than those determined by the analysis of the 3p21 SNPs. Again, the Temne and the PNG sample showed the most differentiation, with an FST of 0.303.
Table 6.
FST values calculated from 17q11-12 Those FST values that did achieve significance with the permutation test [50]
| Mende | Temne | Afr-Amer | Euro-Amer | Hispanics | Mixteca | Mixe | Zapotecs | Chinese | Indonesians | PNG | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mende | 0.000 | ||||||||||
| Temne | -0.004 | 0.000 | |||||||||
| Afr-Amer | 0.017 | 0.021 | 0.000 | ||||||||
| Euro-Amer | 0.242 | 0.248 | 0.141 | 0.000 | |||||||
| Hispanics | 0.225 | 0.233 | 0.120 | 0.018 | 0.000 | ||||||
| Mixteca | 0.232 | 0.238 | 0.128 | 0.005 | 0.006 | 0.000 | |||||
| Mixe | 0.248 | 0.256 | 0.144 | 0.028 | 0.001 | 0.021 | 0.000 | ||||
| Zapotecs | 0.233 | 0.241 | 0.133 | 0.065 | 0.049 | 0.053 | 0.060 | 0.000 | |||
| Chinese | 0.140 | 0.147 | 0.061 | 0.043 | 0.026 | 0.043 | 0.051 | 0.054 | 0.000 | ||
| Indonesians | 0.149 | 0.156 | 0.066 | 0.028 | 0.011 | 0.017 | 0.027 | 0.049 | 0.002 | 0.000 | |
| PNG | 0.297 | 0.303 | 0.222 | 0.258 | 0.154 | 0.216 | 0.153 | 0.235 | 0.208 | 0.163 | 0.000 |
Those population comparisons that did not yield significant differences with the exact test of population differentiation [52] are indicated with a box.
Abbreviations: Afr-Amer = African-Americans; Euro-Amer = European-Americans; PNG = Papua New Guineans.
Discussion
Interpretations of haplotype structure on 3p21
A haplotype block can be defined as a region in which a small proportion of pairwise comparisons of markers show evidence for historical recombination [57]. Under this definition, often greater than 90 per cent of the sample possesses only two to five haplotypes within a block [57,63]. Using the methods described by Gabriel et al.,[56] allelic association between pairs of SNPs is estimated by the D'-statistic and haplotype blocks consist of regions of contiguous SNPs with adjacent pairwise D' values over a threshold value (set at D' > 0.8 for this analysis). Others propose a haplotype block definition that describes the distribution of recombination crossovers between loci using a modification of the FGT [56].
These different 'philosophies' underlying haplotype block definitions can result in ambiguity in the reconstruction of haplotype structure in a region of interest, such as the chemokine receptor genes on chromosome 3p21. For the 14 polymorphisms scattered over the 150 kb region of 3p21, the variation in the total population sample is explained by 11 haplotypes above 1 per cent in the 11 population samples. All of these haplotypes are found in more than one population. There is evidence that long-range linkage disequilibrium exists between SNPs in the extremes of the 150 kb region, and that linkage disequilibrium has remained intact on one haplotype (haplotype 4) long enough to be present (at relatively low frequencies) in several populations on different continents. Moreover, linkage disequilibrium has remained intact between two SNPs, one in the CCR2 promoter (-5983 G/A) and the other in CCRL2(1137 C/G), approximately 50 kb from each other, long enough for the haplotype bearing both these mutations haplotype 10 to reach nearly 30 per cent in the African population sample.
Despite what appears to be long-range linkage disequilibrium, a potential past recombination event between CCR5 and CCR2 is evident in ten geographically distinct populations. The 'global' presence of this break between CCR2 and CCR5 could indicate a relatively old recombination event between two common haplotypes (haplotypes 4 and 7), or recurrent recombination in this region. Under both definitions used here (the minimum-linkage disequilibrium and the FGT), the 150 kb on 3p21 consists of at least two haplotype blocks in each of the 11 population samples. Where the results differed between the two methods, the FGT consistently generated a greater number of haplotype blocks. While the resolution of the analysis of 3p21 presented here is low (14 SNPs in 150 kb), these results indicate that definitions of haplotype blocks are very dependent on the method used to detect them -- even in a region of relatively strong linkage disequilibrium. Additionally, the population-specific patterns of haplotype blocks observed here in 3p21 echoes a previous cautionary note about using a single population sample to describe the structure of variation and haplotypes in a disease candidate region [33]. The variation in haplotype structure between populations can affect which SNPs are chosen as haplotype 'tags'. As indicated from the results presented here, using the htSNPs determined from the CEPH grandparent sample (Europeans) would not be sufficient to describe the overall haplotype structure of the Temne of Sierra Leone, particularly in detecting population-specific haplotypes (such as haplotype 10).
Root haplotypes and ascertainment bias
Fullerton et al. found that the 'root' haplotype of SNPs in the APOE gene, derived from a chimpanzee sequence, was found at a low copy number (only two copies in a total of 196 chromosomes) in their sample, one in a sample of African-Americans from Jackson, Mississippi, and one in a sample of non-Hispanic Caucasians from Rochester, New York [37]. In our analysis of 3p21 haplotypes, only one copy of the 'root' haplotype (11111121211111) was present, and it was found in the CEPH grandparent sample (included in the European-Americans). Unlike this present survey of chemokine receptor SNPs, however, the study of APOE by Fullerton et al. found at least one copy of the 'root' haplotype in a population sample of African origin (African-Americans from Jackson, Missis-sippi)[37]. Their finding is consistent with other studies that have found 'root' haplotypes in populations of African origin [64,65].
It is possible that the 'root' haplotype on chromosome 3p21 is not found in the African, or African-derived, population samples in the present analysis simply due to sampling error. As no sub-Saharan populations were included in this analysis (indeed, the sampling is limited to Sierra Leone in West Africa and African-Americans from metropolitan centres in the eastern USA), the 'root' haplotype could have been missed in Africa by this particular analysis. Hacia et al. found that in a survey of 397 SNP sites throughout the genome, the most frequent allele at an SNP is generally the ancestral allele; however, there were exceptions [66]. Of course, determining which allele is the 'most common' at the SNP site is subject to ascertainment bias in sampling of human populations [67]. In our study of 3p21 haplotypes, the 'root' haplotype (11111121211111) differs from the most common haplotype (11111111111111) by two SNPs [CCR2(N260N) and CCR5(303)] which are present at an appreciable frequency in every population sample.
In an analysis of 9 biallelic loci in 20kb on 3p21 (8 loci in CCR5 and V64I in CCR2), Gonzalez et al. find that the ancestral haplotype is at high frequency in African populations. This current analysis, which includes loci spread over 150 kb, does not include some of the SNPs genotyped in the aforementioned study, namely CCR5 -29 A/G, CCR5-627 T/C, CCR5 -630 C/T and CCR5-927 C/T. However, those SNPs that were typed in both studies CCR2 -V641 CCR5 -208 G/T, CCR5-303 G/A, CCR5-676 A/G and the CCR5 -32bp deletion, present the same ancestral allele.
Gonzalez et al. demonstrate that the ancestral haplotype has a high frequency in Africans and African-Americans (71 per cent in Pygmies, 24 per cent in non-Pygmy Africans and 21 per cent in African-Americans) [29]. Likewise, when the distribution of haplotypes that includes only the SNPs common to the Gonzalez et al. study and the analysis presented here is assessed, the frequency of the ancestral haplotype is high, particularly in the African (34 per cent) and African-derived samples (27 per cent). The scarcity of the 'root' haplotype with all 14 SNPs in the total sample may be a result of the relative age of CCR2 (N260N) and CCR5(303). 'Allele 2' at both of these SNPs, CCR2 (N260N_C) and CCR5 (303_A), has a wide frequency distribution, at 39 per cent and 71 per cent in African and 9 per cent and 15 per cent in PNGs, for example. Population genetics theory states that the oldest neutral SNP sites will generally involve cases where the derived allele is drifting toward fixation, and the ancestral allele is drifting to extinction [66,68]. The observed frequency distribution, and composition of the ancestral haplotype at 3p21, could be a result of drift acting upon these SNPs. Another possibility is the effects of balancing selection at the CCR5 promoter region [30].
Selection in chemokine and chemokine receptor genes
Bamshad et al. resequenced 1.1 kb of the CCR5 promoter region in 124 chromosomes from four populations in the 'Old World': Africans, non-Indian Asians, Europeans and South Indians [30]. Based on comparisons of nucleotide diversity (π, a direct estimate of per-site heterozygosity derived from the average pairwise sequence difference) in the CCR5 promoter with other regions in the genome, the authors conclude that the higher-than-average π values and SNP density in the CCR5 promoter are probably a result of balancing selection. As balancing selection increases the intra-population nucleotide diversity relative to the diversity in the total sample, loci under balancing selection are expected to have very different patterns compared with those loci under no selection (neutral loci) [69]. As the analysis presented here includes no sequences from the CCR5 promoter (only previously discovered single SNPs), we cannot assess comparable statistics in our own study.
Bamshad et al. also report much lower than average FST values (an average of 0.016 for the pooled continental populations, although this was not significant, at p > 0.09), however, which they interpret as being further evidence of the effects of balancing selection at CCR5 [30]. The estimates of FST for the CCR5 promoter are about five-fold lower than estimates of FST generated by genotype data in Alu sequences in these same individuals [30]. Our estimates of the proportion of total haplotype variation over 150 kb of 3p21 that is between population samples (as described by the FST values) are generally higher than those reported in Bamshad et al. [30] It would be expected that any effect balancing selection has on polymorphisms in the CCR5 promoter would be diluted by variation in the other genes included in the entire 150 kb haplotype. It should be noted, however, that, overall, the FST estimates determined by haplotypes in 3p21 are lower than those from haplotypes in the chemokine genes on chromosome 17q12.
Our analysis of SNPs in the 17q12 chemokine gene region yields an interesting result that warrants further study. The non-synonymous change (M106V) in the MPIF-1 chemokine gene has a great difference in frequency between African and non-African populations (75 per cent between populations in Sierra Leone and European-Americans). It is also evident that the most frequent allele at this site (106V) in the African sample is nearly fixed (95-96 per cent) and is not the ancestral allele (M106), based on sequence data from non-human primates. The ancestral allele (M106) is found at a much greater frequency in non-Africans, from 56 per cent in Indonesians to 80 per cent in European-Americans. The lowest frequency of M106 (allele 1) outside of Africa is 28 per cent in the PNGs. While there are not enough data here to make any definitive assessments about the effects of selection at MPIF-1, the pattern observed at this locus is intriguing. Genotype data at more SNPs within and around the gene will be necessary for further analyses of the patterns of variation, and possible effects of selection on MPIF-1.
Supplementary materials
Acknowledgements
The authors would like to thank Cherie Winkler, William Klitz, Mark Stoneking, Tim Garvey, David McLean and Stephen O'Brien for DNA samples. Bert Gold, Raymond Peterson, Bill Modi, George Nelson and Jim Lautenberger were always available for discussion about haplotype analysis. Special thanks to Jeff Long for MLOCUS and associated programs, and to Carrie Pfaff for modifications to the software. Ken Weiss, Andy Clark, Mark Shriver, Steve Schaeffer, Linda Frisse, Carrie Grimsley and, especially, Malia Fullerton provided comments on earlier drafts of this manuscript.
References
- Vaddi K, Keller M, Newton RC. 'The Chemokine Facts Book'. Academic Press, San Diego, CA; 1997. [Google Scholar]
- Walz A, Kunkel SL, Strieter RM. In: Chemokines in Diseas. Koch, A.E. and Strieter, R.M, editor. Chapman and Hall, New York, NY; 1996. 'C-X-C chemokines: An overview'. [Google Scholar]
- Bazan JF, Bacon KB, Hardiman G. et al. 'A new class of membrane-bound chemokine with a CX3C motif'. Natur. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- Pan Y, Lloyd C, Zhou H. et al. 'Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation'. Natur. 1997;387:611–617. doi: 10.1038/42491. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Imai T, Kakizaki M. et al. 'Molecular cloning of a novel C or gamma type chemokine, SCM-1'. FEBS Lett. 1995;360:155–159. doi: 10.1016/0014-5793(95)00093-O. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Imai T, Kakizaki M. et al. 'Identification of single C motif-1/lymphotactin receptor XCR1'. J Biol Chem. 1998;273:16551–16554. doi: 10.1074/jbc.273.26.16551. [DOI] [PubMed] [Google Scholar]
- Dragic T, Litwin V, Allaway GP. et al. 'HIV-1 entry into CD4 + cells is mediated by the chemokine receptor CC-CKR-5''. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- Alkhatib G, Combadiere C, Broder CC. et al. 'CC CKR5: A RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1'. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- Berger EA, Murphy PM, Farber JM. 'Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease'. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- Dean M, Carrington M, Winkler C. et al. 'Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study'. Science. 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- Liu R, Paxton WA, Choe S. et al. 'Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection'. Cell. 1996;86:367–377. doi: 10.1016/S0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- Samson M, Libert F, Doranz BJ. et al. 'Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene'. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- Martin MP, Dean M, Smith MW. et al. 'Genetic acceleration of AIDS progression by a promoter variant of CCR5'. Science. 1998;282:1907–1911. doi: 10.1126/science.282.5395.1907. [DOI] [PubMed] [Google Scholar]
- Kostrikis LS, Huang Y, Moore JP. et al. 'A chemokine receptor CCR2 allele delays HIV-1 disease progression and is associated with a CCR5 promoter mutation'. Nat Med. 1998;4:350–353. doi: 10.1038/nm0398-350. [DOI] [PubMed] [Google Scholar]
- Dean M, Jacobson LP, McFarlane G. et al. 'Reduced risk of AIDS lymphoma in individuals heterozygous for the CCR5-delta32 mutation'. Cancer Res. 1999;59:3561–3564. [PubMed] [Google Scholar]
- Rabkin CS, Yang Q, Goedert JJ. et al. 'Chemokine and chemokine receptor gene variants and risk of non-Hodgkin's lymphoma in human immunodeficiency virus-1-infected individuals'. Blood. 1999;93:1838–1842. [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE. et al. 'HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor'. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- Smith MW, Dean M, Carrington M. et al. 'Contrasting genetic influence of CCR2 and CCR5 variants on HIV-1 infection and disease progression. Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC), ALIVE Study'. Science. 1997;277:959–965. doi: 10.1126/science.277.5328.959. [DOI] [PubMed] [Google Scholar]
- Cocchi F, DeVico AL, Garzino-Demo A. et al. 'Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8 + T cells'. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- Cocchi F, DeVico AL, Yarchoan R. et al. 'Higher macrophage inflammatory protein (MIP)-1alpha and MIP-1beta levels from CD8 + T cells are associated with asymptomatic HIV-1 infection'. Proc Natl Acad Sci USA. 2000;97:13812–13817. doi: 10.1073/pnas.240469997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler C, Modi W, Smith MW. et al. 'Genetic restriction of AIDS pathogenesis by an SDF-1 chemokine gene variant. ALIVE Study, Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC)'. Science. 1998;279:389–393. doi: 10.1126/science.279.5349.389. [DOI] [PubMed] [Google Scholar]
- An P, Martin MP, Nelson GW. et al. 'Influence of CCR5 promoter haplotypes on AIDS progression in African-Americans'. AIDS. 2000;14:2117–2122. doi: 10.1097/00002030-200009290-00007. [DOI] [PubMed] [Google Scholar]
- Strieter RM, Polverini PJ, Arenberg DA. et al. 'Role of C-X-C chemokines as regulators of angiogenesis in lung cancer'. J Leukoc Biol. 1995;57:752–762. doi: 10.1002/jlb.57.5.752. [DOI] [PubMed] [Google Scholar]
- Arenberg DA, Polverini PJ, Kunkel SL. et al. 'The role of CXC chemokines in the regulation of angiogenesis in non-small cell lung cancer'. J Leukoc Biol. 1997;62:554–562. doi: 10.1002/jlb.62.5.554. [DOI] [PubMed] [Google Scholar]
- Moore BB, Arenberg DA, Addison CL. et al. 'Tumor angiogenesis is regulated by CXC chemokines'. J Lab Clin Med. 1998;132:97–103. doi: 10.1016/S0022-2143(98)90004-X. [DOI] [PubMed] [Google Scholar]
- Muller A, Homey B, Soto H. et al. 'Involvement of chemokine receptors in breast cancer metastasis'. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- Mummidi S, Ahuja SS, Gonzalez E. et al. 'Genealogy of the CCR5 locus and chemokine system gene variants associated with altered rates of HIV-1 disease progression'. Nat Med. 1998;4:786–793. doi: 10.1038/nm0798-786. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Bamshad M, Sato N. et al. 'Race-specific HIV-1 disease-modifying effects associated with CCR5 haplotypes'. Proc Natl Acad Sci USA. 1999;96:12004–12009. doi: 10.1073/pnas.96.21.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, Dhanda R, Bamshad M. et al. 'Global survey of genetic variation in CCR5, RANTES, and MIP-1alpha: impact on the epidemiology of the HIV-1 pandemic'. Proc Natl Acad Sci USA. 2001;98:5199–5204. doi: 10.1073/pnas.091056898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Mummidi S, Gonzalez E. et al. 'A strong signature of balancing selection in the 5" cis-regulatory region of CCR5'. Proc Natl Acad Sci USA. 2002;99:10539–10544. doi: 10.1073/pnas.162046399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M, Carrington M, O'Brien SJ. 'Balanced polymorphism selected by genetic versus infectious human disease'. Annu Rev Genomics Hum Genet. 2002;3:263–292. doi: 10.1146/annurev.genom.3.022502.103149. [DOI] [PubMed] [Google Scholar]
- Modi WS, Goedert JJ, Strathdee S. et al. 'MCP-1-MCP-3-Eotaxin gene cluster influences HIV-1 transmission'. AIDS. 2003;17:2357–2365. doi: 10.1097/00002030-200311070-00011. [DOI] [PubMed] [Google Scholar]
- Goldstein DB. 'Islands of linkage disequilibrium'. Nat Genet. 2001;29:109–111. doi: 10.1038/ng1001-109. [DOI] [PubMed] [Google Scholar]
- Johnson GC, Esposito L, Barratt BJ. et al. 'Haplotype tagging for the identification of common disease genes'. Nat Genet. 2001;29:233–237. doi: 10.1038/ng1001-233. [DOI] [PubMed] [Google Scholar]
- Nickerson DA, Taylor SL, Weiss KM. et al. 'DNA sequence diversity in a 9.7-kb region of the human lipoprotein lipase gene'. Nat Genet. 1998;19:233–240. doi: 10.1038/907. [DOI] [PubMed] [Google Scholar]
- Clark AG, Weiss KM, Nickerson DA. et al. 'Haplotype structure and population genetic inferences from nucleotide sequence variation in human lipoprotein lipase'. Am J Hum Genet. 1998;63:595–612. doi: 10.1086/301977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton SM, Clark AG, Weiss KM. et al. 'Apolipoprotein E variation at the sequence haplotype level: Implications for the origin and maintenance of a major human polymorphism'. Am J Hum Genet. 2000;67:881–900. doi: 10.1086/303070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning AM, Durocher F, Healey CS. et al. 'The extent of linkage disequilibrium in four populations with distinct demographic histories'. Am J Hum Genet. 2000;67:1544–1554. doi: 10.1086/316906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anon. 'dbSNP'. National Center for Biotechnology Information, National Institutes of Health; 2001. http://www.ncbi.nlm.nih.gov/SNP [Google Scholar]
- Nickerson DA, Tobe VO, Taylor SL. 'PolyPhred: Automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing'. Nucleic Acids Res. 1997;25:2745–2751. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark VJ, Dean M. 'Characterisation of SNP haplotype structure in chemokine and chemokine receptor genes with CEPH pedigrees and statistical estimation'. Hum Genomics. 2004;1:195–207. doi: 10.1186/1479-7364-1-3-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin PA, Saiz R, Monjazeb A. 'High-throughput single nucleotide polymorphism genotyping by fluorescent 5' exonuclease assay'. Biotechniques. 1999;27:538–540. doi: 10.2144/99273rr02. [DOI] [PubMed] [Google Scholar]
- Clark VJ, Metheny N, Dean M. et al. 'Statistical estimation and pedigree analysis of CCR2-CCR5 haplotypes'. Hum Genet. 2001;108:484–493. doi: 10.1007/s004390100512. [DOI] [PubMed] [Google Scholar]
- O'Connell JR, Weeks DE. 'PedCheck: A program for identification of genotype incompatibilities in linkage analysis'. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JC, Williams RC, Urbanek M. 'An E-M algorithm and testing strategy for multiple-locus haplotypes'. Am J Hum Genet. 1995;56:799–810. [PMC free article] [PubMed] [Google Scholar]
- Long JC. 'Multiple locus haplotype analysis (MLOCUS, OBSHAP, PAIRWISE)'. 2. Software and documentation distributed by the author, Section on Population Genetics and Linkage, Laboratory of Neurogenetics. NIAAA, National Institutes of Health, Bethesda, MD; 1999. [Google Scholar]
- Dempster AP. 'Maximum-likelihood from incomplete data via the EM algorithm'. J R Stat Soc B. 1977;39:1–38. [Google Scholar]
- Peterson RJ, Goldman D, Long JC. 'Effects of worldwide population subdivision on ALDH2 linkage disequilibrium'. Genome Res. 1999;9:844–852. doi: 10.1101/gr.9.9.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. Columbia University Press, New York, NY; 1987. [Google Scholar]
- Schneider S, Roessli D, Excoffier L. Arlequin: A software for population genetics data analysis. 2. Genetics and Biometry Laboratory, University of Geneva, Switzerland; 2000. [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. 'Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data'. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond M, Rousset F. 'An exact test for population differentiation'. Evolution. 1995;49:1280–1283. doi: 10.2307/2410454. [DOI] [PubMed] [Google Scholar]
- Zhang K, Jin L. 'HaploBlockFinder: Haplotype block analyses'. Bioinformatics. 2003;19:1300–1301. doi: 10.1093/bioinformatics/btg142. [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL. 'Statistical properties of the number of recombination events in the history of a sample of DNA sequences'. Genetics. 1985;111:147–164. doi: 10.1093/genetics/111.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Akey JM, Zhang K. et al. 'Distribution of recombination crossovers and the origin of haplotype blocks: The interplay of population history, recombination, and mutation'. Am J Hum Genet. 2002;71:1227–1234. doi: 10.1086/344398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H. et al. 'The structure of haplotype blocks in the human genome'. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S. et al. 'Linkage disequilibrium in the human genome'. Natur. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- Clayton D. 'SNPHap tool'. Innate Immunity in Heart, Lung and Blood Disease; Heart, Lung and Blood Institute, National Institutes of Health; 2003. http://www.innateimmunity.net [Google Scholar]
- Bandelt HJ, Forster P, Sykes BC. et al. 'Mitochondrial portraits of human populations using median networks'. Genetics. 1995;141:743–753. doi: 10.1093/genetics/141.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. 'Median-joining networks for inferring intraspecific phylogenies'. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Network 3.1.1.1. Life Sciences and Engineering Technology Solutions. 2003. http://www.fluxus-engineering.com/sharenet.htm
- Prettybase Formatter. 'Innate Immunity in Heart, Lung and Blood Disease'. Heart, Lung and Blood Institute, National Institutes of Health; 2003. http://www.innateimmunity.net [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF. et al. 'High-resolution haplotype structure in the human genome'. Nat Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- Harding RM, Fullerton SM, Griffiths RC. et al. 'Archaic African and Asian lineages in the genetic ancestry of modern humans'. Am J Hum Genet. 1997;60:772–789. [PMC free article] [PubMed] [Google Scholar]
- Harris EE, Hey J. 'X chromosome evidence for ancient human histories'. Proc Natl Acad Sci USA. 1999;96:3320–3324. doi: 10.1073/pnas.96.6.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacia JG, Fan JB, Ryder O. et al. 'Determination of ancestral alleles for human single-nucleotide polymorphisms using high-density oligonucleotide arrays'. Nat Genet. 1999;22:164–167. doi: 10.1038/9674. [DOI] [PubMed] [Google Scholar]
- Nickerson DA, Taylor SL, Fullerton SM. et al. 'Sequence diversity and large-scale typing of SNPs in the human apolipoprotein E gene'. Genome Res. 2000;10:1532–1545. doi: 10.1101/gr.146900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, UK; 1983. [Google Scholar]
- Schierup MH, Charlesworth D, Vekemans X. 'The effect of hitch-hiking on genes linked to a balanced polymorphism in a subdivided population'. Genet Res. 2000;76:63–73. doi: 10.1017/S0016672300004547. [DOI] [PubMed] [Google Scholar]