

Abstract
Objective:
To assess the available evidence for the diagnostic accuracy of CSF testing for protein 14-3-3 in patients with suspected sporadic Creutzfeldt-Jakob disease (sCJD).
Methods:
The authors performed a systematic review of the available literature from 1995 to January 1, 2011, to identify articles involving patients who were suspected of having sCJD and who had CSF analysis for protein 14-3-3. Studies were rated according to the American Academy of Neurology classification of evidence scheme for diagnostic studies, and recommendations were linked to the strength of the evidence. A pooled estimate of sensitivity and specificity was obtained for all studies rated Class II or higher. The question asked is “Does CSF 14-3-3 protein accurately identify Creutzfeldt-Jakob disease (CJD) in patients with sCJD?”
Results:
The analysis was conducted on the basis of samples of 1,849 patients with suspected sCJD from 9 Class II studies. Assays for CSF 14-3-3 protein are probably moderately accurate in diagnosing sCJD: sensitivity 92% (95% confidence interval [CI] 89.8–93.6), specificity 80% (95% CI 77.4–83.0), likelihood ratio of 4.7, and negative likelihood ratio of 0.10.
Recommendation:
For patients who have rapidly progressive dementia and are strongly suspected of having sCJD and for whom diagnosis remains uncertain (pretest probability ∼20%–90%), clinicians should order CSF 14-3-3 assays to reduce the uncertainty of the diagnosis (Level B).
Sporadic Creutzfeldt-Jakob disease (sCJD) is a rare disease caused by prion proteins that undergo conformational changes, resulting in an invariably fatal transmissible spongiform encephalopathy. The worldwide annual incidence is estimated to be 1:1,000,000.1 Affected individuals usually manifest a rapidly progressing dementing illness in the sixth or seventh decade of life. Confirming the diagnosis of sCJD has always been challenging. Whereas various diagnostic methods are commonly used, none matches the accuracy of a histopathologic diagnosis obtained by brain biopsy or at autopsy. Table e-1 on the Neurology® Web site at www.neurology.org presents various diagnostic criteria for sCJD diagnosis. The differential diagnosis of a patient presenting with rapidly progressive dementia is broad and includes primary neurodegenerative, inflammatory, metabolic, vascular, and malignant conditions. The wide spectrum of clinical manifestations in sCJD2,3 makes determining the diagnosis yet more difficult. Cognitive, emotional, sleep, motor, balance, and sensory abnormalities can all occur in sCJD. This inconsistent clinical picture can lead to a delay in diagnosis and in providing the necessary social and therapeutic interventions. Table e-2 presents conditions often mistaken for sCJD.
The test for the presence of protein 14-3-3 in the CSF of patients with suspected sCJD has been the subject of much debate. Initial studies demonstrated that the test was quite sensitive and specific in an appropriate clinical setting, particularly that of rapidly progressing cognitive decline.4,5 This sensitivity has been disputed, however.6 Variation in sensitivity has also been demonstrated on the basis of the duration of illness,7 Creutzfeldt-Jakob disease (CJD) subtype,8 and laboratory techniques.9,10,11 False-positive results occur in numerous disorders in which there is rapid neuronal loss, including acute stroke, encephalitis, and other dementing disorders.12 ELISA and Western blot (WB) techniques are the most commonly used techniques to detect 14-3-3 protein.
Before its discovery in humans, 14-3-3 protein was thought to be a brain-specific protein and was first described in studies of bovine brains. Investigators performed a starch gel-electrophoresis of various bovine brain proteins and found the one with the fastest mobility rate was that of 15-4-1 protein (S-100) followed by mobility rates overlapping between that of 14-3-2 (neuron-specific enolase [NSE]) and that of 14-3-3 protein.13 The unique migration pattern displayed on the starch gel-electrophoresis is also the source of the proteins' names.13,14 Boston et al.15 described 14-3-3 immunoreactivity in different body organs, and although highest levels were found in the brain, 14-3-3 protein was also detected in the liver, testis, spleen, intestines, adrenal glands, prostate, and lungs.15,16 The small 28–30 kDa 14-3-3 proteins have a growing list of postulated functions, including signal transduction, cell cycle modulation, and apoptosis.17,18 In 1996, Hsich et al.12 aimed to characterize 2 proteins, known as 130 and 131, which were found to be elevated in patients with sCJD; these 2 proteins turned out to be 14-3-3 proteins. Numerous studies followed examining the usefulness of CSF 14-3-3 protein assays in diagnosing sCJD.
The purpose of this review is to answer the following question: For patients with rapidly progressive dementia, does the presence of CSF 14-3-3 protein accurately identify patients who have sCJD?
DESCRIPTION OF THE ANALYTIC PROCESS
The PubMed/Medline, Cochrane Library, and EMBASE databases were searched using the terms 14-3-3 protein, CSF analysis, Creutzfeldt-Jakob disease, prion disease, dementia, and rapidly progressive dementia (exploded terms where appropriate) (appendix e-3). In addition, the reference lists of the articles identified were hand searched to identify articles that may have been missed in the initial search. Duplicates, reviews without original data, meeting abstracts, and case reports/series were excluded. The search included English-language articles and covered publications ranging from 1995 to January 1, 2011.
Studies in human subjects above 18 years of age were included. Nonsporadic cases from growth hormone use, genetic, iatrogenic (postsurgical), and new-variant (mad cow disease) prion diseases were excluded. Also excluded were non-CJD prion disorders and animal studies. In studies that looked at a mix of sCJD and other CJD subtypes, only the data on patients with sCJD were extracted for the analysis.
The articles were rated for their risk of bias according to the American Academy of Neurology classification of evidence criteria for diagnostic testing (appendix e-4), and recommendations were linked to the level of evidence (appendix e-5). In accordance with these criteria, studies with incorporation bias (the results of the 14-3-3 protein assay influenced the determination of the presence of sCJD) are rated Class IV, and studies with spectrum bias (which excluded a priori patients with uncertain diagnoses of sCJD) are rated Class III.
The number of patients, study design, data collection methods, patient population, diagnostic reference standard, and type of 14-3-3 assay used were collected. Other data extracted included the raw numbers of patients who tested positive vs negative for CSF 14-3-3 and their clinical or pathologic diagnosis, or both.
We constructed 2 × 2 tables with the presence or absence of 14-3-3 CSF protein as the independent variable and the presence or absence of sCJD as the dependent variable. We calculated sensitivities and specificities as the primary measures of diagnostic accuracy. Ninety-five percent confidence intervals (CIs) of the sensitivities and specificities were used as the measure of statistical precision.
CSF assays of 14-3-3 protein were dichotomized as either present or absent according to the authors' method of interpretation. For studies in which 14-3-3 analyses were performed with more than one technique,9,19 sensitivities and specificities were calculated for each technique used. To determine whether varying definitions of abnormal 14-3-3 (i.e., varying cutpoints for abnormal) introduced a threshold effect, we calculated the correlation between sensitivities and specificities (Spearman rank correlation coefficient).
We anticipated that the presence or absence of sCJD would be determined with varying levels of diagnostic certainty: definite sCJD—pathologic confirmation by biopsy or autopsy; probable sCJD—typical clinical presentation combined with confirmatory test results (e.g., periodic pattern on EEG, restricted diffusion on diffusion-weighted MRI); possible sCJD—typical clinical presentation without confirmatory ancillary tests; confirmed negative sCJD—pathologic confirmation or a prolonged course not compatible with sCJD.
For the primary calculation of sensitivity and specificity, we categorized patients meeting definite and probable criteria as sCJD present. Possible and confirmed negative sCJD were categorized as sCJD absent. To estimate a single value for the sensitivities and specificities we pooled the results of the identified studies with the lowest risk of bias in a meta-analysis. When studies appeared to acquire their patients from the same surveillance centers, signaling that the same population may have been used in more than one study,4,7,20 only the study enrolling the largest number of patients was considered in the pooled analysis.
To determine the dependence of the estimated sensitivities and specificities on the level of diagnostic certainty, we performed 2 sensitivity analyses. In these analyses, we categorized patients as either sCJD present or sCJD absent, using varying criteria as follows: primary analysis: sCJD present—definite and probable; sCJD absent—possible and confirmed negative; sensitivity analysis 1: sCJD present—definite; sCJD absent—probable, possible, and confirmed negative; sensitivity analysis 2: sCJD present—definite, probable, and possible; sCJD absent—confirmed negative.
The sensitivities and specificities resulting from these different analyses were compared through plotting of the data on a receiver operator characteristic (ROC) graph.
ANALYSIS OF EVIDENCE
Our search strategy identified 11,165 articles (3,488 from Medline, 5,254 from EMBASE, 59 from Cochrane Library, 2,364 from PubMed). After the primary screening in which the titles and abstracts were reviewed, 80 studies were deemed potentially relevant, and their full text was reviewed in the secondary screening process. Thirty-eight articles met inclusion criteria.
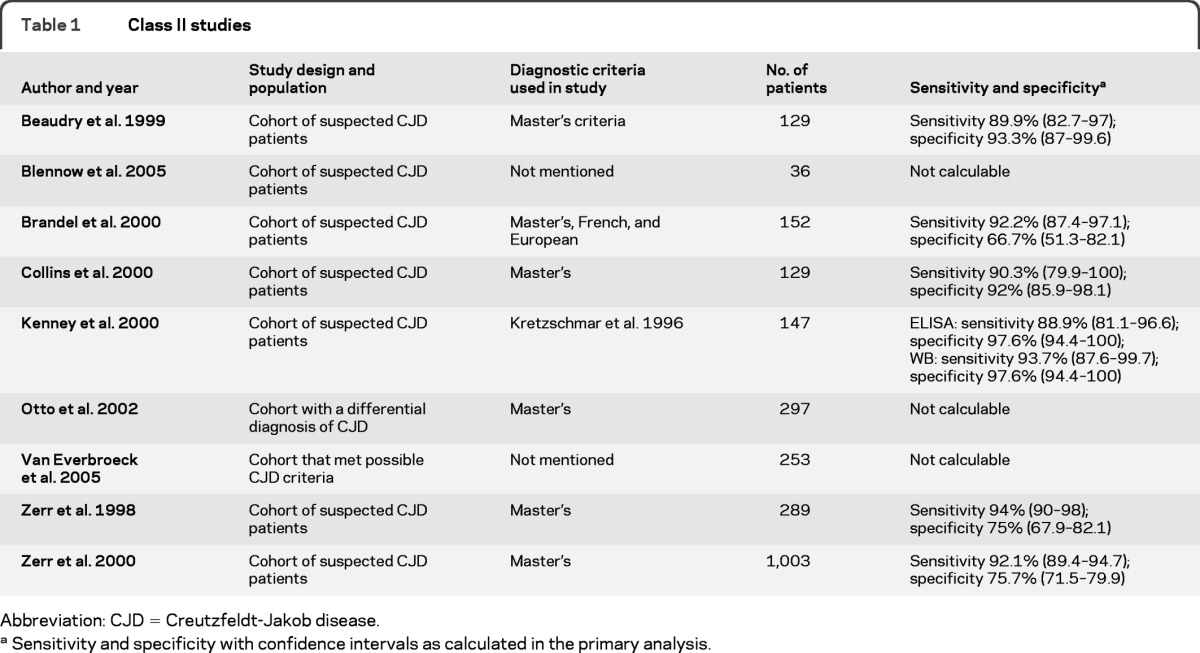
Of the 38 articles included in the study, 9 were deemed Class II; no Class I studies were identified. The majority of the Class II studies were cohort studies with mixed retrospective and prospective data collection (table 1). Fifteen studies were rated Class III, and another 14 were rated Class IV (table e-3). Downgrading was done if the cohort was incomplete. Spectrum bias in patients or controls was also a concern in a number of studies. All Class II studies enrolled patients with rapidly progressive dementia suspected of having CJD, and only these studies will be discussed further.
Table 1.
Class II studies
Abbreviation: CJD = Creutzfeldt-Jakob disease.
Sensitivity and specificity with confidence intervals as calculated in the primary analysis.
The most commonly referenced criteria the study authors used to identify patients with CJD vs patients without CJD was Masters et al. (1979)21 and the World Health Organization.22 Four authors used other criteria whereas 9 did not specify criteria.
Among the Class II studies, one small study followed a cohort of 36 patients with suspected sCJD and performed 9 autopsies, all of which showed definite sCJD. Surviving patients were followed for 2 years without manifesting a clinical picture of sCJD. The authors reported a sensitivity of 44% (95% CI 12–76.9) and specificity of 74% (95% CI 57.5–90.6) when WB analysis for 14-3-3 β isoform was used.23 Another Class II study, which had an objective of comparing 3 sets of diagnostic criteria (Master's, French, and European)24 for diagnosing CJD, included 236 autopsied patients who were classified antemortem into 3 case groups: suspected, probable, or possible. Then, on the basis of the autopsy results that were obtained from 61.2% of the cohort, the percentage of definite cases found in each of the groups was used to estimate the percentage of definite cases in the nonautopsied patient group. The authors concluded that the CJD diagnosis might be underestimated by 12% when European or French criteria were used and overestimated by 7% when the Master's criteria were used.24 Table 1 shows sensitivity and specificity for sCJD cases that met definite and probable criteria.
A Class II study comparing a novel ELISA technique with WB in detecting 14-3-3 β isoform showed that, after combining definite and probable cases as a reference, the sensitivities were 88.9% (95% CI 81.1–96.6) for ELISA and 93.7% (95% CI 87.6–99.7) for WB, with a specificity of 97.6% (95% CI 94.4–100) for both methods. The authors argued that ELISA might yield more-consistent results that are less likely to confound the assessment seen in WB techniques.9 Another Class II study aimed to compare a CSF analysis for tau by ELISA with a 14-3-3 immunoblot in a cohort of 297 patients with the differential diagnosis of CJD. The authors concluded that the 2 assays were quite comparable when used to assess for sCJD. The 14-3-3 immunoblot sensitivity and specificity were 90% and 88%, respectively.25 Data on definite sCJD cases were available for extraction with sensitivity of 86.2% (95% CI 79.8–92.7) and specificity of 83.5% (95% CI 75.6–91.4). An early Class II study that prospectively followed a cohort of 484 patients with suspected CJD investigated the results of 4 different interpretation strategies of the WB analysis for the β isoform of protein 14-3-3. Their interpretation strategy ranged from one that provided maximum avoidance of false-positive results (both raters unanimously agreed on the presence of 14-3-3) to one that provided maximum avoidance of false-negative results (both raters unanimously agreed on the absence of 14-3-3). The sensitivity ranged from 67.7% (95% CI 54.9–78.8) for the first strategy to 100% (95% CI 95.5–100) for the fourth in definite cases, with specificities ranging from 99% (95% CI 94.8–100) to 85.6% (95% CI 77.3–9 1.7), respectively.10
A Class II study by the Australian Creutzfeldt-Jakob Disease Registry looked at a cohort of 124 patients in which spongiform encephalopathy was considered in the differential diagnosis. Thirty-nine patients were considered 14-3-3 positive whereas 72 were considered negative. Thirteen CSF samples are unaccounted for in the article. The sensitivity and specificity are shown in table 1. The authors also clearly identified the potential of false-positive results occurring with CSF samples containing blood.26 This was found with red blood cell counts as low as 600/μL, and these were interpreted as either “weak” or “atypical.”
In another Class II study, the ability to determine whether the detection of antibodies against the γ isoform of 14-3-3 protein was able to improve on the differential diagnosis of sCJD was assessed from samples of 253 patients. Tests for the presence of 3 different antibodies in the CSF were conducted to detect the γ isoform of the protein. The most sensitive and specific antibody yielded a sensitivity of 96.2% (95% CI 90.9–100) and a specificity of 92.5% (95% CI 88.9–96.2), and no significant difference was found regardless of whether the WB technique or the ELISA technique was used.19
A Class II study recruited patients from multiple national registries and carried out CSF analysis with minor modifications in 1,003 patients with suspected CJD in each surveillance center. The non-CJD cases were identified as those that did not meet Master's criteria for CJD, and the mean follow-up time for these patients was 1.8–6.5 months. The CSF analysis for 14-3-3 protein yielded a sensitivity of 94% and a specificity of 84%. Extracted data on definite and probable sCJD are shown in table 1. The authors simultaneously compared the diagnostic accuracy of the 14-3-3 results with the presence of periodic sharp wave complexes on EEG testing and found that a positive 14-3-3 test was more sensitive and specific.20
The last Class II study looked at 129 CSF samples of patients with suspected CJD for 14-3-3, NSE, and S-100 protein and found all 3 biomarkers to be elevated. Extracted data on definite and probable sCJD cases showed a sensitivity of 89.9% (95% CI 82.7–97) and specificity of 93.3% (95% CI 87–99.6). Among the 3 biomarkers, the highest specificity was for protein 14-3-3 at 100% when only definite cases were considered.27
Measures of diagnostic accuracy.
Figure 1 plots on an ROC graph the sensitivity and specificity derived from each of the Class II studies.
Figure 1. Sensitivities and specificities for testing for presence of 14-3-3 for Creutzfeldt-Jakob disease (CJD).

A sensitivity analysis was performed to determine the dependence of the sensitivities and specificities on the varying diagnostic criteria for CJD. The squares in figure 1 indicate the diagnostic accuracy of the presence of 14-3-3 protein when only patients with definite CJD (biopsy or autopsy proven) were considered to have CJD (probable, possible, and confirmed negative CJD considered not to have CJD), the diamonds indicate diagnostic accuracy when definite and probable cases were considered to have CJD, and the triangles indicate the diagnostic accuracy when definite, probable, and possible were considered to have CJD. The analysis demonstrated that, despite the varying case definitions, the sensitivity remained relatively stable whereas the specificity decreased. We thus conclude that the estimate of the sensitivity of CSF 14-3-3 assays derived from this review is more reliable than the specificity.
There was significant correlation between sensitivity and specificity (coefficient of determination 0.89, p = 0.035), indicating that some of the heterogeneity in the results was potentially attributable to the varying cutpoints used to define an abnormal 14-3-3 level.
To estimate a single value for the sensitivities and specificities, results of the Class II studies were pooled in a meta-analysis. For this meta-analysis the sensitivities and specificities were derived from considering patients with definite and probable CJD to have CJD. The pooled sensitivity estimate was 91.9% (95% CI 89.8–93.6), and the pooled specificity estimate was 80.4% (95% CI 77.4–83.0). These values correspond to a positive likelihood ratio of 4.7 and a negative likelihood ratio of 0.10 (moderate diagnostic accuracy).
Conclusion
For patients with suspected CJD, CSF 14-3-3 assays are probably moderately accurate in diagnosing CJD: sensitivity ∼92%, specificity ∼80% (multiple consistent Class II studies). Whereas a negative 14-3-3 assay may be helpful in reducing the suspicion of sCJD, a positive CSF 14-3-3 assay may be found in a potentially treatable case of dementia.
Clinical context
The 14-3-3 assay, although of moderately high diagnostic accuracy, is an imperfect test. The test lacks the diagnostic accuracy either to include a CJD diagnosis as a possibility or to rule out CJD. It is important to realize that the test will not importantly change the probability of CJD in patients who are unlikely to have CJD to begin with. A positive result in such patients should not distract the investigator from considering a different dementing illness, or more important, a reversible cause of dementia. The relationship of this pretest probability of having CJD and the implications of a CSF 14-3-3 result are illustrated in figure 2. The upper curve in figure 2 indicates the degree to which the probability of sCJD will change if the CSF 14-3-3 analysis is positive. The lower curve indicates the degree to which the probability of sCJD will change if the CSF 14-3-3 analysis is negative.
Figure 2. Relationship of pretest probability of having Creutzfeldt-Jakob disease and the implications of a CSF 14-3-3 result.

The method by which the pretest probability is determined is usually implicit, and, as an example, can be based on the specialist's experience or knowledge about the incidence of the disease among individuals presenting with a suggestive clinical picture in a particular population.28 The dependence of the usefulness of CSF 14-3-3 assay on the pretest probability of sCJD can be illustrated by considering the case of a 72-year-old patient with dementia progressing over 18 months. Such a patient would appropriately be judged to have a very low pretest probability of CJD (<1%). A positive 14-3-3 test for this patient would most likely represent a false-positive result (posttest probability of CJD <5%). Likewise, the 14-3-3 assay would provide minimal information for patients with a high probability of having the disease. In the case of a 54-year-old with dementia progressing rapidly over 3 months and with symmetric MRI diffusion-weighted imaging (DWI) changes in the basal ganglia, the person is very likely to have CJD (>95%). A negative 14-3-3 test in this situation would most likely represent a false-negative result (posttest probability of CJD >80%).
The usefulness of the 14-3-3 assay will thus largely depend on a clinician's judgment of the pretest probability of CJD for a given patient. Such judgments will reasonably consider the rarity of CJD (incidence 1 per million per year), the practice setting (community hospital vs tertiary referral center), the patient's clinical presentation, and the results of already obtained ancillary tests such as head MRI. Figure 2 illustrates that the test is most useful when the pretest probability of CJD ranges from 20% to 90%. The use of other ancillary tests may affect this pretest probability. Periodic sharp and slow wave complexes (PSWC) on EEG testing have a sensitivity of 66% and specificity of 74%.20 PSWC tend to occur in older patients, and in half of the patients 6 months or more into the disease course, and then to decrease as the condition progresses.5 Despite the low sensitivity, in the right clinical setting EEG can be highly suggestive of sCJD; however, it is not entirely specific and is seen in various toxic or metabolic conditions or even during the late stages of Alzheimer disease or Lewy body dementia.29
Imaging studies are emerging as valuable tools in diagnosing CJD. DWI and fluid-attenuated inversion recovery (FLAIR) MRI sequences are more useful than EEG.29 The sensitivity and specificity of DWI and FLAIR in diagnosing CJD was found to be 91%–92.3% and 93.8%–95%, respectively.30,31 Reduced diffusivity of apparent diffusion coefficient images in the basal ganglia and cortical regions has been demonstrated and can last as long as 2 months.32
Finally, when considering the results of this analysis, it is important to highlight that there is significant variation in the way the 14-3-3 protein assays are performed.6,33 Most of the earlier studies used the WB technique, which is subjective and interpreted qualitatively. Later studies started to employ the quantitative ELISA technique, where sensitivity and specificity may vary according to the laboratory's determined cutoff.11,33 There also are various isoforms of the 14-3-3 protein, and tests have been conducted on the β isoform mostly; newer studies have started to test for the γ isoforms. The time in the course of the illness when the test is carried out and the disease subtype are also important factors that can contribute to variation in test results.33,34
PRACTICE RECOMMENDATION
For patients who have rapidly progressive dementia and are strongly suspected of having CJD, and for whom diagnosis remains uncertain (pretest probability ∼20%–90%), clinicians should order CSF 14-3-3 assays to reduce the uncertainty of the diagnosis (Level B).
RECOMMENDATIONS FOR FUTURE RESEARCH
Standardization of the definition of a positive 14-3-3 result and validation in well-designed cohort studies would be useful. Issues to be resolved include the question of which specific isoform (β or γ) of protein 14-3-3 is most useful and the need for standardization of the norms for the lab technique used (ELISA or WB).
Investigation for the presence of a combination of multiple biomarkers such as t-tau, p-tau, S-100, or NSE in the CSF is needed in addition to, or in lieu of, protein 14-3-3.
Integration of the recent advancement in MRI technology and DWI with CSF studies examining the presence or absence of protein 14-3-3 or another biomarker is needed.
Further investigation is needed into the utility of CSF biomarkers in subgroups of patients on the basis of demographics, time of presentation, duration of illness, specific etiologies of prion disease, or genetic factors.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Laura Moses Woodroffe and Julie Cox for their contributions in constructing the manuscript.
GLOSSARY
- CI
confidence interval
- CJD
Creutzfeldt-Jakob disease
- DWI
diffusion-weighted imaging
- FLAIR
fluid-attenuated inversion recovery
- NSE
neuron-specific enolase
- PSWC
periodic sharp and slow wave complexes
- ROC
receiver operator characteristic
- sCJD
sporadic Creutzfeldt-Jakob disease
- WB
Western blot.
Footnotes
Appendices e-1 through e-5 and tables e-1 through e-3 are available on the Neurology® Web site at www.neurology.org.
Approved by the Guideline Development Subcommittee on November 19, 2011; by the Practice Committee on February 17, 2012; and by the AAN Board of Directors on July 3, 2012.
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Statistical analysis performed by Gary Gronseth, MD, FAAN. All authors made substantive intellectual contribution to the submitted manuscript.
DISCLOSURE
T. Muayqil reports no disclosures. G. Gronseth serves as an editorial advisory board member of Neurology Now, serves on a speakers' bureau for Boehringer Ingelheim, and receives honoraria from Boehringer Ingelheim and the American Academy of Neurology. R. Camicioli reports no disclosures. Go to Neurology.org for full disclosures.
DISCLAIMER
This statement is provided as an educational service of the American Academy of Neurology. It is based on an assessment of current scientific and clinical information. It is not intended to include all possible proper methods of care for a particular neurologic problem or all legitimate criteria for choosing to use a specific procedure. Neither is it intended to exclude any reasonable alternative methodologies. The AAN recognizes that specific patient care decisions are the prerogative of the patient and the physician caring for the patient, based on all circumstances involved. The clinical context section is made available in order to place the evidence-based guideline(s) into perspective with current practice habits and challenges. No formal practice recommendations should be inferred.
CONFLICT OF INTEREST
The American Academy of Neurology is committed to producing independent, critical and truthful clinical practice guidelines (CPGs). Significant efforts are made to minimize the potential for conflicts of interest to influence the recommendations of this CPG. To the extent possible, the AAN keeps separate those who have a financial stake in the success or failure of the products appraised in the CPGs and the developers of the guidelines. Conflict of interest forms were obtained from all authors and reviewed by an oversight committee prior to project initiation. AAN limits the participation of authors with substantial conflicts of interest. The AAN forbids commercial participation in, or funding of, guideline projects. Drafts of the guideline have been reviewed by at least three AAN committees, a network of neurologists, Neurology peer reviewers and representatives from related fields. The AAN Guideline Author Conflict of Interest Policy can be viewed at www.aan.com.
REFERENCES
- 1. Ladogana A, Puopolo M, Croes EA, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 2005; 64: 1586– 1591 . [DOI] [PubMed] [Google Scholar]
- 2. Appleby BS, Appleby KK, Crain BJ, Onyike CU, Wallin MT, Rabins PV. Characteristics of established and proposed sporadic Creutzfeldt-Jakob disease variants. Arch Neurol 2009; 66: 208– 215 . [DOI] [PubMed] [Google Scholar]
- 3. Meissner B, Westner IM, Kallenberg K, et al. Sporadic Creutzfeldt-Jakob disease: clinical and diagnostic characteristics of the rare VV1 type. Neurology 2005; 65: 1544– 1550 . [DOI] [PubMed] [Google Scholar]
- 4. Sanchez-Juan P, Green A, Ladogana A, et al. CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 2006; 67: 637– 643 . [DOI] [PubMed] [Google Scholar]
- 5. Collins SJ, Sanchez-Juan P, Masters CL, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 2006; 129: 2278– 2287 . [DOI] [PubMed] [Google Scholar]
- 6. Geschwind MD, Martindale J, Miller D, et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol 2003; 60: 813– 816 . [DOI] [PubMed] [Google Scholar]
- 7. Van Everbroeck B, Quoilin S, Boons J, Martin JJ, Cras P. A prospective study of CSF markers in 250 patients with possible Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 2003; 74: 1210– 1214 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Castellani RJ, Colucci M, Xie Z, et al. Sensitivity of 14-3-3 protein test varies in subtypes of sporadic Creutzfeldt-Jakob disease. Neurology 2004; 63: 436– 442 . [DOI] [PubMed] [Google Scholar]
- 9. Kenney K, Brechtel C, Takahashi H, Kurohara K, Anderson P, Gibbs CJ., Jr An enzyme-linked immunosorbent assay to quantify 14-3-3 proteins in the cerebrospinal fluid of suspected Creutzfeldt-Jakob disease patients. Ann Neurol 2000; 48: 395– 398 . [PubMed] [Google Scholar]
- 10. Zerr I, Bodemer M, Gefeller O, et al. Detection of 14-3-3 protein in the cerebrospinal fluid supports the diagnosis of Creutzfeldt-Jakob disease. Ann Neurol 1998; 43: 32– 40 . [DOI] [PubMed] [Google Scholar]
- 11. Aksamit AJ, Jr, Preissner CM, Homburger HA. Quantitation of 14-3-3 and neuron-specific enolase proteins in CSF in Creutzfeldt-Jakob disease. Neurology 2001; 57: 728– 730 . [DOI] [PubMed] [Google Scholar]
- 12. Hsich G, Kenney K, Gibbs CJ, Lee KH, Harrington MG. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N Engl J Med 1996; 335: 924– 930 . [DOI] [PubMed] [Google Scholar]
- 13. Moore BW, Perez VJ. Specific acidic proteins of the nervous systems. In: Physiological and Biochemical Aspects of Nervous Integration. Englewood Cliffs, NJ: Prentice Hall; 1967 [Google Scholar]
- 14. Moore BW, McGregor D. Chromatographic and electrophoretic fractionation of soluble proteins of brain and liver. J Biol Chem 1965; 240: 1647– 1653 . [PubMed] [Google Scholar]
- 15. Boston PF, Jackson P, Kynoch PA, Thompson RJ. Purification, properties, and immunohistochemical localisation of human brain 14-3-3 protein. J Neurochem 1982; 38: 1466– 1474 . [DOI] [PubMed] [Google Scholar]
- 16. Fu H, Subramanian RR, Masters SC. 14-3-3 proteins: structure, function, and regulation. Annu Rev Pharmacol Toxicol 2000; 40: 617– 647 . [DOI] [PubMed] [Google Scholar]
- 17. Burbelo PD, Hall A. 14-3-3 proteins: hot numbers in signal transduction. Curr Biol 1995; 5: 95– 96 . [DOI] [PubMed] [Google Scholar]
- 18. Shaw A. The 14-3-3 proteins. Curr Biol 2000; 10: R400 . [DOI] [PubMed] [Google Scholar]
- 19. Van Everbroeck BR, Boons J, Cras P. 14-3-3 γ-isoform detection distinguishes sporadic Creutzfeldt-Jakob disease from other dementias. J Neurol Neurosurg Psychiatry 2005; 76: 100– 102 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zerr I, Pocchiari M, Collins S, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000; 55: 811– 815 . [DOI] [PubMed] [Google Scholar]
- 21. Masters CL, Harris JO, Gajdusek DC, Gibbs CJ, Jr, Bernoulli C, Asher DM. Creutzfeldt-Jakob disease: patterns of worldwide occurrence and the significance of familial and sporadic clustering. Ann Neurol 1979; 5: 177– 188 . [DOI] [PubMed] [Google Scholar]
- 22. Brown P, Budka H, Cervenakova L, et al. WHO manual for surveillance of human transmissible spongiform encephalopathies including variant Creutzfeldt-Jakob disease. World Health Organization, Communicable Disease Surveillance and Response; 2003 [Google Scholar]
- 23. Blennow K, Johansson A, Zetterberg H. Diagnostic value of 14-3-3beta immunoblot and T-tau/P-tau ratio in clinically suspected Creutzfeldt-Jakob disease. Int J Mol Med 2005; 16: 1147– 1149 . [PubMed] [Google Scholar]
- 24. Brandel JP, Delasnerie-Laupretre N, Laplanche JL, Hauw JJ, Alperovitch A. Diagnosis of Creutzfeldt-Jakob disease: effect of clinical criteria on incidence estimates. Neurology 2000; 54: 1095– 1099 . [DOI] [PubMed] [Google Scholar]
- 25. Otto M, Wiltfang J, Cepek L, et al. Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 2002; 58: 192– 197 . [DOI] [PubMed] [Google Scholar]
- 26. Collins S, Boyd A, Fletcher A, et al. Creutzfeldt-Jakob disease: diagnostic utility of 14-3-3 protein immunodetection in cerebrospinal fluid. J Clin Neurosci 2000; 7: 203– 208 . [DOI] [PubMed] [Google Scholar]
- 27. Beaudry P, Cohen P, Brandel JP, et al. 14-3-3 protein, neuron-specific enolase, and S-100 protein in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord 1999; 10: 40– 46 . [DOI] [PubMed] [Google Scholar]
- 28. Richardson WS, Wilson MC. The process of diagnosis. In: Users' Guides to the Medical Literature: A Manual for Evidence-Based Clinical Practice, 2nd ed New York, NY: McGraw-Hill; 2008 [Google Scholar]
- 29. Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol 2008; 64: 97– 108 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Young GS, Geschwind MD, Fischbein NJ, et al. Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol 2005; 26: 1551– 1562 . [PMC free article] [PubMed] [Google Scholar]
- 31. Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology 2004; 63: 443– 449 . [DOI] [PubMed] [Google Scholar]
- 32. Murata T, Shiga Y, Higano S, Takahashi S, Mugikura S. Conspicuity and evolution of lesions in Creutzfeldt-Jakob disease at diffusion-weighted imaging. AJNR Am J Neuroradiol 2002; 23: 1164– 1172 . [PMC free article] [PubMed] [Google Scholar]
- 33. Aksamit AJ. Cerebrospinal fluid 14-3-3 protein: variability of sporadic Creutzfeldt-Jakob disease, laboratory standards, and quantitation. Arch Neurol 2003; 60: 803– 804 . [DOI] [PubMed] [Google Scholar]
- 34. Pennington C, Chohan G, Mackenzie J, et al. The role of cerebrospinal fluid proteins as early diagnostic markers for sporadic Creutzfeldt-Jakob disease. Neurosci Lett 2009; 455: 56– 59 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.