

Abstract
Atmospheric pressure matrix assisted laser desorption/ionization (AP-MALDI)-derived tryptic peptide ions have been subjected to ion/ion reactions with doubly deprotonated 4-formyl-1,3-benzenedisulfonic acid (FBDSA) in the gas phase. The ion/ion reaction produces a negatively charged electrostatic complex composed of the peptide cation and reagent dianion, whereupon dehydration of the complex via collision-induced dissociation (CID) produces a Schiff base product anion. Collisional activation of modified lysine-terminated tryptic peptide anions is consistent with a covalent modification of unprotonated primary amines (i.e. N-terminus and ε-NH2 of lysine). Modified arginine-terminated tryptic peptides have shown evidence of a covalent modification at the N-terminus and a non-covalent interaction with the arginine residue. The modified anions yield at least as much sequence information upon CID as the unmodified cations for the small tryptic peptides examined here and more sequence information for the large tryptic peptides. This study represents the first demonstration of gas phase ion/ion reactions involving MALDI-derived ions. In this case, covalent modification upon charge inversion is shown to enhance MALDI tandem mass spectrometry of tryptic peptides.
Keywords: Ion/ion, gas phase covalent modification, tryptic peptides, AP-MALDI
INTRODUCTION
Peptide sequencing for identification and structural characterization of proteins is a longstanding activity in molecular biology research.1 Matrix-assisted laser desorption/ionization (MALDI)2,3 and electrospray ionization (ESI)4, two very successful means for the derivation of gas phase ions from proteins and peptides, coupled with tandem mass spectrometry, have become the dominant tools for generating sequence information from mixtures of peptides.5,6,7,8 While there is significant overlap, ESI and MALDI have each found applications for which they are best suited.9 For example, ESI has been widely used to interface on-line liquid separations with mass spectrometry, whereas MALDI is widely used for sampling surfaces, as in imaging applications. MALDI has been adapted to several tandem MS instrument geometries, e.g. tandem time-of-flight (i.e., TOF/TOF), 10 quadrupole/time-of-flight (i.e., Q/TOF), 11 and ion trap12,13,14 analyzers. The appeal of ion trap technology is that it permits MSn experiments to be conducted on MALDI-derived biomolecule-ions.15,16
From the standpoint of sequencing, a significant difference between MALDI and ESI is their propensities for generating multiply charged ions. Under commonly used conditions, MALDI generates ions of lower charge than does ESI. In fact, for peptides, singly charged ions usually dominate. Low energy collisional activation of singly protonated tryptic peptides, however, often does not produce extensive sequence information.17,18 Dissociation behavior is dominated by low energy fragmentation pathways, e.g. cleavage C-terminal to an aspartic acid residue or N-terminal to a proline residue.19 Furthermore, MALDI-derived tryptic peptides may generate limited sequence information due to the basic residue present at the C-terminus.20,21,22 Tryptic peptides often generate fragment ions associated with the C-terminus (e.g. y-ions) as a result of the C-terminal lysine or arginine sequestering the excess proton.20 A variety of approaches have been attempted to improve peptide sequencing in conjunction with MALDI. For example, condensed-phase peptide modification, such as fixed-charged derivatization of the N-terminus, has been attempted with mixed success.23,24 A noteworthy approach, developed by Keough et al., to increase sequence information from the dissociation of MALDI-derived tryptic peptides involved conjugating a sulfonic acid group to the N-terminus via solution phase chemistry.25,26 The objective was to alter fragmentation pathways by introducing a highly acidic group into the peptide. Fragmentation was dramatically improved in derivatized tryptic peptides compared to the underivatized tryptic peptides. However, the N-terminal position of this modification precluded the presence of N-terminal fragments due to the anionic sulfonate group.26
We and others have been developing approaches to modify ions in the gas phase and within the context of a tandem mass spectrometry experiment (i.e., modification of mass-selected ions) to expand the capabilities of tandem mass spectrometry for ion structure characterization using ion/ion reactions. 27 Ion/ion reactions can be used to decouple the ion-type initially generated by the ionization method from the ion-type subjected to tandem mass spectrometry.28 Structural information derived from fragmentation is highly dependent upon the nature of the ion (e.g., positive ion vs. negative ion, even-electron ion versus odd-electron, etc.).29 Therefore, ion/ion reactions can expand the range of ion-types that can be subjected to tandem mass spectrometry relative to the ion-types available from the ionization method. For example, ion/ion reactions have been used to manipulate peptide ion charge state30,31 and polarity32,33,34 via the transfer of one or more protons, generate odd-electron ions from even-electron ions via electron transfer,35,36 and insert metal ions into polypeptides.37 Recent studies have demonstrated the alteration of peptide dissociation behavior via the gas phase electrostatic attachment of reagents34, 38 and via the gas phase covalent modification of peptide ions.39, 40, 41 All such examples to date have been restricted to peptide ions generated via ESI.
In this work, we demonstrate the gas phase modification of peptide ions generated by atmospheric pressure MALDI (AP-MALDI) 42 using a dual source interface developed by Schneider et al.43 attached to a hybrid triple quadrupole/linear ion trap tandem mass spectrometer and the structural characterization of the modified and unmodified versions of the ions via ion trap CID. The formation of imine bonds (i.e. Schiff base formation) using an aldehyde containing dianion, 4-formyl-1,3-benzenedisulfonic acid (FBDSA)44,45 has been demonstrated with peptide ions generated via ESI. Possible sites of peptide covalent modification are the N-terminus and the ε-NH2 of lysine. Strong non-covalent interactions (i.e. electrostatic modification) have also been observed between the sulfonate groups of FBDSA and peptides containing an arginine residue and one or more carboxyl groups.38 Covalent bond cleavage is competitive with disruption of this strong electrostatic interaction under collisional activation conditions. In either case (i.e., covalent modification or strong electrostatic binding), the reaction product is a singly charged anion. The overall approach described here differs from the Keough et al. approach in that singly charged anions are subjected to CID, rather than singly charged cations, but is similar in that sulfonate is introduced into the peptide to alter dissociation behavior. Ion/ion reactions have been performed on tryptic peptides of ubiquitin and two synthetic peptides, APPGFSPFR and GLSDGEWQQVLNVWGK. Collisional activation of modified tryptic peptide anions demonstrates modification of the primary amine at the N-terminus and the C-terminal basic residue. Such modifications produce both modified N- and C-terminal fragment ions compared to the solely C-terminal information observed by Keough. Ion trap CID of the modified anions is shown to generate more sequence information than the unmodified ion, especially in the form of b-type ions. This study represents the first demonstration of gas phase modification of MALDI-derived ions via ion/ion reactions.
EXPERIMENTAL SECTION
Materials
Trifluoroacetic acid (TFA) was purchased from Pierce (Rockford, IL). Ubiquitin from bovine erythrocytes, TPCK treated trypsin from bovine pancreas, alpha-cyano-4-hydroxycinnamic acid (CHCA), FBDSA, and ammonium bicarbonate were purchased from Sigma-Aldrich (St. Louis, MO). Synthetic peptides APPGFSPFR and GLSDGEWQQVLNVWGK were purchased from SynPep (Dublin, CA). Peptides MQIFVK, TITLEVEPSDTIENVK, EGIPPDQQR, LIFAGK, TLSDYNIQK, and ESTLHLVLR were generated from a tryptic digestion of ubiquitin.
Methods
The MALDI matrix, CHCA, was prepared in 39.5/59.5/1 (v/v/v) water/acetonitrile/TFA at a concentration of 10 mg/mL. Peptide analytes were prepared in a ~100 μM aqueous solution prior to a 50/50 (v/v) mixture with the MALDI matrix solution. 2.0 μL of the matrix/analyte solution was spotted onto the MALDI stage plate and allowed to air-dry prior to attachment to the AP-MALDI source. The anion reagent was composed of a 3.5 mM solution of 50/50 (v/v) water/acetonitrile for negative micro-ionspray.
Tryptic Digest
The procedure for the tryptic digest of ubiquitin has been described previously. 46 Separation of the tryptic peptides was performed on a reverse-phase HPLC (Agilent 1100, Palo Alto, CA) using an Aquapore RP-300 (7μm pore size, 100 × 4.6 mm i.d.) column (Perkin-elmer, Wellesley, MA). The gradient for the HPLC separation has been described previously.46 Following HPLC separation, the collected fractions were concentrated and reconstituted in 200 μL of water.
Mass Spectrometry
Experiments were performed on a 4000 AB Sciex QTRAP (Concord, ON, Canada), which has been modified to perform ion/ion reactions.47 The ion source is equipped with an AP-MALDI source (MassTech, Columbia, MD) and with a micro-ionspray emitter (New Objective Inc, Waburn, MA) orthogonal to the sample inlet (Figure 1).43,48 The micro-ionspray emitter, remote to the MS inlet and MALDI stage plate, allows for the introduction of opposite polarity ions into the MS. Briefly, the AP-MALDI source has a circular opening, where a conductive transfer tube penetrates into the ion source and attaches to the curtain plate. The micro-ionspray is directed towards the transfer tube and upon ionization/nebulization, ions travel into the transfer tube and the ion source.
Figure 1.
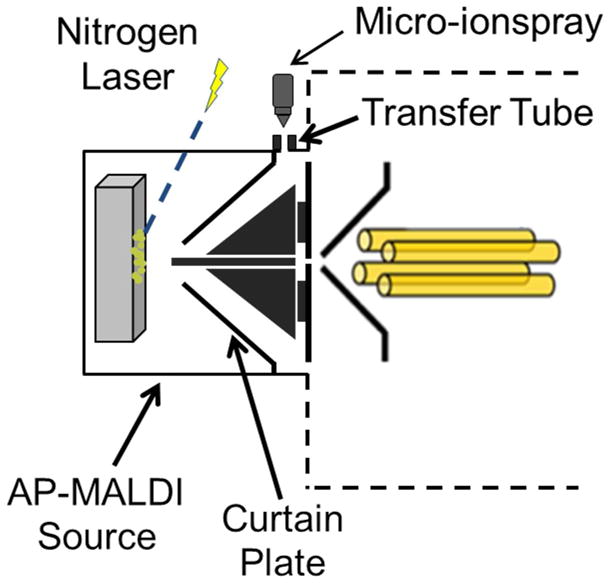
Modified AP-MALDI source, attached to a QTRAP 4000, capable of multisource injection.
The reagent anion, doubly deprotonated FBDSA, was generated via micro-ionspray at a continuous flow rate of 8.0 μL/min from a silica sprayer (364 μm outer dimension, 48 μm inner dimension, Polymicro Technologies, Phoenix, AZ). N2 acts as the nebulizing gas, which helps guide the reagent anions into the ion source. The nebulizing gas was triggered/pulsed via MS Expo software (AB Sciex, Concord, ON, Canada) during negative ion injection conditions. Once doubly deprotonated FBDSA entered the MS, it was transferred to the q2 collision cell and stored. Tryptic peptides, individually spotted in CHCA, were ablated via a 337-nm nitrogen laser (AP-MALDI Ion Source 110, MassTech, Columbia, MD) and transferred to the q2 collision cell to undergo ion/ion reactions with the reagent anions.49,50 In the peptide mixture study, multiple peptides from the reconstituted solutions were spotted on the MALDI stage plate with CHCA. All cations produced via MALDI in the mixture study were transferred to q2 without mass discrimination and reacted with FBDSA dianions. Product ions generated in q2 were transferred to Q3 for further collisional activation and subsequent mass analysis.51
RESULTS AND DISCUSSION
Ubiquitin was subjected to tryptic digestion and the tryptic peptides MQIFVK, TITLEVEPSDTIENVK, EGIPPDQQR, LIFAGK, TLSDYNIQK, and ESTLHLVLR were collected. The MALDI-derived tryptic peptide ions were subjected to ion/ion reactions with doubly deprotonated FBDSA. The main product of the ion/ion reaction is a negatively charged, long-lived complex comprised of the reagent dianion and tryptic peptide cation. The complex is formed when a negatively charged sulfonate interacts strongly with a protonated site on the peptide. The aldehyde group of the reagent undergoes nucleophilic attack by a neutral primary amine of the peptide during collisional activation of the complex leading to the formation of an imine bond (i.e., Schiff base) and loss of a water molecule. The Schiff base product anion is represented in spectra by [M+◆]−. The process of forming a Schiff base via gas phase ion/ion reactions is depicted in process (1):
| (1) |
The diamond symbol (◆) represents the mass addition of the reagent anion following a water loss. Fragments that are consistent in mass with those that retain the covalent modification are labeled with the diamond. The negative ion mode post-ion/ion reaction spectrum (Figure 2(a)) and positive MALDI mass spectrum of a mixture of three tryptic peptides (viz., LIFAGK, EGIPPDQQR, and TLSDYNIQK) are compared in Figure 2. Figure 2(a) was generated by exposing peptide cations of Figure 2(b) to reaction with doubly deprotonated FBDSA. The ion/ion reaction generated a product spectrum containing primarily the intact complex (i.e., [M+FBDSA]−) and signals due to [M+◆]− of much lesser abundance. The [M+◆]− ions were likely generated by CID upon transfer from q2 to Q3. Subsequent isolation and activation of the [M+FBDSA]− ion generates abundant [M+◆]− for subsequent interrogation. Contributions from strong electrostatic binding of the reagent were much more apparent in the data for the arginine terminated peptides than for the lysine terminated peptides (vide infra). For this reason, results from lysine and arginine terminated peptides are presented separately.
Figure 2.

a) Ion/ion product spectrum of peptide cation mixture and [FBDSA-2H]−2 b) Positive ion spectrum of peptide mixture LIFAGK, EGIPPDQQR, and TLSDYNIQK
Lysine Terminated Tryptic Peptides
Ion trap CID spectra of protonated TITLEVEPSDTIENVK and the modified peptide anion (i.e. [M+H]+ and [M+◆]−, respectively) are compared in Figure 3. Collisional activation of [M+H]+ leads to nine of fifteen amide bond cleavages, producing eleven fragment ions (Figure 3(b)). Fragmentation of the singly protonated peptide produces limited sequence informative ions as well as non-informative neutral losses. The product ion spectrum produces relatively large contributions from the fragment ions y6 and y9. The fragment ion y6 arises from the well-established C-terminal cleavage of aspartic acid,52 while the fragment ion y9 originates from the well-established N-terminal cleavage of the proline residue.53 Dominance of low-energy fragmentation pathways has been frequently observed in tandem MS experiments with MALDI-derived peptide cations.19 When compared to b-ions, the y-ions clearly dominate the fragmentation spectrum of the unmodified cation. Collisional activation of the peptide cation produced only four b-ions compared to seven y-ions. Limited N-terminal information (e.g. b-ions) from the collisional activation of [M+H]+ can be explained by the presence of a C-terminal basic lysine residue as reported by Biemann.20
Figure 3.

Ion trap CID product ion spectra of (a) [M+◆]− (b) [M+H]+ derived from M= TITLEVEPSDTIENVK
Collisional activation of modified TITLEVEPSDTIENVK (Figure 3(a)) produces distinct dissociation behavior compared to the unmodified spectrum. In the product ion spectrum, the most abundant peaks are associated with modified b- and y-ions, which are represented as b◆ or y◆. The nomenclature of modified fragment anions is adapted from the peptide literature for protonated peptides.54 The b◆-ions are interpreted to arise from a covalent modification of the N-terminus, while the y◆-ions are interpreted to arise from the covalent modification of the ε-NH2 of lysine. Collisional activation of the modified peptide anion produces a higher degree of fragmentation than the unmodified peptide cation, where twelve of fifteen amide bonds are cleaved, generating a total of sixteen fragment ions and a significant increase in N-terminal information. In the spectrum of the modified ion, seven b◆-ions are produced compared to four b-ions in the spectrum of the unmodified peptide. We note that previous studies have clearly shown that [M+◆]− ions yield significantly more sequence information upon CID than the corresponding [M−H]− ions.38,44,45 The incorporation of the two sulfonate groups into the peptide is expected to have a significant impact on proton mobility within the anion, which influences the favored dissociation pathways. This may favor charge remote processes55,56 or it may require the ions to be elevated to higher energies in order to promote intramolecular transfer. In any case, the disulfonic acid modification to the peptide has been shown to lead to consistently greater sequence coverage than singly charged versions of the unmodified peptide (i.e., [M+H]+ or [M−H]−).38,44,45 Furthermore, modification at the N-terminus gives rise to much greater contributions from anionic b◆-ions, which are not readily observed without the modification. Ion trap CID of [M+◆]− also appears to have less selective cleavage along the peptide backbone compared to the peptide cation, which has been observed in previous studies.45
Another ubiquitin tryptic peptide with a C-terminal lysine, TLSDYNIQK, produced results similar to those noted for the peptide TITLEVEPSDTIENVK. Ion trap CID of modified TLSDYNIQK (i.e. [M+◆]−) produced a high degree of sequence information, where a cleavage at every amide bond is observed (Figure 4(a)). The presence of many b◆- and y◆-ions is consistent with the covalent modification of the N-terminus and ε-NH2 of lysine. The high relative contribution of b4◆ in the product ion spectrum is attributed to the labile C-terminal cleavage of aspartic acid.52 The presence of b1◆ is particularly noteworthy because the b1-ion is not typically observed in the collisional activation of unmodified protonated and unprotonated peptides.57,58 Ion trap CID of the singly protonated peptide generates a product ion spectrum with seven of eight amide bond cleavages (Figure 4(b)); however, many less informative fragment ions are also observed in the spectrum (e.g., internal fragments and neutral losses). The product spectrum of the unmodified peptide cation is dominated by the C-terminal cleavage of aspartic acid, generating the highly abundant y5-ion. Conversely, the modified anion appears to fragment more uniformly. Dissociation of the peptide cation is dominated by the generation of y-ions, while the presence of b-ions is limited. The modified peptide anion produces a significant number of b◆-ions and an even higher number of y-type ions due to the presence of y1◆ and y2◆. With a near doubled increase of y-and b-type ions (i.e. thirteen versus seven fragment ions), the modified peptide ion clearly produced more sequence information compared to the unmodified peptide.
Figure 4.

Ion trap CID product ion spectra of (a) [M+◆]− (b) [M+H]+ derived from M= TLSDYNIQK
The peptides TITLEVEPSDTIENVK and TLSDYNIQK are illustrative of the synthetic peptide GLSDGEWQQVLNVWQK. Collisional activation of the modified and unmodified GLSDGEWQQVLNVWQK is provided in the Supplementary data (Figure S-1). Ion trap CID of [M+◆]− produces a high order of b◆- and y◆-ions (Figure S-1(a)), while the protonated peptide produces limited b- and y-ions (Figure S-1(b)). The modified peptide anion again produced a higher order of sequence information than the unmodified peptide cation. However, the aforementioned lysine containing peptides are not necessarily illustrative of low m/z peptides, such as MQIFVK (Figure 5) and LIFAGK (Figure S-2).
Figure 5.

Ion trap CID product ion spectra of (a) [M+◆]− (b) [M+H]+ derived from M= MQIFVK
Ion trap CID of modified MQIFVK (i.e. [M+◆]−) produces mainly y◆-ions at four of the five amide bond cleavages (Figure 5(a)) and only one ion in the b◆-ion series (i.e., the b◆5-ion), which is distinct from the observation of many more b◆-ions from the large modified peptide ions (i.e. TITLEVEPSDTIENVK, TLSDYNIQK, and GLSDGEWQQVLNVWQK). Ion trap CID of the unmodified peptide cation produces many b- and y-ions, which account for four of the five amide bond cleavages (Figure 5(b)). Smaller peptide ions tend to yield more extensive sequence information in general.17 While similar amide bonds are cleaved in the modified and unmodified peptide ions, there is a degree of complementarity from the two peptide ion-types. The modified anion produces lower m/z y◆-ions, while the [M+H]+ produces mainly b-ions. Also, the relative contributions of the fragment ions in the product ion spectra are markedly different. The modified peptide ion produces a highly abundant y3◆-ion, and the peptide cation generates a highly abundant internal fragment ion, showing the distinct dissociation behavior. The results from the peptide MQIFVK are illustrative of the peptide LIFAGK (Figure S-2). Ion trap CID of [LIFAGK+◆]− produces mainly modified y◆-ions, which is consistent with the covalent modification of the ε-NH2 of lysine. Collisional activation of the unmodified peptide cation produces a-, b-, and y-ions. Ion trap CID of both the modified and unmodified versions of LIFAGK displays a degree of complementarity, much like MQIFVK.
Arginine Terminated Tryptic Peptides
Previous studies of the ion trap CID of modified arginine containing peptides (i.e. angiotensin II (DRVYIHPF) and ubiquitin tryptic peptides) demonstrated product ion spectra consistent with two product populations: Schiff base formation and non-covalent binding of the reagent to the guanidinium side chain of arginine.39,45 Product ion spectra showed the appearance of y◆-ions; however, the only primary amine in the peptide capable of engaging in Schiff base formation was the N-terminus. The y◆-ions were determined to arise from [M+◆]− ions composed of an electrostatic interaction with the arginine guanidinium side chain. The water loss leading to the nominal [M+◆]− originated from elsewhere in the peptide (i.e., it was unrelated to the reagent). The presence of y+FBDSA-ions also provided evidence of an electrostatic rather than a covalent interaction. Model systems (i.e. YGGFLX peptides) further demonstrated strong interactions between the sulfonate groups and peptides containing an arginine residue and one or more carboxyl groups (e.g. C-terminus) in the gas phase.38 These non-covalent interactions with FBDSA dianions are sufficiently strong that covalent bond cleavage can compete with reagent detachment upon collisional activation.38,45
Ion trap CID of modified EGIPPDQQR is displayed in Figure 6(a). Collisional activation of the modified anion generated fragment ions consistent with covalent and electrostatic modifications. The presence of b◆-ions is consistent with the covalent modification of the N-terminus, while y◆ and y-FBDSA-ions are consistent with an electrostatic attachment to the peptide (e.g. reagent interaction with arginine and C-terminus). The presence of y◆-ions can be attributed to the electrostatic attachment of the FBDSA dianion to the C-terminal arginine and a water loss within the fragment ion (e.g. aspartic acid residue). The loss of [FBDSA-H]− also indicates an electrostatic interaction between the analyte and reagent. The loss of [FBDSA-H]− generates a neutral dehydrated peptide due to proton transfer within the complex, as seen in process (2).
Figure 6.

Ion trap CID product ion spectra of (a) [M+◆]− (b) [M+H]+ derived from M= EQIPPDQQR
| (2) |
The high relative abundances from [FBDSA-H]− and y3+FBDSA are particularly noteworthy. The significantly higher relative abundance of y3+FBDSA than [FBDSA-H]− suggests that the electrostatic interaction can compete with cleavage of covalent bonds upon collisional activation. Such a competition gives rise to the y◆ and y-FBDSA-ions.
Collisional activation of the modified anion produces a higher order of sequence information than the peptide cation. The modified anion produces a mixture of ten y+FBDSA and y◆-ions, and when combined, the mixture produces seven of the eight possible C-terminal amide bond cleavages. The modified peptide also produces more N-terminal fragments, i.e., b3◆, b6◆, and b8◆, relative to the unmodified peptide cation. Collisional activation of the singly protonated EGIPPDQQR produces limited sequence information in comparison to the modified version, as seen previously. The unmodified cation produces mainly y-ions, while only a single b-ion was produced. Dissociation of the peptide cation is dominated by cleavage C-terminal to the acidic residues, i.e., the y3- and y8-ions, as seen in many MALDI-derived MS/MS experiments. Activation of the unmodified peptide cation produces similar y-type ions to the modified version, while the modified anion produces lower m/z y-type ions that the unmodified version lacks.
Ion trap CID of the modified and unmodified synthetic tryptic peptide, APPGFSPFR, is illustrated in Figure 7. The modified anion produces cleavages at all eight amide bonds, and the most notable observation in the product ion spectrum is the generation of a b◆-ion at each peptide bond (Figure 7(a)), including the b1◆-ion. Collisional activation of [M+◆]− produces a combination of y◆- and y+FBDSA-ions. The product ion spectrum shows a high contribution from the well-established N-terminal cleavage of the proline residue (i.e. b6◆-H2O); however, more uniformity of cleavage among amide bonds is observed compared to the unmodified cation. While the ion trap CID of [M+H]+ (Figure 7(b)) and [M+◆]− produces similar higher m/z y-type product ions, the modified anion also produces lower m/z modified y-ions (i.e. y1+FBDSA). The most noteworthy observation of the product ion spectra is the drastic difference between the N-terminal fragment ion information. Collisional acitivation of the peptide cation produces only three b-ions, while the modified anion produces eight b◆-ions. The presence of the b◆-ions significantly increases the observed sequence information. The results of EGIPPDQQR and APPGFSPFR are illustrative of ubiquitin tryptic peptide ESTHLVLR (S-3). Ion trap CID of [ESTHLVLR+◆]− produces a mixture of y◆- and y+FBDSA-ions along with higher m/z b◆-ions. The unmodified peptide cation produces similar b- and y-type ions; however, the modified anion additionally produces lower m/z modified y-ions, i.e. y1+FBDSA.
Figure 7.

Ion trap CID product ion spectra of (a) [M+◆]− (b) [M+H]+ derived from M= APPGFSPFR
We note that many of the singly protonated peptides derived by MALDI and subjected to ion/ion reactions in this work have also been derived via electrospray and subjected to reactions with the same reagent.45 Very similar product ion spectra were derived from the same peptide ions derived by these two ionization methods. Not surprisingly, the main difference in the two approaches was that singly protonated peptides dominated in all cases in the MALDI experiment whereas multiply protonated peptides were the major ions noted for the larger peptides in the electrospray experiment.
CONCLUSIONS
Covalent and electrostatic modification of MALDI-derived tryptic peptide cations via gas phase ion/ion reactions with doubly deprotonated FBDSA has been demonstrated. Covalent modification is observed at the N-terminus and ε-NH2 of lysine for lysine terminated tryptic peptides. Covalent and electrostatic modification is observed for arginine terminated tryptic peptides, where the covalent modification occurs at the N-terminal primary amine and electrostatic modification occurs at the C-terminal arginine residue. Both modified lysine and arginine terminated tryptic peptides have shown to result in increased sequence information upon collisional activation compared to the unmodified version in high m/z peptides (>1000 Th). Ion trap CID of the modified anions generally shows an increase in the relative contribution of b-type ions, where unmodified peptide cations have shown limited b-ions. In many cases, the modified anion produces sequence-informative y-ions of lower m/z that are often not observed from the unmodified peptide cation. In general, modified anions have shown more uniform fragmentation compared to the unmodified cations, which often show dominant cleavages due to low energy CID pathways. This study demonstrates the first example of gas phase ion/ion reactions involving MALDI-derived ions (other than those that may occur inherently in the MALDI process). In this case, we demonstrate the gas phase modification of the ions to improve the MS/MS performance of MALDI-derived tryptic peptides without recourse to solution phase chemistry.
Supplementary Material
Acknowledgments
The AP-MALDI/electrospray interface for the instrument was generously provided by AB Sciex. Dr. Brad Schneider, Dr. James Hager, and Dr. Larry Campbell of AB Sciex are gratefully acknowledged for advice and assistance in adapting this hardware for ion/ion reactions. This research was supported by AB Sciex and the National Institutes of Health under Grant GM 45372.
References
- 1.Allen G. Sequencing of Proteins and Peptides. Elsevier/North-Holland; New York: 1981. [Google Scholar]
- 2.Karas M, Bachman D, Bahr U, Hillenkamp F. Int J Mas Spectrom Ion Processes. 1987;78:53–68. [Google Scholar]
- 3.Tanaka K, Ido Y, Akita S, Yoshida Y, Yoshida T. Rapid Commun Mass Spectrom. 1988;2:151–153. [Google Scholar]
- 4.Yamashita M, Fenn JB. J Phys Chem. 1984;88:4451–4459. [Google Scholar]
- 5.Bieman K, Martin SA. Mass Spectrom Rev. 1987;6:1–75. [Google Scholar]
- 6.Aebersold R, Mann M. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 7.Hunt DF, Yates J, Shabanowitz J, Winston S, Hauer CR. Proc Natl Acad Sci USA. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dongre AR, Jones JL, Somogyi A, Wysocki VH. J Am Chem Soc. 1996;118:8365–8374. [Google Scholar]
- 9.Cole RM. Electrospray and MALDI Mass Spectrometry: Fundamentals, Instrumentation, Practicalities, and Biological Applications. John Wiley and Sons; Hoboken, NJ: 2010. [Google Scholar]
- 10.Medzihradszky KF, Campbell JM, Baldwin MA, Falick AM, Juhasz P, Vestal ML, Burlingame AL. Anal Chem. 2000;72:552–558. doi: 10.1021/ac990809y. [DOI] [PubMed] [Google Scholar]
- 11.ShevchenkoALoboda A, Shevchenko A, Ens W, Standing KG. Anal Chem. 2000;72:2132–2141. doi: 10.1021/ac9913659. [DOI] [PubMed] [Google Scholar]
- 12.Schwartz JC, Bier ME. Rapid Commun Mass Spectrom. 1993;7:27–32. [Google Scholar]
- 13.Chambers DM, Goeringer DE, McLuckey SA, Glish GL. Anal Chem. 1993;65:14–20. [Google Scholar]
- 14.Jonscher K, Currie G, McCormack AL, Yates JR. Rapid Commun Mass Spectrom. 1993;7:20–26. doi: 10.1002/rcm.1290070106. [DOI] [PubMed] [Google Scholar]
- 15.Qin J, Steenvoorden RJJM, Chait BT. Anal Chem. 1996;68:1784–1791. doi: 10.1021/ac9511612. [DOI] [PubMed] [Google Scholar]
- 16.Laiko VV, Moyer SC, Cotter RJ. Anal Chem. 2000;72:5239–5243. doi: 10.1021/ac000530d. [DOI] [PubMed] [Google Scholar]
- 17.Qin J, Chait BT. Anal Chem. 1996;86:2108–2112. doi: 10.1021/ac951163m. [DOI] [PubMed] [Google Scholar]
- 18.Kaufmann R, Spengler B, Lutzenkichen F. Rapid Commun Mass Spectrom. 1993;7:902–910. doi: 10.1002/rcm.1290071010. [DOI] [PubMed] [Google Scholar]
- 19.Qin J, Fenyo D, Zhao Y, Hall WW, Chao DM, Wilson CJ, Young RA, Chait BT. Anal Chem. 1997;69:3995–4001. doi: 10.1021/ac970488v. [DOI] [PubMed] [Google Scholar]
- 20.Biemann K. In: Methods in Enzymology. McCloskey JA, editor. Academic Press; San Diego, CA: 1990. p. 445. [Google Scholar]
- 21.Biemann K. Biomed Environ Mass Spectrom. 1988;16:99–111. doi: 10.1002/bms.1200160119. [DOI] [PubMed] [Google Scholar]
- 22.Keough T, Lacey MP, Strife RJ. Rap Commun Mass Spectrom. 2001;15:2227–2239. doi: 10.1002/rcm.499. [DOI] [PubMed] [Google Scholar]
- 23.Huang Z, Wu J, Roth KDW, Yang Y, Gage DA, Watson JT. Anal Chem. 1997;69:137–144. doi: 10.1021/ac9608578. [DOI] [PubMed] [Google Scholar]
- 24.Bartlet-Jones M, Jeffery WA, Hanesen HF, Pappin DJC, Cottrell J. Rapid Commun Mass Spectrom. 1994;8:737–742. doi: 10.1002/rcm.1290080916. [DOI] [PubMed] [Google Scholar]
- 25.Keough T, Youngquist R, Lacey MP. Proc Natl Acad Sci USA. 1999;96:7131–7136. doi: 10.1073/pnas.96.13.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keough T, Youngquist R, Lacey MP. Anal Chem. 2003;75:156A–165A. doi: 10.1021/ac031274i. [DOI] [PubMed] [Google Scholar]
- 27.McLuckey SA, Huang TY. Anal Chem. 2009;81:8669–8676. doi: 10.1021/ac9014935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McLuckey SA. Eur J Mass Spectrom. 2010;16:429–436. doi: 10.1255/ejms.1031. [DOI] [PubMed] [Google Scholar]
- 29.McLuckey SA, Mentinova M. J Am Soc Mass Spectrom. 2011;22:3–12. doi: 10.1007/s13361-010-0004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stephenson JL, Jr, McLuckey SA. J Am Chem Soc. 1996;118:7390–7397. [Google Scholar]
- 31.Stephenson JL, Jr, McLuckey SA. Anal Chem. 1996;68:4026–4032. doi: 10.1021/ac9605657. [DOI] [PubMed] [Google Scholar]
- 32.He M, McLuckey SA. J Am Chem Soc. 2003;125:7756–7757. doi: 10.1021/ja0354521. [DOI] [PubMed] [Google Scholar]
- 33.He M, McLuckey SA. Anal Chem. 2004;76:4189–4192. doi: 10.1021/ac496087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He M, Emory JF, McLuckey SA. Anal Chem. 2005;77:3173–3182. doi: 10.1021/ac0482312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coon JJ, Shabanowitz J, Hunt DF, Syka JEP. J Am Soc Mass Spectrom. 2005;16:880–882. doi: 10.1016/j.jasms.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 37.Newton KA, Amunugama R, McLuckey SA. J Phys Chem A. 2005;109:3608–3616. doi: 10.1021/jp04416i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stutzman JR, Luongo CA, McLuckey SA. J Mass Spectrom. 2012;47:669–675. doi: 10.1002/jms.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han H, McLuckey SA. J Am Chem Soc. 2009;131:12884–12885. doi: 10.1021/ja904812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mentinova M, McLuckey SA. J Am Chem Soc. 2010;132:18248–18257. doi: 10.1021/ja107286p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGee WM, Mentinova M, McLuckey SA. J Am Chem Soc. 2012;134:11412–11414. doi: 10.1021/ja304778j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laiko VV, Baldwin MA, Burlingame AL. Anal Chem. 2000;72:652–657. doi: 10.1021/ac990998k. [DOI] [PubMed] [Google Scholar]
- 43.Schneider BB, Covey TR. J Am Soc Mass Spectrom. 2007;18:991–996. doi: 10.1016/j.jasms.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Hassell KM, Stutzman JR, McLuckey SA. Anal Chem. 2010;82:1594–1597. doi: 10.1021/ac902732v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stutzman JR, Hassel KM, McLuckey SA. Int J Mass Spectrom. 2012;312:195–200. doi: 10.1016/j.ijms.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han H, Xia Y, McLuckey SA. J Proteome Res. 2007;6:3062–3069. doi: 10.1021/pr070177t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia Y, Wu J, Londry FA, Hager JW, McLuckey SA. J Am Soc Mass Spectrom. 2005;16:71–81. doi: 10.1016/j.jasms.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 48.Schneider BB, Covey TR. US Patent 7,679,053 B2. Multiple Sample Sources for use with Mass Spectrometers, and Apparatus, Devices, and Methods Therefor. 2010 Mar 16;
- 49.Liang X, Xia Y, McLuckey SA. Anal Chem. 2006;78:3208–3212. doi: 10.1021/ac052288m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang X, Han H, Xia Y, McLuckey SA. J Am Soc Mass Spectrom. 2007;18:369–376. doi: 10.1016/j.jasms.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 51.Londry FA, Hager JW. J Am Soc Mass Spectrom. 2003;14:1130–1147. doi: 10.1016/S1044-0305(03)00446-X. [DOI] [PubMed] [Google Scholar]
- 52.Gu C, Tsaprailis G, Breci L, Wysocki V. Anal Chem. 2000;72:5804–5813. doi: 10.1021/ac000555c. [DOI] [PubMed] [Google Scholar]
- 53.Harrison AG, Young AB. J Mass Spectrom. 2000;40:1176–1183. doi: 10.1002/jms.891. [DOI] [PubMed] [Google Scholar]
- 54.Roepstorff P, Fohlman J. Biomed Mass Spectrom. 1984;11:601–602. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 55.Gross ML. Int J Mass Spectrom. 1992;118/119:137–165. [Google Scholar]
- 56.Cheng C, Gross ML. Mass Spectrom Rev. 2000;19:398–420. doi: 10.1002/1098-2787(2000)19:6<398::AID-MAS3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 57.Yalcin T, Khouw C, Csizmadia IG, Peterson MR, Harrison AG. J Am Soc Mass Spectrom. 1995;6:1165–1174. doi: 10.1016/1044-0305(95)00569-2. [DOI] [PubMed] [Google Scholar]
- 58.Yalcin T, Csizmadia IG, Peterson MR, Harrison AG. J Am Soc Mass Spectrom. 1996;7:233–242. doi: 10.1016/1044-0305(95)00677-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.