

Abstract
The identification of targets whose interaction is likely to result in the successful treatment of a disease is of growing interest for natural product scientists. In the current study we performed an exemplary application of a virtual parallel screening approach to identify potential targets for 16 secondary metabolites isolated and identified from the aerial parts of the medicinal plant Ruta graveolens L. Low energy conformers of the isolated constituents were simultaneously screened against a set of 2208 pharmacophore models generated in-house for the in silico prediction of putative biological targets, i. e., target fishing. Based on the predicted ligand-target interactions, we focused on three biological targets, namely acetylcholinesterase (AChE), the human rhinovirus (HRV) coat protein and the cannabinoid receptor type-2 (CB2). For a critical evaluation of the applied parallel screening approach, virtual hits and non-hits were assayed on the respective targets. For AChE the highest scoring virtual hit, arborinine, showed the best inhibitory in vitro activity on AChE (IC50 34.7 μM). Determination of the anti-HRV-2 effect revealed 6,7,8-trimethoxycoumarin and arborinine to be the most active antiviral constituents with IC50 values of 11.98 μM and 3.19 μM, respectively. Of these, arborinine was predicted virtually. Of all the molecules subjected to parallel screening, one virtual CB2 ligand was obtained, i.e., rutamarin. Interestingly, in experimental studies only this compound showed a selective activity to the CB2 receptor (Ki of 7.4 μM) by using a radioligand displacement assay. The applied parallel screening paradigm with constituents of R. graveolens on three different proteins has shown promise as an in silico tool for rational target fishing and pharmacological profiling of extracts and single chemical entities in natural product research.
Keywords: Ruta graveolens L, Rutaceae, pharmacophore modelling, virtual parallel screening, acetylcholinesterase, cannabinoid receptor 2, human rhinovirus coat protein
Introduction
Natural product molecules, which are characterized by delicate, complex scaffolds bearing many different functional groups, are progressively regarded as biologically validated drug leads and as promising sources for current drug development. In an early stage of the complex drug discovery process the identification of a single macromolecular target whose inhibition or stimulation is likely to result in the successful treatment of a disease of interest is a crucial step. This process is largely based on trial and error, and is risk-, time-, and cost-intensive. Several studies confirmed the high success rate of computer-assisted tools, such as virtual screening (VS), to increase the efficiency and efficacy of discovering lead structures in medicinal chemistry [1], [2], [3] and natural product science [4], [5]. One major point is that in silico modelling originally has not been developed for natural products, and computer models can only model pre-existing experimental knowledge. Furthermore, 3D-databases used for VS are restricted to already known metabolites; ab initio, novel, untapped chemical entities cannot be captured for in silico prediction.
In this study we tried to overcome this problem by subjecting a limited number of isolated constituents of a specific plant material to VS filtering experiments against a high number of generated pharmacophore models. This so-called 3 D pharmacophore-based parallel screening approach [6], [7] should (i) enable the researcher to focus specifically on those secondary metabolites which are characteristic for the natural material under investigation including potentially novel constituents, and (ii) help to characterize the pharmacological profile of the investigated extract on a molecular level by virtual prediction of the constituents’ biological functions, i. e., target fishing.
In pharmacophore-based parallel screening, each pharmacological target is represented by one or more pharmacophore models. Each model consists of a 3 D arrangement of essential chemical features that are responsible for the interaction of a compound and is the binding site of the pharmacological target [8]. Screening of compounds against a set of models which represent a large number of targets aims to predict the pharmacological profiles of these molecules including desirable activities (e. g., acetylcholinesterase inhibition), metabolism (e. g., via cytochrome P450 enzymes), and undesirable effects (e. g., hERG potassium channel block). The concept is shown in Fig. 1.
Fig. 1.
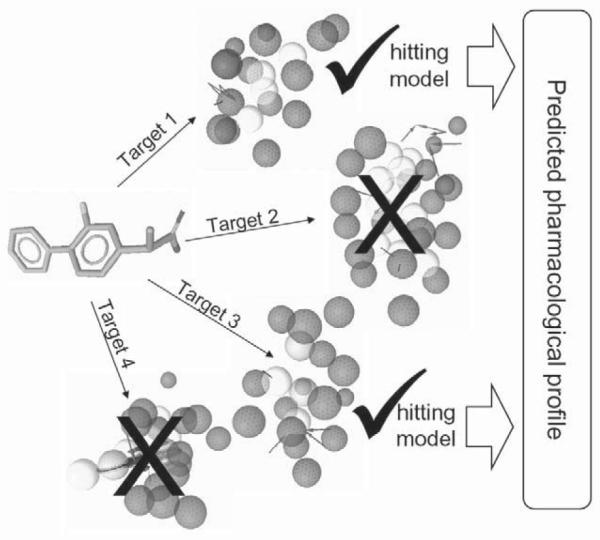
The concept of pharmacophore-based parallel screening mimicking the encounter of a ligand with different pharmacological targets.
Herein we present an exemplary application employing a virtual parallel screening approach using Discovery Studio (Accelrys Inc.) with a collection of 2208 in-house generated pharmacophore models on constituents of the aerial parts of the medicinal plant Ruta graveolens L.
R. geaveolens L. (Rutaceae) or rue is a small, yellow-flowering, evergreen shrub native to the Mediterranean region and the Balkans. The leaves or aerial parts of this aromatic plant have been used since ancient times to prevent contagion, to repel insects and to heal their bites [9]. In folk medicine rue has long been used as an antispasmodic, an emmenagogue, and an abortifacient [10]. Among its number of active constituents are quinoline, furanoquinoline, and acridone alkaloids, flavonoids, and coumarins, especially furocoumarins, which are responsible for the photosensitizing effect of rue on skin. German health authorities (European Scientific Cooperative on Phytotherapy) have concluded that neither rue nor any of its preparations should be utilized in medicine due to an unfavourable risk-benefit ratio and its unproven utility so far.
In the course of an in vitro extract screening of plants native to and cultivated in Tyrol [11], [12] a significant acetylcholinesterase (AChE) inhibiting activity was determined for the dichloromethane and methanol extracts of Rutae herba. This prompted us to phytochemically scrutinize these extracts. In the subsequent analytical and phytochemical investigations 16 constituents were isolated, identified, and subjected to a 3 D pharmacophore-based parallel screening (Fig. 2). Based on the predicted ligand-target interactions, we focused on three biological proteins, namely AChE, the human rhinovirus (HRV) coat protein, and the cannabinoid receptor type-2 (CB2). For a critical assessment of the parallel screening approach, virtual hits and non-hits were evaluated for their interactions on the selected target proteins. Based on the obtained pharmacological results exemplified by constituents of R. graveolens, the applied paradigm showed to be a highly promising computer-assisted tool for rational target fishing in pharmacognostic research.
Fig. 2.
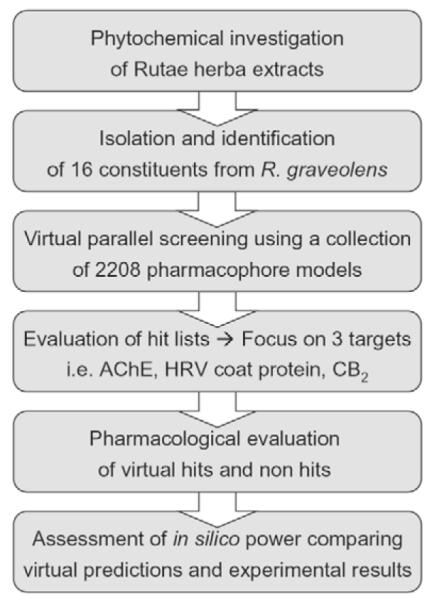
Workflow of the virtual parallel screening approach performed in this study.
Materials and Methods
General
Melting points were recorded on a Köfler hot-stage microscope and are uncorrected. Optical rotations were measured on a Perkin Elmer 341 polarimeter at 25 °C. FT-IR spectra were recorded on a Bruker IFS 25, FT-IR spectrometer in transmission mode within the range of 4000 to 600 cm−1. Samples were rolled on a ZnSe disk of 2 mm thickness. 1 D and 2 D NMR spectra were recorded on a Bruker DRX300 operating at a proton frequency of 300.13 MHz and a carbon frequency of 75.47 MHz. All spectra were recorded at 300 K in CDCl3 and calibrated to the residual solvent signal (δH = 7.26 ppm; δC = 77.0 ppm). Upon request, NMR spectra can be obtained from the corresponding author. LCMS: HPLC parameter: data were obtained on an Agilent 1100 system, equipped with a photodiode array detector and auto sampler. The LC was fitted with a Zorbax SB C-18 column, 150×4.6 mm i. d., 3.5 μm particle size (Agilent). HPLC method: column temperature 45 °C, a flow rate of 1.0 mL/min, injection volume 10 μL. The mobile phases consisted of A: water bidest. and B: acetonitrile (gradient grade; Merck); composition: start 20% B; 20 min 30% B; 30 min 98% B; stop 40 min. The photodiode array detector was set to detection at 210, 230, 275, and 320 nm. MS parameters: The mass spectrometer, Finnigan MAT SSQ 7000, was equipped with a Digital DEC 3000 data station (Digital Equipment Corporation); ESI (in negative or positive mode): LC flow split 1:5; capillary temperature: 200 °C; negative mode: spray voltage −4.5 kV; positive mode: 4.5 kV; CID (collision induced dissociation) 0 V in both cases; nebulizer 40 psi. For semipreparative HPLC a Dionex system with a P580 pump, ASI-100 autosampler, UVD 170U detector, and a Gilson 206 fraction collector was used. The system was fitted with a Phenomenex Synergi 10 μm Max-RP column (10×2500 mm for compounds 5, 6, 7) or XTerra® Prop MSC18 5 μm (7.8×100 mm, for compounds 9, 10, 11, 13) at a column temperature of 25 °C and a flow rate of 2.7 mL/min. The photodiode array detector was set to detection at 210 and 275 nm. All chemicals were analytical grade. Solvents were either of analytical grade or puriss. grade and distilled before use.
Plant material
Top plants of Ruta graveolens L. were cultivated and provided by Gartenbau Strillinger (Söll, Austria), and authenticated by A. Univ.-Prof. Dr. Christian Zidorn. A voucher specimen (JR-20 051009-A1) is deposited in the Herbarium of the Institute of Pharmacy/Pharmacognosy, University of Innsbruck, Austria.
Extraction and isolation
1190 g dried aerial parts of R. graveolens were ground to a fine powder, defatted with petroleum ether (3.6 L, room temperature), and consecutively macerated at room temperature with CH2Cl2 and MeOH (3.6 L CH2Cl2 and 4.8 L MeOH, twice for 3 days, respectively). Upon removal of the solvent under vacuum, the CH2Cl2 extract yielded 20.7 g, the MeOH extract 78.1 g. The former extract was suspended in n-hexane and fractionated by flash silica gel CC (6.0×50 cm, Merck silica gel 60, 0.040–0.063 mm, 230–400 mesh, 450 g) using a step gradient of n-hexane-CH2Cl2-MeOH (n-hexane; n-hexane/CH2Cl2 98:2; 95 :5; 90:10; 80:20; 60:40; 30:70; CH2Cl2; CH2Cl2/MeOH 98:2; 95:5; 90:10; 70:30; 60:40; 40:60; MeOH; 400 mL each) to give 13 fractions (A1 - A13) by using a TLC monitoring (SiO2, toluene/diethyl ether 1:1, saturated with 10% acetic acid, Dragendorff’s reagent). 1.44 g of fraction A13 (elution volume 4855–5170 mL, 2.03 g) was further separated by Sephadex® LH 20 CC (Pharmacia Biotech; 2.5×89 cm) with MeOH as mobile phase yielding 10 fractions (B1–10). Fractions B8 and B9 were combined (72 mg; elution volume 606–697 mL) and rechromato-graphed by Sephadex® LH 20 CC (2.5 × 89 cm) with the mobile phase CH2Cl2/acetone (v/v; 85:15) obtaining seven fractions (C1–7). C4 yielded compound 1 (27.0 mg, elution volume 350–368 mL; Rf: 0.24), and C2 compound 2 (9.8 mg, elution volume 320–338 mL; Rf: 0.23). 227 mg of fraction A10 (elution volume 4580–4640 mL, 364 mg) were subjected to Sephadex® LH 20 CC (2.5 × 99 cm) with CH2Cl2/acetone (v/v; 85:15) as mobile phase yielding 8 fractions (D1–8). D3 gave pure compound 3 (32.9 mg, elution volume 330–350 mL; Rf: 0.71), D5 yielded 18.2 mg of compound 4 (elution volume 370–390 mL; Rf: 0.73). Fraction D7 (71.4 mg, elution volume 405–425 mL) enriched with compounds 6 and 7 was subjected to semipreparative HPLC using a mobile phase of water/acetonitrile (v/v; 70:30 to 65:35 in 10 min, to 55:45 within further 10 min) to yield 21.8 mg of compounds 6 (22.9–28.3 mL elution volume; Rf: 0.49) and 9.7 mg of compound 7 (41.6–50.2 mL elution volume; Rf: 0.53). D8 (31.1 mg, elution volume 425–460 mL) enriched with compounds 5 was also subjected to semipreparative HPLC using a mobile phase of water/acetonitrile (v/v; 67:33 to 62:38 in 13 min) to give 11.1 mg of pure compound 5 (29.2–31.1 mL elution volume; Rf: 0.51).
For alkaloid enrichment, 69.0 g of the MeOH crude extract were suspended in 500 mL water/MeOH (v/v; 4:1) and acidified with 10% HCl to pH 2.5. The aqueous solution was extracted three times with 200 mL of CH2Cl2, to afford 22.0 g of an organic phase I. The remaining aqueous solution was alkalized with 10% aqueous NaOH to pH 9.0 and again extracted four times with 200 mL of CH2Cl2, to afford 1.2 g of a Dragendorff-positive organic phase II. The latter was fractionated by Sephadex® LH 20 CC (2.0×95 cm) with a step gradient of CH2Cl2/acetone (v/v; 85:15, 1000 mL; 50:50. 300 mL, and 600 mL acetone) yielding 13 fractions (E1–13). E3 (48.4 mg; elution volume 408–427 mL) was further separated by Sephadex® LH 20 CC (2.5×85 cm) with MeOH as mobile phase affording 17.3 mg of compound 15 (elution volume 256–264 mL; Rf: 0.77). E4 (425.1 mg; elution volume 472–544 mL) was subjected to semipreparative HPLC using a mobile phase of water/acetonitrile (v/v; 72:28 to 71.5:28.5 in 12 min) to yield compounds 10 (18.3 mg; elution volume 18.1–21.1 mL; Rf: 0.64) and 13 (19.4 mg; elution volume 25.9–29.7 mL; Rf: 0.79). Fraction E6 afforded pure compound 14 (11.5 mg; elution volume 608–680 mL; Rf: 0.72) and E11 yielded compound 8 (14.2 mg; elution volume 1208–1368 mL; Rf: 0.64). Combined fractions E8 and E9 (70.8 mg; elution volume 840–1064 mL) enriched with compounds 9 and 12, were subjected to semipreparative HPLC using a mobile phase of water/acetonitrile (v/v; 75:25 to 73:27 in 5 min, to 50:50 in further 5 min) to yield 15.5 mg of 9 (6.5–11.6 mL elution volume; Rf: 0.52) and 11.9 mg of 12 (24.3–27.0 mL elution volume; Rf: 0.74). E12 (48.4 mg; elution volume 1368–1800 mL) was further purified by Sephadex® LH 20 CC (2.5×85 cm) with MeOH as mobile phase yielding pure compound 16 (10.4 mg; elution volume 277–288 mL; Rf: 0.47) and compound 11 (1.6 mg; elution volume 329–465 mL; Rf: 0.59). Physical and spectroscopic data measured for compounds 1–13 are in accordance with the literature cited (see Results secton).
Methyl 3-hydroxy-3-(4-hydroxy-3,5-dimethoxyphenyl)propanoate (14): yellow amorphous powder (MeOH); glass transition point 35–38 °C; UV (MeOH): λmax = 207, 238, 269, 282 nm; FTI-R (on ZnSe): νmax = 3378, 2937, 2853, 1727, 1613, 1518, 1461, 1436, 1367, 1330, 1216, 1116 cm−1. : −1.03 (c 0.484, MeOH); LC-MS (ESI, pos. ion mode): m/z = 278.8 [M + Na]+, 534.9 [2M + Na]+; HR-FAB-MS: m/z = 256.13 (calcd. for C12H16O6:256.09); NMR data are given in Table 1.
Table 1.
1H-NMR (300 MHz) and 13C-NMR (75 MHz) data of compounds 14, 15, and 16; measured at 300 K; solvent: CDCl3 (δ in ppm, J in Hz)
| 14 | 15 | 16 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Position | δH | δC | HMBC | δH | δC | HMBC | δH | δC | HMBC |
| 1 | – | 133.5 | – | – | 132.1 | – | – | 130.3 | – |
| 2 | 6.62 (s) | 101.8 | C-3, 4, 6, 7 | 6.51 (s) | 103.4 | C-7, 3, 4, 6 | – | 142.9 | – |
| 3 | – | 147.1 | – | – | 146.8 | – | – | 137.2 | – |
| 4 (16 : 3a) | – | 133.9 | – | – | 133.8 | – | – | 144.6 | – |
| 5 (16 : 5a) | – | 147. 1 | – | – | 146.8 | – | – | 125.9 | – |
| 6 | 6.62 (s) | 101.8 | C-2, 4, 5, 7 | 6.51 (s) | 103.4 | C-7, 2, 4, 5 | 7.05 (s) | 114.3 | C-1, 2, 3a, 5, 5a |
| 7 | 5.07 (dd, 8.8, 4.1) | 69.6 | C-2, 6, 9 | 3.54 (d, 8.8) | 48.1 | C-2, 6, 8, 9 | 3.10 (m) 3.21 (m) |
26.3 | C-1, 2, 6, 8, 9 |
| 8 | 2.73 (dd, 8.8, 4.1) | 43.0 | C-1, 7, 9 | 3.43 (d, 8.8) | 44.1 | C-1, 7 | 2.65 (m) 2.77 (m) |
34.5 | C-1, 7, 9 |
| 9 | – | 172.7 | – | – | 172.8 | – | – | 174.2 | – |
| 16 : 4 | – | – | – | – | – | – | 7.58 (d, 2.1) | 145.3 | C-3a, 5, 5a |
| 16 : 5 | – | – | – | – | – | – | 6.68 (d, 2.1) | 106.5 | C-3a, 4, 5a |
| 3-OCH3 | 3.90 (s) | 55.6 | C-3 | 3.85 (s) | 56.1 | C-3 | 4.22 (s) | 60.6 | C-3 |
| 5-OCH3 | 3.90 (s) | 55.6 | C-5 | 3.85 (s) | 56.1 | C-5 | – | – | – |
| 9-OCH3 | 3.74 (s) | 50.7 | C-9 | 3.75 (s) | 52.0 | C-9 | 3.69 (s) | 51.5 | C-9 |
Methyl 3-(4-hydroxy-3,5-dimethoxyphenyl)oxirane-2-carboxylate (15): white microcrystalline powder (prisms; MeOH); m.p. 101.5–104.0 °C; UV (MeOH): λmax = 210, 242, 273, 284 nm; FTIR (on ZnSe): νmax = 3448, 2955, 2844, 1732, 1612, 1520, 1461, 1339, 1218, 1116 cm−1; : + 8.05 (c 0.795, MeOH); LC-MS (ESI, pos. ion mode): m/z = 276.9 [M + Na]+; HR-FAB-MS: m/z = 254.23 (calcd. for C12H14O6:254.08); NMR data are given in Table 1.
Methyl 3-(6-hydroxy-7-methoxybenzofuran-5-yl)propanoate (16): light yellow amorphous powder (MeOH); glass transition point 35–38 °C; UV (MeOH): λmax = 211, 250, 276, 288 nm; FT-IR (on ZnSe): νmax = 3376, 2938, 2880, 1725, 1656, 1596, 1514, 1467, 1439, 1078 cm−1; LC-MS (ESI, pos. ion mode): m/z = 250.7 [M + H]+; HR-FAB-MS: m/z = 250.12 (calcd. for C13H14O5: 250.08); NMR data are given in Table 1.
Pharmacophore modelling
A pharmacophore is the ensemble of steric and electronic features that is necessary to ensure optimal supramolecular interactions with a specific biological target and to trigger or block its biological response [13]. The pharmacophore models used for virtual parallel screening were developed employing structure-based approaches using the software LigandScout (Inte:-Ligand GmbH [14]). In cases where no 3 D structure of the biological target was available, ligand-based approaches were performed using the Catalyst software (Catalyst Vers. 4.5–4.11; Accelrys Inc.). All pharmacophore models were evaluated for their ability to correctly identify known active compounds among inactive ones represented by a set of low-energy conformers.
Parallel screening
The concept of pharmacophore-based parallel screening has recently been introduced by our working group [6], [7]. The technology to perform simultaneous, parallel screening of one or more compounds against a multitude of pharmacophore models is available as a Pipeline Pilot-based program protocol included in Discovery Studio 2.01 (Accelrys; SciTegic). Together with this software, the Inte:Ligand pharmacophore model collection (Inte:Ligand GmbH) can be used for parallel screening. This collection currently comprises 2208 pharmacophore models covering over 280 unique pharmacological targets.
Molecular structures of the 16 Ruta constituents isolated were submitted to Monte Carlo-based conformational analysis (Discovery Studio’s FAST algorithm) allowing maximal 255 conformers with less than 20 kcal/mol above the energy minimum. VS was performed in RIGID mode not allowing the omitting of any features. The interblob distance parameter was set to 0.01. For each compound, a result sheet was returned indicating which models were hits and how well the compound mapped into each model (fit value), respectively.
Pharmacological assays
Spectrophotometric assay for determination of AChE inhibitory activity
The AChE inhibitory activity was determined using a modified Ellman’s method [15], [16] with AChE EC 3.1.1.7, acetylthiocholiniodide, and 5,5′-dithiobis-(2-nitrobenzoic acid) (Sigma-Aldrich Chemie Gmbh); galanthamine.HBr (Tocris; Cookson Ltd; purity > 98%) served as the positive control in our assay (IC50 of 3.2 ± 1.0 μM) using a 96-well microplate assay as previously described [17]. The percentage of enzyme inhibition was calculated by determining the rate in the presence of inhibitor and the vehicle (containing 1% DMSO) compared to the rate in the control sample (n = 4) and analyzed with Student’s t-test.
Evaluation of cytotoxicity and antiviral activity
Both assays were described previously [18], [19]. Briefly, the 50% cytotoxic concentration (CC50) was determined on two day-old confluent HeLa cell monolayers, which were incubated with serial two-fold compound dilutions for 72 h (37 °C, 5% CO2). Then, the cells were fixed and stained with a crystal violet formalin solution. After dye extraction, the optical density of individual wells was quantified spectrophotometrically at 550/630 nm with a microplate reader. Cell viability of individual compound-treated wells was evaluated as the percentage of the mean value of optical density resulting from six mock-treated cell controls, which was set as 100%. The 50% cytotoxic concentration (CC50) was defined as the compound concentration reducing the viability of untreated cell cultures by 50%.
Cytopathic effect (CPE) inhibitory assays
CPE assays have been performed with rhinovirus 2 (HRV-2) in one-day-old confluent HeLa cell monolayers. After removal of the culture medium, drug solutions (dilution factor 2) and virus (multiplicity of infection of 0.01 of HRV-2) were added to the cells. Pleconaril synthesized by Makarov and co-workers [19] (purity 98 %) was used as reference compound. By using the crystal violet uptake assay described for cytotoxic investigations, the inhibition of the virus-induced CPE was scored 72 h after infection.
Radioligand displacement assays on CB1 and CB2 receptors
For the CB1 receptor, binding experiments were performed in the presence of 0.39 nM of the radioligand [3H]CP-55,940 at 30 °C in siliconized glass vials together with 7.16 μg of membrane recombinantly overexpressing CB1 receptor (RBHCB1M; Perkin Elmer Life Sciences), which was resuspended in 0.2 mL (final volume) of binding buffer (50 mM Tris-HCl, 2.5 mM EGTA, 5 mM MgCl2, 0.5 mg/mL fatty acid free bovine serum albumin, pH 7.4). CB1 receptor concentration (Bmax) was 2.5 pmol/mg protein. Rutamarin and the dichloromethane crude extract of R. graveolens were present at varying concentrations, and the non-specific binding of the radioligand was determined in the presence of 10 μM CP-55,940. After 90 min of incubation, the suspension was rapidly filtered through 0.05% polyethyleneimine pre-soaked GF/C glass fiber filters on a 96-well cell harvester and washed nine times with 0.5 mL of ice-cold washing buffer (50 mM Tris-HCl, 2.5 mM EGTA, 5 mM MgCl2, 2% bovine serum albumin, pH 7.4). Radioactivity on filters was measured with a Beckman LS 6500 scintillation counter in 3 mL of Ultima Gold scintillation liquid. Data collected from three independent experiments performed in triplicate were normalized between 100 and 0% specific binding for [3H]CP-55,940. These data were fitted in a sigmoidal curve and graphically linearized by projecting Hill plots, which for both cases allowed the calculation of IC50 values. Derived from the dissociation constant (KD) of [3H]CP-55,940 (0.18 nM for CB1 receptor and 0.39 nM for CB2 receptor) (vide infra) and the concentration-dependent displacement (IC50 value), inhibition constants (Ki) of competitor compounds were calculated using the Cheng-Prusoff equation [Ki = IC50/(1 + L/KD)] [40]. For CB2 receptor binding studies, 3.8 μg of membrane recombinantly overexpressing CB2 receptor (RBXCB2M; Perkin Elmer Life Sciences) were resuspended in 0.6 mL of binding buffer (see above) together with 0.11 nM of the radioligand [3H]CP-55,940. The CB2 receptor radioligand binding assay was conducted in the same manner as for the CB1 receptor. CB2 receptor concentration (Bmax) was 4.7 pmol/mg protein. Bmax and KD values of [3H]CP-55,940 were determined by Perkin Elmer, Life and Analytical Sciences. Cannabiniol (95 % purity) from Lipomed AG was used as positive control.
Supporting information
Fig. 1S Hitting pharmacophore models from the parallel screen of compounds 1–16 for the targets AChE, HRV coat protein, and CB2 receptor. Fig. 2S Ligand-based pharmacophore model for CB2 agonists. Chemical features of the model are colour-coded: hydrogen bond acceptor – green; hydrophobic – cyan; hydrophobic aromatic – dark blue; hydrophobic aliphatic – light blue; shape – grey. Fig. 3S Alignment of selected bioactive hits to a hitting pharmacophore model. A: Tacrin-based AChE inhibitor pharmacophore model. B: Compound 1 fitted into the tacrin-based model. C: R 61837-based model for HRV coat protein inhibitors. D: Compound 1 fitted into the R 61837-based model. E: The highly potent CB2 agonist JWH-267 fitted into the ligand-based CB2 pharmacophore model. F: Compound 3 fitted into the ligand-based CB2 model. Chemical features of the models are colour-coded. Structure-based models (Fig. 3S A – D; LigandScout): hydrogen bond acceptor: red; hydrophobic: yellow; exclusion volumes: grey. Ligand-based models (Fig. 3S E, F; Discovery Studio): hydrogen bond acceptor: green; hydrophobic: cyan; hydrophobic aromatic: dark blue; hydrophobic aliphatic: light blue; shape: grey. For a clearer depiction of the fitting conformations, no excluded volume spheres are shown for the virtual hits.
Results
In a previously performed in vitro screening on acetylcholinesterase (AChE) inhibitory activity of some 130 dichloromethane and methanol extracts of various angiosperms native to and cultivated in Tyrol, Austria [11], [12], the aerial parts of Ruta graveolens L. were revealed as promising starting material for the isolation of natural AChE inhibitors. By using a spectrophotomentric enzyme assay with Ellman’s reagent [15] the dichloromethane and methanol extracts showed AChE IC50 values of 96.7 (70.0–124.5) μg/mL and 138.0 (105.4–175.8) μg/mL, respectively. An alkaloid fraction of the latter extract showed an IC50 of 39.4 (33.5–46.1) μg/mL. These preliminary results prompted us to phytochemically investigate the dichloromethane extract and the alkaloid fraction of the methanol extract of R. graveolens and to isolate their constituents for pharmacological testing.
From the dichloromethane crude extract one alkaloid and six coumarin derivatives were isolated and their structures elucidated by mass spectrometry, 1 D and 2 D NMR as arborinine (1), daphnoretin methyl ether (2), rutamarin (3), isoimperatorine (4), psoralen (5), bergapten (6), and 8-methoxy psoralene (7) (Fig. 3).
Fig. 3.

Chemical structures of isolated constituents from R. graveolens.
From the alkaloid fraction of the methanol extract nine compounds were isolated (Fig. 3). They could be assigned as four alkaloids, namely S-ribalinine (8), isoplatydesmin (9), (−)-edulinine (10), and norgraveoline (11); furthermore, two simple coumarins were identified as 7-methoxycoumarin (12) and 6,7,8-trimethoxycoumarin (13). Compounds 1–7, 12 and 13 have been previously identified as constituents from R. graveolens and other Ruta species Their physical and spectroscopic properties are in accordance with those reported previously [20], [21], [22], [23], [24], [25], [26], [27], [28], [29].
Until now S-ribalinine (8), isoplatydesmine (9) and norgraveoline (11) have never been isolated before from the genus Ruta, although they are well known from other Rutaceae genera; compound 8, e. g., is reported as constituent of Haplophyllum [30] or Evodia [31]; compound 9, e. g., from Melicope semecarpifolia [32], and compound 11 from different Haplophyllum species [33], [34]. The chinolinone alkaloid edulinine (10) has been isolated before from R. graveolens [27], however the authors did not specify the optical rotation. Our isolate (10) showed a negative optical rotation (: −32.8, MeOH, c 0.775). According to the absolute stereochemistry assignment of Boyd and coauthors [20], we state compound 10 to be (−)-S-edulinine with a reported optical rotation of : −32.0 (MeOH, c 0.52).
In addition, three phenylpropionic acid methyl esters (14–16) were isolated, all representing novel natural products. Compound 14 showed a molecular mass of 256.09 by HR-MS (calculated for C12H16O6:256.09). The FT-IR spectrum exhibited a sharp band at νmax = 2937 cm−1 indicating intramolecular hydrogen bonds, a band at νmax = 2853 cm−1 due to methoxy groups, and a carbonyl stretching vibration at νmax = 1727 cm−1 characteristic for a saturated ester. The 1H-NMR spectrum (Table 1) is characterized by a singlet at δ = 6.62 ppm integrating for two protons (H-2 and H-6) and two overlapping methoxy groups at H-3 and H-5 (δ = 3.90 ppm) thus suggesting a symmetrically substituted phenyl ring; further signals could be assigned to a high shifted aliphatic proton (H-7 at δ = 5.07 ppm) adjacent to a methylene group (H-8 at δ = 2.73 ppm) and a methoxy group at δ = 3.74 ppm. Interpretation of the 2 D spectra correlations (HSQC, HMBC, COSY) revealed compound 14 as a sinapic acid derivative, i. e., methyl 3-hydroxy-3-(4-hydroxy-3,5-dimethoxyphenyl)propanoate. Based on its negative optical rotation, the chiral centre at H-7 could be assigned as S according to the absolute stereochemical assignments given by Schöpf and Wüst [35].
HR-MS analysis of compound 15 revealed a molecular formula of C12H14O6; its FT-IR spectrum resembled that of compound 14, as also showing well-characterized hydrogen stretching vibrations for hydroxy (νmax = 2955 cm−1) and methoxy (νmax = 2844 cm−1), and the saturated ester carbonyl stretching at νmax = 1732 cm−1, but with distinct differences in the fingerprint region. The 1H-NMR spectrum of compound 15 shows a similar pattern to that of 14. A two-proton singlet at δ = 6.51 ppm (H-2 and H-6) and two overlapping methoxy groups (δ = 3.85 ppm) point to a symmetrically substituted phenyl ring; a third methyl group (δ = 3.75 ppm) was again assigned to a methyl ester. The difference, however, is evident in two down-field shifted methine groups (AB system) at δ = 3.54 and 3.43 ppm (d, J = 8.8 Hz) indicating an epoxide ring system. As a result of the correlations observed in the 2 D NMR experiments (Table 1), compound 15 could be assigned as methyl 3-(4-hydroxy-3,5-dimethoxyphenyl)oxirane-2-carboxylate with a positive optical rotation. Until now, the isolates 14 and 15 have neither been synthesized, nor have they been isolated from natural material, although related methyl sinapates also occur in other Rutaceae species, e. g., in Philotheca deserti var. deserti [36].
Compound 16 revealed a mass of 250.12 by HR-MS (calculated for C13H14O5). Besides the similarities in its FT-IR spectrum to those of compounds 14 and 15, the most striking difference is due to the additional double bond stretching vibration of the conjugated heterocycle at νmax = 1656 cm−1 max represented by the furano ring system. The 1H-NMR spectrum is characterized by two one-proton doublets of an aromatic AB pattern, which are assigned to H-4 (δ = 7.58) and H-5 (δ = 6.68, d, J = 2.1 Hz) of a furano ring system, and one aromatic singlet (δ = 7.05) of the condensed benzo-ring, indicating that 16 is a 1,2,3 tri-substituted benzofuran. 1 D and 2 D NMR data (Table 1) revealed the substituents to be a methoxy group, a hydroxy group, and two linked methylenes neighbouring a carbonic acid methyl ester. By comparing and contrasting observations from the HMBC experiment with related water-soluble glycosides isolated from R. graveolens [37], the structure of 16 was assigned to methyl 3-(6-hydroxy-7-methoxybenzofuran-5-yl)propanoate. This substance was obtained as a semisynthetic degradation product of xanthotoxin by Spencer et al. [38], but has never been isolated before from any natural material. In contrast to compounds 14 and 15, isolate 16 could not be detected in the original methanol extract. Thus, we assume compound 16 to be an artefact generated during the isolation procedure.
Target fishing
In order to reveal the potential bioactivities of the 16 isolated compounds from Rutae herba, conformational models were generated for these compounds and their 3 D structures were virtually screened against 2208 pharmacophore models. All 16 parallel screening reports were studied, focussing on interesting pharmacological actions of each compound. The number of hitting models varied extensively between compounds; so, compound 12 could only fit ten models, whereas the parallel screening of compound 14 returned 278 hitting models. For compounds from the same chemical scaffold, e.g., compounds 5, 6, and 7, the predicted pharmacological profiles were very similar. In contrast, compounds from other scaffolds, e.g., compound 2, were predicted to be active on other targets. With the exception of the cytochrome P450 enzyme family, no target group was present in all predicted profiles.
Among the number of targets identified by virtual parallel screening, we focussed on the following biological targets: AChE, the HRV coat protein, and the CB2 receptor. This selection was taken according to (i) the predicted ligand-target interactions from the parallel screening, (ii) the previously detected AChE inhibiting effect of the Rutae herba extracts, (iii) available test systems and collaborations for pharmacological evaluation, and (iv) the heterogeneity of the macromolecular targets in terms of their molecular function and their human-pathological relevance (AChE: hydrolase, dementia [39], [40]; HRV coat protein: viral protein, virus infection [41]; CB2: G-protein coupled receptor, inflammation [42], analgesia [43]).
Hitting models for the three focused targets
AChE
The first hitting model for AChE inhibitors was based on the interactions of the natural product galanthamine (GNT) with the AChE active site (PDB entry 1qti, see Supporting Information, Fig. 1S). This model has already been applied successfully as a VS tool for natural compounds as AChE inhibitors [13]. The GNT-derived model predicted compounds 1, 9, 10, 14, 15, and 16 to be active. The second model was based on the potent and prominent AChE inhibitor tacrin (PDB entry 1acj; Supporting Information, Fig. 3SA). This alkaloid shows few, but essential interactions with the enzyme. In order to find known active AChE inhibitors, the hydrogen bond donor from the amino group to an active site water molecule was exchanged with a hydrogen bond acceptor feature (Supporting Information, Fig. 1S: 2 D versus 3 D pharmacophore model depiction). An exclusion volumes coat that represents the binding pocket size was added to the model to increase its restrictiveness. This model predicted compounds 1, 2, 7, 10, 11, 13, 14, 15, and 16 to fit into the AChE binding site.
HRV coat protein
In the Inte:Ligand pharmacophore model database, 62 structure-based models are present that describe possible binding modes between a ligand and the HRV coat protein binding site in a canyon-shaped pocket on the surface of the virus. A subset of these models has already been applied to the VS of commercial [44] as well as natural products databases [45]. Three models from the pharmacophore model database were returned as hitting models. The first model was based on the interactions of the highly active ligand WIN 61209 with the HRV coat protein canyon (PDB entry 1qju, Supporting Information, Fig. 1S). This model was also used for our previous studies as described above. The second and the third models were derived from the complexes of the molecule R 61837 with the binding site (Supporting Information, Fig. 1S, PDB entries 1r09 and 2hwf; Fig. SC). All three models contained a steric shape restriction. In summary, five of the 16 compounds isolated were predicted to bind to the HRV coat protein: compounds 1 (2hwf-model), 2 (2hwf and 1r09 models), 3 (2hwf and 1r09 models), 15 (1qju-model), and 16 (1qju and 2hwf models).
CB2
The only hitting model for CB2 ligands was a ligand-based model that has already been successfully applied to VS of commercial libraries [46]. This model is based on a CB2 training set comprising the five selective agonists AM1241, GW405833, HU-308, JWH-133, and JWH-267 (Supporting Information, Fig. 2S and Fig. 3SE).
Pharmacological evaluation
For a critical assessment of the predictive power of the applied computer-assisted paradigm, compounds 1–16 including virtual hits and non-hits were tested for their ability to inhibit AChE using a spectrophotometric enzyme assay [15]. From all isolated compounds, the acridone alkaloid arborinine (1) exhibited the strongest AChE inhibiting activity. Modest effects were further measured for the quinolone alkaloids 9 and 11, and for the simple coumarin 13 (Table 2).
Table 2.
In vitro AChE inhibiting activity of the isolated constituents from Rutae herba and of the positive control (galanthamine HBr)
| Compound | IC50 ± SD [μM] |
Compound | IC50 ± SD [μM] |
|---|---|---|---|
| Galanthamine | 3.20 ± 1.02 | ||
| 1 | 34.7 ± 7.1 | 9 | 205.6 ± 16.3 |
| 2 | > 500 | 10 | > 500 |
| 3 | > 500 | 11 | 197.3 ± 18.0 |
| 4 | > 500 | 12 | > 500 |
| 5 | > 500 | 13 | 395.8 ± 68.5 |
| 6 | > 500 | 14 | > 500 |
| 7 | > 500 | 15 | > 500 |
| 8 | > 500 | 16 | > 500 |
For evaluation of the HRV antiviral activity, nine Ruta constituents (5 virtual hits and 4 non-hits) were subjected to cytopathic effect (CPE) inhibitory assays performed with the pleconarilsensitive HRV serotype 2 (HRV-2) on HeLa cells [18], [19]. To exclude unspecific compound actions, the cytotoxicity of the tested isolates was determined beforehand. The 50% cytotoxic concentration (CC50) was calculated from the mean dose-response curve of two independent assays. Determination of the CPE inhibitory effect (derived from three independent tests) using non-cytotoxic concentrations revealed arborinine (1) and 6,7,8-trimethoxycoumarin (13) as the most active antiviral constituents. Both protect HeLa cells in the low micromolar range against HRV-2 infection. Weak, but significant and dose-dependent antiviral activities were further recorded for 2, 8 and 16, which all belong to different chemical scaffolds (Table 3, Fig. 3). A potential antiviral effect of compound 3 could not be determined owing to its cytotoxicity.
Table 3.
Cytotoxicity and anti-HRV-2 activity in non cytotoxic concentrations of virtual hits, selected non virtual hits, and the positive control (pleconaril)
| Compound | CC50 [μg/mL]a | Anti-HRV-2 IC50 ± SD [μM]b |
|---|---|---|
| Pleconaril | 12.6 | 0.03 ± 0.01 |
| 1 | > 50 | 3.19 ± 2.24 |
| 2 | > 50 | 97.08 ± 33.85 |
| 3 | 0.9 | cytotoxic |
| 8 | > 50 | 82.95 ± 43.34 |
| 11 | 32.2 | inactive |
| 13 | > 50 | 11.98 ± 7.53 |
| 14 | > 50 | inactive |
| 15 | > 50 | inactive |
| 16 | > 50 | 91.21 ± 40.27 |
Results from two independent tests.
Results from three independent tests.
Compounds 1, 3, 8, 9, and 10 were investigated for their potential to act as CB2 ligands using a radioligand displacement assay as described previously [47]. Among the tested compounds, rutamarin (3) revealed a selective affinity to the CB2 receptor with a Ki of 2.64 ± 0.2 μg/mL or 7.4 ± 0.6 μM ( Table 4). None of the other compounds tested showed significant binding affinities to the CB receptors. Displacement data obtained with the Rutae herba dichloromethane extract yielded a Ki value of 16.8 ± 0.9 μg/mL. Given the approximate percentage of rutamarin (15–20%) in the dichloromethane crude extract, this suggests that rutamarin is the constituent responsible for the effect determined from the extract.
Table 4.
Binding affinities on CB2 (Ki values) of four selected non virtual hits, the virtual hit 3, and the positive control (cannabinol)
| Compound | hCB2Ki ± SD [μM] | hCB1Ki/hCB2Ki |
|---|---|---|
| Cannabinol | 0.12 ± 0.01 μM | 0.42 ± 0.13 μM |
| 1 | > 100 | > 100 |
| 3 | 7.40 ± 0.60 μM | > 100 |
| 8 | > 100 | > 100 |
| 9 | > 100 | > 100 |
| 10 | > 100 | > 100 |
In Table 5 all experimental results determined from the isolated constituents are summarized. Additionally, those compounds with a virtually-predicted interaction to the binding site of one or more of the focused macromolecular targets are labelled in grey. Thus, a comparison between the experimental results obtained from virtual hits and non-hits is provided, which enables the evaluation of the computer-assisted target-fishing approach underlying this study. For pharmacological evaluation, all isolated compounds were assayed on AChE inhibition. For antiviral tests and binding studies on CB2, all virtually predicted ligands were assayed, but only a selection of unpredicted hits was tested due to cost reasons and substance availability. Although the test set subjected to virtual and pharmacological screening consisted of a limited number of compounds, it is obvious from the virtual hit list of the corresponding targets that the selectivity and the predictive power of the individual pharmacophore models vary (Table 5). This fact strongly depends on the accuracy and quantity of input information available as the starting point for the pharmacophore model generation.
Table 5.
Overview of virtual and experimental results obtained from compounds 1–16 on the targets AChE, HRV, and CB2. Virtual predictions: white background, virtual hit; grey grey background, background virtual non hit. Experimental results: – no activity; + weak activity; ++ medium activity; +++ high activity; empty fields: activity not determined.
| No | Compounds | AChE | HRV | CB2 |
|---|---|---|---|---|
| 1 | Arborinine | +++ | +++ | − |
| 2 | Daphnoretin methyl ether | − | + | |
| 3 | Rutamarin | − | cytotoxic | +++ |
| 4 | Isoimperatorin | − | ||
| 5 | Psoralen | − | ||
| 6 | Bergapten | − | ||
| 7 | 8-Methoxypsoralen | − | ||
| 8 | S-Ribalinine | − | + | − |
| 9 | Isoplatydesmine | ++ | − | |
| 10 | Edulinine | − | − | |
| 11 | Norgraveoline | ++ | − | |
| 12 | 7-Methoxycoumarin | − | ||
| 13 | 6,7,8-Trimethoxycoumarin | + | ++ | |
| 14 | Methyl 3-hydroxy-3-(4-hydroxy-3,5-dimethoxy-phenyl)- propanoate |
- | -15 | 3-(4-Hydroxy-3,5-di-methoxyphenyl) oxirane-2- carboxylate |
| 16 | Methyl 3-(6-hydroxy-7-methoxybenzofuran-5-yl) propanoate |
- | + |
AChE inhibition: +++, IC50 < 50 μM; ++, 50–200 μM; +, 200–500 μM; −, > 500 μM.
Anti-HRV activity: +++, IC50 < 10 μM; ++, 10–20 μM; +, 20–100 μM; −, > 100 μM.
CB2 affinity: +++, Ki < 10 μM; −, > 100 μM.
For the target AChE, the two hitting models (one GNT-based, one tacrin-based) predicted 10 out of 16 compounds (62.5 % virtual hits) as putative AChE inhibitors. All four compounds for which we registered an AChE inhibiting effect of > 70 % at a concentration of 200 μg/mL (IC50 < 500 μM) were found in the hit list. However, further six compounds were virtually predicted which did not fulfil these criteria. A high-scored fitting conformation of the virtual hit and most potent AChE-inhibitor arborinine (1) is depicted in Fig. 3SB (Supporting Information).
The three hitting models for the HRV coat protein predicted 5 of 16 compounds (31.2 %). Among the virtual hits, the most potent antiviral constituent, arborinine (1) was correctly identified (fitting conformation shown in the Supporting Material, Fig. 3SD) as well as the two moderately active compounds 2 and 16. We failed to determine the antiviral effect of the virtual hit 3, because of its pronounced cytotoxicity. With the exception of the definitive blank 15, since this compound is inactive, the virtual prediction was successful. From the four compounds not predicted, but assayed (8, 11, 13, and 14), the pharmacophore models failed to map the antiviral 6,7,8-trimethoxycoumarin (13) and the modestly active S-ribalinine (8). The question remains open whether these two compounds reveal their antiviral effect via a capsid binding mechanism as proposed for the virtual hits by the hitting pharmacophore models.
Only one compound in the entire dataset, i. e., rutamarin (3), was predicted by the ligand-based hitting model for CB2 ligands (fitting conformation shown in the Supporting Information, Fig. 3SF). Strikingly, from the five R. graveolens-constituents subjected to experimental validation, exactly this natural product showed a moderate, but selective CB2 binding interaction in the radioligand displacement assay. Concerning this target we could accordingly achieve a 100% prediction accuracy, although with a unrepresentative number of virtual hits and test candidates. This will be scrutinized in future investigations using larger natural product datasets. Moreover, rutamarin may serve as a new CB2 ligand scaffold, which could be optimized by synthetic means. Further studies will have to determine whether this compound acts as an agonist or antagonist. Overall, there seems to be a promising relationship between plant natural products acting on CB2 receptors and the endocannabinoid system [48].
Discussion
A virtual parallel screening paradigm is herein presented for three hitting targets of high human-pathological relevance, i.e., AChE, HRV coat protein, and CB2. Following our protocol only a limited number of experiments has been required for the identification of ligands of the focused targets and for fishing the most active compounds from the dataset under investigation. The applied methodology has the capacity of catalysing drug discovery profoundly for all of those diseases where molecular targets or molecular ligands are well defined to create reliable pharmacophore models. Worth mentioning for natural product scientists is the clear benefit of this approach by handling with compounds already available for pharmacological testing. A characteristic feature of many natural products is their modulating and multi-target oriented biological effect, often implicating only moderate effects on individual targets. In our study, the anticipated bioactivities of the virtually identified natural products are only in the micro molar range; however, the virtual parallel screening provides an estimate of a putative pharmacological profile based on those constituents which characterise the focussed extract, including metabolites that are so far unknown for a specific natural material. Thus, the presented approach promises to significantly enhance the identification of relevant targets for bioactive natural compounds, and thus be a very valuable tool for pharmacognosists.
Supplementary Material
Acknowledgements
This work was granted by the Austrian Science Fund (FWF No P18379) and by the “Nachwuchsförderung” of the Leopold Franzens-University of Innsbruck for J.M. Rollinger. The authors thank A. Univ.-Prof. Ernst Ellmerer for NMR measurements (Institute of Organic Chemistry, University of Innsbruck), Inte:Ligand for providing the LigandScout software and the pharmacophore model collection free of charge, and Johann Strillinger (Gartenbau Strillinger, Söll, Austria) for providing the plant material free of charge.
Abbreviations
- AChE
acetylcholinesterase
- CD2
cannabinoid receptor type-2
- CC50
50% cytotoxic concentration
- CPE
cytopathic effect
- 3D
three dimensional
- GNT
galanthamine
- HRV
human rhinovirus
- PDB
protein databank
- VS
virtual screening
Footnotes
Supporting information available online at http://www.thieme-connect.de/ejournals/toc/plantamedica
References
- 1.Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br J Pharmacol. 2007;152:9–20. doi: 10.1038/sj.bjp.0707305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: Applications to targets and beyond. Br J Pharmacol. 2007;152:21–37. doi: 10.1038/sj.bjp.0707306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirchmair H, Distinto S, Schuster D, Spitzer G, Langer T, Wolber T. Enhancing drug discovery through in-silico screening: Strategies to increase true positives retrieval rates. Curr Med Chem. 2008;51:7021–40. doi: 10.2174/092986708785132843. [DOI] [PubMed] [Google Scholar]
- 4.Rollinger JM, Langer T, Stuppner H. Integrated in silico tools to exploit the natural products’ bioactivity. Planta Med. 2006;72:671–8. doi: 10.1055/s-2006-941506. [DOI] [PubMed] [Google Scholar]
- 5.Rollinger JM, Stuppner H, Langer T. Virtual screening for the discovery of bioactive natural products. In: Petersen F, Amstutz R, editors. Natural compounds as drugs. Vol. I. Birkhäuser Verlag; Basel: 2008. pp. 212–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steindl TM, Schuster D, Wolber G, Laggner C, Langer T. High-throughput structure-based pharmacophore modelling as a basis for successful parallel virtual screening. J Comput Aid Mol Des. 2006;20:703–15. doi: 10.1007/s10822-006-9066-y. [DOI] [PubMed] [Google Scholar]
- 7.Steindl TM, Schuster D, Laggner C, Langer T. Parallel screening: a novel concept in pharmacophore modeling and virtual screening. J Chem Inf Model. 2006;46:146–57. doi: 10.1021/ci6002043. [DOI] [PubMed] [Google Scholar]
- 8.Langer T, Hoffmann RD, Mannhold R, Kubinyi H. Pharmacophores and pharmacophore searches (Methods and Principles in Medicinal Chemistry) Wiley-VCH; Weinheim: 2006. [Google Scholar]
- 9.Moldenke HM, Moldenke AL. Plants of the Bible. Chronica Botanica Co; Waltham (Massachusettes): 1952. p. 208. [Google Scholar]
- 10.Foster S, Tyler VE. Tyler’s honest herbal: a sensible guide to the use of herbs and related remedies. 4th edition The Haworth Herbal Press; Binghampton: 1999. pp. 325–6. [Google Scholar]
- 11.Rollinger JM, Mock P, Zidorn C, Ellmerer EP, Langer T, Stuppner H. Application of the in combo screening approach for the discovery of non-alkaloid acetylcholinesterase inhibitors from Cichorium intybus. Curr Drug Discov Technol. 2005;2:185–93. doi: 10.2174/1570163054866855. [DOI] [PubMed] [Google Scholar]; 2006;3:89. erratum: [Google Scholar]
- 12.Rollinger JM, Schuster D, Baier E, Ellmerer EP, Langer T, Stuppner H. Taspine: bioactivity-guided isolation and molecular ligand-target insight of a potent acetylcholinesterase inhibitor from Magnolia×soulangiana. J Nat Prod. 2006;69:1341–6. doi: 10.1021/np060268p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wermuth CG, Ganellin CR, Lindberg P, Mitscher LA. Glossary of terms used in medicinal chemistry (IUPAC recommendations 1997) Ann Rep Med Chem. 1998;33:385–95. [Google Scholar]
- 14.Ellman GL, Courtney D, Andres V, Featherstone RM. A new and rapid calorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 15.Ingkaninan K, de Best CM, van der Heijden R, Hofte AJP, Karabatakb B, Irth H. High-performance liquid chromatography with on-line coupled UV, mass spectrometric and biochemical detection for identification of acetylcholinesterase inhibitors from natural products. J Chromatogr A. 2000;872:61–73. doi: 10.1016/s0021-9673(99)01292-3. [DOI] [PubMed] [Google Scholar]
- 16.Rollinger JM, Hornick A, Langer T, Stuppner H, Prast H. Acetylcholinesterase inhibitory activity of scopolin and scopoletin discovered by virtual screening of natural products. J Med Chem. 2004;47:6248–54. doi: 10.1021/jm049655r. [DOI] [PubMed] [Google Scholar]
- 17.Wolber G, Langer T. LigandScout: 3 D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45:160–9. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 18.Schmidtke M, Schnittler U, Jahn B, Dahse HM, Stelzner A. A rapid assay for evaluation of antiviral activity against Coxsackie virus B3, influenza virus A, and Herpes simplex virus type 1. J Virolog Methods. 2001;95:133–43. doi: 10.1016/s0166-0934(01)00305-6. [DOI] [PubMed] [Google Scholar]
- 19.Makarov VA, Riabova OB, Granik VG, Wutzler P, Schmidtke M. Novel [(biphenyloxy)propyl]isoxazole derivatives for inhibition of human rhinovirus 2 and Coxsackievirus B3 replication. J Antimicrob Chemother. 2005;55:483–8. doi: 10.1093/jac/dki055. [DOI] [PubMed] [Google Scholar]
- 20.Boyd DR, Sharma ND, Barr SA, Carroll JG, Mackerracher D, Malone JF. Synthesis and absolute stereochemistry assignment of enantiopure dihydrofuro- and dihydropyrano-quinoline alkaloids. J Chem Soc [Perkin I] 2000:3397–405. [Google Scholar]
- 21.Novak I, Buzas G, Minker E, Koltai M, Szendrei K. Active constituents of Ruta graveolens. Pharmazie. 1965;20:738. [PubMed] [Google Scholar]
- 22.Novak I, Buzas G, Minker E, Koltai M, Szendrei K. Active principles of Ruta graveolens. Planta Med. 1965;13:226–33. [Google Scholar]
- 23.Novak I, Buzas G, Minker E, Koltai M, Szendrei K. Isolation of some components from the herb of Ruta graveolens. Acta Pharm Hung. 1967;37:131–42. [PubMed] [Google Scholar]
- 24.Reisch J, Novak I, Szendrei K, Minker E. Chemistry of natural substances. V. Isoimperatorin, a component of Ruta graveolens. Pharmazie. 1966;21:628–9. [Google Scholar]
- 25.Reisch J, Novak I, Szendrei K, Minker E. Chemistry of natural substances. XIX. Additional C3-substituted coumarin derivatives from Ruta graveolens: daphnoretin and its methyl ether. Planta Med. 1968;16:372–6. doi: 10.1055/s-0028-1099923. [DOI] [PubMed] [Google Scholar]
- 26.Reisch J, Adesina SK, Bergenthal D. Constituents of Zanthoxylum leprieurii fruit pericarps. Part 103: Natural product chemistry. Pharmazie. 1985;40:811–2. [Google Scholar]
- 27.Steck W, Bailey BK, Shyluk JP, Gamborg O. Coumarins and alkaloids from cell cultures of Ruta graveolens. Phytochemistry. 1971;10:191–4. [Google Scholar]
- 28.Wolters B, Eilert U. Antimicrobial substances in callus cultures of Ruta graveolens. Planta Med. 1981;43:166–74. doi: 10.1055/s-2007-971494. [DOI] [PubMed] [Google Scholar]
- 29.Del Castillo JB, Rodriguez LF, Secundino M. Angustifolin, a coumarin from Ruta angustifolia. Phytochemistry. 1984;23:2095–6. [Google Scholar]
- 30.Puricelli L, Innocenti G, Delle Monache G, Caniato R, Filippini R, Cappelletti EM. In vivo and in vitro production of alkaloids by Haplophyllum patavinum. Nat Prod Lett. 2002;16:95–100. doi: 10.1080/10575630290019985. [DOI] [PubMed] [Google Scholar]
- 31.Gunawardana YAGP, Cordell GA, Ruangrungsi N, Chomya S, Tantivatana P. Traditional medicinal plants of Thailand. VII. Alkaloids of Evodia lepta and Evodia gracilis. J Sci Soc Thailand. 1987;13:107–12. [Google Scholar]
- 32.Chen JJ, Chang YL, Teng CM, Su CC, Chen IS. Quinoline alkaloids and anti-platelet aggregation constituents from the leaves of Melicope semecarpifolia. Planta Med. 2002;68:790–3. doi: 10.1055/s-2002-34412. [DOI] [PubMed] [Google Scholar]
- 33.Razakova DM, Bessonova IA, Yunusov SY. Alkaloids from Haplophyllum perforatum. Khim Prir Soedin. 1976;5:682. [Google Scholar]
- 34.Akhmedzhanova VI, Bessonova IA, Yunusov SY. Alkaloids of Haplophyllum foliosum. Khim Prir Soed. 1980;6:803–5. [Google Scholar]
- 35.Schöpf C, Wüst W. The absolute configuration of optically active β-hydroxy-β-phenylpropionic acids. Ann. 1959;626:150–4. [Google Scholar]
- 36.Chlouchi A, Girard C, Tillequin F, Bevalot F, Waterman PG, Muyard F. Coumarins and furoquinoline alkaloids from Philotheca deserti var. deserti (Rutaceae) Biochem Syst Ecol. 2006;34:71–4. [Google Scholar]
- 37.Chen CC, Huang YL, Huang FI, Wang W, Ou JC. Water-soluble glycosides from Ruta graveolens. J Nat Prod. 2001;64:990–2. doi: 10.1021/np000582y. [DOI] [PubMed] [Google Scholar]
- 38.Spencer GF, Desjardins AE, Plattner RD. 5-(2-Carboxylethyl)-6-hydroxy-7-methoxybenzofuran, a fungal metabolite of xanthotoxin. Phytochemistry. 1990;29:2495–7. [Google Scholar]
- 39.Relkin NR. Beyond symptomatic therapy: a re-examination of acetylcholinesterase inhibitors in Alzheimer’s disease. Exp Rev Neurother. 2007;7:735–48. doi: 10.1586/14737175.7.6.735. [DOI] [PubMed] [Google Scholar]
- 40.Mukherjee PK, Kumar V, Mal M, Houghton PJ. Acetylcholinesterase inhibitors from plants. Phytomedicine. 2007;14:289–300. doi: 10.1016/j.phymed.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 41.Patick AK. Rhinovirus chemotherapy. Antivir Res. 2006;71:391–6. doi: 10.1016/j.antiviral.2006.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iwamura H, Suzuki H, Kaya T, Inaba T. In vitro and in vivo pharmacological characterization of JTE-907, a novel selective ligand for cannabinoid CB2 receptor. J Pharmacol Exp Ther. 2001;296:420–5. [PubMed] [Google Scholar]
- 43.Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci USA. 2005;102:3093–8. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steindl TM, Crump CE, Hayden FG, Langer T. Pharmacophore modeling, docking, and principal component analysis based clustering: Combined computer-assisted approaches to identify new inhibitors of the human rhinovirus coat protein. J Med Chem. 2005;48:6250–60. doi: 10.1021/jm050343d. [DOI] [PubMed] [Google Scholar]
- 45.Rollinger JM, Steindl TM, Schuster D, Kirchmair J, Anrain K, Ellmerer EP. Structure-based virtual screening for the discovery of natural inhibitors for human rhinovirus coat protein. J Med Chem. 2008;51:842–51. doi: 10.1021/jm701494b. [DOI] [PubMed] [Google Scholar]
- 46.Markt P. Discovery of novel CB2 receptor ligands by a pharmacophore-based virtual screening workflow. J Med Chem. doi: 10.1021/jm801044g. in press. [DOI] [PubMed] [Google Scholar]
- 47.Raduner S, Majewska A, Chen JZ, Xie XQ, Hamon J, Faller B. Alkylamides from Echinacea are a new class of cannabinomimetics: cannabinoid type 2 receptor-dependent and -independent immunomodulatory effects. J Biol Chem. 2006;281:14192–206. doi: 10.1074/jbc.M601074200. [DOI] [PubMed] [Google Scholar]
- 48.Gertsch J, Leonti M, Raduner S, Racz I, Chen JZ, Xie XQ. Beta-caryophyllene is a dietary cannabinoid. Proc Natl Acad Sci USA. 2008;105:9099–104. doi: 10.1073/pnas.0803601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.