

Abstract
In pregnant women, the diabetic condition results in a three- to fivefold increased risk for fetal cardiac malformations as a result of elevated glucose concentrations and the resultant osmotic stress in the developing embryo and fetus. Heart development before septation in the chick embryo was studied under two hyperglycemic conditions. Pulsed hyperglycemia induced by daily administration of glucose during 3 days of development caused daily spikes in plasma glucose concentration. In a second model, sustained hyperglycemia was induced with a single injection of glucose into the yolk on day 0. The sustained model raised the average plasma glucose concentration from 70 mg/dL to 180 mg/dL and led to decreased gene expression of glucose transporter GLUT1. Both models of hyperglycemia reduced embryo size, increased mortality, and delayed development. Within the heart outflow tract, reduced proliferation of myocardial and endocardial cells resulted from the sustained hyperglycemia and hyperosmolarity. The cell cycle inhibitor p21 was significantly increased, whereas cyclin D1, a cell cycle promoter, decreased in sustained hyperglycemia compared with controls. The evidence suggests that hyperglycemia-induced developmental delays are associated with slowed cell cycle progression, leading to reduced cellular proliferation. The suppression of critical developmental steps may underlie the cardiac defects observed during late gestation under hyperglycemic conditions.
Structural malformations of the heart affect 1% of newborn infants in the U.S. and are the leading cause of noninfectious death among children. Maternal diabetes is associated with a three- to five-fold increased risk for cardiac and valve malformations, with hyperglycemia acting as a major teratogen. This is more alarming because of the rising incidence of diabetes in the population. In 2010, an estimated 50 million people worldwide had type 1 diabetes and 171 million people had type 2 diabetes (1). An estimated 7% of U.S. pregnancies in 2010 had gestational diabetes, (2) resulting in 21% fetal mortality (3). Many pregnant women are unaware of both their diabetes status during early pregnancy and the increased risk of morbidity and mortality that diabetes has on their unborn child (4). Most pregnancies are not recognized clinically until ≥2 weeks after conception, with the test for gestational diabetes typically performed at 24–28 weeks. Frequently, diabetes is diagnosed and strict glycemic control begins after the critical periods of embryogenesis and organogenesis. Thus, even blood glucose management started in early pregnancy, but after organogenesis, may be inadequate to prevent adverse pregnancy outcomes (5). Although high maternal glucose concentration and diabetes are associated with heart defects, the cellular and subcellular effects of maternal diabetes on embryonic heart development remain unknown.
Fetuses from diabetic mothers show a range of neural tube (6) and cardiac malformations, including those of the outflow tract (OFT) (7). To investigate the origins of OFT malformations, we used a chick embryo model that is commonly used to study heart development (8). In this model, hyperglycemia during organogenesis induced cardiac malformations that are apparent in late-stage fetuses (9). Glucose-induced malformations in animal models, including retarded growth and abnormal heart development (10), depend on the developmental stage of hyperglycemic exposure and glucose concentration (11). Previous studies found an increase in apoptosis and a decreased proliferation in the endothelial cells of 10-day-old chick embryos in which a 30% (wt/vol) glucose solution was injected into a chorioallantoic membrane vessel at day 8 (12). Embryonic stage delay as a result of maternal hyperglycemia has been noted in mouse (11) and zebrafish (13). Previous studies of pregnant mice with insulin-deficient streptozotocin-induced diabetes exhibited a developmental delay in preimplantation embryos during their progression to a blastocyst stage both in vivo and in vitro (11). Additionally, in vitro cellular studies found that varying concentrations of exogenous d-glucose (20 and 40 mmol/L) caused a decrease in endothelial cell proliferation after 8 days of exposure (14). Embryonic cell viability has been shown to decrease when exposed to 33 mmol/L d-glucose for 96 h, whereas 44 mmol/L of d-glucose exposure for 24 h decreased myoblast proliferation (15). A clinically relevant model of maternal hyperglycemia during cardiac organogenesis, a critical stage of pregnancy, is needed. The current study examined developmental delay, stage delay, and differences between sustained and pulsatile glycemic stress on cardiac development.
Using chick embryos, we developed and tested two in vivo models of hyperglycemia: 1) sustained hyperglycemia, mimicking the exposure of embryos of pregnant women with poor glycemic control, and 2) pulsatile hyperglycemia, representing embryonic conditions of diabetic mothers after meal-associated increases in glucose. The models tested the hypothesis that hyperglycemia slows embryonic development through an increase in osmotic stress, a decrease of glucose transport, and cell cycle suppression. We investigated the underlying causes of developmental delay and growth suppression in the chick embryo by examining cell cycle regulators p21 and cyclin D1, the glucose transport gene GLUT1, and OFT myocardial and endocardial cell proliferation.
RESEARCH DESIGN AND METHODS
Preparation of chick embryos.
The chick embryo was used as a model system of cardiac development for several reasons: 1) Early development of human hearts is similar to that of chick embryos but with a faster time course (21 days in chicks compared with 9 months in humans) (16); 2) congenital heart defects have been well documented in the chick embryo (17,18); 3) the chick embryo is transparent, easy to access, and easily observed without affecting cardiac function (19); and 4) the chick OFT is a key structure that connects the ventricle to the arterial system and is the site where a number of serious septal and valve anomalies originate in human hearts (20).
A random mix of nonincubated, fertilized, White Leghorn chicken eggs were obtained from a hatchery (Featherland Farms Inc., Coburg, Oregon). Two models of hyperglycemia, pulsed and sustained, were developed. To induce a pulsed elevation in plasma glucose concentration, a 2-cm window was made in the blunt end of fertilized chicken eggs before incubation and 50 μL of 30 or 50 mmol/L d-glucose, l-glucose (osmotic control), or chick Ringer saline solution (vehicle control) was dropped in the egg air sac directly on top of the embryo. Drops were added on days 0, 1, 2, and 3 of incubation. For a sustained elevation in plasma glucose concentration, a 2-cm window was made above the air sac of the egg and using a 30-gauge needle, 600 μL of d-glucose in 0.7% saline was slowly injected directly into the egg yolk before incubation; care was taken to avoid injecting bubbles and damaging the blastocyst. To establish a clinically relevant elevation of plasma glucose, d-glucose was added at concentrations of 30, 50, 100, 250, 500, and 750 mmol/L. Control embryos received an injection of l-glucose or vehicle. After yolk or air sac injections, all chicken eggs were sealed with plastic wrap and incubated at 38°C with the blunt end up in a humid atmosphere.
Plasma glucose measurements.
Plasma glucose measurements were taken when embryos reached Hamburger-Hamilton developmental stage 24 (HH24) (21). Using a dissecting microscope, the ventricle was punctured with a 30-gauge needle; blood was withdrawn into 3.8% sodium citrate and stored on ice. At this early embryonic age, 30–40 μL of blood was obtained from each HH24 embryo. The blood was centrifuged (2,500 rpm for 15 min) to isolate plasma. The plasma was frozen at −80°C until biochemical analyses were performed. Plasma glucose concentrations were measured using a glucose assay kit (Sigma) according to the manufacturer’s instructions. Briefly, glucose oxidase/peroxidase reagent was added to the isolated plasma and incubated for 30 min at 37°C. The addition of N2SO4 stopped the reaction. Using a spectrophotometer set at 540 nm, the absorbance of each solution was measured against a standard curve to measure glucose concentration.
Viability and growth measurements.
To assess the physiological effects of sustained and pulsed hyperglycemia on the chick embryos, viability and growth measurements were made. After 96 h of incubation, the embryonic developmental stage was determined based on developmental markers and the Hamburger-Hamilton chick embryonic developmental scale (21). Embryos were removed from their eggs and weighed to quantify differences in embryonic size. Percent mortality was measured by counting the number of living versus dead embryos at 96 h.
Proliferation.
Proliferating cells in the OFT were DNA labeled with a Click-iT EdU kit (Invitrogen) by detection of a thymidine analog in the intact double-stranded DNA. Four hundred microliters of 10 mmol/L EdU antibody diluted in chick saline (0.75 g/L NaCl), was dropped on each living HH24 embryo through the egg window and incubated for 3 h. The embryos were removed and fixed in 4% paraformaldehyde overnight. After fixation, the OFTs were dissected from the embryonic hearts, cut longitudinally, and stained according to the manufacturer’s instructions. Briefly, OFTs were permeabilized in Triton X-100 and incubated in the reaction cocktail (reaction buffer, CuSO4, Alexa Fluor azide, and buffer additive) for 30 min. The OFTs were blocked with 1% BSA and incubated in anti-myosin heavy chain antibody (1:10) (MF20; Developmental Studies Hybridoma Bank) for 1 h. The OFTs were incubated in secondary antibody (1:100 goat anti-mouse IgG Alexa Fluor 564; Invitrogen) for 1 h. The DNA nuclear counterstain Hoechst (1:1000) was applied followed by mounting in SlowFade (Invitrogen). The stained OFTs were imaged using confocal laser scanning microscopy (Bio-Rad MRC500 on a Zeiss Axiovert inverted microscope). To analyze proliferation in all layers of the OFT wall, images were collected in three dimensions as a z-series of ∼3 μm per step. The z-series was analyzed using ImageJ (National Institutes of Health) software and reconstructed as a three-dimensional map of the OFT (22–24); images were taken at five sections along the length of the OFT from the proximal to the distal end. Proliferating cells were counted in each section of the z-series to get a ratio of proliferating cells versus nonproliferating cells.
Quantitative PCR.
For quantitative PCR analysis, the dissected OFTs from HH24 embryos were placed in a lysate solution and homogenized. Total RNA was isolated using an RNeasy kit (Qiagen) and reverse-transcribed using SuperScript III Reverse Transcriptase (Invitrogen). Quantitative PCR was performed with a 96-well Agilent Mx3005P QPCR System and Power SYBR Green. Forward and reverse primers (spanning at least 2 exons) were made using Ensembl genome browser and Primer3 software. Gene expression was determined relative to a calibrator, which comprised pooled OFTs from unopened and untreated eggs at HH24, and normalized to the housekeeping gene PPIA using the standard curve method and taking primer efficiencies into account. Forward and reverse primers for p21, cyclin D1, GLUT1, and PPIA were as follows:
1. p21: TCCTCCTCCTACCAGAGATG, TGTACCTGAGGCTCCTTGTC;
2. cyclin D1: TCGGTGTCCTACTTCAAGTG, GGAGTTGTCGGTGTAAATGC;
3. GLUT1: TCTCTGTCGCCATCTTCTCG, TGGTGAGGCCAGAATACAGG; and
4. PPIA: TGACAAGGTGCCCATAACAG, TTCTCGTCGGCAAACTTCTC.
Protein analysis.
Western blot protein quantification was performed to determine whether the transcription and protein levels correlated for p21 and cyclin D1. Protein was extracted from three pooled OFTs using a lysis solution of 5 mmol/L Tris-HCl, 5 mmol/L EGTA, 5 mmol/L EDTA, and 0.06% SDS (RIPA buffer; Millipore, Temecula, CA) and protease inhibitor (Roche, Indianapolis, IN) and was quantified using a bicinchoninic acid protein assay (Pierce). Protein (15 μg) was separated by SDS-PAGE and transferred onto reinforced nitrocellulose membranes before incubation with primary antibodies, mouse monoclonal p21, mouse monoclonal cyclin D1, and mouse monoclonal α-tubulin (Santa Cruz Biotechnology) overnight followed by incubation with horseradish peroxidase goat anti-mouse IgG (Cell Signaling). Protein was visualized by chemiluminescence (SuperSignal; Pierce), and digitally quantified (ImageJ) to detect endogenous levels of total protein with respect to α-tubulin.
Statistical analysis.
Data are presented as mean ± SD. The effects of glucose treatment were analyzed by ANOVA with Fisher least significant difference (LSD) post hoc. Results were considered significant at P < 0.05.
RESULTS
Plasma glucose.
To determine whether the addition of exogenous glucose increased the blood glucose concentration in embryonic chicks, plasma glucose concentrations were measured. Normal untreated embryos had a plasma glucose concentration of 70 ± 15 mg/dL. The single addition of 50 mmol/L d-glucose into the air sac on day 4 (HH24) into a previously unopened egg led to a steep transient increase in circulating plasma glucose concentration, which peaked at 176 ± 20 mg/dL at 60 min and returned to a normal concentration of 76 ± 15 mg/dL by 120 min (Fig. 1A). The 30 mmol/L l- and d-glucose–treated embryos followed the same pattern. Repeating the glucose addition into the air sac on days 0, 1, 2, and 3 of incubation did not alter the blood glucose level on day 4 (data not shown). To induce sustained hyperglycemia, a higher concentration of glucose with continuous delivery was needed. Glucose additions of 30–500 mmol/L to the yolk did not raise plasma glucose concentrations to a clinically significant level (Fig. 1B). The injection of 750 mmol/L d-glucose raised plasma glucose concentration in HH24 embryos to 177 ± 20 mg/dL, a clinically significant level compared with 72 ± 9 mg/dL in the stage-matched unopened chick embryos (Fig. 1B). To determine whether the sustained hyperglycemic state was maintained later in development, plasma glucose concentrations of d-glucose-injected embryos on day 6 of incubation were measured and found to be 165 ± 30 mg/dL compared with vehicle-treated embryos with a plasma glucose concentration of 80 ± 8 mg/dL. Determination of the expression of GLUT1 was used to evaluate the role of glucose transporters during hyperglycemia. The gene expression level of GLUT1 (Fig. 1C) was decreased by twofold for d-glucose-treated embryos compared with vehicle.
FIG. 1.

The addition of exogenous glucose to chick embryos increased plasma glucose concentration. A: Transient plasma glucose concentration in chick embryos at day 4 (HH24) for embryos treated with a single pulse of 30 mmol/L d-glucose, 50 mmol/L d-glucose, or 30 mmol/L l-glucose with an enlargement of the critical area of glucose spike for 0–90 min. B: Plasma glucose concentration for HH24 embryos after yolk injection of varying concentrations of d-glucose at day 0. A clinically significant concentration of plasma glucose resulted from the 750 mmol/L d-glucose injection. C: Under sustained hyperglycemia, expression of GLUT1 levels in the OFT for vehicle-, l-glucose–, and d-glucose–treated embryos. Data are mean ± SD with trend lines for n = 10. *Difference from vehicle (P < 0.05) using ANOVA with Fisher LSD post hoc.
Viability and growth.
By measuring the viability and embryonic growth of treated and untreated embryos at 96 h, the effects of exogenous glucose on embryonic development were determined. Pulsed hyperglycemia caused a small developmental stage delay, with an average embryonic age of HH23 or a 4- to 6-h delay in their development compared with vehicle controls at HH24 (Table 1). A decreasing trend in embryonic weight was found in these pulsed hyperglycemic embryos because the average weights of d- and l-glucose-treated embryos decreased 20 and 10%, respectively, compared with vehicle controls (Table 1). Pulsed d-glucose embryos had 10% mortality, whereas l-glucose– and vehicle-treated embryos had 7 and 1% mortality, respectively.
TABLE 1.
The effects of pulsed and sustained hyperglycemia on 96-h chick embryos
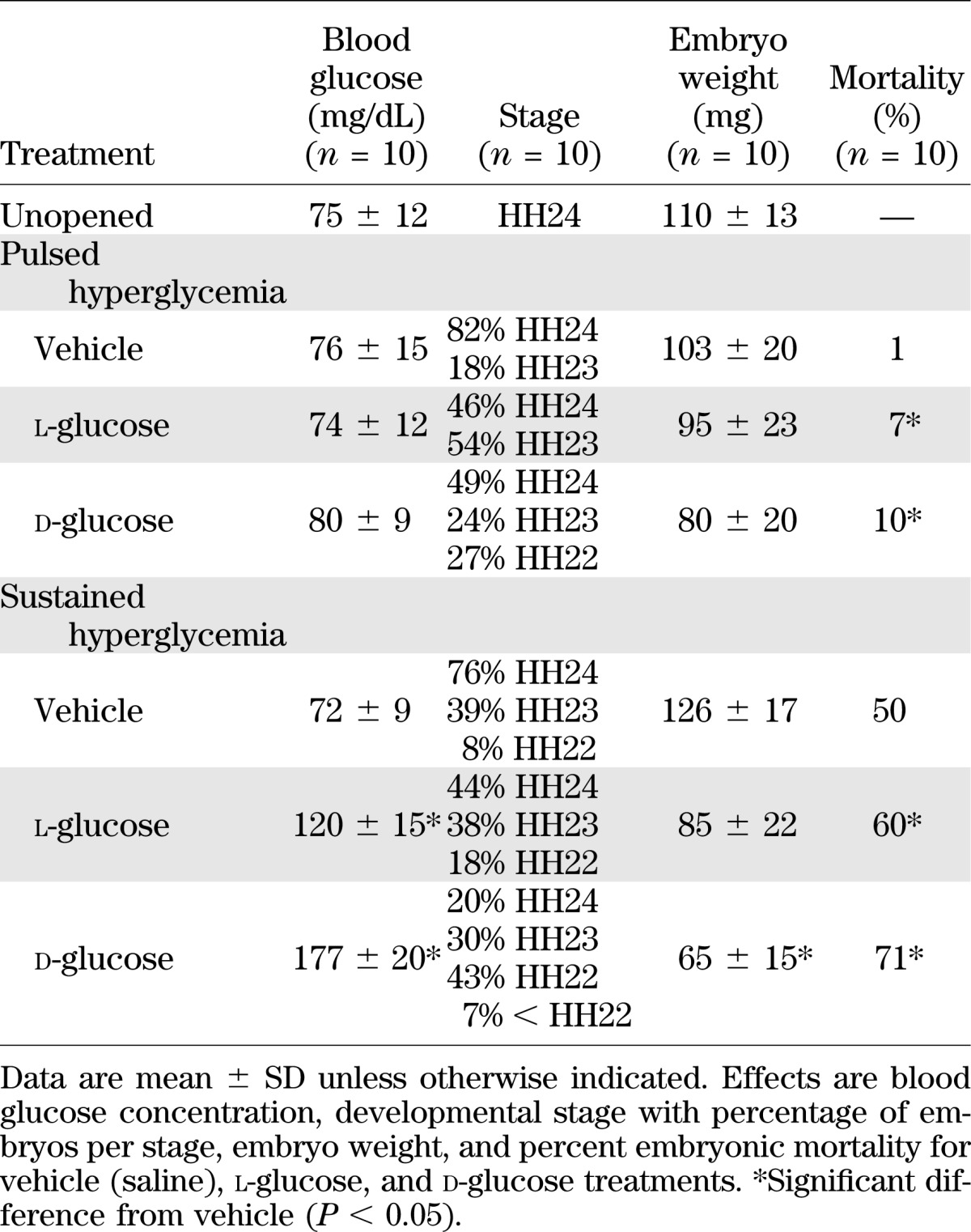
Although the pulsed hyperglycemic condition resulted in a slight reduction in growth and embryo viability, the sustained hyperglycemic model induced larger changes in the embryos. Sustained hyperglycemia produced a stage delay, equivalent to a 12-h delay in development, in the embryos, with an average stage of HH22 compared with that for the l-glucose and vehicle control embryos of HH23 and HH24, respectively (Table 1). The average weight of d- and l-glucose–treated embryos decreased by 50 and 10%, respectively, compared with vehicle controls. Percent mortality was high for all the yolk-injected embryos because the insertion of a syringe alone into the yolk of the eggs causes a 50% mortality rate for embryos (25). Using vehicle-injected embryos as a baseline, the sustained hyperglycemic condition significantly increased mortality by 30% compared with vehicle control and 11% compared with l-glucose.
OFT cellular proliferation.
A decrease in size and stage delay was found in both pulsed and sustained hyperglycemic embryos. To examine a potential cause of this delay, the degree of cell proliferation in stage-matched HH24 OFTs was determined. There were no significant differences in the number of proliferating endocardial or myocardial cells in the OFT for the pulsed model (Fig. 2). Sustained hyperglycemia resulted in a 10% average decrease in OFT cell proliferation for endocardial and myocardial cells for both the d- and l-glucose–treated embryos compared with the vehicle controls (Fig. 3). Proliferation did not vary with location in the OFT.
FIG. 2.

Confocal images indicating the proliferating cells for the pulsed hyperglycemic condition. The nuclei of the proliferating endocardial cells for the vehicle-treated (A), l-glucose–treated (B), and d-glucose–treated (C) OFTs are indicated by the green stain. Myocardial cell proliferation for vehicle (D), l-glucose (E), and d-glucose (F) is designated by the green nuclear stain (Alexa Fluor 488) with the red myosin stain (Alexa Fluor 564) to differentiate the cell type. Original magnification 400×. Scale bar represents 20 µm. The percentage of volumetric (G), endocardial cell (H), and myocardial cell (I) proliferation in the OFTs of HH24 embryos for pulsed additions of vehicle, l-glucose, and d-glucose are shown. Data are mean ± SD for n = 10.
FIG. 3.

Confocal images indicated the proliferating cells for the sustained hyperglycemic condition. The nuclei of the proliferating endocardial cells for the vehicle-treated (A), l-glucose–treated (B), and d-glucose–treated (C) OFTs are indicated by the green stain. Myocardial cell proliferation for vehicle (D), l-glucose (E), and d-glucose (F) is designated by the green nuclear stain (Alexa Fluor 488) with the red myosin stain (Alexa Fluor 564) to differentiate the cell type. Original magnification 400×. Scale bar represents 20 µm. The percentage of volumetric (G), endocardial cell (H), and myocardial cell (I) proliferation in the OFTs of HH24 embryos for sustained additions of vehicle, l-glucose, and d-glucose are shown. Data are mean ± SD for n = 10. *Difference from vehicle (P < 0.05) using ANOVA with Fisher LSD post hoc.
Cell cycle–regulated gene and protein expression.
The expression levels of p21 and cyclin D1 in the OFT were evaluated in sustained hyperglycemic embryos to determine whether cell cycle dysregulation contributed to the reduction of cell proliferation and embryonic developmental delay. Gene expression for p21 significantly increased by twofold in d-glucose–treated embryos, whereas no significant difference was found between the vehicle and l-glucose–treated embryos (Fig. 4A). The gene expression level of cyclin D1 (Fig. 4B) was decreased by threefold for d-glucose–treated embryos compared with vehicle and l-glucose control embryos. Similarly, the p21 protein expression was significantly increased for d-glucose–treated embryos, and there was a trend toward decreased protein expression for cyclin D1 in d-glucose-treated embryos compared with vehicle and l-glucose controls (Fig. 5).
FIG. 4.

Sustained hyperglycemia caused changes in expression of genes regulating the cell cycle. The fold changes of gene expression were compared with an OFT from an unopened calibrator for p21 (A) and cyclin D1 (B) for the embryo yolk injected with vehicle, l-glucose, or d-glucose. Data are mean ± SD for n = 10. *Difference from vehicle (P < 0.05) and †difference from l-glucose (P < 0.05) using ANOVA with Fisher LSD post hoc.
FIG. 5.

Protein levels of p21 and cyclin D1 in sustained hyperglycemic OFTs. p21 significantly increased in d-glucose–treated OFTs, whereas cyclin D1 trended toward decreased protein expression compared with l-glucose and vehicle. Data are mean ± SD for n = 5. *Difference from vehicle (P < 0.05) and †difference from vehicle (P < 0.05) using ANOVA with Fisher LSD post hoc.
DISCUSSION
The offspring of mothers with unrecognized type 1, type 2, or gestational diabetes have a high risk for being born with congenital anomalies, macrosomia, and neonatal, childhood, and adult complications (5). Congenital malformations found in the children of diabetic mothers include neural tube defects, which can lead to anencephaly, spina bifida aperta, meningocele, and encephalocele, and cardiovascular defects, such as septal defects and malformations of the heart and great vessels (26). Although these complications are known, few models exist to determine the effects and underlying causes of varying concentrations of glycemic stress on the development of congenital malformations during organogenesis. Thus, we hypothesized that hyperglycemia slows embryonic development through an increase in osmotic stress and the suppression of the glucose transporters and the cell cycle. To test this hypothesis, we developed and characterized two in vivo embryonic chick models to mimic embryonic conditions during maternal hyperglycemia: 1) pulsed hyperglycemia (associated with meal-induced hyperglycemia) and 2) sustained hyperglycemia (associated with poorly controlled maternal hyperglycemia). The embryos in this study were exposed to either a pulse or a sustained increase in blood glucose concentration. The plasma glucose concentrations in the pulsed hyperglycemic model were characterized by a peak at 60 min with a return to normal by 120 min. Although measurements at earlier developmental stages were not possible, we speculate that each daily glucose treatment caused a temporary spike in embryonic plasma glucose concentration followed by a return to normal. A longer-term exposure to glucose pulses (injections on days 0, 1, 2, and 3 of incubation), which has previously been shown at day 18 to both result in a significant increase in plasma glucose and lead to embryonic malformation (23), did not result in increased circulating glucose concentrations at day 4. An increase in the plasma glucose concentration of HH24 chicks at day 4 was accomplished in the sustained hyperglycemic model, which maintained an elevated blood glucose concentration until at least day 6 of development. In both models, the addition of d- and l-glucose resulted in clinically relevant plasma glucose concentrations in the chick embryos, elevating the average circulating glucose concentration to 180 mg/dL compared with the normal plasma glucose level of 70 mg/dL. To create these high concentrations, large quantities of exogenous glucose were added as follows: 50 and 750 mmol/L d-glucose in the pulse and sustained models, respectively, which affected the osmolarity of the blood. Hyperglycemic conditions in humans and in the present model induce osmotic stress through increased activity in the polyol pathway and excess generation of sorbitol (27). Thus, l-glucose was used as an osmotic control because cells do not readily uptake l-glucose, whereas glucose transporters such as GLUT1 facilitate the transport of d-glucose across the cell membrane. The l-glucose controls enabled us to distinguish between the effects of hyperglycemia and hyperosmolarity.
Glucose is essential for normal fetal growth and metabolism, but excess amounts are detrimental to fetal development. Facilitated glucose transport across the plasma membrane of a cell is mediated by glucose transporters. The distribution of glucose transporters varies in organ systems based on differing tissue requirements for glucose. GLUT1 is the most prominent glucose transporter in embryonic development, particularly in the embryonic chick heart, yet it is insulin independent (28). GLUT4 is an insulin-dependent glucose transporter in human hearts (28), but it is not present in the avian population, and the existence of an insulin-dependent glucose transporter in birds remains unknown. In the current study, sustained hyperglycemic conditions resulted in a decrease in gene expression of GLUT1 in the OFT. Similarly, in vivo preimplantation embryo studies with mouse models have shown a decrease in GLUT1, GLUT2, and GLUT3 as a result of maternal hyperglycemia (29). The downregulation of GLUT1 as a result of maternal hyperglycemia could lead to low levels of intracellular glucose, depriving cells of nutrition and predisposing embryos to developmental delays and other malformations (30). GLUT1-deficent embryonic mice have been shown to slow growth, reduce body weight, and lead to other malformations (31) similar to that of the hyperglycemic chick embryos. Therefore, the downregulation of GLUT1 in the current model facilitates excess amounts of circulating glucose while reducing intracellular glucose concentrations, which may play a role in embryonic malformations from maternal diabetes-induced hyperglycemia.
Embryonic stage delay as a result of hyperglycemia has been demonstrated in zebrafish (13), mice (11), and rats (32). Both models in the current study showed a developmental delay, increased embryonic mortality, and a smaller embryo size, similar to fetuses of diabetic mothers during early pregnancy (33). However, sustained hyperglycemia had a more pronounced effect on embryonic development. These outcomes are in agreement with others that have shown an increase in embryo mortality and abnormal growth as a result of a concentration-dependent effect of d-glucose (34). Osmotic stresses or a predominance of female embryos (male chicks hatch earlier than female chicks [35]) are possible explanations for the stage delay and decrease in weight of l-glucose embryos. Thus, a random sample of both male and female embryos was used. The high concentration of circulating glucose and osmotic stress in the embryo that accompany maternal diabetes results in embryonic delay and impairs all organ systems (36,37), particularly the cardiovascular system. Therefore, we examined the mechanisms of this delay in the heart OFT to determine whether cellular dysfunction in the OFT contributes to the development of cardiac malformations.
In the OFT, a decrease in proliferation of both endocardial and myocardial cells was demonstrated in the sustained hyperglycemic model. The development of the embryonic heart and OFT depends on the functions of the endocardial, myocardial, and neural crest cells. Reduced proliferation in neural crest cells has been shown under conditions of high glucose concentrations in both cells cultured in 50 mmol/L d-glucose and mouse embryos with streptozotocin-induced diabetes (38,39). In the current model, neural crest cells are likely a factor in the hyperglycemia-induced OFT changes from the onset of incubation; however, the effect of hyperglycemia on neural crest cell proliferation remains to be determined in this model. The decrease in cellular proliferation in the OFT may contribute to OFT underdevelopment and produce a malformed structure. Zhao (40) found a decrease in mitosis in the endocardial cushions of the OFT in embryos of diabetic mice, changing the structure of the cushions. Thus, the decrease in proliferation may be instrumental in the formation of similar malformations seen at late stages of human development, such as semilunar valve malformations. The proliferation for both endocardial and myocardial cells significantly decreased for both d- and l-glucose–treated embryos, suggesting a role for osmotic stress in the reduction of cell proliferation. This finding is in agreement with others who have shown decreased endothelial cell proliferation because of high glucose concentration and hyperosmolarity (41). Because a significant decrease in proliferation was found in the sustained hyperglycemic case, cell cycle gene signaling was investigated.
In the current study, we found that regulators of the cell cycle were altered in cardiac cells located in the OFT wall of hyperglycemic embryonic chick hearts. Cell proliferation is the end point of an ordered set of phases in the cell cycle, which are regulated by cyclins and cyclin-dependent kinases. Cyclins drive cells from the G1 phase to the S phase and from the G2 phase to the mitosis phase by binding to cyclin-dependent kinases at critical points during the cell cycle. This binding leads to the phosphorylation of target proteins, which enables progression to the next phase. The cyclin-dependent kinase inhibitor p21 is associated with cell cycle arrest in the G1 phase and, thus, is a critical mediator of growth arrest (42) by inhibiting mitosis, DNA replication, and repair (43). During the cell cycle, the G1/S phase depends on cyclin D1 and is negatively regulated by p21. Previous studies in an ovine model of cardiomyocytes demonstrated that p21 and cyclin D1 are reliable indicators of cell proliferation (44,45), with an increase in p21 and a decrease in cyclin D1 contributing to reduced cell proliferation. Under sustained hyperglycemic conditions, embryonic chick cardiac cells may have arrested in the G1 phase because of the suppression of cyclin D1 and the upregulation of p21, which would lead to a reduction of endocardial and myocardial cell proliferation. Although the decrease in cardiac cell proliferation was similar in the OFTs of embryos treated with d- and l-glucose, there were significant differences in the expression of p21 and cyclin D1 between the d-glucose–treated embryos and the l-glucose controls. High osmolarity can also contribute to decreased cell proliferation by arresting cells in their G1/S and G2 phases. For example, hyperosmolarity can activate the G2/M checkpoint through p38 activation, which causes a drop in the cyclin-dependent kinase 2 activity (46). Thus, the inhibition of cell proliferation that we found in the d- and l-glucose–treated embryos may be through different mechanisms of cell cycle arrest or another mechanism associated with proliferation. Although not examined here, apoptosis is another potential mechanism leading to maternal diabetes-induced stage and developmental delay. Hyperglycemia can lead to apoptosis during embryogenesis (47). The apoptotic pathway may be triggered by reactive oxygen species generation, and tissue damage from this apoptosis could lead to abnormalities (48). However, more research needs to be conducted on this pathway.
Maternal diabetes has detrimental effects on embryos, increasing the rates of embryonic malformations. In this avian model, elevated glucose decreased the glucose transporter GLUT1 and resulted in embryonic stage delay, development retardation, and increased embryonic mortality. Additionally, sustained hyperglycemia reduced endocardial and myocardial cell proliferation if the heart OFT, which may be the associated with the downregulation of the cell cycle by p21 and cyclin D1. This change in cellular proliferation is likely to affect the development of the OFT and contribute to malformations later in development. Importantly, this model enables the study of concentration-dependent effects of hyperglycemia on cardiac development during early organogenesis and determination of the underlying mechanisms of hyperglycemia-induced cardiac malformations.
ACKNOWLEDGMENTS
A portion of this work was funded by National Institutes of Health Grant R01-HL-094570.
No potential conflicts of interest relevant to this article were reported.
The funder played no role in the conduct of the study, collection of data, management of the study, analysis of data, interpretation of data, or preparation of the manuscript.
D.E.S.-D. conducted experiments, researched data, and wrote the manuscript. S.R. and K.L.T. contributed to discussion and reviewed and edited the manuscript. D.L.M. contributed to discussion and reviewed the manuscript. M.T.H. researched data, contributed to discussion, and reviewed and edited the manuscript. D.E.S.-D. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Natasha Chattergoon (Oregon Health & Science University, Heart Research Center) for providing assistance with the Western blot protein analysis.
Footnotes
See accompanying commentary, p. 27.
REFERENCES
- 1.Roman MA. Preconception care for women with preexisting type 2 diabetes. Clin Diabetes 2011;29:10–16 [Google Scholar]
- 2.Chu SY, Callaghan WM, Kim SY, et al. Maternal obesity and risk of gestational diabetes mellitus. Diabetes Care 2007;30:2070–2076 [DOI] [PubMed] [Google Scholar]
- 3.Madri JA, Enciso J, Pinter E. Maternal diabetes: effects on embryonic vascular development—a vascular endothelial growth factor-A-mediated process. Pediatr Dev Pathol 2003;6:334–341 [DOI] [PubMed] [Google Scholar]
- 4.Evers IM, de Valk HW, Visser GH. Risk of complications of pregnancy in women with type 1 diabetes: nationwide prospective study in The Netherlands. BMJ 2004;328:915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eriksson UJ. Congenital anomalies in diabetic pregnancy. Semin Fetal Neonatal Med 2009;14:85–93 [DOI] [PubMed] [Google Scholar]
- 6.Eriksson UJ, Borg LA, Cederberg J, et al. Pathogenesis of diabetes-induced congenital malformations. Ups J Med Sci 2000;105:53–84 [DOI] [PubMed] [Google Scholar]
- 7.Loffredo CA, Wilson PD, Ferencz C. Maternal diabetes: an independent risk factor for major cardiovascular malformations with increased mortality of affected infants. Teratology 2001;64:98–106 [DOI] [PubMed] [Google Scholar]
- 8.Li P, Yin X, Shi L, Liu A, Rugonyi S, Wang RK. Measurement of strain and strain rate in embryonic chick heart in vivo using spectral domain optical coherence tomography. IEEE Trans Biomed Eng 2011;58:2333–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hughes AF, Freeman RB, Fadem T. The teratogenic effects of sugars on the chick embryo. J Embryol Exp Morphol 1974;32:661–674 [PubMed] [Google Scholar]
- 10.Roest PA, van Iperen L, Vis S, et al. Exposure of neural crest cells to elevated glucose leads to congenital heart defects, an effect that can be prevented by n-acetylcysteine. Birth Defects Res A Clin Mol Teratol 2007;79:231–235 [DOI] [PubMed] [Google Scholar]
- 11.Moley KH, Chi MM, Manchester JK, McDougal DB, Lowry OH. Alterations of intraembryonic metabolites in preimplantation mouse embryos exposed to elevated concentrations of glucose: a metabolic explanation for the developmental retardation seen in preimplantation embryos from diabetic animals. Biol Reprod 1996;54:1209–1216 [DOI] [PubMed] [Google Scholar]
- 12.Larger E, Marre M, Corvol P, Gasc JM. Hyperglycemia-induced defects in angiogenesis in the chicken chorioallantoic membrane model. Diabetes 2004;53:752–761 [DOI] [PubMed] [Google Scholar]
- 13.Liang J, Gui Y, Wang W, Gao S, Li J, Song H. Elevated glucose induces congenital heart defects by altering the expression of tbx5, tbx20, and has2 in developing zebrafish embryos. Birth Defects Res A Clin Mol Teratol 2010;88:480–486 [DOI] [PubMed] [Google Scholar]
- 14.Varma S, Lal BK, Zheng R, et al. Hyperglycemia alters PI3k and Akt signaling and leads to endothelial cell proliferative dysfunction. Am J Physiol Heart Circ Physiol 2005;289:H1744–H1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar SD, Yong SK, Dheen ST, Bay BH, Tay SSW. Cardiac malformations are associated with altered expression of vascular endothelial growth factor and endothelial nitric oxide synthase genes in embryos of diabetic mice. Exp Biol Med (Maywood) 2008;233:1421–1432 [DOI] [PubMed] [Google Scholar]
- 16.Martinsen BJ. Reference guide to the stages of chick heart embryology. Dev Dyn 2005;233:1217–1237 [DOI] [PubMed] [Google Scholar]
- 17.Hove JR, Köster RW, Forouhar AS, Acevedo-Bolton G, Fraser SE, Gharib M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 2003;421:172–177 [DOI] [PubMed] [Google Scholar]
- 18.Clark EB, Rosenquist GC. Spectrum of cardiovascular anomalies following cardiac loop constriction in the chick embryo. Birth Defects Orig Artic Ser 1978;14:431–442 [PubMed] [Google Scholar]
- 19.Liu A, Wang R, Thornburg KL, Rugonyi S. Efficient postacquisition synchronization of 4-D nongated cardiac images obtained from optical coherence tomography: application to 4-D reconstruction of the chick embryonic heart. J Biomed Opt 2009;14:044020 [DOI] [PubMed]
- 20.Rothenberg F, Fisher SA, Watanabe M. Sculpting the cardiac outflow tract. Birth Defects Res C Embryo Today 2003;69:38–45 [DOI] [PubMed] [Google Scholar]
- 21.Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. 1951. Dev Dyn 1992;195:231–272 [DOI] [PubMed] [Google Scholar]
- 22.Pentecost JO, Silva C, Pesticelli MJ, Jr, Thornburg KL. Modeling cardiogenesis: the challenges and promises of 3D reconstruction. Curr Top Dev Biol 2003;56:115–143 [DOI] [PubMed] [Google Scholar]
- 23.Miller RR, Burum A.L, Leithard ME, Hart JD. Hyperglycemia-induced changes in hepatic membrane fatty acid composition correlate with increased caspase-3 activities and reduced chick embryo viability. Comp Biochem Physiol B Biochem Mol Biol 2005;141:323–330 [DOI] [PubMed] [Google Scholar]
- 24.Ahnfelt-Rønne J, Jørgensen MC, Hald J, Madsen OD, Serup P, Hecksher-Sørensen J. An improved method for three-dimensional reconstruction of protein expression patterns in intact mouse and chicken embryos and organs. J Histochem Cytochem 2007;55:925–930 [DOI] [PubMed] [Google Scholar]
- 25.Gebhardt DOE. The teratogenic action of propylene glycol (propanediol-1,2) and propanediol-1,3 in the chick embryo. Teratology 1968;1:153–161 [DOI] [PubMed] [Google Scholar]
- 26.Kalter H. Teratology in the 20th century: environmental causes of congenital malformations in humans and how they were established. Neurotoxicol Teratol 2003;25:131–282 [PubMed] [Google Scholar]
- 27.Mizisin AP, Li L, Perello M, et al. Polyol pathway and osmoregulation in JS1 Schwann cells grown in hyperglycemic and hyperosmotic conditions. Am J Physiol 1996;270:F90–F97 [DOI] [PubMed] [Google Scholar]
- 28.Carver FM, Shibley IA, Jr, Pennington JS, Pennington SN. Differential expression of glucose transporters during chick embryogenesis. Cell Mol Life Sci 2001;58:645–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moley KH, Chi MM, Mueckler MM. Maternal hyperglycemia alters glucose transport and utilization in mouse preimplantation embryos. Am J Physiol 1998;275:E38–E47 [DOI] [PubMed] [Google Scholar]
- 30.Moley KH. Diabetes and preimplantation events of embryogenesis. Semin Reprod Endocrinol 1999;17:137–151 [DOI] [PubMed] [Google Scholar]
- 31.Heilig CW, Saunders T, Brosius FC, 3rd, et al. Glucose transporter-1-deficient mice exhibit impaired development and deformities that are similar to diabetic embryopathy. Proc Natl Acad Sci U S A 2003;100:15613–15618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Svensson AM, Borg LA, Eriksson UJ. Glucose metabolism in embryos of normal and diabetic rats during organogenesis. Acta Endocrinol (Copenh) 1992;127:252–257 [DOI] [PubMed] [Google Scholar]
- 33.Pedersen JF, Mølsted-Pedersen L. Early growth retardation in diabetic pregnancy. BMJ 1979;1:18–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Datar S, Bhonde RR. Shell-less chick embryo culture as an alternative in vitro model to investigate glucose-induced malformations in mammalian embryos. Rev Diabet Stud 2005;2:221–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reis LH, Gama LT, Soares MC. Effects of short storage conditions and broiler breeder age on hatchability, hatching time, and chick weights. Poult Sci 1997;76:1459–1466 [DOI] [PubMed] [Google Scholar]
- 36.Becerra JE, Khoury MJ, Cordero JF, Erickson JD. Diabetes mellitus during pregnancy and the risks for specific birth defects: a population-based case-control study. Pediatrics 1990;85:1–9 [PubMed] [Google Scholar]
- 37.Schaefer-Graf UM, Buchanan TA, Xiang A, Songster G, Montoro M, Kjos SL. Patterns of congenital anomalies and relationship to initial maternal fasting glucose levels in pregnancies complicated by type 2 and gestational diabetes. Am J Obstet Gynecol 2000;182:313–320 [DOI] [PubMed] [Google Scholar]
- 38.Suzuki N, Svensson K, Eriksson UJ. High glucose concentration inhibits migration of rat cranial neural crest cells in vitro. Diabetologia 1996;39:401–411 [DOI] [PubMed] [Google Scholar]
- 39.Molin DGM, Roest PAM, Nordstrand H, et al. Disturbed morphogenesis of cardiac outflow tract and increased rate of aortic arch anomalies in the offspring of diabetic rats. Birth Defects Res A Clin Mol Teratol 2004;70:927–938 [DOI] [PubMed] [Google Scholar]
- 40.Zhao Z. Cardiac malformations and alteration of TGFbeta signaling system in diabetic embryopathy. Birth Defects Res B Dev Reprod Toxicol 2010;89:97–105 [DOI] [PubMed] [Google Scholar]
- 41.Mizutani M, Okuda Y, Yamaoka T, et al. High glucose and hyperosmolarity increase platelet-derived growth factor mRNA levels in cultured human vascular endothelial cells. Biochem Biophys Res Commun 1992;187:664–669 [DOI] [PubMed] [Google Scholar]
- 42.Rosso A, Balsamo A, Gambino R, et al. p53 Mediates the accelerated onset of senescence of endothelial progenitor cells in diabetes. J Biol Chem 2006;281:4339–4347 [DOI] [PubMed] [Google Scholar]
- 43.Chang BD, Watanabe K, Broude EV, et al. Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: implications for carcinogenesis, senescence, and age-related diseases. Proc Natl Acad Sci U S A 2000;97:4291–4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chattergoon NN, Giraud GD, Thornburg KL. Thyroid hormone inhibits proliferation of fetal cardiac myocytes in vitro. J Endocrinol 2007;192:R1–R8 [DOI] [PubMed] [Google Scholar]
- 45.Chattergoon NN, Giraud GD, Louey S, Stork P, Fowden AL, Thornburg KL. Thyroid hormone drives fetal cardiomyocyte maturation. FASEB J 2012;26:397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Nadal E, Alepuz PM, Posas F. Dealing with osmostress through MAP kinase activation. EMBO Rep 2002;3:735–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moley KH. Hyperglycemia and apoptosis: mechanisms for congenital malformations and pregnancy loss in diabetic women. Trends Endocrinol Metab 2001;12:78–82 [DOI] [PubMed] [Google Scholar]
- 48.Dhanasekaran N, Wu YK, Reece EA. Signaling pathways and diabetic embryopathy. Semin Reprod Endocrinol 1999;17:167–174 [DOI] [PubMed] [Google Scholar]