

Abstract
Aim
Loss of superoxide dismutase (SOD) activity is a defining biochemical feature of asthma. However, mechanisms for the reduced activity are unknown. We hypothesized that loss of asthmatic SOD activity is due to greater susceptibility to oxidative inactivation. Result: Activity assays of blood samples from asthmatics and healthy controls revealed impaired dismutase activity of copper-zinc SOD (CuZnSOD) in asthma. CuZnSOD purified from erythrocytes or airway epithelial cells from asthmatic was highly susceptible to oxidative inactivation. Proteomic analyses identified that inactivation was related to oxidation of cysteine 146 (C146), which is usually disulfide bonded to C57. The susceptibility of cysteines pointed to an alteration in protein structure, which is likely related to the loss of disulfide bond. We speculated that a shift to greater intracellular reducing potential might account for the change. Strikingly, measures of reduced and oxidized glutathione confirmed greater reducing intracellular state in asthma, compared with controls. Similarly, greater free thiol in CuZnSOD was confirmed by ∼2-fold greater N-ethylmaleimide binding to C146 in asthma as compared with controls. Innovation: Greater reducing potential under a chronic inflammatory state of asthma, thus, leads to susceptibility of CuZnSOD to oxidative inactivation due to cleavage of C57-C146 disulfide bond and exposure of usually unavailable cysteines. Conclusion: Vulnerability of CuZnSOD influenced by redox likely amplifies injury and inflammation during acute asthma attacks when reactive oxygen species are explosively generated. Overall, this study identifies a new paradigm for understanding the chemical basis of inflammation, in which redox regulation of thiol availability dictates protein susceptibility to environmental and endogenously generated reactive species. Antioxid. Redox Signal. 18, 412–423.
Introduction
Asthma, a disease of chronic airway inflammation, is characterized by reversible airflow obstruction and bronchial hyper-responsiveness (22). Spontaneous asthma attacks and models of experimental antigen challenge are associated with immediate release of high levels of reactive oxygen species (ROS) (11), for example, superoxide (O2•−) and hydrogen peroxide (H2O2), which persist throughout the late asthmatic response (3, 4, 8, 41). Experimental models of asthma and clinical studies of asthma indicate a mechanistic link between intermittent excessive oxidative processes and the progressive airway injury and remodeling, which defines severe asthma pathology (39).
The overall effect of ROS is highly dependent on the locally available antioxidant defenses, including enzymatic systems, catalase, and superoxide dismutases (SODs) (3, 4, 8, 11). SODs dismutate O2•− to H2O2, and catalase converts H2O2 to water and oxygen (5). Three isoforms of SOD proteins are present in eukaryotic cells: cytosolic copper-zinc SOD (CuZnSOD), mitochondrial manganese SOD (MnSOD), and extracellular SOD (EcSOD). CuZnSOD, the 32 kDa homodimeric protein, involves cyclic reduction and reoxidation of copper during the dismutation reaction of O2•− (40). Each monomer of CuZnSOD binds one copper and one zinc ion. Structurally, CuZnSOD is described as an eight-stranded Greek β-barrel with three extended loop regions. Four cysteine residues (C6, C57, C111, and C146) are present in CuZnSOD monomer; C57 and C146 form a disulfide bond within each monomer, which is important for the maintenance of the tertiary structure and stability of the dimer enzyme. The C111, along with histidine and tryptophan, has been described as being available for oxidation and inactivation of the CuZnSOD enzyme (14, 32, 43, 46).
Innovation.
Greater reducing potential under the chronic inflammatory state of asthma leads to loss of copper zinc super oxide dismutase (CuZnSOD) activity in human asthma. Susceptibility to oxidative inactivation is due to cleavage of the disulfide bond and exposure of usually unavailable cysteines. The distortion and loss of CuZnSOD activity likely amplifies injury and inflammation during acute asthma attacks when reactive oxygen species are explosively generated. This study has significance for understanding a previously unsuspected impact of physiological redox regulation of a potent antioxidant CuZnSOD, where redox regulation of thiol availability dictates protein susceptibility to environmental and endogenously generated reactive species.
The inactivation of antioxidants has been described in the asthmatic airways and linked to the pathophysiology of airway reactivity (7, 16). Total SOD activity in asthmatic lungs and blood is lower than normal and has been previously suggested to be related to CuZnSOD activity (8). Although MnSOD in the asthmatic airway has oxidative modifications (7), the majority of activity in the airway and blood is primarily accounted for by CuZnSOD, which is typically highly resistant to oxidation. The cause for the greater than 50% reduction of SOD activity in asthma is unknown.
We hypothesized that the widespread loss of SOD activity in asthma is due to the oxidative modification and inactivation of CuZnSOD. To test this, in-gel SOD activity assays were performed to evaluate CuZnSOD activity in a wide variety of samples from asthmatics and healthy controls. Subsequently, CuZnSOD was purified from erythrocytes and airway epithelial cells that were freshly obtained from human asthmatics and healthy controls. Targeted proteomic analyses were used to identify oxidized peptides and quantify relative abundance in asthmatic samples as compared with control samples. Unexpectedly, although SOD activity in asthma was lower than in normal, proteomic analyses of CuZnSOD freshly obtained from asthmatics, it had no greater oxidative modification than healthy control protein. Rather, CuZnSOD from asthmatic samples was susceptible to oxidative modifications and inactivation only on ex-vivo H2O2 exposure or reaction during kinetic dismutase activity assay, in which O2•− is exogenously generated by xanthine/xanthine oxidase (X/XO) and H2O2 is generated by the SOD enzyme activity. The susceptibility of asthmatic CuZnSOD to oxidation was due to the availability of cysteine 146, which is usually inaccessible for reaction due to the tertiary structure and participation in a disulfide bond. The availability of cysteine to oxidation reactions was accounted for by a global shift toward a greater intracellular reducing state as determined by the measuring of reduced and oxidized glutathione (GSH/GSSG) in asthma.
Result
Loss of CuZnSOD activity accounts for the low serum SOD activity in asthma
Asthmatics were similar to controls [age, asthma: 34.6±1.9, healthy control: 37.6±1.7; asthma: n=25 (female=13), healthy control: n=37 (female=19)] but had lower lung functions (predicted forced expiratory volume in 1 s, asthmatic 82.5±3.9, control 95.2±2.4; *p=0.04). All asthma subjects were nonsevere, as defined by criteria of the American Thoracic Society (45)(Table 1). None of the participants were current users of tobacco products. Not all samples were available for use in all assays due to limitations of sample amount; numbers evaluated are identified in each result. Similar to previous reports (3, 6–8), total SOD activity in serum of asthmatics was approximately 50% of normal activity, as determined by kinetic assay in which SOD activity units are determined from the inhibition of cytochrome C reduction by xanthine/xanthine oxidase (X/XO)-O2•− producing system [control: 23±3 U/ml, asthma: 12±2 U/ml, *p=0.003]. To determine the contribution of SOD isoforms to loss of activity, total serum proteins were separated by nondenaturing, nonreducing polyacrylamide gel electrophoresis followed by nitroblue tetrazolium in-gel activity assessment. CuZnSOD was identified as the major contributor to total serum activity based on the mobility and size of the gel (Fig. 1A). Asthmatic serum CuZnSOD in-gel activity was ∼75% less than control serum [Fig. 1B, SOD in-gel activity as% of positive control: Control: 80%±11%, Asthma 21%±5%, *p=0.0007]. Serum did not have detectable EcSOD activity. MnSOD in-gel activity was present at low but similar levels among control and asthmatic serum (data not shown).
Table 1.
Asthma and Healthy Control Population
| Control (n=37) | Asthma (n=25) | |
|---|---|---|
| Age | 37.6 (1.7) | 34.6 (1.9) |
| Gender (female/male) | 18/19 | 12/13 |
| Race (C/AA/A/other) | 23/11/2/1 | 13/8/1/3 |
| %FEV1 | 95.2 (2.4) | 82.5 (3.9)a |
| %FVC | 97.5 (2.5) | 91.8 (3.0) |
| FEV1/FEC | 0.81 (0.01) | 0.75 (0.02)a |
| Inhaled corticosteroid | 0% | 36% (9)a |
| Oral or injectable corticosteroid | 0% | 0% |
Mean (SEM) shown for continuous variables (ap<0.05).
%FEV1, predicted forced expiratory volume in 1 s; %FVC, forced percentual vital capacity.
FIG. 1.

Decreased copper zinc super oxide dismutase (CuZnSOD) activity in asthma. (A) Control (lanes: 1–3) and asthma (lanes: 4–9) serum was electrophoresed on nondenaturing, nonreducing gels and exposed to nitroblue tetrazolium (NBT), riboflavin, and TEMED. SOD activity was detected by clear zones where there was an inhibition of reduction of NBT to dark blue precipitate. All samples have apparent CuZnSOD. Lane 10 represents in-gel activity for pure CuZnSOD. (B) CuZnSOD activity of control (n=13) and asthma serum (n=16) by densitometry (*p=0.0007). (C) Control (Ctrl, lanes: 1–3) and asthma (lanes: 4–6) erythrocyte samples evaluated for CuZnSOD in-gel activity and protein expression by Western analysis. Pure CuZnSOD is shown as a positive control for in-gel activity assay. (D) Erythrocyte CuZnSOD activity by densitometry (*p=0.04). (E) Activity of purified CuZnSOD determined by inhibition of cytochrome C reduction by superoxide generated by Xanthine-Xanthine oxidase (X/XO). The kinetic assay shows a change in optical density at 550 nm for X/XO alone (Blank), X/XO with positive control (CuZnSOD from human erythrocyte; Sigma-Aldrich), or equal amounts (14 μg) of CuZnSOD purified from control (Ctrl) or asthmatic (Asthma) erythrocytes. The asthma sample has less activity than the control as shown by less inhibition of cytochrome C reduction. (F) Control (Ctrl, n=10) and asthma (n=10) platelet samples evaluated for CuZnSOD activity by spectrophotometric analysis (mean±SE; *p=0.001).
CuZnSOD activity in asthmatic platelets and erythrocytes is less than in healthy control
Asthmatic platelets also had significantly lower SOD activity in kinetic assays as compared with controls [Fig. 1F, SOD activity units/mg protein: control: 21±2, asthma 11±3, *p<0.001]. More than one-third of the asthma group was on inhaled corticosteroid (Table 1). In order to assess a potential effect of inhaled corticosteroids, we evaluated platelet SOD activity in the asthmatics on inhaled corticosteroid in comparison to SOD activity in subjects not on inhaled corticosteroid. The SOD activity in platelets between the two asthmatic groups was similar [SOD U/mg protein, (−) inhaled corticosteroid 10.6±3.1; (+) inhaled corticosteroid 7.8±2.9; p=0.5], and each group was lower than in control samples [control SOD U/mg protein 21±2; *p<0.01 for all comparisons]. Since the color of hemoglobin interferes with spectrophotometric kinetic assay, erythrocyte lysates were evaluated for activity by the in-gel method. Asthmatic erythrocytes also had low activity as compared with controls (Fig. 1C), but western analysis for CuZnSOD (Fig. 1C) did not reveal a difference in protein amounts between control and asthmatic erythrocytes. A densitometric analysis of erythrocyte CuZnSOD activity relative to CuZnSOD protein confirmed the loss of specific activity [Fig. 1D: CuZnSOD in-gel activity/protein in erythrocyte: control 0.8±0.09, asthma 0.47±0.06, *p=0.04].
Given lower in-gel activity, activities of purified CuZnSOD from control and asthmatic erythrocytes were quantitatively determined via a kinetic measure of cytochrome C reduction by O2•− generated from xanthine and xanthine oxidase (X/XO). The purification process removed hemoglobin, making it suitable for the spectrophotometric assay in equal amounts of protein from asthma and control samples. Integrity of pure protein before use in the assay was checked by Western analysis (inset, Fig. 2). The protein was also analyzed by mass spectrometry (MS). Coverage of CuZnSOD (Accession number: AAB05661.1) identified more than 90% (number of peptide matches: 17). A tryptic peptide of CuZnSOD, 10Gly-Lys23 (Fig. 2) was identified in all the CuZnSOD digestions and was used as a normalization factor in all quantitative analyses. Complete lists of peptides identified in proteomic analyses are given in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/ars) (Supplementary data). Quantitative analysis of peptides from CuZnSOD freshly purified from erythrocytes from asthmatics was similar to control samples. Specifically, tryptic digests containing cysteine and tryptophan had similarly very low levels of oxidative modifications in freshly purified protein. However, purified CuZnSOD from asthmatic erythrocytes had lower specific enzyme activity as compared with control samples in kinetic activity assay (U/mg protein; control: 27.2±5.9, asthmatic: 14.1±1.6). A greater change in optical density with asthmatic samples as compared with controls shows the lower dismutase activity of asthmatic CuZnSOD (Fig. 1E).
FIG. 2.

Purity of CuZnSOD from human samples. CuZnSOD was partially purified from healthy control and asthma erythrocytes and airway epithelial cells. Each purification was tested for CuZnSOD in sodium dodecyl sulfate (SDS)- poly acrylamide gel electrophoresis (PAGE) and Western analysis. Inset shows a representative Coomassie-stained CuZnSOD and corresponding Western analysis (Lane 1, partially purified lysate, lane 2, positive control for CuZnSOD). Collision induced dissociation (CID) spectrum for the [M+2H]+2 tryptic peptide GDGPVQGIINFEQK (10Gly-Lys23) is presented. The 751.7 Da ions contain a series of C-terminal y ions from y2 to y12 along with a series of N-terminal b ions from b4 to b13. The peptide was identified in all of the CuZnSOD tryptic digests and was used as a normalization factor in all of the quantitative analysis. All data are representative of at least three different experiments.
Modification at C146 of CuZnSOD derived from human asthmatic erythrocyte and airway epithelial cells, in response to oxidative stress
In order to reconcile the lower level of activity of CuZnSOD purified from asthma, and our lack of discovery of modifications in the isolated protein from asthmatic samples, we considered that in-gel and cytochrome C reduction kinetic assays are performed over finite times during which reactive species are generated. Specifically, all activity assays are performed under conditions of O2•− generation and lead to the formation of H2O2. During kinetic activity assays, xanthine oxidase generates O2•−. that not only reduces cytochrome C, but is also spontaneously converted to H2O2. If SOD activity is present in the reaction, O2•− conversion to H2O2 is accelerated, and cytochrome c reduction is inhibited. CuZnSOD is susceptible to H2O2 oxidative inactivation but is relatively resistant to O2•− (1). Hence, we questioned whether there might be greater susceptibility to the inactivation of asthmatic CuZnSOD during the kinetic assays.
To directly address whether asthmatic CuZnSOD had greater susceptibility to reactive species separate from its catalytic activity, CuZnSOD protein modification was interrogated by quantitative proteomic liquid chromatography (LC)-tandem MS analysis after xanthine/xanthine oxidase treatment or direct exposure to exogenous H2O2. Consistent with a previous report (1), on 50 mM H2O2 treatment, CuZnSOD from control samples lost ∼30% of its activity; however, under similar condition, asthma samples lost ∼90% activity (SOD U/mg protein: control 16.2±3, asthma 1.3±0.1). To identify modified peptides, CuZnSOD purified from control and asthmatic erythrocytes and bronchial epithelial cell lysates were treated with 50 mM H2O2, then separated on sodium dodecyl sulfate (SDS)-poly acrylamide gel electrophoresis (PAGE) gel along with corresponding untreated samples. A series of mass spectrometric-based proteomic experiments were conducted to identify the site(s) of oxidative modification in CuZnSOD. Several oxidized sites were identified in the analysis of the H2O2-treated CuZnSOD, including the oxidation of tryptophan (W), histidine (H), and cysteine (C) containing peptides (Table 2). The amount of the unmodified and oxidized forms of W was similar among control and asthmatic samples. While a peptide containing oxidized histidine (2-oxo-histidine) was identified, its low abundance as well as low abundance of C57 prohibited quantitative analysis. Surprisingly, C146 was found to be modified in control and asthmatic samples on 50 mM H2O2 treatment. The C146 residue was identified in the 144Leu-Gln153–tryptic peptide in both alkylated (Fig. 3A, left panel) and cysteic acid forms (Fig. 3A, right panel). The ratio of the oxidized and alkylated C146 peptides was subsequently determined in selected reaction-monitoring (SRM) analysis. Strikingly, CuZnSOD from asthma airway erythrocyte had more than twice the amount of oxidized peptide at C146 (144Leu-Gln153) after 50 mM H2O2 treatment as compared with untreated samples (Fig. 3C, right panel:% change in C146ox/C146, untreated: 100±16.8, 50 mM H2O2 treated: 231±18.1, *p=0.04). A significant increase in asthmatic C146 oxidation was also noticed even at lower doses of H2O2 exposure (Fig. 3C, right panel, *p<0.05). Different doses of H2O2 exposure did not increase amounts of this oxidized peptide in healthy control samples (Fig. 4C left panel, p>0.6). Post-treatment in gel activity and corresponding densitometric analyses also revealed that asthmatic CuZnSOD is more susceptible to the loss of activity even on a low dose of oxidant exposure (Fig. 3C).
Table 2.
Identification of Unmodified and Oxidized Tryptic Peptides in Copper Zinc Super Oxide Dismutase
| Peptide sequence | [M+H]+ | m/z |
|---|---|---|
| 10GDGPVQGIINFEQK23,a | 1501.7 | 751.7 |
| 144LACGVIGIAQ153 | 1001.5 | 1001.4 |
| 144LACoxGVIGIAQ153 | 992.5 | 992.5 |
| 4AVCVLKGDGPVQGIINFEQK23 | 2172.1 | 725.3 |
| 4AVCoxVLKGDGPVQGIINFEQK23 | 2163.1 | 1082.4 |
| 116TLVVHEK122 | 825.5 | 413.2 |
| 116TLVVHoxEK122 | 841.5 | 421.5 |
| 31VWGSIK36 | 689.4 | 345.3 |
| 31VW16GSIK36 | 705.4 | 353.3 |
| 31VW14GSIK36 | 703.5 | 352.3 |
| 31VW32GSIK36 | 721.4 | 361.2 |
| 31VW30GSIK36 | 719.4 | 360.2 |
represents the reference peptide for this analysis.
FIG. 3.

Identification of C146 oxidation in CuZnSOD on oxidant exposure. (A) CID spectra of the unoxidized (left panel) and oxidized (right panel) peptide LACGVIGIAQ. The CID spectra were acquired by liquid chromatography (LC)-tandem mass spectroscopy (MS) analysis of CuZnSOD purified from control and asthmatic samples before and after oxidant exposure. Spectrum on left represents unoxidized peptide in which the cysteine residue is alkylated with iodoacetamide and to which 57 Da is added to give a peptide with a molecular mass of 1002 Da (M+H). Spectrum on right represents the oxidized peptide in which the cysteine residue is converted to a cysteic acid. This modification adds 48 kDa to give a peptide with a molecular mass of 993 Da (M+H). The CID spectrum is informative and places the observed 160 Da and 151 Da mass differences at the C146 position through the y8 and y7 ions. (B) The % change in C146ox/C146 in asthma samples indicates significantly higher oxidation at C146 on xanthine/xanthine oxidase treatment compared with respective untreated samples (mean±SE; *p=0.02), whereas % change in C146ox/C146 is not significantly altered between oxidant treated and untreated healthy control samples (p=0.6). (C) Greater oxidation was observed in all asthma samples exposed to different concentrations (50, 0.1, 0.05 mM) H2O2 (closed square,% change in C146ox/C146: mean±SE; all *p<0.05), whereas % change in C146ox/C146 remained unchanged in control samples under similar conditions (all p>0.06). Loss of CuZnSOD activity (open square) at respective doses of H2O2 was observed in asthmatic samples. Asthmatic CuZnSOD had greater susceptibility to oxidant inactivation. SOD activity in respective untreated samples served as a negative control (†p<0.05).
Fig. 4.

Inflammatory and intracellular redox status in asthma. (A) Redox environment in asthma erythrocyte. GSH/GSSG ratio in asthmatic erythrocyte reveals a more intracellular reducing environment than in control samples (p=0.01). (B) Increased inflammation in asthmatic population. Plasma IL-13 level was increased in asthma (n=7) samples compared with controls (n=11; *p<0.0007).
SRM analysis also revealed that on xanthine/xanthine oxidase treatment, asthmatic CuZnSOD had a significantly increased amount of oxidized peptide at C146 compared with the untreated samples (Fig. 3B,% change in C146ox/C146, untreated: 100±15, X/XO treated: 158±25, *p=0.02). The % change in C146ox/C146 in CuZnSOD from controls was not affected on similar oxidant exposure (Fig. 3B). The susceptibility of C146 to oxidation indicated that it was present in the reduced sulfhydryl state rather than participating in the disulfide bond with C57. This suggested a possible change in the intracellular reducing state of asthma.
Increased intracellular redox potential and evidence of inflammation in asthma as compared with controls
Intracellular redox potential was assessed by the ratio of GSH/GSSG in erythrocytes. The GSH/GSSG ratio was ∼1.6-fold higher in freshly obtained asthmatic erythrocyte as compared with controls (Fig. 4A; *p=0.01). Inflammatory status of the subjects was evaluated by measuring the T helper cell Type 2 (Th2) cytokine, Interleukin 13 (IL-13) in control and asthmatic plasma. Plasma IL-13 level was ∼6-fold higher in asthma compared with control (IL-13 pg/mL, control: 4.6±3, Asthma: 31.3±9, *p<0.007), confirming the presence of Th2-mediated inflammation in asthmatic subjects that also had greater reducing potential (Fig. 4B).
Identification of reduced thiol in asthmatic CuZnSOD
Given the higher reducing environment of asthmatic cells, the presence of free thiol in asthmatic CuZnSOD was tested by SRM analysis. To address this, erythrocyte lysates from control and asthmatic samples were first treated with 50 mM N-ethylmaleimide (NEM), which irreversibly binds all free thiols. CuZnSOD was then partially purified from NEM treated samples and tested by SRM analysis. In these results, the NEM modified residues (CNEM) represent free cysteines, and the iodoacetamide-modified residues (C) correspond to disulfide bound cysteines. As a routine analysis, these samples were run using two different LC-MS/MS methods. The first is the survey analysis, which is used to identify CuZnSOD in these bands; the second is the MS/MS analysis, which looks for specific cysteine-containing peptides. C146 residue was identified in 144Leu-Gln153–tryptic peptide in both NEM modified (Fig. 5A) and alkylated forms. The ratio of the NEM modified and alkylated peptides was subsequently determined in a quantitative targeted proteomic analysis. The CNEM/C ratio for C111-containing peptide, which is known to be available in the free thiol state, was equally modified by NEM in control and asthma samples (Fig. 5B, right panel; p=0.8). In contrast, CNEM/C ratio for the C146 peptide in asthma samples was more than 2-fold greater than controls (Fig. 5B, left panel; *p=0.0001). Although alkylated C6 peptide was found in this analysis, we could not identify NEM-modified C6 peptide. Two different forms of the C6 peptide 4Ala-Lys9 (AVCVLK) and 4Ala-Lys23 (AVCVLKGDGPVQGIINFEQK) were found in these experiments, of which the latter includes the missed cleavage form. The presence of lysine (K9) next to C6 may have interfered in the detection of NEM-modified C6 peptide.
FIG. 5.

Identification and quantitation of free cysteine in CuZnSOD. (A) CID spectra of the N-ethylmaleimide (NEM) modified C146 peptide LACNEMGVIGIAQ. The modification of a cysteine residue with NEM results in the addition of 125 Da to give a cysteine with a residue mass of 228 Da and a peptide with a molecular mass of 1069 Da (M+H). The observed 228 Da mass difference between the y8 and y7 ions is consistent with a cysteine modified by N-ethylmaleimide (CNEM) residue at position C146. (B) The % change in C146NEM/C146in asthma samples indicates significantly higher free cysteine at C146 compared with healthy controls (*p=0.0001), whereas free cysteine at C111 is similar between asthmatic and healthy controls (p=0.8).
Modification at C6 of asthmatic CuZnSOD in response to 50 mM H2O2 oxidant exposure
On high levels of oxidant exposure (50 mM H2O2), proteomic analysis identified oxidation at the C6 residue of CuZnSOD. C6 was found in 4Ala-Lys23-tryptic peptide in both alkylated (represents free C residues, Fig. 6A) and cysteic acid forms (represents oxidized cysteine residues, Fig. 6B). The ratio of the oxidized and alkylated C6 was subsequently determined in quantitative targeted proteomic analysis as described earlier. Strikingly, CuZnSOD from asthmatics had more than twice the amount of oxidized peptide at C6 after 50 mM H2O2 treatment as compared with untreated samples (Fig. 6C; p=0.02). However, CuZnSOD from controls did not exhibit a significant increase in the amount of oxidized peptide at C6 after H2O2 exposure as compared with corresponding untreated samples (p=0.2). The susceptibility of C6 of asthmatic CuZnSOD toward oxidation also indicated that the structure of the protein is labile and the enzymic core is disrupted in asthma.
FIG. 6.

Identification of C6 oxidation in asthmatic CuZnSOD upon 50 mM oxidant exposure. (A and B) CID spectra of the unoxidized and oxidized peptide AVCVLKGDGPVQGIINFEQK. The CID spectra were acquired by LC-tandem MS analysis of CuZnSOD purified from control and asthmatic erythrocytes before and after exposure to 50 mM H2O2. Spectrum A represents unoxidized peptide in which the cysteine residue is alkylated with iodoacetamide and to which 57 Da is added to give a peptide with a molecular mass of 1086.8 Da (M+2H+2). Spectrum B represents the oxidized peptide in which the cysteine residue is converted to a cysteic acid. This modification adds 48 Da to give a peptide with a molecular mass of 1082.4 Da (M+2H+2). The observed 160 Da and 151 Da mass difference at the C6 position through the y18+2 and y17+2 ions. (C) The % change in C6ox/C in asthma samples indicates a significantly higher oxidation at C6 compared with respective untreated samples (*p=0.02), whereas % change in C6ox/C is similar between oxidant treated and untreated healthy controls (p=0.2).
To directly assess whether the reduction of CuZnSOD leads to the loss of activity and greater susceptibility to oxidation at C146, in-gel activity analyses were performed for recombinant CuZnSOD on treatment with different doses of Dithiothreitol (DTT) (0–25 mM). Loss of activity was observed at 10 mM DTT treatment, which further significantly declined at the dose of 25 mM DTT treatment (Fig. 7A). The 25 mM DTT-treated recombinant CuZnSOD was subsequently exposed to 50 mM H2O2, followed by SRM analyses for evaluation of oxidative modification at C146. H2O2 exposure led to greater cysteic acid formation at C146 in the 25 mM DTT-treated CuZnSOD as compared with untreated CuZnSOD (Fig. 7B).
FIG. 7.

Analyses of in-gel CuZnSOD activity and oxidation at C146 on dithiothreitol (DTT) treatment and oxidant exposure. (A) Recombinant CuZnSOD was treated with different doses of DTT (0–25 mM) followed by in-gel SOD activity analysis. Dose-dependent loss of activity was observed starting at 10 mM DTT with a further significant decline with 25 mM DTT. (B) 25 mM DTT-treated CuZnSOD had significantly more cysteic acid formation at C146 on H2O2 treatment as compared with the DTT-treated, H2O2-untreated, as well as DTT- and H2O2-untreated sample (*p<0.0001).
Discussion
Here, we identify CuZnSOD as the major isoform accounting for the global loss of SOD activity in asthma. Previous reports suggested that the decrease in SOD activity in asthmatic airways was due to loss of CuZnSOD activity (3, 7), but the mechanism for the loss of SOD activity in asthma was unknown. Although protein levels of CuZnSOD are similar between asthma and controls, with no significant difference in protein modifications at baseline, the CuZnSOD from asthma samples is more susceptible to inactivation during enzyme catalysis. Since CuZnSOD generates H2O2 in the presence of O2•−, it is not possible to separate the primary oxidant cause of SOD inactivation during enzyme catalysis. CuZnSOD is typically highly resistant to O2•− but is susceptible to H2O2 inactivation (1). We found profound loss of CuZnSOD activity in asthma samples when exposed to different concentrations of H2O2 and also on exposure to X/XO at levels used in kinetic assay. These data suggest that the oxidative modification of CuZnSOD likely occurred by its own end product, H2O2 (1). The inactivation of CuZnSOD by H2O2 has been described to occur but is unusual at rates generated by the enzyme reaction (19). Loss of activity with mM levels of H2O2 is generally attributed to the fragmentation of enzyme and/or loss of the metal ion. In this context, control CuZnSOD did not exhibit a significant increase in the amount of the oxidized peptide at C6 after 50 mM H2O2 exposure. In contrast, the susceptibility of C6 in asthma CuZnSOD to oxidation indicates that the structure of the protein is labile and CuZnSOD enzymic core is disrupted in asthma samples (Fig. 8). The difference in the susceptibility of cysteine oxidation indicates a change in the dithiol/disulfide state of the CuZnSOD in asthma. Overall, mechanisms underlying the loss of SOD activity in asthma are consistent with previous in vitro studies of purified recombinant protein, which identify that the loss of CuZnSOD activity occurs due to changes in apoenzyme stability and/or conformational changes in dimer (12, 24). There may be accumulation of H2O2 at the site of inflammation in asthma, in part related to the low level of catalase activity (16). In support that catalytic inactivation may be relevant to the pathophysiologic state of asthma, numerous studies report that high levels of O2•− and H2O2 are generated in the airways and found in the exhaled breath condensate of asthmatic patients, particularly during asthma exacerbations (Supplementary Table S2) (41). To our knowledge, this is the first report which identifies that a more reducing intracellular environment causes disulfide bond disruption in CuZnSOD, making the protein susceptible to oxidation inactivation.
FIG. 8.
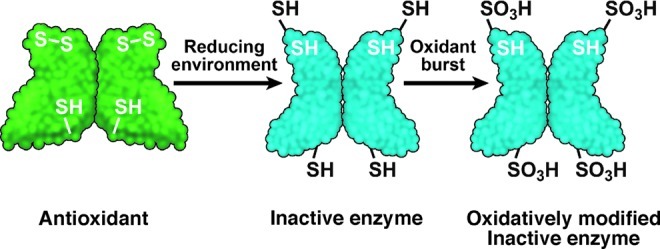
Redox effects on Cys 6, 146 in human CuZnSOD. Cys57 and Cys146 are usually involved in the intra-subunit disulfide bond (S-S) in CuZnSOD and are critical for antioxidant activity. Under the greater cellular reducing environment in asthma, the C57-146 disulfide bond becomes labile, causing distortion in structure and C6 is exposed, contributing to loss of activity. On extreme oxidant exposure, Cys146 and Cys6 of asthmatic CuZnSOD are then readily oxidized to cysteic acid (-SO3H). The findings suggest that the structurally distorted enzyme is incapable of performing dismutase activity. (To see this illustration in color the reader is referred to the web version of this article at www.liebertpub.com/ars.)
High levels of H2O2 can cause inactivation and fragmentation of the normal enzyme (35, 37). Uchida and Kawakishi (43) showed that histidine residues, located at active sites of the enzyme, are selective targets of O2•− and H118 (corresponding to human H120), which are selectively oxidized to 2-oxo-histidine. Several other studies also confirm that histidines present in CuZnSOD are prone to oxidation on exposure to ascorbic acid/Copper (II) Chloride (32) or peroxynitrite (1). Consistent with these reports, in our study, a peptide-containing modified histidine (2-oxo histidine at H120) was found, but at very low abundance in asthma and control samples, indicating that nonspecific oxidation or fragmentation was not a primary cause of activity loss. Structurally, human CuZnSOD does not have a tyrosine residue, a potential target of nitrative modification; rather, it has a single tryptophan residue (W32), the modification of which has been described to cause up to 15% loss of activity (48). In our study, several peptides with oxidative modification of tryptophan were identified (Table 2); however, again the amount was similar among control and asthmatic samples, indicating that nonspecific oxidation was not the mechanism of CuZnSOD inactivation.
Human CuZnSOD has 4 cysteine residues: C6, C57, C111, and C146. An intra-monomeric disulfide bond exits between C57 and C146 and is responsible for protein stability (40). Consistent with the findings in this article, a recent study has shown significant loss of CuZnSOD activity in the C146S mutant (18). The crystal structure of the cysteine-depleted human CuZnSOD reveals that loss of the C57-C146 disulfide bond frees the interface loop IV in the apoprotein (21). C6 is well buried in the β-barrel motif and is usually inaccessible to the solvent unless the SOD β-barrel core structure is severely disrupted (42). In contrast, C111 is exposed on the protein surface near the dimer interface and may be oxidized or modified (33). The sulfur atom of cysteine can undergo different types of oxidation: (i) reversible to disulfide (S-S) or sulfenic acid (-SOH) or (ii) irreversible oxidation to sulfinic acid (-SO2H) or sulfonic acid (-SO3H) (34). Earlier studies identified that C111 in human CuZnSOD may be oxidized to corresponding sulfinic acid and sulfonic acid even by oxygen in air (14). It was also demonstrated that CuZnSOD isolated from human erythrocytes can be glutathionylated at C111 (46), and this modification can induce dimer dissociation in CuZnSOD (33). However, this particular modification at C111 was not found in our study. All cysteine modification studies were performed in reducing conditions, which might contribute to the absence of detection of modifications at C111. The very low abundance of the C57 peptide also precluded quantitation in this study. On the other hand, C146 of asthmatic CuZnSOD was found to be selectively oxidized to corresponding sulfonic acid in different types and amounts of oxidant exposure.
Greater susceptibility of C146 in asthma samples to oxidation identifies that it is present in a reduced state and, therefore, more available to reactions. We reasoned that this could occur if the redox in asthmatic cells was set to a greater reduced (-SH) environment. This appears counterintuitive at first, because asthma is a disease that is characterized by increased production of reactive oxygen and nitrogen species. However, the general cellular response to oxidative inflammation is to increase reducing potential in order to guard against oxidative stress. The induction of intracellular GSH is a known response to oxidative stress in many systems and is the primary determinant of tolerance and survival to oxidizing environments (4, 9, 31). In a previous study of airway epithelial cells exposed to chronic oxidative stress, intracellular GSH levels increased and the ratio of GSH/GSSG increased more than 1.6-fold (4). Previous studies also identify that oxidative stress increases GSH and the ratio of GSH/GSSG via induction of the rate-limiting enzyme in GSH biosynthesis, gamma-glutamylcysteine synthetase (31), as well as via the increased cell uptake of GSH and cell export of GSSG (9). It is interesting to speculate that asthmatic tissues/cells may use similar strategies in response to the inflammation and oxidative stress of asthma in vivo (9). In support of this possibility, intracellular GSH/GSSG ratio was significantly increased in asthma as compared with control samples, that is, intracellular redox potential in asthma is skewed to a more reducing state. Consistent with these findings, a recent study by Fitzpatrick et al. have shown that the extracellular airway epithelial lining fluid of asthmatic children is characterized by higher GSSG and a lower GSH/GSSG ratio than nonasthmatic samples (13). Taken together, asthma may be characterized by greater GSH synthesis and greater GSSG extrusion from cells (3, 4). In addition to the differential regulation of redox in intracellular and extracellular compartments in the airway, the assessment of multiple types of samples (plasma, erythrocyte) reveals that the shift to a more reducing intracellular state is systemic, even though asthma is localized to airways. This causes us to consider the notion that patients with localized inflammation have systemic changes in intracellular redox that influence proteins throughout the body, for example, via effects on disulfide bonds. Although several studies suggest that GSH or its precursor may benefit asthma (23), other studies conducted on mild asthma suggest that GSH worsens asthma (26). Further studies are needed to assess the potential role of redox-modulating therapies in asthma.
Although disulfide bonds usually maintain stability and integrity of proteins (20), a few proteins exhibit functions that are regulated by disulfide bond cleavage to dithiol state (15, 20, 44). Yang et al. established that the formation of protein disulfides are redox regulated in mammalian cells (49). Peterson et al. showed that the EcSOD can have different primary structures due to alternate disulfide bridge arrangements which occur when EcSOD is synthesized in cells; indeed, the disulfide bridge formation of EcSOD profoundly affects the activity of the final extruded protein (30). Studies conducted by Erle's group found that the anterior gradient homologue 2, an unique member of protein disulfide isomerase family, plays a role in intracellular mucin processing through mixed disulfide bond formation and may contribute to mucin overproduction in asthma (38). Disulfide bond cleavage in disease pathogenesis has been previously described in human immunodeficiency virus (HIV) infection, where disulfide exchange in cluster of differentiation 4 (CD4) is exploited by HIV-1 to enter T-cell. (27). This study extends the concept of disulfide bond loss in pathophysiologic protein function; the dithiol CuZnSOD is highly vulnerable to oxidant inactivation and has the potential to propogate oxidative events (19, 36, 50). The exact mechanism for disulfide cleavage is still not clear. C146 in the disulfide bond is located in the β-sheet, pointing outward from β-barrel and is not under high strain as in the disulfide bond of CD4 (20). Acid-assisted hydrolysis could be a mechanism of cleavage; sputum and exhaled breath condensate of asthmatics identify acidity in contrast to the neutral pH of healthy individuals (17). Disulfide cleavage might be particularly favored in the acidic and reducing conditions of the asthmatic airway, where aspartic acid (D55), located close to C57, may facilitate hydrolysis via ammonium cation formation (20).
The instability of the CuZnSOD dimer is known to change the enzyme's tertiary structure and expose C6 to oxidation (42). Significant oxidation at C6 on H2O2 exposure was identified in asthma samples in this article. However, we could not identify free C6 residue, which may be due to the reactivity of the adjacent lysine (K9) amine group toward NEM or its modification by other processes. On the other hand, NEM equally modified the C111 residue in healthy control and asthmatic CuZnSOD, suggesting that it is not involved in the loss of SOD activity in asthma. In summary, patients with asthma have loss of CuZnSOD antioxidant activity. Although further studies are needed to evaluate structure-function mechanisms of loss, specific proteomic findings in this study uncover a destabilization of CuZnSOD due to instability of the disulfide bond in cellular conditions of asthma. Others and we have shown that asthmatics have inflammation that is typified by oxidative and nitrative stress (7, 16). In this article, the intracellular environment in asthma is revealed to be globally skewed toward a reducing state, perhaps in response to intermittent bursts of inflammation and oxidative stress that accompany asthma exacerbations. A greater intracellular reducing environment in asthmatic cells contributes to the lability of the normally rigid intra-monomeric C57-146 disulfide (Fig. 8), which weakens dimeric interface of the protein and subsequently disrupts the enzyme core (42). Given that loss of dismutase activity is closely linked to asthma severity and airway reactivity (6), further studies conducted on the redox environment underlying structure-function changes in asthma, and in particular in severe asthma where oxidative burden is high and unremitting, are needed to define the biochemical pathophysiology of asthma. If this paradigm holds, it will be important for the design of therapies, which may apply existing therapies to protect free thiols (27, 29).
Materials and Methods
Samples of blood and airway from individuals with asthma or healthy controls
The study population is described in table 1. Control and asthmatic individuals underwent bronchoscopy to obtain fresh bronchial airway epithelial cells as previously described (8) and were processed for SOD activity assays or proteomic studies as described.
Peripheral blood was obtained from study subjects on the same day as bronchoscopy. Serum and packed erythrocytes were extracted by centrifugation of the whole blood and were frozen at −80°C. Platelets were isolated by differential centrifugation from human venous blood collected in citrate-containing tubes. Briefly, whole blood was centrifuged (150 g, 10 min) in the presence of Prostacyclin (PGI2),; Sigma-Aldrich) to obtain platelet-rich plasma. Platelets were subsequently pelleted from platelet-rich plasma (1500 g, 10 min). Platelet pellets were washed with “Erythrocyte Lysis Buffer” (Qiagen) containing PGI2 and were resuspended in modified tyrode's buffer. The study was approved by the Cleveland Clinic Institutional Review Board, and written informed consent was obtained from all individuals who had enrolled in the study.
Sample preparation for SOD activity analysis
Erythrocytes from normal and asthmatic blood were lysed with an equal volume of activity lysis buffer (50 mM Tris.Cl (pH 7.5), 50 mM sodium chloride, 2 mM ethylenediaminetetraacetic acid, 0.5% Triton X-100, 5% glycerol, and protease inhibitors) for 20 min on ice with occasional vortexing. Lysates were centrifuged (13,000 g, 20 min, 4°C), and the supernatant was used for CuZnSOD activity analysis. Aliquots of serum samples were treated with an equal volume of activity lysis buffer and concentrated (2×) through a spin column (10 kDa cut off). Platelets were lysed in SOD activity assay buffer (Cayman Chemicals).
SOD activity assay
In-gel SOD activity for serum and erythrocytes was determined in a nondenaturing polyacrylamide gel (PAGE) (2). The assay is based on the inhibitory effect of SOD on the reduction of tetrazolium by O2•− that is generated by photochemically reduced riboflavin (8). A densitometric analysis of serum in-gel SOD assay was performed using the CuZnSOD band of the healthy control in lane 1 as 100% of activity.
Total SOD activity in partially purified SOD of healthy control and asthmatic erythrocytes was also spectrophotometrically measured, where the rate of cytochrome c reduction with one unit (U) of SOD activity was defined as the amount of SOD which was required to inhibit the rate of cytochrome c reduction by 50% (7). For a proteomic analysis, the reaction was scaled down to 200 μl. SOD activity in platelets was spectrophotometrically measured by using a commercially available SOD activity assay (Cayman Chemicals).
Partial purification of CuZnSOD
Cell lysates were diluted by adding 3×volume of ice-cold deionized water; for red cells, hemoglobin was precipitated by using Tsuchihasi's choloroform–ethanol treatment (28). Ice-cold ethanol and chloroform were added to diluted lysates to a final concentration of 25% and 12% (v/v) respectively. Then, the mixture was rotated for 30 min (4°C) followed by centrifugation at 13,000 g (10 min). The supernatant obtained was allowed to warm at room temperature, and solid dipotassium phosphate (300 g/L) was added to it, resulting in the separation of two liquid phases. The top lighter phase containing ethanol and CuZnSOD was collected and centrifuged; the pale yellow supernatant was cooled at 4°C, and 0.75 volume of cold acetone was added to precipitate CuZnSOD. After 20 min of incubation at −20°C, the suspension was centrifuged to collect the precipitate and finally dissolved in 50 mM potassium phosphate buffer or 100 μl in-gel-activity lysis buffer.
Susceptibility of CuZnSOD to oxidation modifications
In specified experiments that evaluate the susceptibility of CuZnSOD to H2O2 and protein modifications, partially purified CuZnSOD were treated with different doses of H2O2 (0.05, 0.1, and 50 mM) in phosphate buffer (pH 7.2). Immediately after the reaction, half of the reaction mixture was evaluated for in-gel SOD activity; the remainder was heated to 100°C with gel-loading buffer for SDS-PAGE and SRM analysis. Untreated CuZnSOD served as the corresponding negative control.
Treatment of partially purified CuZnSOD with NEM
To test the presence of free thiols in CuZnSOD, erythrocyte lysates from control and asthmatic samples were first treated with 50 mM NEM (15 min) (10), followed by the partial purification procedure as described earlier. The final precipitate was dissolved in SDS-PAGE gel-loading buffer and tested by SRM analysis.
Protein digestion and quantitation of modified peptide by proteomic analysis
The protein bands were digested according to an in-gel digestion procedure previously described (16). The quantitation of the modified peptides was carried out using an SRM experiment to acquire the full MS/MS spectrum for each ion of interest. In these experiments, a series of m/z ratios that correspond to peptides of interest are isolated and fragmented in an ion trap mass spectrometer over the entire course of the LC experiment. The formation of specific fragment ions along with chromatographic elution times is used to construct chromatograms. Chromatographic peaks from each peptide are integrated using the instrument software Qual Browser (Thermo Scientific, San Jose, CA), and resulting peak areas are used in quantitative calculations (47). Sequest program was used to compare Collision Induced Dissociation spectra of CuZnSOD to the database (National Center for Biotechnology Information, accession no. AAB05661.1), using the appropriate mass change for the different amino acid oxidation products.
Treatment of CuZnSOD with DTT and H2O2
Recombinant CuZnSOD was treated with different doses of DTT (0–25 mM) followed by in-gel CuZnSOD activity analysis. 25 mM DTT-treated recombinant CuZnSOD was further exposed to 50 mM H2O2, followed by oxidative modification analyses at C146 by SRM.
Western blot analysis
Partially purified CuZnSOD was evaluated by Western analysis as previously described (16). The primary antibody was polyclonal anti-CuZnSOD (Santa Cruz Biotechnology, Inc.).
Measurement of GSH/GSSG
GSH/GSSG was measured in control and asthmatic erythrocytes as previously described (25).
Measurement of IL-13
IL-13 was measured in control and asthmatic plasma by ELISA (R&D system).
Statistical analysis
All data were expressed as the mean and standard error of the mean. The comparisons between the groups were performed using analysis of variance or Student's t-test.
Supplementary Material
Abbreviations Used
- AGR2
anterior gradient homologue 2
- Ala
alanine
- C
cysteine
- C6
cysteine at 6th residue
- C57
cysteine at 57th residue
- C111
cysteine at 111th residue
- C146
cysteine at 146th residue
- C146ox
cysteic acid oxidation at 146th residue
- C146S
mutation of cysteine at 146th residue to serine
- C57-146
disulfide bond between C57 and C146
- CD4
cluster of differentiation 4
- CID
collision induced dissociation
- Cu
copper
- CuCl2
copper (II) chloride
- CuZnSOD
copper zinc super oxide dismutase
- CNEM
cysteine modified by N-ethylmaleimide
- D
aspartic acid
- D55
aspartic acid at 55th residue
- DTT
dithiothreitol
- EcSOD
extracellular super oxide dismutase
- %FEV1
predicted forced expiratory volume in 1 s
- %FVC
forced percentual vital capacity
- Gln
glutamine
- Gly
glycine
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- H
histidine
- H118
histidine at 118th residue
- H120
histidine at 120th residue
- HIV
human immunodeficiency virus
- H2O2
hydrogen peroxide
- IL-13
interleukin 13
- K2HPO4
dipotassium phosphate
- K/Lys
lysine
- LC
liquid chromatography
- Leu
leucine
- MnSOD
manganese super oxide dismutase
- MS
mass spectrometry
- NBT
nitroblue tetrazolium
- NEM
N-ethylmaleimide
- O2•−
superoxide
- PAGE
poly acrylamide gel electrophoresis
- PGI2
prostacyclin
- ROS
reactive oxygen species
- SDS
sodium dodecyl sulfate
- -SH
reduced sulfydral
- SOD
super oxide dismutase
- -SOH
sulfenic acid
- -SO2H
sulfinic acid
- -SO3H
sulfonic acid
- SRM
selected reaction-monitoring
- S-S
disulfide
- Th2
T helper cell Type 2
- Tris
Tris(hydroxymethyl)amino methane,
- W
tryptophan
- X/XO
xanthine/xanthine oxidase
- Zn
zinc
Acknowledgments
The authors thank J. Lang for photography and D. Schumick for artwork. This work is supported by HL69170, HL081064, and UL1RR024989 from the National Center for Research Resources. Dr. Erzurum is a senior fellow of the American Asthma Foundation.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Alvarez B. Demicheli V. Duran R. Trujillo M. Cervenansky C. Freeman BA. Radi R. Inactivation of human Cu,Zn superoxide dismutase by peroxynitrite and formation of histidinyl radical. Free Radic Biol Med. 2004;37:813–822. doi: 10.1016/j.freeradbiomed.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Beauchamp C. Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 3.Comhair SA. Bhathena PR. Dweik RA. Kavuru M. Erzurum SC. Rapid loss of superoxide dismutase activity during antigen-induced asthmatic response. Lancet. 2000;355:624. doi: 10.1016/S0140-6736(99)04736-4. [DOI] [PubMed] [Google Scholar]
- 4.Comhair SA. Bhathena PR. Farver C. Thunnissen FB. Erzurum SC. Extracellular glutathione peroxidase induction in asthmatic lungs: evidence for redox regulation of expression in human airway epithelial cells. Faseb J. 2001;15:70–78. doi: 10.1096/fj.00-0085com. [DOI] [PubMed] [Google Scholar]
- 5.Comhair SA. Erzurum SC. Redox control of asthma: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2010;12:93–124. doi: 10.1089/ars.2008.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comhair SA. Ricci KS. Arroliga M. Lara AR. Dweik RA. Song W. Hazen SL. Bleecker ER. Busse WW. Chung KF. Gaston B. Hastie A. Hew M. Jarjour N. Moore W. Peters S. Teague WG. Wenzel SE. Erzurum SC. Correlation of systemic superoxide dismutase deficiency to airflow obstruction in asthma. Am J Respir Crit Care Med. 2005;172:306–313. doi: 10.1164/rccm.200502-180OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Comhair SA. Xu W. Ghosh S. Thunnissen FB. Almasan A. Calhoun WJ. Janocha AJ. Zheng L. Hazen SL. Erzurum SC. Superoxide dismutase inactivation in pathophysiology of asthmatic airway remodeling and reactivity. Am J Pathol. 2005;166:663–674. doi: 10.1016/S0002-9440(10)62288-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Raeve HR. Thunnissen FB. Kaneko FT. Guo FH. Lewis M. Kavuru MS. Secic M. Thomassen MJ. Erzurum SC. Decreased Cu,Zn-SOD activity in asthmatic airway epithelium: correction by inhaled corticosteroid in vivo. Am J Physiol. 1997;272:L148–L154. doi: 10.1152/ajplung.1997.272.1.L148. [DOI] [PubMed] [Google Scholar]
- 9.Deneke SM. Fanburg BL. Regulation of cellular glutathione. Am J Physiol. 1989;257:L163–L173. doi: 10.1152/ajplung.1989.257.4.L163. [DOI] [PubMed] [Google Scholar]
- 10.Dowal L. Yang W. Freeman MR. Steen H. Flaumenhaft R. Proteomic analysis of palmitoylated platelet proteins. Blood. 2011;118:e62–e73. doi: 10.1182/blood-2011-05-353078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erzurum SC. Lemarchand P. Rosenfeld MA. Yoo JH. Crystal RG. Protection of human endothelial cells from oxidant injury by adenovirus-mediated transfer of the human catalase cDNA. Nucleic Acids Res. 1993;21:1607–1612. doi: 10.1093/nar/21.7.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ezure T. Suzuki T. Ando E. Nishimura O. Tsunasawa S. Expression of human Cu, Zn-superoxide dismutase in an insect cell-free system and its structural analysis by MALDI-TOF MS. J Biotechnol. 2009;144:287–292. doi: 10.1016/j.jbiotec.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 13.Fitzpatrick AM. Teague WG. Holguin F. Yeh M. Brown LA. Airway glutathione homeostasis is altered in children with severe asthma: evidence for oxidant stress. J Allergy Clin Immunol. 2009;123:146–152. doi: 10.1016/j.jaci.2008.10.047. e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujiwara N. Nakano M. Kato S. Yoshihara D. Ookawara T. Eguchi H. Taniguchi N. Suzuki K. Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J Biol Chem. 2007;282:35933–35944. doi: 10.1074/jbc.M702941200. [DOI] [PubMed] [Google Scholar]
- 15.Gelderman KA. Hultqvist M. Holmberg J. Olofsson P. Holmdahl R. T cell surface redox levels determine T cell reactivity and arthritis susceptibility. Proc Natl Acad Sci U S A. 2006;103:12831–12836. doi: 10.1073/pnas.0604571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh S. Janocha AJ. Aronica MA. Swaidani S. Comhair SA. Xu W. Zheng L. Kaveti S. Kinter M. Hazen SL. Erzurum SC. Nitrotyrosine proteome survey in asthma identifies oxidative mechanism of catalase inactivation. J Immunol. 2006;176:5587–5597. doi: 10.4049/jimmunol.176.9.5587. [DOI] [PubMed] [Google Scholar]
- 17.Greenwald R. Fitzpatrick AM. Gaston B. Marozkina NV. Erzurum S. Teague WG. Breath formate is a marker of airway S-nitrosothiol depletion in severe asthma. PLoS One. 2010;5:e11919. doi: 10.1371/journal.pone.0011919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He C. Murthy S. McCormick ML. Spitz DR. Ryan AJ. Carter AB. Mitochondrial Cu,Zn-superoxide dismutase mediates pulmonary fibrosis by augmenting H2O2 generation. J Biol Chem. 2011;286:15597–15607. doi: 10.1074/jbc.M110.187377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodgson EK. Fridovich I. The interaction of bovine erythrocyte superoxide dismutase with hydrogen peroxide: inactivation of the enzyme. Biochemistry. 1975;14:5294–5299. doi: 10.1021/bi00695a010. [DOI] [PubMed] [Google Scholar]
- 20.Hogg PJ. Disulfide bonds as switches for protein function. Trends Biochem Sci. 2003;28:210–214. doi: 10.1016/S0968-0004(03)00057-4. [DOI] [PubMed] [Google Scholar]
- 21.Hornberg A. Logan DT. Marklund SL. Oliveberg M. The coupling between disulphide status, metallation and dimer interface strength in Cu/Zn superoxide dismutase. J Mol Biol. 2007;365:333–342. doi: 10.1016/j.jmb.2006.09.048. [DOI] [PubMed] [Google Scholar]
- 22.Horwitz RJ. Busse WW. Inflammation and asthma. Clin Chest Med. 1995;16:583–602. [PubMed] [Google Scholar]
- 23.Koike Y. Hisada T. Utsugi M. Ishizuka T. Shimizu Y. Ono A. Murata Y. Hamuro J. Mori M. Dobashi K. Glutathione redox regulates airway hyperresponsiveness and airway inflammation in mice. Am J Respir Cell Mol Biol. 2007;37:322–329. doi: 10.1165/rcmb.2006-0423OC. [DOI] [PubMed] [Google Scholar]
- 24.Lindberg MJ. Normark J. Holmgren A. Oliveberg M. Folding of human superoxide dismutase: disulfide reduction prevents dimerization and produces marginally stable monomers. Proc Natl Acad Sci U S A. 2004;101:15893–15898. doi: 10.1073/pnas.0403979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mansoor MA. Svardal AM. Ueland PM. Determination of the in vivo redox status of cysteine, cysteinylglycine, homocysteine, and glutathione in human plasma. Analytical biochemistry. 1992;200:218–229. doi: 10.1016/0003-2697(92)90456-h. [DOI] [PubMed] [Google Scholar]
- 26.Marrades RM. Roca J. Barbera JA. de Jover L. MacNee W. Rodriguez-Roisin R. Nebulized glutathione induces bronchoconstriction in patients with mild asthma. Am J Respir Crit Care Med. 1997;156:425–430. doi: 10.1164/ajrccm.156.2.9611001. [DOI] [PubMed] [Google Scholar]
- 27.Matthias LJ. Yam PT. Jiang XM. Vandegraaff N. Li P. Poumbourios P. Donoghue N. Hogg PJ. Disulfide exchange in domain 2 of CD4 is required for entry of HIV-1. Nat Immunol. 2002;3:727–732. doi: 10.1038/ni815. [DOI] [PubMed] [Google Scholar]
- 28.McCord JM. Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 29.Nguyen C. Teo JL. Matsuda A. Eguchi M. Chi EY. Henderson WR., Jr. Kahn M. Chemogenomic identification of Ref-1/AP-1 as a therapeutic target for asthma. Proc Natl Acad Sci U S A. 2003;100:1169–1173. doi: 10.1073/pnas.0437889100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petersen SV. Oury TD. Valnickova Z. Thogersen IB. Hojrup P. Crapo JD. Enghild JJ. The dual nature of human extracellular superoxide dismutase: one sequence and two structures. Proc Natl Acad Sci U S A. 2003;100:13875–13880. doi: 10.1073/pnas.2436143100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rahman I. Antonicelli F. MacNee W. Molecular mechanism of the regulation of glutathione synthesis by tumor necrosis factor-alpha and dexamethasone in human alveolar epithelial cells. J Biol Chem. 1999;274:5088–5096. doi: 10.1074/jbc.274.8.5088. [DOI] [PubMed] [Google Scholar]
- 32.Rakhit R. Cunningham P. Furtos-Matei A. Dahan S. Qi XF. Crow JP. Cashman NR. Kondejewski LH. Chakrabartty A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277:47551–47556. doi: 10.1074/jbc.M207356200. [DOI] [PubMed] [Google Scholar]
- 33.Redler RL. Wilcox KC. Proctor EA. Fee L. Caplow M. Dokholyan NV. Glutathionylation at Cys-111 induces dissociation of wild type and FALS mutant SOD1 dimers. Biochemistry. 2011;50:7057–7066. doi: 10.1021/bi200614y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rudolph TK. Freeman BA. Transduction of redox signaling by electrophile-protein reactions. Sci Signal. 2009;2:re7. doi: 10.1126/scisignal.290re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salo DC. Pacifici RE. Lin SW. Giulivi C. Davies KJ. Superoxide dismutase undergoes proteolysis and fragmentation following oxidative modification and inactivation. J Biol Chem. 1990;265:11919–11927. [PubMed] [Google Scholar]
- 36.Sampson JB. Beckman JS. Hydrogen peroxide damages the zinc-binding site of zinc-deficient Cu,Zn superoxide dismutase. Arch Biochem Biophys. 2001;392:8–13. doi: 10.1006/abbi.2001.2418. [DOI] [PubMed] [Google Scholar]
- 37.Sato K. Akaike T. Kohno M. Ando M. Maeda H. Hydroxyl radical production by H2O2 plus Cu,Zn-superoxide dismutase reflects the activity of free copper released from the oxidatively damaged enzyme. J Biol Chem. 1992;267:25371–25377. [PubMed] [Google Scholar]
- 38.Schroeder BW. Verhaeghe C. Park SW. Nguyenvu LT. Huang X. Zhen G. Erle DJ. AGR2 is Induced in Asthma and Promotes Allergen-Induced Mucin Overproduction. Am J Respir Cell Mol Biol. 2012;47:178–185. doi: 10.1165/rcmb.2011-0421OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sedgwick JB. Calhoun WJ. Vrtis RF. Bates ME. McAllister PK. Busse WW. Comparison of airway and blood eosinophil function after in vivo antigen challenge. J Immunol. 1992;149:3710–3718. [PubMed] [Google Scholar]
- 40.Tainer JA. Getzoff ED. Richardson JS. Richardson DC. Structure and mechanism of copper, zinc superoxide dismutase. Nature. 1983;306:284–287. doi: 10.1038/306284a0. [DOI] [PubMed] [Google Scholar]
- 41.Teng Y. Sun P. Zhang J. Yu R. Bai J. Yao X. Huang M. Adcock IM. Barnes PJ. Hydrogen peroxide in exhaled breath condensate in patients with asthma: a promising biomarker? Chest. 2011;140:108–116. doi: 10.1378/chest.10-2816. [DOI] [PubMed] [Google Scholar]
- 42.Tiwari A. Hayward LJ. Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J Biol Chem. 2003;278:5984–5992. doi: 10.1074/jbc.M210419200. [DOI] [PubMed] [Google Scholar]
- 43.Uchida K. Kawakishi S. Identification of oxidized histidine generated at the active site of Cu,Zn-superoxide dismutase exposed to H2O2. Selective generation of 2-oxo-histidine at the histidine 118. J Biol Chem. 1994;269:2405–2410. [PubMed] [Google Scholar]
- 44.Usuki F. Yamashita A. Fujimura M. Post-transcriptional defects of antioxidant selenoenzymes cause oxidative stress under methylmercury exposure. J Biol Chem. 2011;286:6641–6649. doi: 10.1074/jbc.M110.168872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wenzel S. Fahy J. Irvin C. Peters S. Spector S. Szefler S. Proceedings of the ATS workshop on refractory asthma: current understanding, recommendations, and unanswered questions. Am J Respir Crit Care Med. 2000;162:2341–2351. doi: 10.1164/ajrccm.162.6.ats9-00. [DOI] [PubMed] [Google Scholar]
- 46.Wilcox KC. Zhou L. Jordon JK. Huang Y. Yu Y. Redler RL. Chen X. Caplow M. Dokholyan NV. Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J Biol Chem. 2009;284:13940–13947. doi: 10.1074/jbc.M809687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willard BB. Ruse CI. Keightley JA. Bond M. Kinter M. Site-specific quantitation of protein nitration using liquid chromatography/tandem mass spectrometry. Anal Chem. 2003;75:2370–2376. doi: 10.1021/ac034033j. [DOI] [PubMed] [Google Scholar]
- 48.Yamakura F. Matsumoto T. Ikeda K. Taka H. Fujimura T. Murayama K. Watanabe E. Tamaki M. Imai T. Takamori K. Nitrated and oxidized products of a single tryptophan residue in human Cu,Zn-superoxide dismutase treated with either peroxynitrite-carbon dioxide or myeloperoxidase-hydrogen peroxide-nitrite. J Biochem. 2005;138:57–69. doi: 10.1093/jb/mvi095. [DOI] [PubMed] [Google Scholar]
- 49.Yang Y. Song Y. Loscalzo J. Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc Natl Acad Sci U S A. 2007;104:10813–10817. doi: 10.1073/pnas.0702027104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yim MB. Chock PB. Stadtman ER. Copper, zinc superoxide dismutase catalyzes hydroxyl radical production from hydrogen peroxide. Proc Natl Acad Sci U S A. 1990;87:5006–5010. doi: 10.1073/pnas.87.13.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.