

Background: Spinal muscular atrophy (SMA) is a devastating genetic disorder caused by low levels of survival motor neuron (SMN) protein.
Results: Ubiquitin-specific protease 9x (Usp9x) interacts with, deubiquitinates, and stabilizes SMN.
Conclusion: Usp9x likely deubiquitinates SMN to protect it from Ub-dependent degradation.
Significance: Usp9x is a key mediator that regulates the protein levels of SMN and the SMN complex.
Keywords: Deubiquitination, Proteasome, Protein Degradation, Ubiquitin-dependent Protease, Ubiquitination, Spinal Muscular Atrophy, Survival Motor neuron
Abstract
Spinal muscular atrophy (SMA), the leading genetic disorder of infant mortality, is caused by low levels of survival motor neuron (SMN) protein. Currently it is not clear how the SMN protein levels are regulated at the post-transcriptional level. In this report, we find that Usp9x, a deubiquitinating enzyme, stably associates with the SMN complex via directly interacting with SMN. Usp9x deubiquitinates SMN that is mostly mono- and di-ubiquitinated. Knockdown of Usp9x promotes SMN degradation and reduces the protein levels of SMN and the SMN complex in cultured mammalian cells. Interestingly, Usp9x does not deubiquitinate nuclear SMNΔ7, the main protein product of the SMN2 gene, which is polyubiquitinated and rapidly degraded by the proteasome. Together, our results indicate that SMN and SMNΔ7 are differently ubiquitinated; Usp9x plays an important role in stabilizing SMN and the SMN complex, likely via antagonizing Ub-dependent SMN degradation.
Introduction
Spinal muscular atrophy (SMA),3 an autosomal recessive disorder that results in degeneration of α-motor neurons in the spinal cord, is the leading hereditary disease causing infant mortality (1–3). SMA is caused by homozygous deletions or mutations in the survival motor neuron 1 gene (SMN1) (4). The vast majority of SMA (>95%) is caused by SMN1 deletion (4, 5). Humans have two SMN genes, both the telomeric SMN1 and the centromeric SMN2 encode full-length SMN protein (294 amino acids). However, a C to T nucleotide change at the +6 position of exon 7 in SMN2 usually causes exclusion of exon 7 during splicing and only ∼15% of full-length transcript is made (6). SMA patients usually have one or more copies of SMN2. Studies have shown that the copy number of SMN2 inversely correlates with disease severity in human patients (4, 5, 7–9). Mice have a single Smn gene, ablation of which results in early embryonic lethality (10). Introduction of two copies of SMN2 in the Smn-null background rescues embryonic lethality, whereas mice with eight copies of SMN2 show complete rescue (11). Thus, SMA is caused by low protein levels of SMN (12, 13).
SMN is widely expressed in all cell types of vertebrates and distributes in both the cytoplasm and the nucleus of cells (14–17). In the nucleus, SMN concentrates in punctate foci called gems, which usually co-localize with coilin in Cajal bodies (16–18). SMN forms a stable complex with Gemins 2–8 and Unrip, termed the SMN complex (19), in which Gemins 2, 3, and 8 directly interact with SMN (20). A well characterized function of the SMN complex is to help assembly and transport of uridine-rich small nuclear ribonucleoproteins (21–24), core components of the spliceosome. SMN also interacts with a variety of other proteins (25). A group of SMN-interacting proteins including profilin I and II, heterogeneous nuclear ribonucleoprotein-R, and plastin 3 are involved in regulating β-actin mRNA transport or actin dynamics in axons (25), which regulates neuronal growth. Particularly, SMN-deficient motor neurons, derived from severe SMA mice, were found to have reduced localization of β-actin mRNA in axons and reduced protein levels of β-actin in axonal growth cones (26). Accordingly, splicing defects of a group of transcripts that are critical for α-motor neuron survival and impairment of β-actin mRNA transport and localization were hypothesized to explain SMA pathogenesis (25, 27, 28). Currently, direct evidence that supports either hypothesis is still lacking.
SMNΔ7, the protein product of the SMN2 transcript with exon 7 exclusion, lacks the C-terminal 16 amino acids of SMN, but has four extra amino acids (EMLA) because of a frameshift. SMNΔ7 is barely detectable in cells or tissues derived from SMA human patients or mice, despite SMNΔ7 mRNA being present at high levels. In agreement with this, SMNΔ7 has a much shorter half-life (∼2–3 h) than that of wild type SMN (∼6–8 h) (29–31). One potential explanation is that SMNΔ7 cannot self-associate to oligomerize and assemble into the SMN complex (29). In addition, a recent study has shown that the C-terminal region of SMNΔ7 functions as a degron to promote rapid SMNΔ7 degradation (31). Thus, degradation of SMN and SMNΔ7 might be regulated differently in cells. It has been reported that SMN and SMNΔ7 are substrates of the proteasome (29, 31, 32), however, little is known about the mechanism by which the ubiquitin (Ub)-proteasome system degrades SMN and SMNΔ7.
Usp9x (2547 amino acids) is one of the largest DUBs belonging to the Ub-specific protease family (33). Based on a bioinformatics study, Usp9x contains a Ub-like domain (amino acids 873–966) and a Ub C-terminal hydrolase domain (amino acids 1531–1971) that catalyzes deubiquitination (34, 35). Usp9x exhibits diverse DUB activities to deubiquitinate its substrates modified with mono-Ub or poly-Ub chains linked through lysines 29, 33, 48, or 63 of Ub. There are a dozen substrates of Usp9x having been identified including AF-6, β-catenin, NUAK1, MARK4, ErbB2, EFA6, Smad4, Mcl1, ASK1, and survivin (36–44). In this study, we found that SMN is a substrate of Usp9x; Usp9x interacts with, deubiquitinates, and stabilizes SMN that is mostly mono- and di-ubiquitinated. In contrast, Usp9x does not regulate the ubiquitination and stability of SMNΔ7; SMNΔ7 can be polyubiquitinated and it is much more susceptible to proteasomal degradation than SMN.
EXPERIMENTAL PROCEDURES
Antibodies
The following antibodies were purchased: Usp9x, Gemin4, and Gemin5 (Bethyl Laboratories); SMN, Ub (P4D1), and Myc (9E10) (Santa Cruz Biotechnology); SMN, Gemin2, Gemin3, and coilin (BD Biosciences); FLAG (M2), Gemin8 (1F8), and β-actin (AC-15) (Sigma); and HA (16B12) (Covance). The Y12 antibody was a gift from D. Bentley (University of Colorado, Denver, CO).
Mammalian Expression Constructs
The cDNAs of full-length human Usp9x and Usp9x(C1566A) were provided by Dr. D. R. Alessi (University of Dundee) (38). We subcloned both Usp9x and Usp9x(C1556A) into the pRK5, pRK7, or pQCXIP-HTBH (45) vector using the BamHI and NotI sites in the provided plasmids. Expression plasmids of SMN, Gemins 2–8, Unrip, and Ub were constructed by PCR amplification of the corresponding open reading frames from a human 293T cDNA library using Pfu polymerase (Stratagene) and subsequently subcloned into the pRK5 or pRK7 vector containing an N-terminal HA or FLAG tag (46). All plasmids were validated by DNA sequencing.
Cell Culture and Transfection
Fibroblast cells derived from a normal carrier (GM03814) and two type I SMA patients (GM03813 and GM00232) were obtained from Coriell Cell Repositories. Human embryonic kidney (HEK) 293T, HeLa, and fibroblast cells were grown in DMEM supplemented with 10% fetal bovine serum (Sigma) and 100 μg/ml of penicillin and streptomycin at 37 °C in a 5% CO2 atmosphere. Human neuroblastoma SH-SY5Y cells and lung adenocarcinoma epithelial A549 cells were maintained in DMEM/F-12 (1:1) and F-12 medium with 10% fetal bovine serum, respectively. 293T cells were grown to 50–60% confluence and transfected using the standard calcium phosphate precipitation method (46). Typically, 10 μg of plasmid was used for a single-gene transfection of a 100-mm dish of cells, up to 30 μg of plasmids were used for co-transfection of three plasmids. Transfection of HeLa cells was performed using the Lipofectamine 2000 (Invitrogen) or Turbofect (Fisher) transfection reagent according to the manufacturers' instructions. In assays with proteasome inhibition, 10 μm MG132 or epoxomicin was incubated with cells for 16 h.
Establishing Usp9x Stable Knockdown Cell Lines
MISSION shRNAs of Usp9x (TRCN0000007361 and TRCN0000007364) and a control EGFP shRNA (SHC005) in pLKO.1 vector were purchased from the Functional Genomic Facility at the University of Colorado. To produce viruses, 293FT cells at 60–70% confluence were co-transfected with 5 μg of the corresponding shRNA pLKO.1-puro plasmid plus 2 μg of each of the packaging plasmids (Sigma) using Lipofectamine 2000. 16 h post-transfection, medium was changed and cells were cultured for an additional 48 h. The medium was then collected and centrifuged to remove cell debris and the supernatants were filtered through 0.22-μm membranes. The resulting virus-containing supernatants (5 ml) were used to infect the corresponding cells (60-mm dish) in medium supplemented with 8 μg/ml of Polybrene (Sigma) overnight. Cells were then selected with 2 μg/ml of puromycin for at least 5 days.
Immunoprecipitation and Immunoblotting Assays
For nondenaturing immunoprecipitation, cells in a 100-mm dish (90% confluence) were harvested and washed with 1× PBS, then lysed with 1.5 ml of cold cell lysis buffer (20 mm Tris, pH 7.6, 150 mm NaCl, 2 mm EDTA, 0.5% Triton X-100, 10% glycerol) with 1× protease inhibitor mixture (Roche Applied Science). After clearing the lysates by centrifugation, supernatants were incubated with 2 μg of an appropriate antibody or control IgG for 4 h at 4 °C, then supplemented with 20 μl of protein A beads that were preincubated with 2 mg/ml of BSA to reduce nonspecific binding. After overnight rocking, protein A beads were pelleted by centrifugation and washed three times with the cell lysis buffer plus 0.6 m NaCl. Bound proteins were eluted with 50 μl of 1× SDS sample buffer. For denaturing immunoprecipitation, HeLa cells in a 100-mm dish or 293T cells in a 60-mm dish were lysed into 1 ml of cell lysis buffer plus 1% SDS. Cell lysates were collected and then heated at 95 °C for 30 min. After centrifugation, 0.3-ml supernatants were diluted with 1.2 ml of cell lysis buffer to reduce the SDS concentration to 0.2%. The immunoprecipitation assay was performed as described above except that 5 μg of anti-FLAG M2 antibody or mouse IgG was used in each reaction. 20-μl elutions were resolved by SDS-PAGE and transferred to nitrocellulose membranes for immunoblotting assays.
Immunofluorescence Staining and Confocal Microscopy
HeLa cells cultured on glass coverslips were fixed with 100% methanol at −20 °C for 10 min and then blocked with blocking buffer (1% BSA in TTBS) for 1 h. Cells were then incubated with an appropriate primary antibody (1:200–1:1000 dilution) in blocking buffer overnight at 4 °C, followed by incubating with anti-mouse or rabbit IgG (H+L) (Alexa Fluor 488 or 555 conjugate) (Cell Signaling) for 1 h at room temperature. The coverslip was mounted with the ProLong® Gold Antifade kit (Invitrogen) containing the blue fluorescent nuclear counterstain DAPI. The cells were observed and the images were captured using a Zeiss 3I Marianas spinning disk confocal microscope with a ×63 oil immersion objective lens (Light Microscope Core, University of Colorado School of Medicine).
Protein Purification
The cDNA coding region of SMN or SMNΔ7 was subcloned into the NheI and XhoI sites of the pET-28a(+) vector. The plasmids were transformed into Escherichia coli BL21(DE3) cells. E. coli cells containing the expression plasmid were grown to an A600 = 0.6 at 37 °C, protein expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for induction of 5 h. SMN or SMNΔ7 protein was purified from cell pellets followed by refolding. Briefly, cell pellets from a 2-liter culture were resuspended into 40 ml of lysis buffer (20 mm Tris, pH 7.6, 150 mm NaCl, 2 mm β-ME, 1× leupeptin, 1× PMSF), followed by sonication and centrifugation at 30,000 × g for 30 min at 4 °C. Insoluble pellets were rinsed by lysis buffer and then resuspended in a denaturing buffer (10 mm Tris, pH 8.0, 6 m guanidine HCl). After sonication and centrifugation, supernatants were rocked with 5 ml of nickel-nitrilotriacetic acid resins (Invitrogen) overnight. Resins were subsequently washed with a urea-containing denaturing buffer (10 mm Tris, pH 8.0, 6 m urea) plus 25 mm imidazole, and proteins were eluted off nickel-nitrilotriacetic acid resins with the urea denaturing buffer plus 150 mm imidazole. The elutions were then loaded into a 20-ml Q-Sepharose column on an AKTA FPLC system (performed at room temperature). Proteins were eluted using a NaCl gradient from 0 to 250 mm in 20 mm Tris, pH 8.0, 5 m urea. Corresponding fractions with SMN or SMNΔ7 as judged by Coomassie-stained SDS-PAGE were pooled and dialyzed against arginine refolding buffer (1 m arginine, 100 mm Tris, pH 8.0, 100 mm NaCl) at 4 °C. After 36 h dialysis (refolding), insoluble proteins were pelleted by centrifugation. Arginine was removed by dialyzing folded SMN or SMNΔ7 into a storage buffer (20 mm Tris, pH 8.0, 250 mm NaCl, 2 mm β-ME, and 10% glycerol).
The cDNA coding region of Usp9x was subcloned into the pYES-263 vector (47) using the BamHI and NotI sites. Expression plasmids were transformed into yeast strain RSY334 (MATα pep4-3 prb1–1122 ura3–52 leu2–3,112 reg1–501 gal1) (48) by the lithium acetate method by selection for Ura+ colonies. A 5-ml overnight culture (A600 = 0.7) from a single colony was inoculated into 1 liter of −Ura selective media and grown to A600 = 0.25 (4.5 × 106 cells). Protein expression was induced by adding 2% galactose. Cultures were then shaken overnight at 30 °C. Cells were harvested and cell pellets were washed with ice-cold Milli-Q H2O, followed by washing with ice-cold grinding buffer (50 mm Tris, pH 7.6, 200 mm NaCl, 1 mm β-ME, 1 mm EDTA, 1 mm PMSF, 10% glycerol). After centrifugation, cell pellets were resuspended in 15 ml of grinding buffer and frozen with liquid nitrogen in a dropwise manner using a 1-ml pipette to prepare small frozen pellets. Pellets were then ground into dry powder using 6870 Freezer/Mill (SPEX SamplePrep). To purify GST-Usp9x, ground yeast powder from 3-liter cultures were dissolved into 50 ml of lysis buffer (20 mm Tris, pH 7.6, 1 mm β-ME, 1 mm EDTA, 10% glycerol, 1× PMSF, 1× leupeptin) and centrifuged at 30,000 × g for 30 min at 4 °C. Supernatants were then ultracentrifuged at 90,000 × g using a Ti-70.1 rotor for 60 min at 4 °C. The resulting supernatants were then loaded into a 20-ml Q-Sepharose column equilibrated in lysis buffer on an AKTA FPLC system. Proteins were eluted using a NaCl gradient from 0 to 400 mm NaCl. Fractions eluted between 275 and 350 mm NaCl were confirmed to contain Usp9x by immunoblotting. These fractions were pooled and the NaCl concentration was raised to ∼1 m. Next, the protein mixtures were rocked with 2 ml of glutathione-Sepharose at 4 °C for 4 h. Resins were then loaded into a 10-ml open column and washed with 30 ml of washing buffer (20 mm Tris, pH 7.6, 500 mm NaCl, 1 mm β-ME, and 10% glycerol). GST-Usp9X was eluted with 10 mm glutathione and dialyzed into the washing buffer for storage.
Identification of Usp9x-interacting Protein
To identify Usp9x-interacting proteins by mass spectrometry, Usp9x was purified from 293T cells stably expressing Usp9x-HTBH. 30 dishes (100 mm) of 293T cells stably expressing HTBH or Usp9x-HTBH were harvested and washed with 1× PBS. Cells were lysed into 30 ml of lysis buffer (20 mm Tris, pH 7.6, 100 mm NaCl, 2 mm β-ME, 10% glycerol, 0.3% CHAPS, and 1× protease inhibitor mixture) by sonication. Cell lysates were cleared by ultracentrifugation (100,000 × g) for 1 h at 4 °C. The supernatants were then mixed with 100 μl of streptavidin resin (Fisher Scientific) and rocked at 4 °C overnight. Resins were transferred into a 2-ml centrifugation tube and washed twice with 1.5 ml of lysis buffer plus 0.2 m NaCl, followed by one wash with the elution buffer (20 mm Tris, pH 7.6, 100 mm NaCl, 2 mm β-ME). Resins were then transferred into a 0.65-ml centrifugation tube and resuspended into 150 μl of elution buffer with 0.06 μg/μl of TEV protease. After 4 h rocking at room temperature, eluted proteins were assayed by SDS-PAGE with silver staining. 20 μl of eluted proteins were subjected to mass spectroscopic analysis carried out on an optimized proteomics platform as previously reported (49). Briefly, protein samples were resolved on a short SDS gel and stained with Coomassie, followed by in-gel digestion and peptide extraction. The resulting peptides were analyzed by a nanoscale reverse phase liquid chromatography coupled with an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific). The peptides were dissolved in buffer A (0.1% formic acid, 0.005% heptafluorobutyric acid, and 5% acetonitrile), loaded onto a 75-μm inner diameter × 10-cm column (HALO® C18 resin, 90 Å, 2.7 μm; tip size 15 μm; New Objective, MA), and then eluted during a 60-min gradient of 10–40% buffer B (0.1% formic acid, 0.005% heptafluorobutyric acid, 70% acetonitrile, flow rate of 250 nl/min). The eluted peptides were analyzed by a circling instrument method, including one MS survey scan and up to 10 data-dependent MS/MS scans. Acquired MS/MS spectra were searched against a human Uniprot database using the SEQUEST algorithm. Searching parameters included mass tolerance of precursor ions (±20 ppm) and product ion (±0.5 Da), tryptic restriction, dynamic mass shifts for oxidized Met (+15.9949), two maximal modification sites, two maximal missed cleavages, as well as only b and y ions counted. To evaluate the false discovery rate during spectrum-peptide matching, all original protein sequences were reversed to generate a decoy database that was concatenated to the original database (50). Assigned peptides were grouped by charge state and then filtered by matching scores (XCorr and ΔCn) to reduce the protein false discovery rate to ∼1%. If peptides were shared in a protein family, the family proteins were clustered into a single group. Based on the principle of parsimony, the group was represented by the protein with the highest number of assigned peptides, and by other proteins if they were matched by unique peptide(s).
In Vitro Deubiquitination Assay
K48-linked Ub4 was purified as previously described (51). To monitor Usp9x-mediated deubiquitination of K48 Ub4, 200 nm K48 Ub4 was mixed with 10 nm Usp9x in the deubiquitination buffer (20 mm Tris, pH 7.6, 150 mm NaCl, 2 mm DTT, and 10% glycerol) and incubated at 37 °C. 20-μl Reaction mixtures were withdrawn at the designated time points and mixed with 5 μl of 5× SDS sample buffer to stop the reaction. Deubiquitination was assayed by immunoblotting of Ub. To monitor Usp9x-mediated deubiquitination of SMN, cell lysates of three 100-mm dishes of 293T cells expressing FLAG-SMN and HA-Ub were subjected to immunoprecipitation with the anti-FLAG M2 antibody using the nondenaturing immunoprecipitation assay. Proteins immobilized on protein A beads were washed three times with the cell lysis buffer plus 0.5% Triton X-100 and 1.2 m NaCl. Protein A beads were then resuspended into 150 μl of deubiquitination buffer and were equally split into three parts to set up the deubiquitination reactions: without Usp9x, with 10 nm Usp9x, or 10 nm Usp9x + 2.5 μm Ub aldehyde (UBPBio). After a 6-h incubation at 30 °C, reactions were stopped by adding 5× SDS sample buffer. Deubiquitination was monitored by immunoblotting using an anti-HA or FLAG antibody.
Supplemental Materials
Supplemental materials include supplemental Figs. S1–S8.
RESULTS
Usp9x Interacts with the SMN Complex
In an effort to identify Usp9x-interacting proteins, we established 293T cell lines stably expressing FLAG-HTBH or FLAG-Usp9x-HTBH (Fig. 1, A and B). The HTBH tag consists of a His6, a TEV protease sequence, an in vivo biotinylation sequence, and another His6 (45). Usp9x was enriched from cell lysates with streptavidin resin, and then released by TEV protease digestion (Fig. 1C). Proteins co-purified with Usp9x were identified by mass spectrometry. Among the identified proteins, peptides of five components of the SMN complex were identified (Fig. 1D and supplemental Fig. S1). In reminiscence of a recent proteomic study in which Usp9x was found to interact with GFP-SMN (52), we speculated that Usp9x interacts with the SMN complex. Indeed, components of the SMN complex including SMN, Gemins 2, 3, 4, and 8 existed in the Usp9x precipitates, but not in the control as assayed by immunoblotting (Fig. 1E). In addition, components of the Sm complex that interacts with the SMN complex were also detected with the Y12 antibody (Fig. 1E), which recognizes SmB′/B and SmD subunits of the heptameric Sm complex (53). Thus, the SMN/Sm complex co-purified with Usp9x. To determine whether Usp9x interacts with the SMN complex under physiological conditions, we immunoprecipitated endogenous SMN in HeLa cells and found that Usp9x co-immunoprecipitated with SMN and other components of the SMN complex (Fig. 1F). Reciprocally, immunoprecipitation of endogenous Usp9x precipitated components of the SMN complex (Fig. 1G). In control blots, SMN and Usp9x were unable to immunoprecipitate the DUB Usp14 and Usp7, respectively (Fig. 1, F and G). Collectively, these results demonstrate that Usp9x stably interacts with the SMN complex in cells.
FIGURE 1.

Usp9x interacts with the SMN complex. A, diagram of the FLAG-Usp9x-HTBH expression construct. B, immunoblotting (IB) of FLAG or HRP-conjugated streptavidin in cell lysates from 293T cells stably expressing FLAG-HTBH (control) or FLAG-Usp9x-HTBH. C, silver staining of purified Usp9x and its interacting proteins resolved by SDS-PAGE. D, peptides of SMN, Gemins 2–5 identified by mass spectrometry. E, immunoblotting of SMN, Usp9x, and Gemins in mass spectrometric samples shown in C. F, immunoblotting of proteins co-immunoprecipitated with endogenous SMN in HeLa cells. G, immunoblotting of proteins co-immunoprecipitated with endogenous Usp9x in HeLa cells.
Usp9x Directly Interacts with SMN
The SMN complex contains at least nine proteins including SMN, Gemins 2–8, and Unrip. We next sought to identify the protein(s) in the SMN complex that directly binds Usp9x. To do this, we overexpressed HA-Usp9x with a FLAG-tagged component of the SMN complex in 293T cells. Immunoprecipitation, using the anti-FLAG M2 antibody, followed by immunoblotting of HA revealed that FLAG-SMN co-immunoprecipitated with HA-Usp9x under a stringent washing condition containing 1.2 m NaCl (Fig. 2A). Despite all other overexpressed components of the SMN complex being efficiently immunoprecipitated, none of these proteins were able to co-immunoprecipitate HA-Usp9x (Fig. 2A). These results suggest that SMN directly interacts with Usp9x. To further confirm this, we purified recombinant GST-Usp9x from Saccharomyces cerevisiae (Fig. 2B) and His6-SMN from E. coli (Fig. 2C). In vitro GST pulldown assays showed that GST-Usp9x, but not GST, precipitated SMN (Fig. 2D). Thus, Usp9x directly interacts with SMN.
FIGURE 2.
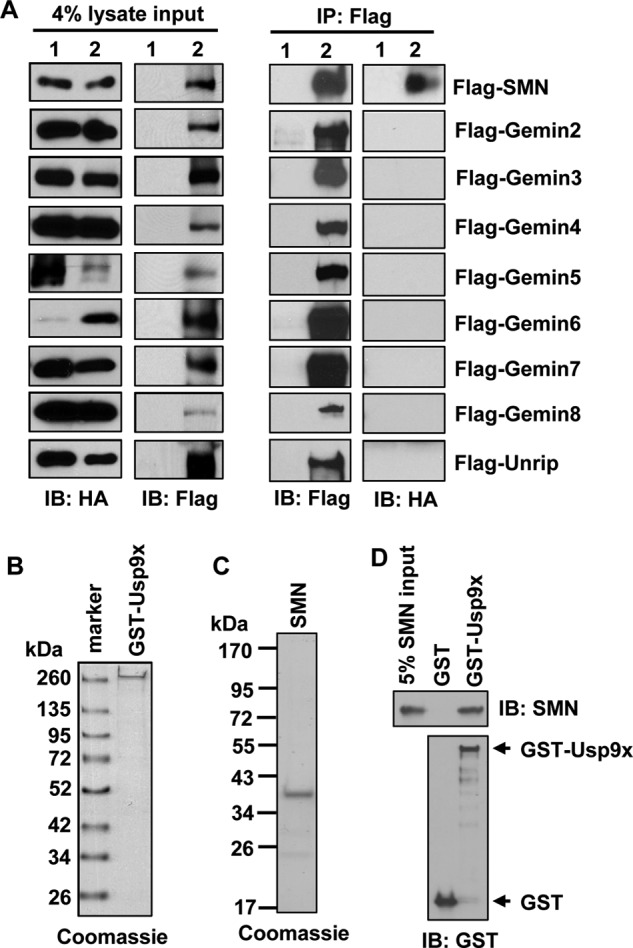
Usp9x directly interacts with SMN. A, immunoblotting (IB) of either FLAG or HA to examine overexpressed HA-Usp9x, FLAG-SMN, Gemins 2–8, or Unrip in 293T cell lysates. FLAG-SMN, Gemins 2–8, or Unrip was immunoprecipitated with the anti-FLAG M2 antibody. Precipitates were immunoblotted with an anti-FLAG or anti-HA antibody. Lane 1, cells overexpressing HA-Usp9x; lane 2, cells overexpressing HA-Usp9x plus a FLAG-tagged one of the proteins of the SMN complex as labeled on the far right. B, Coomassie staining of 1 μg of recombinant GST-Usp9x resolved by SDS-PAGE. C, Coomassie staining of 2 μg of recombinant His6-SMN resolved by SDS-PAGE. D, immunoblotting of SMN (upper blot) and GST (lower blot) contained in GST (control) or GST-Usp9x pulldown assay.
Knockdown of Usp9x Decreases the Protein Levels of SMN and the SMN Complex
To examine whether Usp9x could regulate the protein levels of SMN, we established stable HeLa cell lines expressing either a control shRNA of green fluorescence protein (GFP) or one of the two distinct shRNAs of Usp9x. As shown in Fig. 3A, knockdown of Usp9x caused substantial reduction of protein levels of SMN, Gemins 2 and 8 in whole cell lysates. Protein levels of Gemin 3 had mild decrease. In contrast, protein levels of Gemin 4 and Sm did not change at all. Similar results were obtained when A549 cells (Fig. 3B), fibroblast cells of a type I SMA patient (Fig. 3C), or SH-SY5Y cells (supplemental Fig. S2) were applied for the knockdown experiments. Interestingly, simultaneous reduction of protein levels of SMN, Gemins 2, 3, and 8 were also observed in fibroblast cells derived from SMA patients compared with a normal carrier (54) (supplemental Fig. S3). These results suggest that protein levels of several core components of the SMN complex are tightly co-regulated. Importantly, their protein levels can be regulated by Usp9x. The observed effect is specific to Usp9x because we found that siRNA Usp14, a DUB known to bind the 26 S proteasome and regulate cellular Ub levels (55, 56), had no effect on protein levels of SMN, Gemins 2, 3, and 8. In contrast, siRNA Usp9x caused reduction of their protein levels (supplemental Fig. S4), despite being less efficient compared with shRNA-mediated Usp9x knockdown (Fig. 3A). Together, these results demonstrate that Usp9x regulates the protein levels of several core components of the SMN complex.
FIGURE 3.
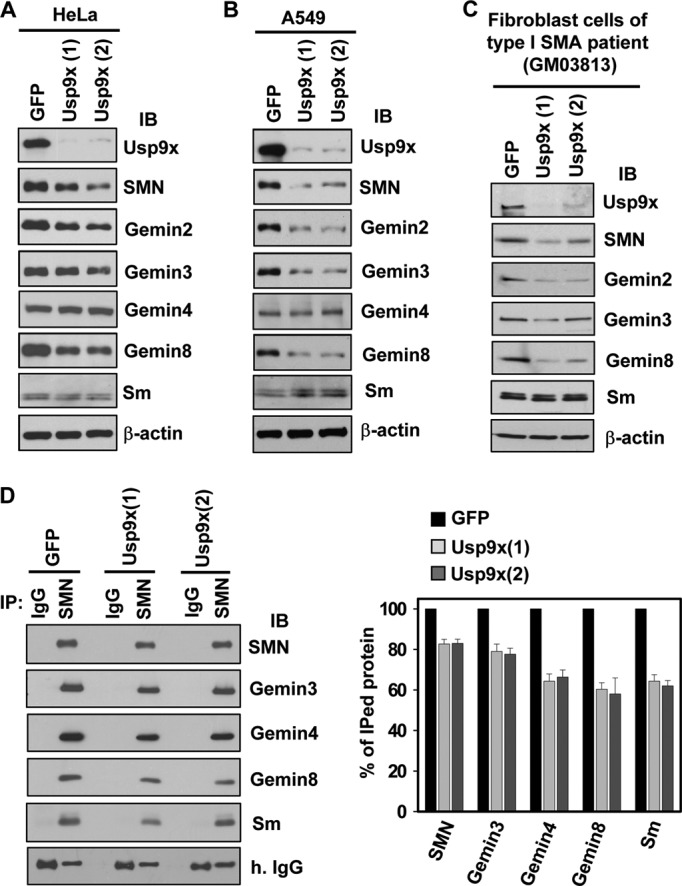
Knockdown of Usp9x decreases the protein levels of SMN, Gemins 2, 3, and 8 in cultured mammalian cells. A-C, HeLa (A), A549 (B), or fibroblast derived from a type I SMA patients (GM03813) (C) cell lines stably expressing a shRNA of GFP or either of the two distinct shRNAs of Usp9x were established. Whole cell lysates were prepared for immunoblotting of Usp9x, Gemins, Sm, and β-actin. D, immunoblotting (IB) of Gemins and Sm co-immunoprecipitated (IP) with endogenous SMN in control (GFP) or Usp9x knockdown HeLa cells. The panel showed the quantitative results of three independent experiments. The protein band intensities were quantitated by densitometry. The ratio between each protein and the IgG in GFP knockdown samples was referenced as 100%. Error bars represent S.D. of three independent experiments.
Because knockdown of Usp9x decreases the total cellular protein levels of several components of the SMN complex, we hypothesized that the protein levels of the SMN complex decrease as well. To test this, we immunoprecipitated endogenous SMN to compare protein levels of the co-immunoprecipitated components of the SMN complex. The immunoprecipitated SMN was less in cell lysates from Usp9x knockdown cells than that from GFP knockdown cells (Fig. 3D). For components of the SMN complex, protein levels of co-immunoprecipitated Gemins 4 and 8 and the Sm proteins were ∼25% less than the SMN protein levels in Usp9x knockdown cells (Fig. 3D), in which the protein levels of Gemin4 and Sm were not affected at all in whole cell lysates (Fig. 3, A–C). Thus, loss of Usp9x decreases protein levels of the SMN complex.
Loss of Usp9x Impairs Gems Formation and SMN/Coilin Co-localization in the Nucleus
Cellular fractionation from previous studies has shown that either no Usp9x or only a small portion of Usp9x was found in the nucleus (39, 52). Immunofluorescence staining of endogenous Usp9x and SMN in HeLa cells revealed that both of them mostly localized in the cytoplasm and partially co-localized in the perinuclear region; no Usp9x/SMN co-localization was observed in gems (Fig. 4A). These results are consistent with a previous study using GFP-SMN (52). Thus, Usp9x might regulate the SMN complex stability in the cytoplasm. The SMN complex assembles in the cytoplasm and imports into the nucleus to form gems (21, 57, 58). We next examined whether knockdown of Usp9x affects gems formation. Immunofluorescence staining of SMN in either GFP or Usp9x knockdown HeLa cells found that loss of Usp9x significantly reduced the number of gems in the nucleus (Fig. 4B). Quantification showed that, ∼40% of Usp9x knockdown cells had no gems at all, whereas only 10% of GFP knockdown cells had no gems; the percentage of cells with one or two gems remained roughly the same between Usp9x and control knockdown cells. However, the percentage of cells with three or more gems was significantly decreased in Usp9x knockdown cells (Fig. 4C). Furthermore, we found that knockdown of Usp9x significantly increased the percentage of cells that did not have SMN/coilin co-localization, ∼50% of Usp9x knockdown cells versus 20% of GFP knockdown cells (Fig. 4, D and E). Thus, loss of Usp9x significantly impairs gems formation and SMN/coilin co-localization in the nucleus.
FIGURE 4.

Loss of Usp9x impairs gems formation and SMN/coilin co-localization in the nucleus. A, immunofluorescence staining of SMN, Usp9x, and the nucleus in HeLa cells. B, immunofluorescence staining of SMN and the nucleus in control (GFP) or Usp9x knockdown HeLa cells. C, quantification of the gems numbers in control (GFP) or Usp9x knockdown HeLa cells as shown in B. D, immunofluorescence staining of coilin and SMN in control (GFP) or Usp9x knockdown HeLa cells. E, quantification of cells with SMN/coilin co-localization in control (GFP) or Usp9x knockdown HeLa cells as shown in D. In both C and E, a total of 200 cells were counted in each area. Error bars represent S.D. from three different areas.
SMN Is Predominantly Mono- and Di-ubiquitinated
SMN was reported to be a ubiquitinated protein and a substrate of the proteasome (29, 32). We performed denaturing immunoprecipitation to determine the ubiquitination status of SMN, which would obviate the possibility that observed ubiquitination comes from the binding partners of SMN. In this assay, FLAG-SMN and HA-Ub were expressed in 293T cells. FLAG-SMN was immunoprecipitated from cell lysates using the anti-FLAG M2 antibody under a denaturing condition (see “Experimental Procedures”). Immunoblotting of HA revealed that FLAG-SMN was predominantly conjugated with mono- and di-Ub, and a small portion of SMN was conjugated with more than two Ub (Fig. 5A). We noticed that monoubiquitination was the major modification of endogenous SMN as well in a previous study (29). Interestingly, inhibition of the proteasome with MG132 did not obviously increase the ubiquitination levels of SMN (Fig. 5A).
FIGURE 5.

Usp9x deubiquitinates and protects SMN from degradation. A, immunoblotting of HA-Ub conjugated on FLAG-SMN in 293T cells. FLAG-SMN was immunoprecipitated (IP) with denaturing IP. The asterisk marks monoubiquitinated SMN as judged by its molecular weight. B, immunoblotting (IB) of Myc-Ub conjugated on FLAG-SMN in 293T cells co-transfected with the indicated plasmids. FLAG-SMN was immunoprecipitated with denaturing IP. C, immunoblotting of HA-Ub conjugated on FLAG-SMN overexpressed in GFP or Usp9x knockdown HeLa cells. Immunoprecipitated FLAG-SMN (denaturing IP) was quantitated by densitometry. The amount of immunoprecipitated FLAG-SMN in GFP cells was referenced as 1. D, immunoblotting of Ub to monitor the time-dependent deubiquitination of K48 Ub4 by purified GST-Usp9x. The lane denoted with 3 + Ubal indicates that 2.5 μm Ub aldehyde (Ubal) was included in the reaction and the reaction was stopped at 3 h. E, deubiquitination of ubiquitinated FLAG-SMN by purified GST-Usp9x. Ubiquitinated FLAG-SMN was immunoprecipitated from 293T cells as described under “Experimental Procedures.” F, immunoblotting of Gemins, Sm, Usp9x, and β-actin in cell lysates prepared from control (GFP) or Usp9x stable knockdown HeLa cells. Cells were harvested at the indicated time points after treating with 75 μg/ml of CHX. The lanes 10 + epo indicate that cells were treated with 10 μm epoxomicin + 75 μg/ml of CHX for 10 h. G and H, degradation of endogenous SMN (G) and Gemin 8 (H) in F were quantitated by densitometry. Error bars in G and H represent S.D. of three independent experiments.
Usp9x Deubiquitinates and Protects SMN from Degradation
We next sought to determine whether Usp9x deubiquitinates SMN. As revealed by denaturing immunoprecipitation (Fig. 5B), overexpression of HA-Usp9x significantly reduced the protein levels of ubiquitinated SMN. In contrast, overexpression of HA-Usp9x(C1556A), a catalytically inactive mutant, had no effect on SMN ubiquitination. Consistent with this result, knockdown of Usp9x caused the accumulation of more monoubiquitinated SMN in HeLa cells compared with GFP knockdown cells, although the immunoprecipitated FLAG-SMN in Usp9x knockdown cells was only 73% of that in GFP knockdown cells (Fig. 5C). The lower amount of immunoprecipitated FLAG-SMN in Usp9x knockdown cells is presumably because loss of Usp9x caused reduction of FLAG-SMN as shown in the cell lysates (Fig. 5C). We further performed an in vitro deubiquitination assay to determine whether Usp9x deubiquitinates SMN. Ubiquitinated SMN was immunoprecipitated from cell lysates of 293T cells expressing FLAG-SMN and HA-Ub. The immunoprecipitated FLAG-SMN was washed with a stringent buffer containing 1% Triton X-100 and 1.35 m NaCl, and then incubated with or without recombinant GST-Usp9x, which exhibited DUB activity in an assay using K48-linked Ub4 as the substrate (Fig. 5D). In the absence of purified Usp9x, no deubiquitination of SMN was observed after incubation; addition of purified GST-Usp9x promoted SMN deubiquitination, which was abolished when the DUB activity of Usp9x was inhibited with Ub aldehyde (Fig. 5E). These results demonstrate that Usp9x deubiquitinates SMN.
We next examined whether Usp9x functions to protect SMN from degradation using the cycloheximide (CHX)-chase assay. As shown in Fig. 5, F–H, stable knockdown of Usp9x in HeLa cells significantly promoted degradation of endogenous SMN and Gemin 8, and their degradation can be greatly inhibited by epoxomicin, a potent proteasome inhibitor (Fig. 5F). In contrast, degradation of Gemins 2, 3, 4, and Sm were not affected in the same assay. Together, these results indicate that Usp9x deubiquitinates SMN and plays a role in mediating SMN degradation.
SMN Ubiquitination Might Occur on Multiple Lysine Residues
In an attempt to identify the lysine residue(s) of SMN that mediates SMN ubiquitination, we found that FLAG-SMN(K0), a mutant in which all 22 lysine residues of SMN were mutated to arginines, was not able to be ubiquitinated (supplemental Fig. S5A). However, introducing any lysine residue back into the SMN(K0) construct, individually or in certain combinations, partially regained SMN monoubiquitination as revealed by analyzing the cell lysates prepared from 293T cells overexpressing one of these constructs together with HA-Ub (data not shown). Thus, SMN ubiquitination is conjugated on lysine residues and the ubiquitination sites are promiscuous. Consistent with this, several large scale proteomics studies identified that endogenous SMN can be ubiquitinated on multiple lysine residues including lysines 41, 51, 186, and 209 (59–62). With confirmation that SMN(K0) abolishes SMN ubiquitination, we examined whether ubiquitination mediates SMN degradation. To this end, we prepared HeLa cells stably expressing HA-SMN or HA-SMN(K0). CHX-chase assays showed that HA-SMN(K0) was resistant to degradation (supplemental Fig. S5B). This result suggests that SMN degradation is Ub-dependent.
SMNΔ7 Is Mostly Polyubiquitinated and Degraded by the Proteasome
SMNΔ7, the main protein product of the SMN2 gene, is an unstable protein and a substrate of the proteasome (29–31). It is unknown whether SMNΔ7 degradation is Ub-dependent. We therefore assessed the ubiquitination status of SMNΔ7. To do this, we expressed FLAG-SMNΔ7 and HA-Ub in 293T cells, which were either treated with MG132 to block proteasomal degradation or DMSO as a control. Denaturing immunoprecipitation assays demonstrated that SMNΔ7 was conjugated with mono-Ub and short Ub chains in cells treated with DMSO, whereas polyubiquitination of SMNΔ7 was apparent when cells were treated with MG132 (Fig. 6A). Also, proteasome inhibition greatly increased protein levels of SMNΔ7 (Fig. 6A). The smeared high molecular weight species of SMNΔ7 was resistant to treatment with 6 m urea plus 2.5% SDS (Fig. 6B), excluding the possibility that they were aggregated SMNΔ7 conjugated with short Ub chains. Thus, in addition to being mono- and di-ubiquitinated, SMNΔ7 can also be highly ubiquitinated and its protein and ubiquitination levels are sensitive to proteasome inhibition.
FIGURE 6.

Usp9x does not regulate SMNΔ7 ubiquitination and degradation. A, immunoblotting (IB) of HA-Ub conjugated on FLAG-SMNΔ7 in 293T cells. FLAG-SMNΔ7 and HA-Ub were expressed in 293T cells. In A–C, FLAG-SMNΔ7 was immunoprecipitated (IP) by denaturing IP. B, FLAG-SMNΔ7 was immunoprecipitated as shown in A and eluted with 6 m urea plus 2.5% SDS. C, immunoblotting of Myc-Ub conjugated on FLAG-SMNΔ7 in 293T cells overexpressing the indicated proteins. D, immunoblotting of HA-SMNΔ7, Usp9x, and β-actin in cell lysates prepared from control (GFP) or Usp9x stable knockdown HeLa cells that stably express HA-SMNΔ7. Cells were harvested at the indicated time points after treating with 75 μg/ml of CHX. Error bars in the right panel represent S.D. of three independent experiments. E, confocal images of immunofluorescence staining of overexpressed FLAG-SMNΔ7 in HeLa cells.
Usp9x Does Not Deubiquitinate SMNΔ7, nor Does It Regulate SMNΔ7 Degradation
Next, we examined whether Usp9x regulates SMNΔ7 degradation. To our surprise, expressing HA-Usp9x did not reduce the ubiquitination levels of SMNΔ7 in 293T cells (Fig. 6C). Nor did Usp9x(1–1971), a Usp9x deletion mutant that was able to deubiquitinate SMN (supplemental Fig. S6A) and the SMA-causing SMN(A2G) mutant (supplemental Fig. S6B), deubiquitinate SMNΔ7 (supplemental Fig. S6C). Thus, Usp9x likely does not deubiquitinate SMNΔ7. Moreover, compared with control shRNA, knockdown of Usp9x had no effect on the protein half-life of HA-SMNΔ7 stably expressed in HeLa cells (Fig. 6D). In contrast, knockdown of Usp9x promoted degradation of HA-SMN stably expressed in HeLa cells (supplemental Fig. S7), similar as the effect on degradation of endogenous SMN (Fig. 5, F and G). Thus, we conclude that Usp9x does not regulate polyubiquitination and the protein half-life of SMNΔ7. One potential explanation for this is that Usp9x and SMNΔ7 have different cellular localizations. Usp9x is mainly a cytoplasmic protein (39, 52). Previous studies have shown that exon 7 in SMN encodes a cytoplasmic localization sequence, and SMNΔ7 is largely confined to the nucleus in chick forebrain neurons (63). Consistent with this, overexpressed FLAG-SMNΔ7 in HeLa cells was found to be predominantly in the nucleus existing as excess foci (Fig. 6E).
DISCUSSION
In this study, we found that Usp9x associated stably with the SMN complex via a direct interaction with SMN. Knockdown of Usp9x decreased protein levels of SMN, Gemins 2, 3, and 8 in several cultured mammalian cell lines. Consequently, protein levels of the SMN complex decreased and SMN nuclear localization was impaired. We also found that Usp9x deubiquitinated SMN, which was predominantly mono- and di-ubiquitinated. Moreover, knockdown of Usp9x decreased the protein half-life of SMN in HeLa cells. Taken together, we propose that Usp9x directly interacts with and deubiquitinates SMN, which protects SMN from Ub-dependent degradation.
The protein levels of SMN, Gemins 2, 3, and 8 seem to be tightly co-regulated as observed by analysis of their protein levels in SMA patient cells (54), as well as in Usp9x knockdown cells shown in this study. Usp9x-mediated regulation is likely exerted post-transcriptionally because RT-PCR analyses showed that the mRNA levels of all gene products of the SMN complex in Usp9x knockdown cells were similar as those in GFP knockdown cells (supplemental Fig. S8). The CHX-chase experiments showed that knockdown of Usp9x promoted degradation of SMN and Gemin 8, but not Gemins 2 and 3, although their protein levels decreased when Usp9x was knocked down in several cell lines. Presumably, loss of SMN and/or Usp9x activates feedback loops to block Gemins 2 and 3 protein translation, and thus, reducing their protein levels without affecting their degradation. The accelerated Gemin 8 degradation in Usp9x knockdown cells might result from the loss of its binding partner SMN. Interestingly, the extent of reduction of SMN and Gemins 2, 3, and 8 protein levels in the examined cell lines was different when Usp9x was knocked down (Fig. 3, A–C and supplemental Fig. S2). This might be due to the fact that their cellular protein levels are different in these cell lines; and/or degradation of these proteins is differentially regulated in these cell lines, such as having different E3 Ub ligases.
SMN was previously found to be a substrate of the proteasome (29, 32). We found that SMN was predominantly mono- and di-ubiquitinated, with a small fraction of SMN was modified with three or more Ub (Fig. 5A). In contrast, SMNΔ7 was able to be highly polyubiquitinated and its protein and ubiquitination levels were significantly increased in response to proteasomal inhibition (Fig. 6A). The 26 S proteasome usually degrades polyubiquitinated proteins conjugated with at least tetra-Ub (64). Mono- and oligo-ubiquitination (multiple mono-ubiquitinations) are also able to target proteins for proteasomal degradation (65–69). However, mono-Ub and short Ub chains have much weaker proteasomal binding affinity than that of poly-Ub chains, this might explain why SMNΔ7 is more rapidly degraded than SMN by the 26 S proteasome. The reason for differential ubiquitination of SMN and SMNΔ7 is not clear. On the one hand, conformational differences between SMN and SMNΔ7 could alter their binding/accessibility/affinity to E3/E4 Ub ligases, which result in differential ubiquitination. Although the crystal structures of SMN and SMNΔ7 are unknown, their structural differences have been clearly demonstrated in the aspect of self-association. The regions encoded by exons 2b and 6 mediate SMN oligomerization, which in turn mediates formation of the SMN complex (54, 70). Oligomerization of SMNΔ7 was found to be significantly impaired (54). On the other hand, cellular localizations of SMN and SMNΔ7 might contribute to their differential ubiquitination. SMN distributes in both the cytoplasm and the nucleus, whereas SMNΔ7 is predominantly located in the nucleus (63), as also shown in HeLa cells overexpressing FLAG-SMNΔ7 (Fig. 6E). It is possible that SMNΔ7 is polyubiquitinated and degraded in the nucleus. Because Usp9x is mainly a cytoplasmic protein, it is not surprising that Usp9x could not deubiquitinate and regulate degradation of nuclear SMNΔ7. Future studies aiming to identify E3/E4 Ub ligases of SMN and SMNΔ7 will provide further insights on why SMN and SMNΔ7 are ubiquitinated and degraded differently.
One well established function of SMN is to help assembly of small nuclear ribonucleoproteins (21–24), core components of the spliceosome. Whether SMA is caused by splicing defects is controversial. For instance, significant reduction of SMN protein levels to ∼15% or less of normal levels caused reduction of snRNA levels, especially the minor snRNAs, and tissue-specific splicing defects in severe SMA mice (71–73). However, the observed splicing defects in severe SMA mice could result from a secondary effect of cell injury as the disease progresses because one recent study found that no significant splicing defects were detected in postnatal days 1 or 7 SMA mice compared with littermate control mice, whereas splicing defects were observed in day 13 severe SMA mice (74). In a Drosophila model, Smn null mutants displayed larval lethality and showed reductions of minor snRNAs. However, no appreciable defects were found in the splicing of mRNAs containing minor-class introns (75). In contrast, increased U12 minor intron retention in two genes in Drosophila expressing loss-of-function SMN73Ao, which produces an unstable SMN protein, was detected. Moreover, Stasimon, encoded by one of these two genes, was found to regulate motor circuit function (76). Apparently, the role of SMN in regulating gene splicing and whether splicing defects in a subset of genes directly cause SMA require further investigation. We have performed mRNA-Seq to probe the transcriptome in Usp9x or control knockdown HeLa and A549 cells. No obvious gene splicing differences were probed when compared the splicing events in Usp9x knockdown cells to those in control knockdown cells. This might be caused by the fact that the SMN protein levels in the Usp9x knockdown cells are not yet reduced to the threshold that would cause splicing defects; and/or the SMN protein levels are not critical for regulating gene splicing in these two cell lines.
SMA is caused by low SMN protein levels. It will be interesting to determine to what extent the SMN protein levels decrease in various tissues when Usp9x is completely ablated in animals. Ablation of FAM, the mouse ortholog of human Usp9x, results in embryonic lethality (41) and FAM was found to play an essential role in embryo development (77). Conditional knock-out of FAM in mice or deletion of Usp9x orthologs in other organisms might be useful to evaluate whether loss of Usp9x leads to SMA development in future studies.
Acknowledgments
We are grateful to D. R. Alessi for providing the Usp9x and Usp9x(C1566A) plasmids, L. G. Xu for providing a 293T cDNA library, and B. Chong for initial Usp9x purification. We thank the members of the Liu lab for insightful discussion and reading of the manuscript. Imaging experiments were performed in the University of Colorado Anschutz Medical Campus Advance Light Microscopy Core supported in part by NIH/NCRR Colorado CTSI Grant UL1 RR025780.
This work was supported, in whole or in part, by National Institutes of Health Grant 5RO1NS72397 (to C. W. L.) and grants from the American-Syrian-Lebanese Associated Charities (to J. P.) and the Cancer League of Colorado (to K. J. H.).

This article contains supplemental Figs. S1–S8.
- SMA
- spinal muscular atrophy
- SMN
- survival motor neuron
- DUB
- deubiquitination enzyme
- CHX
- cycloheximide
- Ub
- ubiquitin
- β-ME
- β-mercaptoethanol.
REFERENCES
- 1. Burghes A. H., Beattie C. E. (2009) Spinal muscular atrophy. Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 10, 597–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Melki J. (1997) Spinal muscular atrophy. Curr. Opin. Neurol. 10, 381–385 [DOI] [PubMed] [Google Scholar]
- 3. Monani U. R. (2005) Spinal muscular atrophy. A deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron 48, 885–896 [DOI] [PubMed] [Google Scholar]
- 4. Lefebvre S., Bürglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165 [DOI] [PubMed] [Google Scholar]
- 5. Burghes A. H. (1997) When is a deletion not a deletion? When it is converted. Am. J. Hum. Genet. 61, 9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Monani U. R., Lorson C. L., Parsons D. W., Prior T. W., Androphy E. J., Burghes A. H., McPherson J. D. (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183 [DOI] [PubMed] [Google Scholar]
- 7. McAndrew P. E., Parsons D. W., Simard L. R., Rochette C., Ray P. N., Mendell J. R., Prior T. W., Burghes A. H. (1997) Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 60, 1411–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mailman M. D., Heinz J. W., Papp A. C., Snyder P. J., Sedra M. S., Wirth B., Burghes A. H., Prior T. W. (2002) Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 4, 20–26 [DOI] [PubMed] [Google Scholar]
- 9. Feldkötter M., Schwarzer V., Wirth R., Wienker T. F., Wirth B. (2002) Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR. Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 70, 358–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schrank B., Götz R., Gunnersen J. M., Ure J. M., Toyka K. V., Smith A. G., Sendtner M. (1997) Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. U.S.A. 94, 9920–9925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Monani U. R., Sendtner M., Coovert D. D., Parsons D. W., Andreassi C., Le T. T., Jablonka S., Schrank B., Rossoll W., Rossol W., Prior T. W., Morris G. E., Burghes A. H. (2000) The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn−/− mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 9, 333–339 [DOI] [PubMed] [Google Scholar]
- 12. Coovert D. D., Le T. T., McAndrew P. E., Strasswimmer J., Crawford T. O., Mendell J. R., Coulson S. E., Androphy E. J., Prior T. W., Burghes A. H. (1997) The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 6, 1205–1214 [DOI] [PubMed] [Google Scholar]
- 13. Lefebvre S., Burlet P., Liu Q., Bertrandy S., Clermont O., Munnich A., Dreyfuss G., Melki J. (1997) Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 16, 265–269 [DOI] [PubMed] [Google Scholar]
- 14. Liu Q., Dreyfuss G. (1996) A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 15, 3555–3565 [PMC free article] [PubMed] [Google Scholar]
- 15. Carvalho T., Almeida F., Calapez A., Lafarga M., Berciano M. T., Carmo-Fonseca M. (1999) The spinal muscular atrophy disease gene product, SMN. A link between snRNP biogenesis and the Cajal (coiled) body. J. Cell Biol. 147, 715–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Young P. J., Le T. T., Dunckley M., Nguyen T. M., Burghes A. H., Morris G. E. (2001) Nuclear gems and Cajal (coiled) bodies in fetal tissues. Nucleolar distribution of the spinal muscular atrophy protein, SMN. Exp. Cell Res. 265, 252–261 [DOI] [PubMed] [Google Scholar]
- 17. Young P. J., Le T. T., thi Man N., Burghes A. H., Morris G. E. (2000) The relationship between SMN, the spinal muscular atrophy protein, and nuclear coiled bodies in differentiated tissues and cultured cells. Exp. Cell Res. 256, 365–374 [DOI] [PubMed] [Google Scholar]
- 18. Matera A. G., Frey M. R. (1998) Coiled bodies and gems. Janus or gemini? Am. J. Hum. Genet. 63, 317–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paushkin S., Gubitz A. K., Massenet S., Dreyfuss G. (2002) The SMN complex, an assemblyosome of ribonucleoproteins. Curr. Opin. Cell Biol. 14, 305–312 [DOI] [PubMed] [Google Scholar]
- 20. Otter S., Grimmler M., Neuenkirchen N., Chari A., Sickmann A., Fischer U. (2007) A comprehensive interaction map of the human survival of motor neuron (SMN) complex. J. Biol. Chem. 282, 5825–5833 [DOI] [PubMed] [Google Scholar]
- 21. Narayanan U., Achsel T., Lührmann R., Matera A. G. (2004) Coupled in vitro import of U snRNPs and SMN, the spinal muscular atrophy protein. Mol. Cell 16, 223–234 [DOI] [PubMed] [Google Scholar]
- 22. Liu Q., Fischer U., Wang F., Dreyfuss G. (1997) The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 90, 1013–1021 [DOI] [PubMed] [Google Scholar]
- 23. Fischer U., Liu Q., Dreyfuss G. (1997) The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 90, 1023–1029 [DOI] [PubMed] [Google Scholar]
- 24. Pellizzoni L., Yong J., Dreyfuss G. (2002) Essential role for the SMN complex in the specificity of snRNP assembly. Science 298, 1775–1779 [DOI] [PubMed] [Google Scholar]
- 25. Rossoll W., Bassell G. J. (2009) Spinal muscular atrophy and a model for survival of motor neuron protein function in axonal ribonucleoprotein complexes. Results Probl. Cell Differ. 48, 289–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rossoll W., Jablonka S., Andreassi C., Kröning A. K., Karle K., Monani U. R., Sendtner M. (2003) Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of β-actin mRNA in growth cones of motoneurons. J. Cell Biol. 163, 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eggert C., Chari A., Laggerbauer B., Fischer U. (2006) Spinal muscular atrophy. The RNP connection. Trends Mol. Med. 12, 113–121 [DOI] [PubMed] [Google Scholar]
- 28. Pellizzoni L. (2007) Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 8, 340–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burnett B. G., Muñoz E., Tandon A., Kwon D. Y., Sumner C. J., Fischbeck K. H. (2009) Regulation of SMN protein stability. Mol. Cell Biol. 29, 1107–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lorson C. L., Androphy E. J. (2000) An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet. 9, 259–265 [DOI] [PubMed] [Google Scholar]
- 31. Cho S., Dreyfuss G. (2010) A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 24, 438–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chang H. C., Hung W. C., Chuang Y. J., Jong Y. J. (2004) Degradation of survival motor neuron (SMN) protein is mediated via the ubiquitin/proteasome pathway. Neurochem. Int. 45, 1107–1112 [DOI] [PubMed] [Google Scholar]
- 33. Jones M. H., Furlong R. A., Burkin H., Chalmers I. J., Brown G. M., Khwaja O., Affara N. A. (1996) The Drosophila developmental gene fat facets has a human homologue in Xp11.4 which escapes X-inactivation and has related sequences on Yq11.2. Hum. Mol. Genet. 5, 1695–1701 [DOI] [PubMed] [Google Scholar]
- 34. Nijman S. M., Luna-Vargas M. P., Velds A., Brummelkamp T. R., Dirac A. M., Sixma T. K., Bernards R. (2005) A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786 [DOI] [PubMed] [Google Scholar]
- 35. Zhu X., Menard R., Sulea T. (2007) High incidence of ubiquitin-like domains in human ubiquitin-specific proteases. Proteins 69, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taya S., Yamamoto T., Kanai-Azuma M., Wood S. A., Kaibuchi K. (1999) The deubiquitinating enzyme Fam interacts with and stabilizes β-catenin. Genes Cells 4, 757–767 [DOI] [PubMed] [Google Scholar]
- 37. Taya S., Yamamoto T., Kano K., Kawano Y., Iwamatsu A., Tsuchiya T., Tanaka K., Kanai-Azuma M., Wood S. A., Mattick J. S., Kaibuchi K. (1998) The Ras target AF-6 is a substrate of the fam deubiquitinating enzyme. J. Cell Biol. 142, 1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Al-Hakim A. K., Zagorska A., Chapman L., Deak M., Peggie M., Alessi D. R. (2008) Control of AMPK-related kinases by USP9X and atypical Lys29/Lys33-linked polyubiquitin chains. Biochem. J. 411, 249–260 [DOI] [PubMed] [Google Scholar]
- 39. Schwickart M., Huang X., Lill J. R., Liu J., Ferrando R., French D. M., Maecker H., O'Rourke K., Bazan F., Eastham-Anderson J., Yue P., Dornan D., Huang D. C., Dixit V. M. (2010) Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463, 103–107 [DOI] [PubMed] [Google Scholar]
- 40. Théard D., Labarrade F., Partisani M., Milanini J., Sakagami H., Fon E. A., Wood S. A., Franco M., Luton F. (2010) USP9x-mediated deubiquitination of EFA6 regulates de novo tight junction assembly. EMBO J. 29, 1499–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nagai H., Noguchi T., Homma K., Katagiri K., Takeda K., Matsuzawa A., Ichijo H. (2009) Ubiquitin-like sequence in ASK1 plays critical roles in the recognition and stabilization by USP9X and oxidative stress-induced cell death. Mol. Cell 36, 805–818 [DOI] [PubMed] [Google Scholar]
- 42. Marx C., Held J. M., Gibson B. W., Benz C. C. (2010) ErbB2 trafficking and degradation associated with K48 and K63 polyubiquitination. Cancer Res. 70, 3709–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dupont S., Mamidi A., Cordenonsi M., Montagner M., Zacchigna L., Adorno M., Martello G., Stinchfield M. J., Soligo S., Morsut L., Inui M., Moro S., Modena N., Argenton F., Newfeld S. J., Piccolo S. (2009) FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell 136, 123–135 [DOI] [PubMed] [Google Scholar]
- 44. Vong Q. P., Cao K., Li H. Y., Iglesias P. A., Zheng Y. (2005) Chromosome alignment and segregation regulated by ubiquitination of survivin. Science 310, 1499–1504 [DOI] [PubMed] [Google Scholar]
- 45. Wang X., Chen C. F., Baker P. R., Chen P. L., Kaiser P., Huang L. (2007) Mass spectrometric characterization of the affinity-purified human 26 S proteasome complex. Biochemistry 46, 3553–3565 [DOI] [PubMed] [Google Scholar]
- 46. Han K. J., Yang Y., Xu L. G., Shu H. B. (2010) Analysis of a TIR-less splice variant of TRIF reveals an unexpected mechanism of TLR3-mediated signaling. J. Biol. Chem. 285, 12543–12550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Melcher K. (2000) A modular set of prokaryotic and eukaryotic expression vectors. Anal. Biochem. 277, 109–120 [DOI] [PubMed] [Google Scholar]
- 48. Hovland P., Flick J., Johnston M., Sclafani R. A. (1989) Galactose as a gratuitous inducer of GAL gene expression in yeasts growing on glucose. Gene 83, 57–64 [DOI] [PubMed] [Google Scholar]
- 49. Xu P., Duong D. M., Peng J. (2009) Systematical optimization of reverse-phase chromatography for shotgun proteomics. J. Proteome. Res. 8, 3944–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peng J., Elias J. E., Thoreen C. C., Licklider L. J., Gygi S. P. (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis. The yeast proteome. J. Proteome. Res. 2, 43–50 [DOI] [PubMed] [Google Scholar]
- 51. Raasi S., Pickart C. M. (2005) Ubiquitin chain synthesis. Methods Mol. Biol. 301, 47–55 [DOI] [PubMed] [Google Scholar]
- 52. Trinkle-Mulcahy L., Boulon S., Lam Y. W., Urcia R., Boisvert F. M., Vandermoere F., Morrice N. A., Swift S., Rothbauer U., Leonhardt H., Lamond A. (2008) Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 183, 223–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hirakata M., Craft J., Hardin J. A. (1993) Autoantigenic epitopes of the B and D polypeptides of the U1 snRNP. Analysis of domains recognized by the Y12 monoclonal anti-Sm antibody and by patient sera. J. Immunol. 150, 3592–3601 [PubMed] [Google Scholar]
- 54. Lorson C. L., Strasswimmer J., Yao J. M., Baleja J. D., Hahnen E., Wirth B., Le T., Burghes A. H., Androphy E. J. (1998) SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 19, 63–66 [DOI] [PubMed] [Google Scholar]
- 55. Chernova T. A., Allen K. D., Wesoloski L. M., Shanks J. R., Chernoff Y. O., Wilkinson K. D. (2003) Pleiotropic effects of Ubp6 loss on drug sensitivities and yeast prion are due to depletion of the free ubiquitin pool. J. Biol. Chem. 278, 52102–52115 [DOI] [PubMed] [Google Scholar]
- 56. Anderson C., Crimmins S., Wilson J. A., Korbel G. A., Ploegh H. L., Wilson S. M. (2005) Loss of Usp14 results in reduced levels of ubiquitin in ataxia mice. J. Neurochem. 95, 724–731 [DOI] [PubMed] [Google Scholar]
- 57. Sleeman J. E., Lamond A. I. (1999) Newly assembled snRNPs associate with coiled bodies before speckles, suggesting a nuclear snRNP maturation pathway. Curr. Biol. 9, 1065–1074 [DOI] [PubMed] [Google Scholar]
- 58. Hebert M. D., Szymczyk P. W., Shpargel K. B., Matera A. G. (2001) Coilin forms the bridge between Cajal bodies and SMN, the spinal muscular atrophy protein. Genes Dev. 15, 2720–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wagner S. A., Beli P., Weinert B. T., Nielsen M. L., Cox J., Mann M., Choudhary C. (2011) A proteome-wide quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell Proteomics 10, M111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Danielsen J. M., Sylvestersen K. B., Bekker-Jensen S., Szklarczyk D., Poulsen J. W., Horn H., Jensen L. J., Mailand N., Nielsen M. L. (2011) Mol. Cell Proteomics 10, M110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Povlsen L. K., Beli P., Wagner S. A., Poulsen S. L., Sylvestersen K. B., Poulsen J. W., Nielsen M. L., Bekker-Jensen S., Mailand N., Choudhary C. (2012) Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 14, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 62. Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang H. L., Pan F., Hong D., Shenoy S. M., Singer R. H., Bassell G. J. (2003) Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J. Neurosci. 23, 6627–6637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Thrower J. S., Hoffman L., Rechsteiner M., Pickart C. M. (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Boutet S. C., Disatnik M. H., Chan L. S., Iori K., Rando T. A. (2007) Regulation of Pax3 by proteasomal degradation of monoubiquitinated protein in skeletal muscle progenitors. Cell 130, 349–362 [DOI] [PubMed] [Google Scholar]
- 66. Carvallo L., Muñoz R., Bustos F., Escobedo N., Carrasco H., Olivares G., Larraín J. (2010) Non-canonical Wnt signaling induces ubiquitination and degradation of Syndecan4. J. Biol. Chem. 285, 29546–29555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dimova N. V., Hathaway N. A., Lee B. H., Kirkpatrick D. S., Berkowitz M. L., Gygi S. P., Finley D., King R. W. (2012) APC/C-mediated multiple monoubiquitylation provides an alternative degradation signal for cyclin B1. Nat. Cell Biol. 14, 168–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kravtsova-Ivantsiv Y., Cohen S., Ciechanover A. (2009) Modification by single ubiquitin moieties rather than polyubiquitination is sufficient for proteasomal processing of the p105 NF-κB precursor. Mol. Cell 33, 496–504 [DOI] [PubMed] [Google Scholar]
- 69. Yin H., Gui Y., Du G., Frohman M. A., Zheng X. L. (2010) Dependence of phospholipase D1 multi-monoubiquitination on its enzymatic activity and palmitoylation. J. Biol. Chem. 285, 13580–13588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Young P. J., Man N. T., Lorson C. L., Le T. T., Androphy E. J., Burghes A. H., Morris G. E. (2000) The exon 2b region of the spinal muscular atrophy protein, SMN, is involved in self-association and SIP1 binding. Hum. Mol. Genet. 9, 2869–2877 [DOI] [PubMed] [Google Scholar]
- 71. Gabanella F., Butchbach M. E., Saieva L., Carissimi C., Burghes A. H., Pellizzoni L. (2007) Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One 2, e921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang Z., Lotti F., Dittmar K., Younis I., Wan L., Kasim M., Dreyfuss G. (2008) SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133, 585–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Boulisfane N., Choleza M., Rage F., Neel H., Soret J., Bordonné R. (2011) Impaired minor tri-snRNP assembly generates differential splicing defects of U12-type introns in lymphoblasts derived from a type I SMA patient. Hum. Mol. Genet. 20, 641–648 [DOI] [PubMed] [Google Scholar]
- 74. Bäumer D., Lee S., Nicholson G., Davies J. L., Parkinson N. J., Murray L. M., Gillingwater T. H., Ansorge O., Davies K. E., Talbot K. (2009) Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 5, e1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Praveen K., Wen Y., Matera A. G. (2012) A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. Cell Rep. 1, 624–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lotti F., Imlach W. L., Saieva L., Beck E. S., Hao le T., Li D. K., Jiao W., Mentis G. Z., Beattie C. E., McCabe B. D., Pellizzoni L. (2012) An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151, 440–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pantaleon M., Kanai-Azuma M., Mattick J. S., Kaibuchi K., Kaye P. L., Wood S. A. (2001) FAM deubiquitylating enzyme is essential for preimplantation mouse embryo development. Mech. Dev. 109, 151–160 [DOI] [PubMed] [Google Scholar]