

Abstract
We have determined the effects of myosin binding protein-C (MyBP-C) and its domains on the microsecond rotational dynamics of actin, detected by time-resolved phosphorescence anisotropy (TPA). MyBP-C is a multidomain modulator of striated muscle contraction, interacting with myosin, titin, and possibly actin. Cardiac and slow skeletal MyBP-C are known substrates for protein kinase-A (PKA), and phosphorylation of the cardiac isoform alters contractile properties and myofilament structure. To determine the effects of MyBP-C on actin structural dynamics, we labeled actin at C374 with a phosphorescent dye and performed TPA experiments. The interaction of all three MyBP-C isoforms with actin increased the final anisotropy of the TPA decay, indicating restriction of the amplitude of actin torsional flexibility by 15–20° at saturation of the TPA effect. PKA phosphorylation of slow skeletal and cardiac MyBP-C relieved the restriction of torsional amplitude but also decreased the rate of torsional motion. In the case of fast skeletal MyBP-C, its effect on actin dynamics was unchanged by phosphorylation. The isolated C-terminal half of cardiac MyBP-C (C5–C10) had effects similar to those of the full-length protein, and it bound actin more tightly than the N-terminal half (C0–C4), which had smaller effects on actin dynamics that were independent of PKA phosphorylation. We propose that these MyBP-C-induced changes in actin dynamics play a role in the functional effects of MyBP-C on the actin–myosin interaction.
Keywords: spectroscopy, site-directed probes, contractility, hypertrophic cardiomyopathy
Regulation of cardiac muscle contraction is mediated in the sarcomere by finely tuned interactions between myosin and actin, with protein phosphorylation playing a critical role. For example, phosphorylated serine or threonine residues in cardiac isoforms of troponin I or myosin binding protein-C (MyBP-C) can influence force and timing of contraction differently in healthy, hypertrophic, and failing hearts (1–6). Cardiac MyBP-C (cMyBP-C) is composed of 8 Ig (Ig I) and 3 fibronectin (Fn) type III domains (Fig. 1B), and its presence and protein kinase A (PKA) phosphorylation status contribute to the modulation of contraction (7, 8). The cardiac isoform has an Ig domain C0 at its N terminus, where both skeletal isoforms have Pro/Ala-rich linkers instead. Cardiac and slow skeletal MyBP-C contain three to four PKA-phosphorylatable serine/threonine residues near their N termini, whereas fast skeletal MyBP-C contains a single PKA site (9–11) (Fig. 1B).
Fig. 1.

(A) Actin filament twisting motions in the μs time range are detected by TPA. (B) Domain organization of MyBP-C isoforms. Domains listed from N-terminal C0 to C-terminal C10, including the Pro/Ala rich linker (LP/A) and phosphorylation motif (m). (C) Effect of 1 μM fast skeletal (red), 1 μM slow skeletal (blue) or 4 μM cMyBP-C (black) on the TPA decay of ErIA-actin (gray). (D) Dependence of final anisotropy on actin binding.
It has been proposed that (i) unphosphorylated cMyBP-C slows contractile kinetics by binding of its N-terminal region to myosin subfragment-2 (S2), restricting myosin subfragment-1 (S1) access to actin, with its C terminus anchored to the thick filament backbone (12), and (ii) phosphorylation of cMyBP-C relieves the constraint on S2 and facilitates myosin’s interaction with actin (13–16). PKA phosphorylation of cMyBP-C in skinned myocardium accelerates cross-bridge cycling and reduces Ca2+-sensitivity of force, probably by accelerating cross-bridge detachment (15–18). Models of MyBP-C phosphorylation to fine-tune the cardiac contractile machinery via its interaction with myosin have been supported by numerous studies (12–22), but there is also evidence for interaction of cMyBP-C with actin (23–28). Most studies suggest that the C-terminal domains interact with the thick filament backbone, whereas the N-terminal domains interact with S2, the regulatory light chain (RLC), and/or actin.
Rybakova et al. (28) used a cosedimentation assay to characterize the actin-binding properties of recombinant full-length mouse cMyBP-C and its fragments, expressed in insect cells, showing that cMyBP-C interacts with F-actin in a saturable manner via a single moderate-affinity site localized in the C-terminal half of cMyBP-C (domains C5–C10), whereas no saturable binding occurred with N-terminal fragments. If actin binding by monomeric MyBP-C via its C-terminal micromolar-affinity binding site were to occur in intact muscle, this would be surprising, as the same C-terminal domains (C8–C10) (29, 30) appear to lie on the thick filament backbone, based on electron microscopy (12). Remarkably, actin binding is not affected by PKA-mediated phosphorylation of cMyBP-C (28), despite PKA-mediated changes in structural and mechanical properties of myosin cross-bridges in skinned myocardium (15, 16). Thus, further studies by structural, physiological, and biochemical approaches are needed to clarify the interaction of actin with MyBP-C.
In the present study, we have used spectroscopy to further define the effects of MyBP-C binding on actin structural dynamics, exploring the dependence on MyBP-C isoform, phosphorylation state, and truncated domains. We performed time-resolved phosphorescence anisotropy (TPA) on F-actin labeled with a phosphorescent dye at C374 (31) to quantitate the microsecond internal rotational dynamics of actin filaments. This time range is dominated by intrafilament torsional (twisting) motions (Fig. 1A) (31) and coincides with the time range of key mechanical transients in muscle fibers (32) and motions of myosin heads bound to actin in solution (33) and in myofibrils (34, 35). We used pH 8.0 to avoid actin bundling (EM observations and ref. 24), thus focusing on MyBP-C interactions with individual actin filaments. We corrected our results for variations in binding, thus determining directly the structural dynamics of bound complexes. Our results support previously reported biochemical data using the same baculovirus-expressed MyBP-C (28) and provide biophysical insight into the dynamic interaction of MyBP-C with actin, with important implications for regulation of actomyosin.
Results
Binding of MyBP-C to Erythrosin Iodoacetamide-Labeled F-Actin (ErIA-actin).
Binding of MyBP-C and fragments to ErIA-actin was measured under the conditions used previously (28), yielding Bmax and Kd values (Eq. S1) within 10% of the values reported previously for unlabeled actin (28). These values were used to obtain values for the fraction of actin protomers that are bound to MyBP-C, so the TPA effects of different proteins can be compared quantitatively.
Effects of Full-Length MyBP-C and Its Isoforms on TPA of Actin.
At saturation, all three isoforms of MyBP-C (Fig. 1B) dramatically reduced the amplitude (increased the final anisotropy, as defined in Materials and Methods, Eq. 2) of ErIA-actin’s TPA decay (Fig. 1C), indicating substantial restriction of the angular amplitude of actin filament twisting dynamics (Fig. 1A). However, both skeletal isoforms are more effective than the cardiac isoform in increasing actin anisotropy at low occupancy (Fig. 1D). Despite similar affinities for actin (Kd ≈ 4 μM) and stoichiometries of binding (1:1 molar ratio with respect to the actin protomer) (28, 36), the effect of skeletal MyBP-C was near maximal when 15–20% of actin protomers were occupied, whereas the effect of cMyBP-C was near maximal only with >40% occupancy (Fig. 2A). We observed no effect on initial anisotropy, indicating that MyBP-C does not significantly change submicrosecond flexibility within actin protomers. Lifetimes and amplitudes of the actin-bound phosphorescent probe were not significantly affected by any isoform of MyBP-C, indicating that the bound proteins had negligible effects on the local environment of the probe, so TPA effects were caused by changes in actin filament rotational dynamics.
Fig. 2.

(A) Final anisotropy of actin (Fig. 1) as a function of the fraction bound to isoforms of MyBP-C. Effects in the absence (white bars) and presence (cyan bars) of PKA-mediated phosphorylation on the angular amplitudes (B, Eq. 3) and rates (C, Eq. 4) of actin torsional dynamics (Fig. 1A) at saturation. *Significant change in absence of PKA treatment compared to actin alone (P < 0.05). #Significant change due to PKA treatment (P < 0.05).
Effects of Phosphorylation.
We tested whether and how phosphorylation of each MyBP-C isoform changes its effects on actin structural dynamics, under conditions where untreated MyBP-C had maximal effects on the amplitudes (Fig. 2A) and rates (Fig. 2B) of intrafilament motions in actin (Table S1). Phosphorylation was catalyzed by PKA and confirmed by the analysis of Sypro-Ruby and Pro-Q Diamond-stained SDS/PAGE of TPA samples. For fast skeletal MyBP-C, the TPA effects were unchanged by phosphorylation; that is, the extent of restriction of amplitude (Fig. 2A) and increase in rate (Fig. 2B) of actin’s intrafilament motions were essentially the same for phosphorylated (cyan) and unphosphorylated (white) MyBP-C. In contrast, phosphorylation of slow skeletal or cardiac MyBP-C had significant effects on both amplitudes and rates of motions in actin. In the case of amplitudes, phosphorylation relieved restrictive effects of both isoforms, so that the amplitudes in the presence of phosphorylated MyBP-Cs became similar to that of actin alone (Fig. 2A). In contrast, the changes in rates were isoform-specific. In the case of slow skeletal MyBP-C, although its unphosphorylated form increased the rates (Fig. 2A), phosphorylation relieved this effect (Fig. 2B). Unphosphorylated cMyBP-C had no significant effect on rate, but phosphorylation decreased the rates by about 30% (Fig. 2B). Thus, phosphorylation effects on actin dynamics were diverse and isoform-specific, suggesting unique mechanisms of actin regulation.
Effects of N- and C-Terminal Fragments.
We examined the effects of truncated cMyBP-C on TPA of ErIA-actin (Table S1) to relate the above effects to particular domains (Fig. 3A). cMyBP-C was split between domains C4 and C5 to yield truncated constructs C0–C4 and C5–C10. This separated the proposed actin-binding sites (24) and the phosphorylation motif located in the C0–C4 segment from the segment C5–C10 containing the moderate-affinity actin-binding site (Fig. 3A) (28). Alternatively, cMyBP-C was partitioned near its N terminus between domains C1 and the phosphorylation motif into C0–C1 and ΔC0–C1 fragments, such that C0–C1 contains the proposed actin-binding sites in C0 and C1 (37), whereas ΔC0–C1 contains the phosphorylation motif and C-terminal actin-binding site (Fig. 3A) (28). Using the same range of concentrations as studied for full-length cMyBP-C, we found that removal of proposed actin-binding domains C0–C1 (i.e., using ΔC0–C1) and removal of domains C0–C4 (i.e., using C5–C10) had no effect on torsional amplitude compared with full-length MyBP-C on (Fig. 3B). These results suggest that the effect of full-length MyBP-C on amplitudes of actin intrafilament motions is localized primarily in its C-terminal domains C5–C10, which is consistent with the observation that the C6–C10 region is indispensible for the full-length cMyBP-C interaction with actin (28). PKA-mediated effects on torsional amplitude differed in that ΔC0–C1 effects were relieved by PKA treatment, as in the case of the full-length protein, whereas PKA had no effect on C5–C10 to restrict actin amplitude, as expected. This suggests that the motif, but not necessarily C0–C1, is required for PKA effects in regulating actin dynamics. On the other hand, N-terminal fragments of the protein (C0–C1 and C0–C4) did not significantly affect actin rotational amplitude (Fig. 3B).
Fig. 3.

Effect of cMyBP-C fragments (A) and PKA treatment on actin twisting dynamics detected by TPA. Effects in the absence (white bars) and presence (cyan bars) of PKA treatment on the amplitudes (B, Eq. 3) and rates (C, Eq. 4) of actin torsional dynamics at saturation. *Significant change in absence of PKA treatment compared to actin alone (P < 0.05). #Significant change due to PKA treatment (P < 0.05). Full-length cMyBP-C (FL) shown in Fig. 1B.
The effect of PKA phosphorylation to decrease the rate of intrafilament motions required the presence of the phosphorylation motif and C-terminal domains (ΔC0–C1), whereas constructs lacking the motif (C5–C10 and C0–C1) or C-terminal domains (C0–C4) were not significantly affected by PKA treatment with respect to rate (Fig. 3C). The only significant effect of N-terminal fragments was the effect of C0–C4 to increase rate without affecting amplitude (Fig. 3 B and C). Although full-length cMyBP-C had no effect on rotational rate when unphosphorylated, the rate was decreased in both cardiac and slow skeletal MyBP-C upon phosphorylation. C-terminal domains in C5–C10 decreased rate regardless of phosphorylation status (Fig. 3C). Thus, the ability of unphosphorylated full-length cMyBP-C to decrease amplitude, or for phosphorylated cMyBP-C to decrease rate, is probably localized to the C-terminal half, as C5–C10 exhibits both of these characteristics simultaneously, regardless of phosphorylation (Fig. 3 B and C). Of course, the phosphorylation motif is the switch required to regulate these PKA-mediated effects. In contrast, fragments C0–C4 and ΔC0–C1, each containing at least the motif and N-terminal domains C2–C4, increased rate (Fig. 3C). Thus, N-terminal domains generally increased rate and C-terminal domains generally decreased rate.
Electron Microscopy.
We observed no evidence of filament aggregation or bundling in electron micrographs when ErIA-actin was mixed with at least equimolar full-length cMyBP-C or fragments in pH 8.0 MyBP-buffer (i.e., 5 μM ErIA-actin: 5 μM MyBP-C in the presence of 1 mM EGTA at physiological ionic strength), which is consistent with earlier reports (24, 38). We conclude that MyBP-C affects the internal dynamics of actin filaments, not interactions between filaments or interaction of MyBP-C with multiple actin filaments. Earlier studies using electron microscopy and fluorescence microscopy have each demonstrated that the length distribution and average filament length are not affected by ErIA labeling (31).
Discussion
Molecular Implications for Muscle Contraction.
The effects of all three isoforms of MyBP-C on actin filament structural dynamics could have profound physiological and pathological significance. The observed restriction of rotational amplitudes but enhancement of rate, along with an isoform-dependent restoration of flexibility with phosphorylation, may be crucial for optimal function of the myosin–actin interaction in the sarcomere. It has previously been shown that actin’s twisting motions, as detected by TPA, facilitate the weak-to-strong disorder-to-order transition that occurs in both myosin cross-bridges and actin filaments (39). These microsecond rotational motions are essential to actomyosin kinetics: if actin’s microsecond torsional flexibility is too rigid or too floppy, myosin activation and force generation are impeded (40, 41). We hypothesize that MyBP-C has the capacity to regulate actomyosin kinetics by modulating actin torsional dynamics in a phosphorylation-dependent manner. Although unphosphorylated MyBP-C restricts actin torsional amplitude to sample a smaller angular range of myofilament space with faster twisting kinetics, phosphorylated MyBP-C allows actin to sample a greater range of myofilament space with slower motions. We hypothesize that phosphorylation of cMyBP-C enhances the probability of actomyosin interaction and thus accelerates cross-bridge attachment. Such a mechanism would be complementary to an earlier proposal that phosphorylation of MyBP-C relieves a constraint on myosin, thereby freeing it to explore more space and increasing its probability of binding actin (14). Thus, MyBP-C could regulate the kinetics of contraction via effects on protein mobility in both thin and thick filaments.
Whether MyBP-C enhances actin elastic properties or limits the probability of actin–cross-bridge interactions, the influence may be particularly relevant at low levels of Ca2+ activation where few cross-bridge-binding sites on actin are available for force generation. Moreover, our TPA results from the ΔC0–C1-actin complex show that phosphorylation propagates a structural change involving domains C-terminal of the motif, suggesting that cMyBP-C acts as a scaffold that allows for communication between thick and thin filaments.
For all three isoforms, changes in actin anisotropy due to MyBP-C saturate well below stoichiometric amounts (Fig. 1D), indicating that one molecule of MyBP-C influences multiple actin protomers, which is possible given its length with respect to the interfilament distance in the sarcomere (23, 42). Similar ideas have been explored for MyBP-C’s interaction with myosin, where its 1:8 stoichiometry probably influences additional myosin molecules by cooperative means (19, 43). Positive cooperativity occurs in all three isoforms, but only in the cMyBP-C is there a substantial lag, indicating negative cooperativity at very low stoichiometry. This could arise from a difference in N-terminal charge, as cMyBP-C contains the basic domain C0 at the N terminus, where both skeletal isoforms contain a less charged Pro/Ala-rich linker. The additional positive charge at the N terminus of cMyBP-C may interact electrostatically with actin’s negatively charged N terminus, thus competing with C-terminal binding to actin at low MyBP-C concentrations (Fig. 1D).
Our TPA results and actin-binding studies by Rybakova et al. (28) using cMyBP-C fragments, share a common theme: the effects most pronounced and most similar to full-length cMyBP-C were elicited by the C-terminal half. If domains C8–C10 lie along the thick filament backbone (12), it is conceivable that domains C5–C7 (∼12–15 nm) could still reach the thin filament in cardiac muscle (44) (11–13 nm interfilament space). Indeed, SAXS studies of human cMyBP-C fragments show that C5–C7 spans ∼14.5 nm and C5–C6 spans ∼12 nm in solution. Many possible configurations cannot yet be ruled out, as even the C0 domain and linker could span the interfilament spacing (44).
Although C-terminal interactions with actin in solution have major effects on actin dynamics (Fig. 3), the C terminus of MyBP-C binds about 10 times tighter to myosin than to actin (30), and the N terminus has been shown to alter the mechanical function of actin in solution and in the myofibril, according to biochemical and biophysical studies (45, 46). An ancestral protein relative of MyBP-C, myosin binding protein-H (MyBP-H), is a component of muscle and nonmuscle myosin II and is structurally homologous to the C-terminal half of MyBP-C (domains Fn3-IgI-Fn3-IgI) but lacks the upstream regulatory residues. Like MyBP-C, MyBP-H has been shown to be involved with both myosin and actin binding (47, 48). Thus, the effects of domains C5–C10 to bind actin (28) and to restrict actin TPA (Fig. 3 B and C) could be related to ancestral functions of MyBP-H to influence cell motility, for example, by affecting nonmuscle actomyosin bundle contractility at points of cell–cell contact (48).
PKA Phosphorylation Mediates Nondissociative Conformational Changes in the MyBP-C-Actin Assembly.
The addition of a bulky negative charge with serine phosphorylation could directly influence the structure of the MyBP-C or its interactions with actin in their assembly to disrupt effects on torsional amplitude. Phosphorylation effects are due both to direct electrostatic effects and to more complex perturbations of structure and stability (49–52). Thus, our results suggest that phosphorylation causes a structural change that propagates at least from the motif toward the C terminus, without changing binding affinity.
Phospholamban (PLB), which regulates the calcium pump (SERCA), plays a role in the heart that is remarkably analogous to that of MyBP-C. PLB is also a substrate for PKA, and PLB phosphorylation activates SERCA without a change in binding affinity. Phosphorylation of PLB causes a transition from order to dynamic disorder that activates SERCA (53–55), and PKA phosphorylation of cMyBP-C may also induce transitions in dynamic disorder, without dissociation from its myofilament binding partners.
Comparison with Other Actin-Binding Proteins.
Effects on actin rotational dynamics have been previously characterized by TPA for other actin-binding proteins, with a wide range of effects. Whereas tropomyosin slightly restricts actin torsional flexibility (56), cofilin actually increases it with strong cooperativity (57). Although strongly bound myosin S1 allosterically and cooperatively restricts actin rotational dynamics (41), the weakly bound acto-S1 complex exhibits actin dynamics intermediate between the free and strongly bound complex and lacks the cooperative changes propagated along the actin filament by strongly bound S1 (58). The effects on actin dynamics depend on the myosin isoform; muscle myosin II decreases rates of intrafilament torsional motion, and nonmuscle myosin V increases these rates (59). Thus, it is not surprising that different MyBP-C isoforms, domains, and phosphorylation can uniquely influence actin dynamics. Utrophin and dystrophin, which are known to interact with actin in the muscle cytoskeleton (not in the myofibrillar array), have been shown to restrict actin filament microsecond twisting amplitude while increasing the rate (60). This combination of effects, similar to those caused by unphosphorylated MyBP-C, has been proposed to enhance the muscle cytoskeleton’s resilience (61), which is lost in Duchenne’s muscular dystrophy due to defects in dystrophin.
Conclusion
We have used TPA to characterize the structural dynamics of F-actin, to detect changes in actin torsional dynamics as affected by MyBP-C and its phosphorylation. All three isoforms of MyBP-C restrict the amplitude of actin’s microsecond torsional dynamics, which is essential for actomyosin kinetics. In cardiac and slow skeletal isoforms, PKA reverses this effect (increases amplitude) without dissociating the complex. PKA also decreases the rate of actin dynamics. Effects of truncated constructs show that both N- and C-terminal domains are needed for full effects on actin dynamics, including the reversal by PKA. Thus, differential phosphorylation and domains of MyBP-C produce novel states of actin dynamics that are likely to impact myosin crossbridge kinetics, allowing for the tuning of torsional amplitude and rate in response to myocardial inotropy and other physiological factors.
Materials and Methods
Preparation and in vitro phosphorylation of MyBP-C, labeling of actin, actin binding analysis, and electron microscopy are described in SI Text.
TPA Experiments.
Phalloidin-stabilized ErIA-actin was diluted in MyBP-buffer (actin buffer plus 10 μM E-64 inhibitor of cysteine proteases) to 1 μM, and increasing concentrations of unlabeled proteins were added. MyBP-C was stored in MyBP-buffer at –80 °C for up to 6 mo. After vortexing, ErIA-actin was incubated with full-length or truncated MyBP-C for 20 min at 25 °C. To prevent photobleaching of the dye, oxygen was removed using glucose oxidase (58, 62). Phosphorescence was measured at 25 °C. ErIA was excited with a vertically polarized 1.2-ns pulse from a FDSS 532–150 laser (CryLas) at 532 nm, operating at a repetition rate of 100 Hz. Phosphorescence emission was selected by a 670 nm glass cutoff filter (Corion), detected by a photomultiplier (R928, Hamamatsu), and digitized by a transient digitizer (CompuScope 14100, GaGe) at a time resolution of 1 μs/channel. The TPA decay is defined by:
where Iv(t) and Ih(t) are the vertically and horizontally polarized components of the detected emission signal, using a single detector at 90° and a rotating sheet polarizer that alternated between the two orientations every 500 laser pulses, and G is a correction factor (58, 60). TPA experiments were recorded with 30 cycles of 1,000 total pulses (500 each in the horizontal and vertical planes) (58, 60).
TPA Data Analysis.
TPA decays were analyzed by fitting to the sum of two exponential terms with the function (58, 60):
varying rotational correlation times, ϕ1 (slow) and ϕ2 (fast); the corresponding amplitudes, r1 and r2; and the final anisotropy r∞. The initial anisotropy was then calculated as r0 = r(0) = r1 + r2 + r∞. This method of analysis was established previously (41, 58) and was validated by comparing residuals and χ2 for fits with one, two, and three exponential terms, with the best fit consistently requiring two exponential terms. The overall angular amplitude of microsecond rotational motion was defined as the radius of a cone, assuming the wobble-in-a-cone model (58, 60):
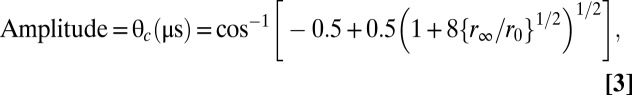 |
Maximal flexibility corresponds to a final anisotropy of r∞ = 0, yielding a cone angle θc = 90°, and maximal rigidity corresponds to r∞ = r0 (i.e., no decay) and θc = 0 (i.e., no detectable rotation).
The mean rate of actin filament torsional motions was defined as the inverse of the mean correlation time:
Each result is reported as mean ± SEM (n > 5), unless indicated otherwise. An unpaired t test was used as a test of significance. P values < 0.05 were taken as indicating significant differences.
Supplementary Material
Acknowledgments
Special thanks to Octavian Cornea for assistance with manuscript preparation and submission. TPA experiments were performed at the Biophysical Spectroscopy Center, University of Minnesota. This study was supported by National Institutes of Health Grants AR032961 and AR057220 Core C (to D.D.T.) and F32 HL107039 (to B.A.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213027109/-/DCSupplemental.
References
- 1.Maughan DW. Kinetics and energetics of the crossbridge cycle. Heart Fail Rev. 2005;10(3):175–185. doi: 10.1007/s10741-005-5248-2. [DOI] [PubMed] [Google Scholar]
- 2.de Tombe PP, Solaro RJ. Integration of cardiac myofilament activity and regulation with pathways signaling hypertrophy and failure. Ann Biomed Eng. 2000;28(8):991–1001. doi: 10.1114/1.1312189. [DOI] [PubMed] [Google Scholar]
- 3.Vandenboom R, Metzger JM. A “wringing” endorsement for myosin phosphorylation in the heart. Mol Interv. 2002;2(7):422–424. doi: 10.1124/mi.2.7.422. [DOI] [PubMed] [Google Scholar]
- 4. Winegrad S (2003) Myosin-binding protein C (MyBP-C) in cardiac muscle and contractility. Adv Exp Med Biol 538(1): 31–40; discussion 40–41. [DOI] [PubMed]
- 5.van Dijk SJ, et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119(11):1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 6.Molenaar P, Chen L, Semmler AB, Parsonage WA, Kaumann AJ. Human heart beta-adrenoceptors: Beta1-adrenoceptor diversification through “affinity states” and polymorphism. Clin Exp Pharmacol Physiol. 2007;34(10):1020–1028. doi: 10.1111/j.1440-1681.2007.04730.x. [DOI] [PubMed] [Google Scholar]
- 7.Winegrad S. Cardiac myosin binding protein C. Circ Res. 1999;84(10):1117–1126. doi: 10.1161/01.res.84.10.1117. [DOI] [PubMed] [Google Scholar]
- 8.Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol. 2010;48(5):866–875. doi: 10.1016/j.yjmcc.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan C, et al. Quantitative comparison of sarcomeric phosphoproteomes of neonatal and adult rat hearts. Am J Physiol Heart Circ Physiol. 2008;295(2):H647–H656. doi: 10.1152/ajpheart.00357.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flashman E, Redwood C, Moolman-Smook J, Watkins H. Cardiac myosin binding protein C: Its role in physiology and disease. Circ Res. 2004;94(10):1279–1289. doi: 10.1161/01.RES.0000127175.21818.C2. [DOI] [PubMed] [Google Scholar]
- 11.Ackermann MA, Kontrogianni-Konstantopoulos A. Myosin binding protein-C slow: An intricate subfamily of proteins. J Biomed Biotechnol. 2010;2010:652065. doi: 10.1155/2010/652065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zoghbi ME, Woodhead JL, Moss RL, Craig R. Three-dimensional structure of vertebrate cardiac muscle myosin filaments. Proc Natl Acad Sci USA. 2008;105(7):2386–2390. doi: 10.1073/pnas.0708912105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weisberg A, Winegrad S. Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle. Proc Natl Acad Sci USA. 1996;93(17):8999–9003. doi: 10.1073/pnas.93.17.8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colson BA, et al. Protein kinase A-mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ Res. 2008;103(3):244–251. doi: 10.1161/CIRCRESAHA.108.178996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stelzer JE, Patel JR, Walker JW, Moss RL. Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ Res. 2007;101(5):503–511. doi: 10.1161/CIRCRESAHA.107.153650. [DOI] [PubMed] [Google Scholar]
- 16. Colson BA, et al. (2012) Myosin binding protein-C phosphorylation is the principal mediator of protein kinase A effects on thick filament structure in myocardium. J Mol Cell Cardiol 53(5):609–16. [DOI] [PMC free article] [PubMed]
- 17.Bardswell SC, et al. Distinct sarcomeric substrates are responsible for protein kinase D-mediated regulation of cardiac myofilament Ca2+ sensitivity and cross-bridge cycling. J Biol Chem. 2010;285(8):5674–5682. doi: 10.1074/jbc.M109.066456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen PP, Patel JR, Rybakova IN, Walker JW, Moss RL. Protein kinase A-induced myofilament desensitization to Ca(2+) as a result of phosphorylation of cardiac myosin-binding protein C. J Gen Physiol. 2010;136(6):615–627. doi: 10.1085/jgp.201010448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moos C, Offer G, Starr R, Bennett P. Interaction of C-protein with myosin, myosin rod and light meromyosin. J Mol Biol. 1975;97(1):1–9. doi: 10.1016/s0022-2836(75)80017-9. [DOI] [PubMed] [Google Scholar]
- 20.Hartzell HC. Effects of phosphorylated and unphosphorylated C-protein on cardiac actomyosin ATPase. J Mol Biol. 1985;186(1):185–195. doi: 10.1016/0022-2836(85)90268-2. [DOI] [PubMed] [Google Scholar]
- 21.Gruen M, Prinz H, Gautel M. cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett. 1999;453(3):254–259. doi: 10.1016/s0014-5793(99)00727-9. [DOI] [PubMed] [Google Scholar]
- 22.Kunst G, et al. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ Res. 2000;86(1):51–58. doi: 10.1161/01.res.86.1.51. [DOI] [PubMed] [Google Scholar]
- 23.Moos C, Mason CM, Besterman JM, Feng IN, Dubin JH. The binding of skeletal muscle C-protein to F-actin, and its relation to the interaction of actin with myosin subfragment-1. J Mol Biol. 1978;124(4):571–586. doi: 10.1016/0022-2836(78)90172-9. [DOI] [PubMed] [Google Scholar]
- 24.Shaffer JF, Kensler RW, Harris SP. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J Biol Chem. 2009;284(18):12318–12327. doi: 10.1074/jbc.M808850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitten AE, Jeffries CM, Harris SP, Trewhella J. Cardiac myosin-binding protein C decorates F-actin: Implications for cardiac function. Proc Natl Acad Sci USA. 2008;105(47):18360–18365. doi: 10.1073/pnas.0808903105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orlova A, Galkin VE, Jeffries CM, Egelman EH, Trewhella J. The N-terminal domains of myosin binding protein C can bind polymorphically to F-actin. J Mol Biol. 2011;412(3):379–386. doi: 10.1016/j.jmb.2011.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ratti J, Rostkova E, Gautel M, Pfuhl M. Structure and interactions of myosin-binding protein C domain C0: Cardiac-specific regulation of myosin at its neck? J Biol Chem. 2011;286(14):12650–12658. doi: 10.1074/jbc.M110.156646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rybakova IN, Greaser ML, Moss RL. Myosin binding protein C interaction with actin: characterization and mapping of the binding site. J Biol Chem. 2011;286(3):2008–2016. doi: 10.1074/jbc.M110.170605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilbert R, Kelly MG, Mikawa T, Fischman DA. The carboxyl terminus of myosin binding protein C (MyBP-C, C-protein) specifies incorporation into the A-band of striated muscle. J Cell Sci. 1996;109(Pt 1):101–111. doi: 10.1242/jcs.109.1.101. [DOI] [PubMed] [Google Scholar]
- 30.Flashman E, Watkins H, Redwood C. Localization of the binding site of the C-terminal domain of cardiac myosin-binding protein-C on the myosin rod. Biochem J. 2007;401(1):97–102. doi: 10.1042/BJ20060500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prochniewicz E, Zhang Q, Howard EC, Thomas DD. Microsecond rotational dynamics of actin: Spectroscopic detection and theoretical simulation. J Mol Biol. 1996;255(3):446–457. doi: 10.1006/jmbi.1996.0037. [DOI] [PubMed] [Google Scholar]
- 32.Brenner B. Rapid dissociation and reassociation of actomyosin cross-bridges during force generation: A newly observed facet of cross-bridge action in muscle. Proc Natl Acad Sci USA. 1991;88(23):10490–10494. doi: 10.1073/pnas.88.23.10490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berger CL, Svensson EC, Thomas DD. Photolysis of a photolabile precursor of ATP (caged ATP) induces microsecond rotational motions of myosin heads bound to actin. Proc Natl Acad Sci USA. 1989;86(22):8753–8757. doi: 10.1073/pnas.86.22.8753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berger CL, Thomas DD. Rotational dynamics of actin-bound myosin heads in active myofibrils. Biochemistry. 1993;32(14):3812–3821. doi: 10.1021/bi00065a038. [DOI] [PubMed] [Google Scholar]
- 35.Berger CL, Thomas DD. Rotational dynamics of actin-bound intermediates of the myosin adenosine triphosphatase cycle in myofibrils. Biophys J. 1994;67(1):250–261. doi: 10.1016/S0006-3495(94)80476-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamamoto K, Moos C. The C-proteins of rabbit red, white, and cardiac muscles. J Biol Chem. 1983;258(13):8395–8401. [PubMed] [Google Scholar]
- 37.Squire JM, Luther PK, Knupp C. Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J Mol Biol. 2003;331(3):713–724. doi: 10.1016/s0022-2836(03)00781-2. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto K. The binding of skeletal muscle C-protein to regulated actin. FEBS Lett. 1986;208(1):123–127. doi: 10.1016/0014-5793(86)81545-9. [DOI] [PubMed] [Google Scholar]
- 39.Thomas DD, Kast D, Korman VL. Site-directed spectroscopic probes of actomyosin structural dynamics. Annu Rev Biophys. 2009;38:347–369. doi: 10.1146/annurev.biophys.35.040405.102118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prochniewicz E, Katayama E, Yanagida T, Thomas DD. Cooperativity in F-actin: Chemical modifications of actin monomers affect the functional interactions of myosin with unmodified monomers in the same actin filament. Biophys J. 1993;65(1):113–123. doi: 10.1016/S0006-3495(93)81057-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prochniewicz E, Thomas DD. Perturbations of functional interactions with myosin induce long-range allosteric and cooperative structural changes in actin. Biochemistry. 1997;36(42):12845–12853. doi: 10.1021/bi971201r. [DOI] [PubMed] [Google Scholar]
- 42.Offer G, Moos C, Starr R. A new protein of the thick filaments of vertebrate skeletal myofibrils. Extractions, purification and characterization. J Mol Biol. 1973;74(4):653–676. doi: 10.1016/0022-2836(73)90055-7. [DOI] [PubMed] [Google Scholar]
- 43.Stelzer JE, Fitzsimons DP, Moss RL. Ablation of myosin-binding protein-C accelerates force development in mouse myocardium. Biophys J. 2006;90(11):4119–4127. doi: 10.1529/biophysj.105.078147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeffries CM, et al. Human cardiac myosin binding protein C: Structural flexibility within an extended modular architecture. J Mol Biol. 2011;414(5):735–748. doi: 10.1016/j.jmb.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 45.Razumova MV, Bezold KL, Tu AY, Regnier M, Harris SP. Contribution of the myosin binding protein C motif to functional effects in permeabilized rat trabeculae. J Gen Physiol. 2008;132(5):575–585. doi: 10.1085/jgp.200810013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weith A, et al. Unique single molecule binding of cardiac myosin binding protein-C to actin and phosphorylation-dependent inhibition of actomyosin motility requires 17 amino acids of the motif domain. J Mol Cell Cardiol. 2012;52(1):219–227. doi: 10.1016/j.yjmcc.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Welikson RE, Fischman DA. The C-terminal IgI domains of myosin-binding proteins C and H (MyBP-C and MyBP-H) are both necessary and sufficient for the intracellular crosslinking of sarcomeric myosin in transfected non-muscle cells. J Cell Sci. 2002;115(Pt 17):3517–3526. doi: 10.1242/jcs.115.17.3517. [DOI] [PubMed] [Google Scholar]
- 48.Hosono Y, et al. MYBPH, a transcriptional target of TTF-1, inhibits ROCK1, and reduces cell motility and metastasis. EMBO J. 2012;31(2):481–493. doi: 10.1038/emboj.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamelberg D, Shen T, McCammon JA. Phosphorylation effects on cis/trans isomerization and the backbone conformation of serine-proline motifs: accelerated molecular dynamics analysis. J Am Chem Soc. 2005;127(6):1969–1974. doi: 10.1021/ja0446707. [DOI] [PubMed] [Google Scholar]
- 50.Groban ES, Narayanan A, Jacobson MP. Conformational changes in protein loops and helices induced by post-translational phosphorylation. PLOS Comput Biol. 2006;2(4):e32. doi: 10.1371/journal.pcbi.0020032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Espinoza-Fonseca LM, Kast D, Thomas DD. Molecular dynamics simulations reveal a disorder-to-order transition on phosphorylation of smooth muscle myosin. Biophys J. 2007;93(6):2083–2090. doi: 10.1529/biophysj.106.095802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Espinoza-Fonseca LM, Kast D, Thomas DD. Thermodynamic and structural basis of phosphorylation-induced disorder-to-order transition in the regulatory light chain of smooth muscle myosin. J Am Chem Soc. 2008;130(37):12208–12209. doi: 10.1021/ja803143g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karim CB, Kirby TL, Zhang Z, Nesmelov Y, Thomas DD. Phospholamban structural dynamics in lipid bilayers probed by a spin label rigidly coupled to the peptide backbone. Proc Natl Acad Sci USA. 2004;101(40):14437–14442. doi: 10.1073/pnas.0402801101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karim CB, Zhang Z, Howard EC, Torgersen KD, Thomas DD. Phosphorylation-dependent conformational switch in spin-labeled phospholamban bound to SERCA. J Mol Biol. 2006;358(4):1032–1040. doi: 10.1016/j.jmb.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 55.Ha KN, et al. Controlling the inhibition of the sarcoplasmic Ca2+-ATPase by tuning phospholamban structural dynamics. J Biol Chem. 2007;282(51):37205–37214. doi: 10.1074/jbc.M704056200. [DOI] [PubMed] [Google Scholar]
- 56.Thomas DD, Seidel JC, Gergely J. Rotational dynamics of spin-labeled F-actin in the sub-millisecond time range. J Mol Biol. 1979;132(3):257–273. doi: 10.1016/0022-2836(79)90259-6. [DOI] [PubMed] [Google Scholar]
- 57.Prochniewicz E, Janson N, Thomas DD, De la Cruz EM. Cofilin increases the torsional flexibility and dynamics of actin filaments. J Mol Biol. 2005;353(5):990–1000. doi: 10.1016/j.jmb.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 58.Prochniewicz E, Walseth TF, Thomas DD. Structural dynamics of actin during active interaction with myosin: Different effects of weakly and strongly bound myosin heads. Biochemistry. 2004;43(33):10642–10652. doi: 10.1021/bi049914e. [DOI] [PubMed] [Google Scholar]
- 59.Prochniewicz E, et al. Myosin isoform determines the conformational dynamics and cooperativity of actin filaments in the strongly bound actomyosin complex. J Mol Biol. 2010;396(3):501–509. doi: 10.1016/j.jmb.2009.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prochniewicz E, Henderson D, Ervasti JM, Thomas DD. Dystrophin and utrophin have distinct effects on the structural dynamics of actin. Proc Natl Acad Sci USA. 2009;106(19):7822–7827. doi: 10.1073/pnas.0812007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin AY, et al. Impacts of dystrophin and utrophin domains on actin structural dynamics: Implications for therapeutic design. J Mol Biol. 2012;420(1-2):87–98. doi: 10.1016/j.jmb.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eads TM, Thomas DD, Austin RH. Microsecond rotational motions of eosin-labeled myosin measured by time-resolved anisotropy of absorption and phosphorescence. J Mol Biol. 1984;179(1):55–81. doi: 10.1016/0022-2836(84)90306-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.