

Abstract
The sudden infant death syndrome (SIDS) causes the sudden death of an apparently healthy infant, which remains unexplained despite a thorough investigation, including the performance of a complete autopsy. The triple risk model for the pathogenesis of SIDS points to the coincidence of a vulnerable infant, a critical developmental period, and an exogenous stressor. Primary electrical diseases of the heart, which may cause lethal arrhythmias as a result of dysfunctioning cardiac ion channels (“cardiac ion channelopathies”) and are not detectable during a standard postmortem examination, may create the vulnerable infant and thus contribute to SIDS. Evidence comes from clinical correlations between the long QT syndrome and SIDS as well as genetic analyses in cohorts of SIDS victims (“molecular autopsy”), which have revealed a large number of mutations in ion channel-related genes linked to inheritable arrhythmogenic syndromes, in particular the long QT syndrome, the short QT syndrome, the Brugada syndrome, and catecholaminergic polymorphic ventricular tachycardia. Combining data from population-based cohort studies, it can be concluded that at least one out of five SIDS victims carries a mutation in a cardiac ion channel-related gene and that the majority of these mutations are of a known malignant phenotype.
1. Sudden Infant Death Syndrome
Unlike other syndromes, the diagnosis of the sudden infant death syndrome (SIDS) is one of exclusion. Another particular feature of the syndrome is that all of its carriers are dead at the time of diagnosis. Not surprisingly, SIDS does not have a clear-cut pathophysiology. A large number of pathophysiological mechanisms have been suggested and investigated, including uncontrolled inflammatory responses, serotonergic abnormalities, and metabolic disorders. Presumably, SIDS is a multifactorial disorder, with several intrinsic and extrinsic factors resulting in or predisposing to its development, as proposed in the “triple risk model.” In the present section, I will present and discuss the various definitions of SIDS, its prevalence, common risk factors, the triple risk model, and some noncardiac genetic predispositions.
1.1. Definition
The sudden and unexplained death of an apparently healthy infant is a tragic event. Typically, the infant is routinely put to sleep and found dead when one of the parents takes a look, which may be the next morning after having put the infant to sleep for the night or only minutes after having put the infant to sleep for a nap in the morning or afternoon. For such death, commonly known as “crib death” or “cot death,” the term “sudden infant death syndrome” may be used, provided that certain definitional conditions are met. The first definition of SIDS was proposed at the Second International Conference on Causes of Sudden Death in Infants, which was held in Seattle in 1969. This definition, which is known as the “Seattle definition,” states that SIDS is “the sudden death of any infant or young child which is unexpected by history and in which a thorough postmortem examination fails to demonstrate an adequate cause of death” [1]. This definition was revised by an expert panel convened by the National Institute of Child Health and Human Development (NICHD) in 1989. This revised definition is known as the “NICHD definition” and states that SIDS is “the sudden death of an infant under one year of age which remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene, and review of the clinical history” [2]. It thus limits the age of the SIDS victim to <1 year and specifies that the thorough examination should include complete autopsy and review of the death scene and clinical history. Of note, in an accompanying statement the expert panel emphasized the necessity for postmortem investigation in the SIDS diagnosis: “cases failing to meet the standards of this definition, including those without a post-mortem investigation, should not be diagnosed as SIDS. Cases that are autopsied and carefully investigated, but which remain unresolved may be designated as ‘undetermined,' ‘unexplained,' or the like” [2].
A further refinement of the definition of SIDS came with the 2004 “San Diego definition” [3]: “SIDS is defined as the sudden unexpected death of an infant <1 year of age, with onset of the fatal episode apparently occurring during sleep, that remains unexplained after a thorough investigation, including performance of a complete autopsy and review of the circumstances of death and the clinical history.” Thus, this definition adds that the death apparently occurred during sleep and that the evaluation of the circumstances of death is included. The San Diego definition is the currently used general definition of SIDS. Apart from this general definition, SIDS categories “IA,” “IB,” “II,” and “Unclassified” were defined for research purposes, where category IA is the most strict and limits the infant's age to more than 21 days and less than 9 months [3]. It is important to note that SIDS remains a diagnosis of exclusion in all definitions and that SIDS is not a syndrome with a single well-defined cause. A comprehensive review of the purposes for and challenges in defining SIDS has recently been provided by Krous [4].
Despite the above attempts in defining SIDS, the definitions and protocols used for diagnosing SIDS have never been fully standardized [4, 5]. This is illustrated by the audit of publications that was undertaken by Byard and Marshall [6]. They reviewed fifty papers dealing with SIDS that were published in 2005. In as many as 29 of these papers (58%), there was either no definition of SIDS or the definition was nonstandard or idiosyncratic. In the remaining 21 (42%) of the papers, one used the original Seattle definition, 15 (30%) used the NICHD definition, and 5 (10%) used the then recently published San Diego definition. As emphasized by Byard and Marshall [6], the evaluation of SIDS research may be severely hampered by the failure to use standard published definitions of SIDS and/or to clearly specify the definition that has been adopted. A further problem comes from the persistent inconsistency in the way pathologists report on sudden death in infants, as demonstrated by surveys among pediatric pathologists in the UK [7, 8].
In clinical practice, SIDS is specified as the cause of death through an International Classification of Diseases (ICDs) code on the death certificate, with its inherent shortcomings [9]. Currently, the Tenth Revision (ICD-10) is in use, with SIDS carrying the code R95, which was 798.0 in the Ninth Revision (ICD-9). A major update is to be realized with the introduction of the separate codes R95.0 (“sudden infant death syndrome with mention of autopsy”) and R95.9 (“sudden infant death syndrome without mention of autopsy”). This update has been approved by the World Health Organization in October 2009, with a suggested implementation date of January 2013.
SIDS is not to be confused with “sudden and unexpected death in infancy” (SUDI), or “sudden unexpected infant death” (SUID), which is a general term referring to all infant deaths that are sudden and unexpected, not just to those that meet the definition of SIDS [4, 10]. Even more than SIDS, SUDI is an umbrella label. As with SIDS, the use of the term SUDI is variable and efforts are undertaken to standardize the definition of SUDI [10–12]. A further term in use is postneonatal mortality (PNM), which simply refers to the death of liveborn infants from 28 through 364 days of age. It has been suggested that, given the variability and inconsistency in the assignment of the cause of infant death within and across countries, that the rate of PNM may be a better indicator of trends in both SIDS and other SUDI for comparative purposes [13].
1.2. Prevalence
The prevalence of SIDS varies considerably among countries, but a common trend is a significant decrease over time in the past decades. In the 1980s the rate of SIDS, expressed as the number of SIDS cases per 1,000 live births, varied around 1.4–1.5 in the United States (Figure 1, orange bars). In the 1990s, a remarkable decrease occurred, after which the SIDS rate stabilized at 0.5–0.6 per 1,000 live births in the 2000s. Nevertheless, SIDS has until today remained a major cause of infant mortality in the United States and other developed countries. Accounting for a total of 2,226 deaths in 2009, which implies that six infants died of SIDS per day, it was the third most important cause of infant mortality in the United States in 2009, only outranked by “congenital malformations, deformations and chromosomal abnormalities” (ICD-10 codes Q00–Q99) and “disorders related to short gestation and low birth weight, not elsewhere classified” (ICD-10 code P07) [14]. It is unlikely that the introduction of the ICD-10 code R95 in reporting SIDS in 2000 contributed to the observed decrease in SIDS rate. If anything, it was to be expected that SIDS would be selected as the underlying cause of death more often under ICD-10 than under ICD-9 [15].
Figure 1.
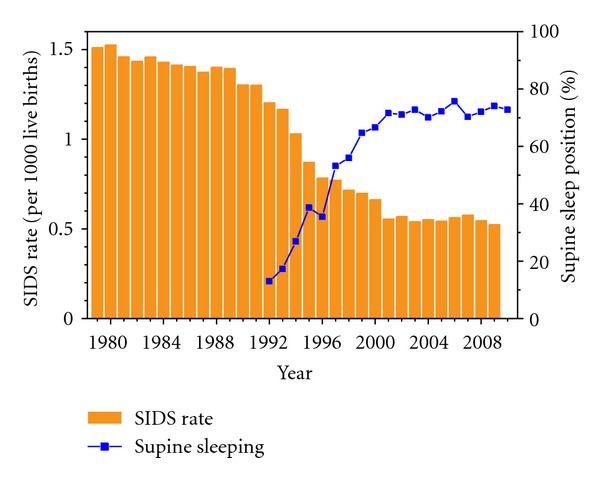
Rate of SIDS (orange bars), expressed as the number of SIDS cases per 1,000 live births, and the percent of infants put to sleep in the supine position (blue line with squares) in the United States over the period from 1979 to 2010. Data on SIDS rate (1979–2009) from the Centers for Disease Control and Prevention (CDC), as provided on the CDC Wide-ranging Online Data for Epidemiologic Research (WONDER) website (http://wonder.cdc.gov/mortSQL.html). For the years 1979–1999, the data refer to all deaths labeled with code 798.0 (“sudden infant death syndrome”) of the Ninth Revision of the International Classification of Diseases (ICD-9) of the World Health Organization. For the years 2000–2009, the data refer to all deaths specified with code R95 (“sudden infant death syndrome”) of the Tenth Revision (ICD-10). Data on supine sleep position (1992–2010) from the National Infant Sleep Position Study (NISP) of the National Institute of Child Health and Human Development (NICHD), as provided on the NISP public access website hosted by Boston University (http://slone-web2.bu.edu/ChimeNisp/Main_Nisp.asp).
The sharp decrease in SIDS rate in the 1990s coincides with a sharp increase in the rate of supine sleeping, as determined from data collected in the National Infant Sleep Position Study (NISP) [16], which stabilized to ≈72% in the 2000s (Figure 1, blue line with squares). This increase is considered a direct effect of the 1992 recommendation of the American Academy of Pediatrics to avoid babies to sleep in the prone position and the subsequent “Back to Sleep” campaign initiated in 1994, although a low adherence to sleep position recommendations among specific demographic groups, particularly among socioeconomically disadvantaged groups, persists [17–21]. The decrease in SIDS rate upon the increase in supine sleeping illustrates the significantly increased risk of SIDS of prone sleeping, as reviewed in several studies [21–24]. However, one should be careful to attribute all of the decrease in SIDS rates to the effects of the “Back to Sleep” campaign, since there is also a declining trend in diagnosing and reporting infant deaths as SIDS [25–27].
A similar or even higher decrease in SIDS rates in the 1990s is observed in other countries than the United States, where campaigns focusing on infants to be placed down for sleep in a nonprone position were initiated around 1990. In England and Wales, for example, where the national risk reduction campaign began in 1991, the SIDS rate dropped from 1.7 per 1,000 live births in 1990 to 0.41 per 1,000 live births in 2000 and stabilized to 0.3–0.4 in the 2000s [13] (Figure 2, blue line with circles). In The Netherlands, where the national risk reduction campaign began in 1987 [13], there was a decrease from 1.1 per 1,000 live births in 1986 to a steady 0.1 in the 2000s (Figure 2, green line with diamonds). This SIDS rate of 0.1 per 1,000 live births is among the lowest worldwide, together with that of Japan [13]. The differences in SIDS rates and PNM rates, between countries, as illustrated in Figure 2, is intriguing and has been discussed in detail elsewhere [13]. An obvious difference between countries is the age of inclusion for SIDS, which may be birth to one year, one week to one year, or three weeks to one year [13]. It is, however, unlikely that this creates large differences in SIDS rates across countries, because the far majority of SIDS cases occur after three weeks of age, as illustrated for the United States in Figure 3. In each of the years selected for Figure 3, >90% of the cases occurred after three weeks of age (Figure 3, blue bars). Although there have been some small shifts in the age of death over the years, the occurrence of SIDS remains rare during the first month of life (cf. Figure 3), increases to a peak between two and three months of age, and then decreases [28], as illustrated by the average age at the time of death of 2.9 ± 1.9 months in the Mayo Clinic cohort of 292 unrelated SIDS cases (see Section 4.2).
Figure 2.
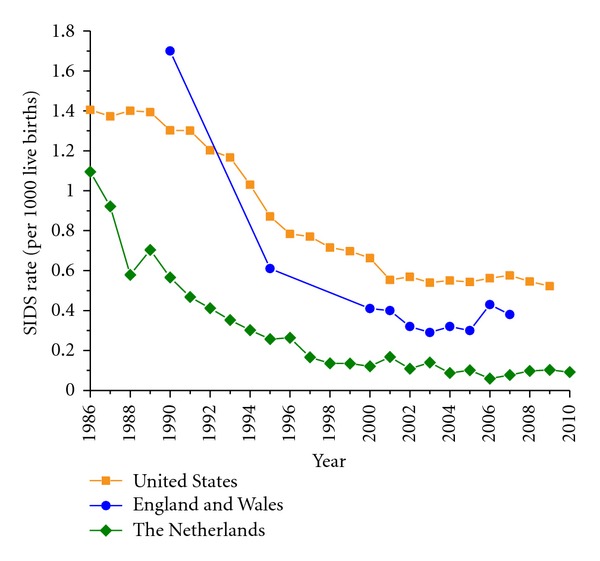
Rate of SIDS, expressed as the number of SIDS cases per 1,000 live births, over the period from 1986 to 2010 in the United States (orange solid line with squares), England and Wales (blue solid line with circles), and The Netherlands (green solid line with diamonds). Data on SIDS rate for the United States as in Figure 1. Data for England and Wales from Hauck and Tanabe [13] (1990–2005), completed with data for 2006 and 2007 from the International Society for the Study and Prevention of Perinatal and Infant Death (ISPID), as provided on the ISPID public access website (http://www.ispid.org/statistics.html). Data for The Netherlands from Statistics Netherlands (Centraal Bureau voor de Statistiek, CBS), as provided on the CBS public access website (http://www.cbs.nl/).
Figure 3.
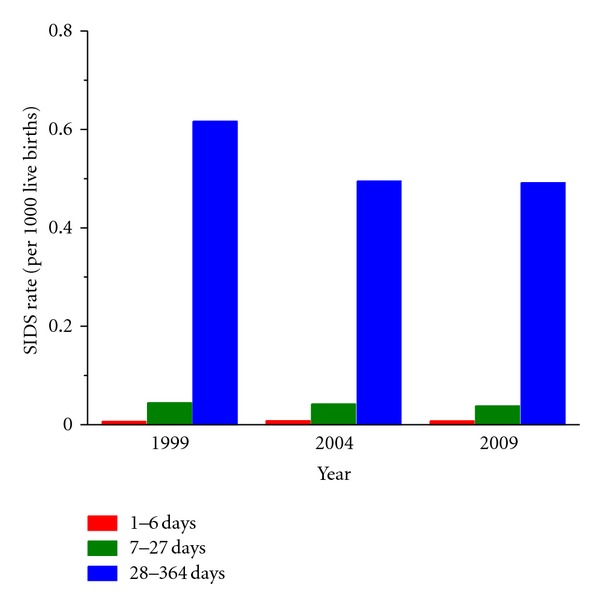
Rate of SIDS, expressed as the number of SIDS cases per 1,000 live births, occurring during week 1 of the infant's life (days 1–6, red bars), during weeks 2–4 (days 7–27, green bars), and during week 5 and later (days 28–364, blue bars) in the United States in 1999, 2004, and 2009. Data from the Compressed Mortality database as provided on the Wide-ranging Online Data for Epidemiologic Research (WONDER) website of the Centers for Disease Control and Prevention (CDC, http://wonder.cdc.gov/mortSQL.html).
1.3. Common Risk Factors
In the previous section, prone sleeping was already addressed as an important, but fortunately avoidable, risk factor for SIDS. Related asphyxia generating risk factors are head covering [29, 30] and bed sharing [31, 32], which emerged as additional prominent risk factors after the “Back to Sleep” campaign had driven back the prone sleeping numbers [33]. Bed sharing is not to be confused with sharing the sleeping room at night with one or more adults, which rather considerably lowers the risk of SIDS [34].
Another important risk factor for SIDS is smoking, both by the mother during pregnancy and in the household after birth [35, 36]. As with bed sharing, the attributed risk associating maternal smoking and SIDS has increased following the “Back to Sleep” campaign [37]. Interestingly, several studies have recently been undertaken in animal models to elucidate the mechanisms by which prenatal exposure to nicotine or postnatal exposure to cigarette smoke may contribute to SIDS [38, 39].
Data on alcohol consumption as a risk factor for SIDS are, perhaps surprisingly, less clear, which may be due to the obscuring effect of its strong correlation with smoking, which makes it difficult to prove any additional independent effect [40]. In the study by Blair et al. [40], the SIDS victim mothers drank slightly more alcohol than controls, but these differences were not significant when adjusted for maternal smoking. McDonnell-Naughton et al. [41], however, recently reported that mothers of SIDS victims consumed significantly more alcohol during pregnancy than control mothers and that, within drinkers, the amount of alcohol consumed was also greater. Alm et al. [42], on the other hand, had found that heavy postnatal but not prenatal intake of alcohol by the mother increased the risk of SIDS.
As with alcohol consumption, data on maternal caffeine consumption throughout pregnancy as a risk factor for SIDS are not unequivocal. Again, there was a strong relation with smoking [42]. Whereas Ford et al. [43] found that, after adjustment for confounders, caffeine consumption of >400 mg/day (equivalent to four or more cups of coffee per day) throughout pregnancy significantly increased the risk of SIDS, such caffeine consumption during or after pregnancy was not found to be an independent risk factor for SIDS after adjustment for confounders by Alm et al. [42].
A further risk factor is found in child care settings [44, 45], with a significantly higher number of SIDS cases occurring in child care than would expected from the time spent there. There are no clear explanations why child care settings are a risk factor for SIDS. The current hypothesis is that changes in the routine infant care and resulting stress play an important role [44, 46]. These might cause sleep deprivation, leading to deeper sleep and impaired arousal [47].
Of note, each of the above risk factors for SIDS, that is, prone sleeping, head covering, bed sharing, smoking, and the consumption of alcohol and caffeine, is avoidable or modifiable. This is unfortunately not the case for all risk factors considered in the “triple risk model.”
1.4. Triple Risk Model
The presumably multifactorial nature of SIDS is reflected in the “triple risk model” that was introduced by Filiano and Kinney [48]. They proposed that SIDS can only occur if a vulnerable infant experiences one or more exogenous stressors during a critical developmental period in homeostatic control. Similar “triple risk hypotheses” had been put forward, as reviewed and discussed by Guntheroth and Spiers [49].
Figure 4(a) illustrates the “triple risk model,” based on the visualization by Courts and Madea [50]. Genetic predisposition and risks during development, such as the aforementioned maternal cigarette smoking, create a vulnerable infant. If environmental triggers occur during a critical developmental period, SIDS may occur, typically between two and three months of age [28]. The “triple risk model” may also be visualized by means of a Venn diagram, as in Figure 4(b), which emphasizes, after Trachtenberg et al. [34], that SIDS tends to occur in the presence of a combination of “intrinsic risk factors” (genetic predisposition, risks during development) and “extrinsic risk factors” during a critical developmental period.
Figure 4.
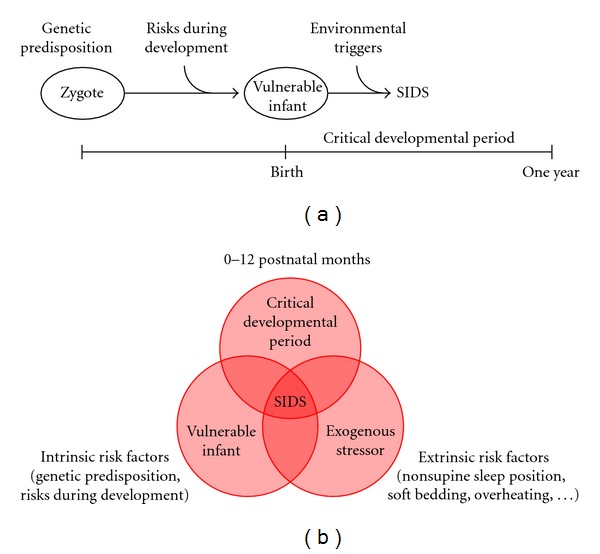
Triple risk model in the sudden infant death syndrome (SIDS). (a) Visualization of the triple risk model after Courts and Madea [50]. Genetic predisposition and risks during development create a vulnerable infant. If this vulnerable infant encounters environmental triggers during a critical developmental period, it may become an SIDS victim. (b) Visualization of the triple risk model after Filiano and Kinney [48] and Trachtenberg et al. [34]. A combination of intrinsic risk factors (genetic predisposition, risks during development) and extrinsic risk factors during a critical developmental period may create an SIDS victim.
In line with the triple risk model, multiple intrinsic and/or extrinsic risk factors are found in the far majority of SIDS cases [34]. Here, an intrinsic risk factor is defined as “a genetic or environmental factor that affects susceptibility, including African American race, male gender, prematurity (<37 gestational weeks at birth), and prenatal maternal smoking or alcohol intake,” whereas an extrinsic risk factor is defined as “a physical stressor around the time of death that may increase the risk of SIDS for an already vulnerable infant,” such as an infection of the upper respiratory tract, which is relatively often observed in SIDS victims [34, 51].
Of note, the triple risk model does not provide a single distinct explanation of SIDS. It rather underscores the multifactorial nature of SIDS, with genetic predispositions, known risk factors in development, and environmental triggers all playing their role. These environmental triggers include viral and bacterial infections [52–54].
1.5. Genetic Predispositions
Over the years, genetic predisposition to SIDS through mutations or (a combination of) polymorphisms has received wide attention. As reviewed by Blackwell et al. [55], several polymorphisms that facilitate uncontrolled inflammatory responses, in particular those resulting in the underproduction of the anti-inflammatory cytokine interleukin-10 (IL-10) or the overexpression of the proinflammatory cytokines IL-1β and IL-6, may be found at a higher proportion in SIDS victims than in controls. A genetic predisposition to elevated vascular endothelial growth factor (VEGF) levels may also play a role in SIDS. An association of SIDS with a VEGF gene polymorphism has been reported [56] and significantly higher VEGF concentrations were found in the cerebrospinal fluid of SIDS victims, pointing to a role for hypoxia in the cascade of events that lead to SIDS [57].
Also, polymorphisms in genes related to the autonomic nervous system have been associated with SIDS. Starting with the study by Narita et al. [58], several studies have demonstrated that SIDS victims are more likely than matched controls to have the long or extra-long allele of the 5HTT serotonin transporter gene, which increases the effectiveness of the promoter and therefore of serotonin re-uptake by the 5-HT serotonin transporter, thus facilitating decreased serotonin concentrations at nerve endings and less effective protective responses to homeostatic challenges during sleep (but see Paterson et al. [59]). The “medullary serotonergic network deficiency hypothesis” and “brainstem hypothesis” have been reviewed elsewhere [60–62], with a particular emphasis on the role of abnormalities in the brainstem serotonin receptor binding, which is also a subject of one of the aforementioned animal studies on SIDS [38].
Further genetic factors related to the autonomic nervous system have been identified in SIDS research. Weese-Mayer et al. [63] analyzed several genes pertinent to early embryologic development of the autonomic nervous system. They identified rare protein-changing polymorphisms in association with SIDS in five genes (PHOX2a, RET, ECE1, TLX3, and EN1). After investigating left ventricular and blood samples from nine SIDS cases, Livolsi et al. [64] reported cholinergic abnormalities in the intracardiac part of the autonomic nervous system. Compared with controls, SIDS cases showed an increase in both the density of cardiac muscarinic receptors and the erythrocyte acetylcholinesterase enzyme activity. A subsequent study demonstrated that the cardiac muscarinic receptor overexpression plays a critical role in the development of vagal hyperreactivity, whereas the acetylcholinesterase hyperactivity appears as a compensatory consequence of it [65].
The above list of genetic factors associated with SIDS is far from complete and, for example, lacks any reference to polymorphisms related to glucose homeostasis that have been associated with SIDS [66–68]. However, it is my aim to provide a comprehensive overview of the genetic predisposition to SIDS that may result from mutations in cardiac ion channel-related genes. This overview will be given in subsequent sections, starting with the clinical associations that emerged in the 1970s.
2. Clinical Associations
In the 1970s and 1980s, possible associations of SIDS with cardiac disorders received a growing attention and were addressed in several studies, albeit with unequivocal results. It took until 1998 for conclusive data on the association of SIDS with a prolonged QT interval on the electrocardiogram (ECG), obtained in a 19-year prospective study, to become available. In the present section, the quest for clinical associations of SIDS with cardiac disorders is reviewed.
2.1. Cardiac Conduction Disorders and SIDS
Already in 1966, Fraser and Froggatt [69] suggested, in a “Letter to the Editor,” that “a proportion, probably small, of sudden unexplained deaths in infancy are due to genetically determined disorders of cardiac conduction.” Evidence for a role for cardiac conduction disorders in SIDS was later provided by Keeton et al. [70], who reported on six cases of “near-miss” sudden infant deaths in which severe conduction disorders where diagnosed and treated just before they became fatal. One may only speculate that in some of these cases the cardiac conduction disorder was genetically determined. In particular, mutations in the SCN5A gene, which encodes the pore-forming α subunit of the cardiac fast sodium channel, may result in the inhibition of the fast sodium current (I Na) and thus underlie conduction disorders. Such genetically determined cardiac conduction disorder (CCD), also designated “cardiac conduction defect” and “cardiac conduction disease,” has been reviewed in detail elsewhere [71–73].
2.2. Imbalance of Autonomic Tone and SIDS
In 1976, Schwartz [74] hypothesized that sympathetic imbalance may underlie SIDS. In particular, impaired right sympathetic activity would create such imbalance and thereby increase the likelihood of ventricular fibrillation. A clinical symptom might be QT interval prolongation on the ECG [74]. An imbalance of autonomic tone was also hypothesized by Montague et al. [75]. However, their data suggested an increase in sympathetic tone in infants at risk for SIDS. These data were obtained in a comparison of 17 infants at risk for SIDS and 17 age- and sex-matched control subjects. Their at-risk group consisted of 11 infants admitted to hospital for the investigation of unexplained apnea and six other infants, of whom five were subsequent siblings of SIDS patients and one was a sibling of a patient with near-miss SIDS. Significant cardiac dysrhythm and QT prolongation were not found in the at-risk infants. Rather, the rate-corrected QT interval (QTc interval) was consistently shorter in the at-risk group, indicative of increased sympathetic or decreased parasympathetic tone, of which an increase in sympathetic tone was considered the most likely [75].
Further evidence for a role of a functional abnormality in the autonomic nervous system came from the study by Haddad et al. [76], who measured the QT interval in 7 near-miss SIDS victims and 12 control infants. Again, in contrast with the hypothesis of Schwartz [74], no QT prolongation was observed. The QT interval was even significantly smaller in the infants with aborted SIDS than in the control infants in both rapid eye movement (REM) and quiet sleep. These observations, together with data on heart rate and heart rate variability during sleep obtained at monthly intervals in 18 control infants and 12 infants with aborted SIDS during the first four months of life, led Leistner et al. [77] to conclude that the infants with aborted SIDS had an increase in sympathetic activity or in circulating levels of catecholamines.
2.3. Long QT Syndrome and SIDS
The long QT syndrome (LQTS) is characterized by QT prolongation on the ECG and a predisposition to syncope, seizures, and sudden cardiac death, caused by episodic polymorphic ventricular tachyarrhythmias such as Torsade de Pointes. These and other (clinical) features of LQTS have been reviewed in detail elsewhere [78, 79]. Especially in young people, LQTS is an important cause of sudden death. Of note, all features of LQTS, including a negative postmortem examination, are compatible with SIDS, which makes LQTS a likely cause of SIDS.
2.3.1. Unequivocal Observations in the 1970s and 1980s
In 1976, Maron et al. [80] proposed inheritable LQTS as a possible cause of SIDS. They obtained electrocardiographic data from 42 sets of parents who had at least one infant with SIDS and found that a considerable proportion of first-degree relatives of infants with SIDS had a prolongation of the QT interval on the ECG. In 27 sets of parents, the ECG was normal for both parents, as illustrated in Figure 5. However, at least one of the parents showed an abnormal ECG in the remaining 15 sets. This included QT prolongation in 11 of these sets, which is in 26% of the 42 sets studied (Figure 5). To investigate the possible genetic transmission of the prolonged QT interval, ECGs were taken from 23 siblings of infants with SIDS. All of these siblings were members of eight families in which at least one of the parents had a prolonged QT interval. The ECGs showed the prolongation of the QT interval in 9 of the 23 siblings (39%), whereas none of the 18 siblings of infants with SIDS from seven families in which neither parent had a prolonged QT interval showed QT interval prolongation. As a further evidence for an association between SIDS and a prolonged QT interval, Maron et al. [80] also described the case of an infant with near-miss SIDS, who survived a cardiorespiratory arrest at seven weeks of age and showed marked prolongation of the QT interval. However, her parents did not show prolongation of the QT interval, nor did 16 other family members except a 10-month-old nephew with a mildly prolonged QT interval. Tragically, having been in apparent excellent health for twelve years, she still died unexpectedly in her sleep, suggesting “the possibility that “near-miss” episodes in infancy may infer a long-term risk that has not been appreciated” [81]. Of note, Maron et al. [80] emphasized that their results were not definitive and that the confirmation of a relation between QT interval prolongation and SIDS would require large prospective investigations.
Figure 5.
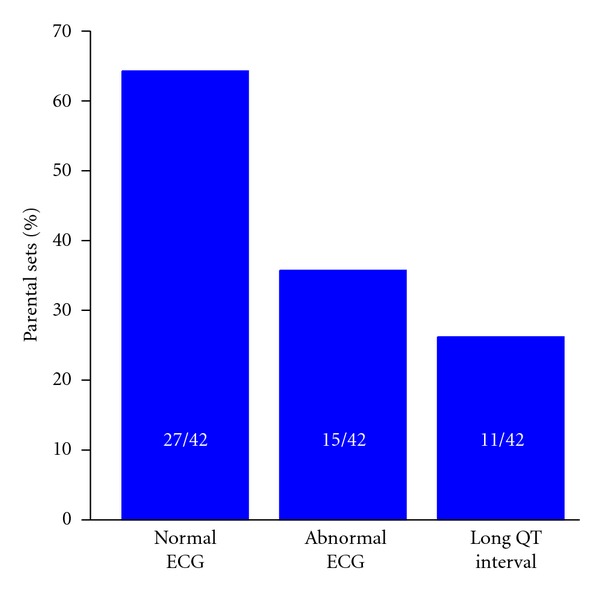
Electrocardiographic data in 42 sets of parents who had infants with SIDS [80]. In 27 sets of parents, both parents had a normal ECG (left bar). In 15 sets (36%, middle bar), at least one parent had an abnormal ECG (middle bar), including a long QT interval in 11 sets (26%, right bar).
Although the results of Maron et al. [80] strongly suggested a role for QT interval prolongation in SIDS, one should realize that no such data were obtained in several other studies, including some discussed in the previous section. In 1973, Froggatt and James [82] already postulated a role for “aberrant ventricular repolarization expressed through a prolonged QT interval” in SIDS, but such a role could not be established from their data collected from a total of 162 SIDS cases over the years. The families were visited in 148 cases and the ECGs obtained from 238 of possible 296 parents. These ECGs revealed no significant arrhythmias and the number with a prolonged QT interval was not enhanced as compared to what would be expected from natural variation. Also, none of the parents had a history of pertinent syncopal attacks. In 1977, Kelly et al. [83] reported the results of their review of the postresuscitation ECGs obtained from 21 near-miss SIDS infants to evaluate the role of QT interval prolongation in the genesis of SIDS. The QT intervals were not significantly different from those of age- and sex-matched controls. Also in 1977, Kukolich et al. [84] reported that ECG studies in a total of 108 first-degree relatives of 26 SIDS victims in comparison with 99 such subjects from 22 control families failed to show any significant differences in the QT interval between these two groups. In 1978, Steinschneider [85] reported data on the QTc interval of 30 neonates whose sibling had died of SIDS in comparison to that of 75 control neonates. The QTc interval of siblings of SIDS victims did not differ from that of the control infants. Also, data were available on the QTc interval of 52 parents of an SIDS victim. No prolongation of the QT interval was apparent in this group. In a subsequent study, Weinstein and Steinschneider [86] found that none of the eight infants who subsequently died of SIDS had a prolonged QTc interval. However, the RR interval during REM sleep was significantly shorter in these future SIDS victims than in control subjects, as in the aforementioned study by Leistner et al. [77] on infants with aborted SIDS. Finally, Southall et al. [87] recorded standard ECGs from a total of 7,254 newly born infants from two maternity hospitals. Fifteen of these infants subsequently suffered SIDS (corresponding to an SIDS rate of 2.1 per 1,000 live births in this cohort). However, no significant differences could be identified in their QTc intervals compared to controls in age matched-groups or after individual matching for postnatal age, hospital of birth, and weight at birth. This was in line with the results of a previous study by Southall et al. [88] on 1,157 preterm and/or low-birth-weight infants within one week prior to discharge from eight neonatal intensive care units. Five of these infants became SIDS victims (corresponding to an SIDS rate of 4.0 per 1,000 live births in this at-risk cohort). Southall et al. [88] made 24-h recordings of beating movement and ECG and found that a proportion of apparently well infants had prolonged apnea, extreme bradycardia, or cardiac arrhythmias. However, none of these disorders was predictive of the five subsequent cases of SIDS. A similar, one-year-later study [89] included 6,914 full-term and 2,337 preterm and/or low-birth-weight infants, of whom 29 (17 full term, 12 preterm) subsequently suffered SIDS (corresponding to SIDS rates of 2.5 and 5.1 per 1,000 live births in the full-term and preterm cohorts, resp.). Again, no abnormal prolongation of the QT interval was observed.
Thus, the initial study by Maron et al. [80] was followed by a series of negative data on an association between LQTS and SIDS [82–89]. At the same time, some supporting evidence was also obtained. In 1979, Southall et al. [90] reported a case of a neonate who had suffered from arrhythmias in utero and bradycardia for the first 9 days of life. A normal heart rate was documented at 10 days and he was discharged from hospital, but died suddenly and unexpectedly three days later, the negative postmortem examination classifying him as an SIDS victim. A retrospective analysis of the perinatal ECG revealed a substantially prolonged QTc interval. A second neonate had also suffered from arrhythmias in utero. She showed a prolonged QTc interval and frequent premature ventricular beats on a 24-hour ECG, but she was successfully treated with the β-blocker propranolol. In 1982, a prospective study by Schwartz et al. [91] was published. This study was specifically designed to test the “QT hypothesis” for SIDS. Standard ECGs were recorded from 4,205 newborns on the fourth day of life and for some at later stages as well. Of the 2,000 infants checked at one year, three had become SIDS victims (corresponding to an SIDS rate of 1.5 per 1,000 live births in this cohort). One had a clearly prolonged QTc interval of 563 ms on the ECG recorded on the fourth day of life, whereas the other two also showed signs of a prolonged QTc interval. Sadeh et al. [92] studied the dependence of the QT interval on the preceding RR interval in 10 infants with SIDS and 29 healthy control infants, analyzing ≈5,000 pairs of QT and RR intervals in each subject over a wide range of RR intervals. They found an impaired dependence, that is, an impaired ability to shorten the QT interval as the heart rate increased, in 5 of 10 infants who subsequently died of SIDS. There are, however, some serious methodological shortcomings in the study by Sadeh et al. [92], as set out by Guntheroth [93] in a critical review of the possibility that SIDS is due to a primary cardiac disorder, in which he concluded—at the end of the 1980s—that “there is no statistical basis, with sound and reproducible methods, to support a cardiac theory for the cause of sudden infant death syndrome.”
2.3.2. Conclusive Data in the 1990s
As set out in the previous section, numerous studies carried out in the 1970s and 1980s addressed the issue of a relationship between LQTS and SIDS. Unfortunately, the data that were available by the end of the 1980s were far from conclusive, as illustrated by the aforementioned critical review by Guntheroth [93]. This changed with the publication in 1998 of a hallmark paper by Schwartz et al. [94], in which they reported results from a 19-year electrocardiographic assessment of Italian neonates at day three or four of life. Of the 34,442 infants enrolled, one-year follow-up data were available for 33,034. In this cohort, 24 infants died of SIDS (corresponding to an SIDS rate of 0.73 per 1,000 live births in this cohort). The mean QTc interval of the SIDS victims was significantly longer than that of infants who survived or died of other causes. More importantly, in 12 of them the QTc interval was considered to be prolonged (>440 ms), whereas it was not in any of the 10 infants who died of other causes. It was found that the odds ratio for SIDS in infants with a prolonged QTc interval in the first week of life was as high as 41.3. It remains to be elucidated why no indications for such strong association had been obtained in earlier prospective studies [87–89].
In Italy, as a followup of the study by Schwartz et al. [94], a large prospective ECG study in a population of as many as 44,596 neonates, in whom an ECG was recorded between the 15th and the 25th day of life to identify infants with LQTS and thus at risk for SIDS, was recently completed [95]. In 28 of the 29 infants with marked QT interval prolongation (QTc interval >470 ms), a molecular screening for mutations in known “LQTS genes” (see Section 3.1 below), was performed. This screening revealed LQTS mutations in 12 neonates. Another four mutation carriers were identified upon genetic analysis of 14 of the 28 neonates with a QTc interval between 461 and 470 ms. All but one, because of parental refusal, of the 29 neonates with a QTc interval >470 ms were successfully treated with a β-blocker (propranolol). It is tempting to speculate that this treatment prevented the occurrence of SIDS in this group.
3. Primary Electrical Cardiac Diseases
As we know now, LQTS is an inheritable disorder, caused by mutations in genes predominantly encoding subunits of cardiac ion channels [96, 97]. If, for example, such mutation causes a “loss of function” of potassium channels that carry an outward current that contributes to the repolarization of the cardiac action potential, a delay in repolarization and prolongation of the QT interval results. LQTS is considered a “primary electrical cardiac disease,” with arrhythmogenic events generated at the cellular or multicellular level due to disturbed ion channels. As such, LQTS and other “ion channelopathies” may well underlie cases of SIDS, because they are not detectable during a standard postmortem examination. This does not only hold for LQTS, but also for other primary electrical cardiac diseases, in particular the short QT syndrome (SQTS), the Brugada syndrome (BrS), and cathecholaminergic polymorphic ventricular tachycardia (CPVT), which share several clinical features with LQTS, such as a predisposition to sudden cardiac death. With their primary electrical nature, they should be taken into account in relation to SIDS.
3.1. Long QT Syndrome
At the cellular level, LQTS is characterized by an increase in action potential duration, which may result from an increase in inward current during the plateau phase of the action potential, for example, due to an increase in the late component of the fast sodium current (I Na; “gain of function”), or a decrease in outward current, for example, due to a decrease in the rapid or slow delayed rectifier potassium current (I Kr and I Ks, resp.; “loss of function”), as illustrated in Figure 6(a). Table 1 provides an overview of the mutations in ion channel-related genes that have been associated with LQTS. For example, mutations in KCNH2 and KCNE2, encoding the α and β subunits of the I Kr channel and classified as LQTS types 2 (LQT2) and 6 (LQT6), respectively, may both result in loss of function of the I Kr channel, thereby prolonging the action potential and the QT interval. LQTS-related mutations have not only been found in α and β subunits of ion channels, but also in genes encoding anchoring and scaffolding proteins that affect the function of ion channels. This is the case in the (rare) LQTS types 4, 9, 11, and 12 (Table 1).
Figure 6.
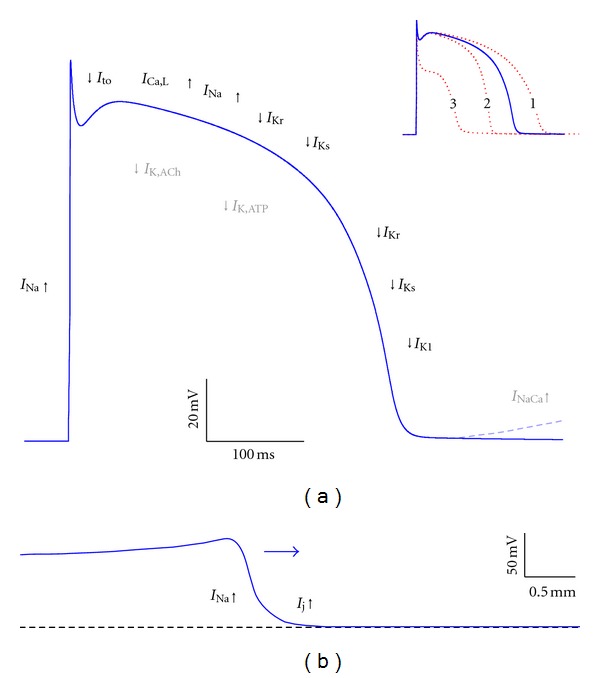
Action potential formation and propagation in ventricular myocytes. (a) Diagram of an action potential in an isolated ventricular myocyte with individual ion currents that “push up” (inward currents, upward arrows) or “pull down” (outward currents, downward arrows) the action potential. I Ca,L: L-type calcium current; I K,ACh: acetylcholine-sensitive potassium current; I K,ATP: ATP-sensitive potassium current; I K1: inward rectifier potassium current; I Kr: rapid delayed rectifier potassium current; I Ks: slow delayed rectifier potassium current; I Na: fast sodium current; I NaCa: sodium-calcium exchange current; I to: transient outward potassium current. Grey symbols refer to currents that become important under special conditions (release of ACh, shortage of ATP and calcium overload for I K,ACh, I K,ATP, and I NaCa, resp.). Loss of function of “pull down” currents may result in action potential lengthening (inset, red dashed trace labeled “1”), whereas loss of function of “push up” currents may result in action potential shortening (inset, red dashed trace labeled “2”) and loss of the action potential dome (inset, red dashed trace labeled “3”). (b) Diagram of action potential propagation. The propagating action potential (horizontal arrow) brings neighboring cells to threshold through the I Na driven gap junctional current I j.
Table 1.
Genes linked to long QT syndrome.
| Type | OMIM | Gene | Protein | Functional role in cardiomyocytes | Effect of mutation |
|---|---|---|---|---|---|
| Autosomal dominant inheritance | |||||
|
| |||||
| LQT1 | #192500 | KCNQ1 (KVLQT1) | Kv7.1 (KvLQT1) | α subunit of I Ks channel | Loss of function of I Ks |
| LQT2 | #613688 | KCNH2 (HERG) | Kv11.1 | α subunit of I Kr channel | Loss of function of I Kr |
| LQT3 | #603830 | SCN5A | Nav1.5 | α subunit of I Na channel | Gain of function of I Na |
| LQT4 | #600919 | ANK2 | Ankyrin-B | Anchoring protein | Loss of function of multiple ion channels and transporters |
| LQT5 | #613695 | KCNE1 | KCNE1 (minK) | β subunit of I Ks channel | Loss of function of I Ks |
| LQT6 | #613693 | KCNE2 | KCNE2 (MiRP1) | β subunit of I Kr channel | Loss of function of I Kr |
| LQT7‡ | #170390 | KCNJ2 | Kir2.1 | α subunit of I K1 channel | Loss of function of I K1 |
| LQT8† | #601005 | CACNA1C | Cav1.2 | α 1C subunit of I Ca,L channel | Gain of function of I Ca,L |
| LQT9 | #611818 | CAV3 | Caveolin-3 | Caveolar coating | Gain of function of I Na |
| LQT10 | #611819 | SCN4B | Nav β4 | β subunit of I Na channel | Gain of function of I Na |
| LQT11 | #611820 | AKAP9 | Yotiao | Anchoring protein for PKA regulatory subunit | Loss of function of I Ks |
| LQT12 | #612955 | SNTA1 | α 1-syntrophin | Scaffolding protein | Gain of function of I Na |
| LQT13 | #613485 | KCNJ5 | Kir3.4 | α subunit of I K,ACh channel | Loss of function of I K,ACh |
|
| |||||
| Autosomal recessive inheritance | |||||
|
| |||||
| JLN1 | #220400 | KCNQ1 (KVLQT1) | Kv7.1 (KvLQT1) | α subunit of I Ks channel | Loss of function of I Ks |
| JLN2 | #612347 | KCNE1 | KCNE1 (minK) | β subunit of I Ks channel | Loss of function of I Ks |
OMIM: Online Mendelian Inheritance in Man compendium of human genes and genetic phenotypes; LQT1– LQT13: long QT syndrome types 1–13; JLN1 and JLN2: Jervell and Lange-Nielsen syndrome types 1 and 2; I Ca,L: L-type calcium current; I K,ACh: acetylcholine-sensitive potassium current; I K1: inward rectifier potassium current; I Kr: rapid delayed rectifier potassium current; I Ks: slow delayed rectifier potassium current; I Na: fast sodium current.
‡Also known as Andersen's syndrome or Andersen-Tawil syndrome
†Also known as Timothy syndrome.
Inheritance of LQTS is autosomal dominant except for the rare autosomal recessive variant that is known as the Jervell and Lange-Nielsen syndrome [98, 99]. In this syndrome, autosomal recessive mutations in KCNQ1 or KCNE1, encoding the α and β subunits of the I Ks channel, respectively, cause severe QT prolongation [100, 101]. The syndrome is also associated with inner ear deafness, which is explained by the critical role of I Ks channels in the secretion of potassium ions into the endolymph [102]. Of note, other rare types of LQTS, in particular LQTS types 7 and 8 (LQT7 and LQT8), are also associated with extracardiac symptoms. In LQT7, which is also known as the Andersen-Tawil syndrome [103, 104], mutations in the KCNJ2 gene result in disturbed Kir2.1 channels, which are expressed in multiple organs. LQT7 is not only characterized by (mild) QT prolongation and other ECG abnormalities, but also by periodic paralysis and dysmorphologies [105–107]. In LQT8, which is also known as the Timothy syndrome [108, 109], mutations in the CACNA1C gene result in disturbed Cav1.2 channels [110]. Expression of these calcium channels is not limited to the heart. As a result, LQT8 is not only characterized by QT prolongation and other cardiac symptoms, but also by syndactyly of fingers and toes, intermittent hypoglycemia, immune deficiency, cognitive abnormalities, and autism [108, 111].
The aforementioned recently completed study by Schwartz et al. [95] revealed 29 infants with marked QT interval prolongation (QTc interval >470 ms). In 28 of these 29 infants, a molecular screening for mutations in any of the LQTS genes KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, CAV3, and SCN4B, associated with the LQTS types 1–3, 5, 6, 9, and 10, respectively (Table 1), was performed. LQTS mutations were identified in 12 neonates. Another four mutation carriers were identified upon genetic analysis of 14 of the 28 neonates with a QTc interval between 461 and 470 ms. From the number of 17 of 43,080 white infants affected by LQTS—16 with disease-causing mutations and one with a clear-cut clinical diagnosis—one arrives at an LQTS prevalence of ≈1 : 2,500 apparently healthy live births [95]. Given that infants with a QTc interval >450 ms were not molecularly screened and that the same holds for silent mutation carriers, that is, infants who carry a disease-causing mutation but showed a normal QT interval, the actual prevalence of LQTS may be around 1 : 2,000 [97]. However, the prevalence of LQTS remains a matter of debate, with on the one hand data suggesting an underestimation [112] and on the other hand data suggesting an overestimation [113].
In the far majority of clinically definite LQTS cases mutations in LQTS genes are found, with KCNQ1 (LQT1),KCNH2 (LQT2), andSCN5A (LQT3) as the most common LQTS genes, accounting for ≈90% of all genotype-positive cases [97, 114]. Gain-of-function mutations in the cardiac sodium channel gene SCN5A cause LQTS, due to a persistent inward sodium current during the plateau phase of the action potential, which is also known as “late” or “sustained” sodium current (Figure 6(a)). Although defects in SCN5A account for only ≈10% of LQTS cases [115, 116], this type (LQT3) plays a rather important role in the etiology of LQTS. It appears that patients with LQT3 have significantly more severe clinical events than patients with LQT1 or LQT2, as the overall number of cardiac deaths is similar for these subgroups while the frequency of events is lower in LQT3 [117]. More specifically, severe symptoms are common in children with LQT3 [118]. This discrepancy between prevalence and severity should be taken into account when considering mutations in LQTS genes in relation to SIDS.
3.2. Brugada Syndrome
Mutations in cardiac ion channel genes may also lead to the Brugada syndrome (BrS) [119–122], which shares several clinical features with LQTS, such as a predisposition to sudden cardiac death. In BrS patients sudden death often occurs during rest or while sleeping, which makes BrS a likely cause for SIDS. BrS is characterized by changes in the ST segment of the ECG rather than QT prolongation, which may result from transmural dispersion in action potential duration, in particular in the right ventricle, due to an early repolarization in the epicardial cell layers. This has been explained on a cellular basis by the loss of the action potential dome in cells with a large transient outward potassium current, like the right ventricular epicardial cells, due to a decrease in the peak component of I Na as a result of a loss-of-function mutation in SCN5A [123]. It should, however, be noted that this explanation of BrS as a repolarization disease is a matter of debate and that it may also, or at the same time, be a depolarization disease [124, 125]. The exact pathophysiological mechanism remains elusive and may involve structural abnormalities that are not easily detectable [126], but as far as ion channel disturbances are involved, these have as a common functional effect that they decrease the net inward current during the rapid depolarization phase of the action potential and/or increase the net outward current during the subsequent early repolarization phase (Table 2, Figure 6(a)). Several genes have been linked to BrS, but in some the associated mutations are not universally considered pathogenic, as reflected in literature where different genes can be found in relation to BrS types 8 and higher.
Table 2.
Genes linked to Brugada syndrome.
| Type | OMIM | Gene | Protein | Functional role in cardiomyocytes | Effect of mutation |
|---|---|---|---|---|---|
| Autosomal dominant inheritance | |||||
|
| |||||
| BrS1 | #601144 | SCN5A | Nav1.5 | α subunit of I Na channel | Loss of function of I Na |
| BrS2 | #611777 | GPD1-L | G3PD1L | Not fully established | Loss of function of I Na |
| BrS3 | #611875 | CACNA1C | Cav1.2 | α 1C subunit of I Ca,L channel | Loss of function of I Ca,L |
| BrS4 | #611876 | CACNB2b | Cav β2b | β 2b subunit of I Ca,L channel | Loss of function of I Ca,L |
| BrS5 | #612838 | SCN1B | Nav β1 | β subunit of I Na channel | Loss of function of I Na |
| BrS6 | #613119 | KCNE3 | KCNE3 (MiRP2) | β subunit of voltage-dependent K+ channels | Gain of function of I to |
| BrS7 | #613120 | SCN3B | Nav β3 | β subunit of I Na channel | Loss of function of I Na |
| NC | #613123 | HCN4 | HCN4 | α subunit of I f channel | Gain of function of I f |
| NC | — | CACNA2D1 | Cav α 2 δ-1 | α 2 δ subunit of I Ca,L channel | Loss of function of I Ca,L |
| NC | — | MOG1 | MOG1 | Regulates trafficking of Nav1.5 to the membrane | Loss of function of I Na |
| NC | — | KCND3 | Kv4.3 | α subunit of I to channel | Gain of function of I to |
| NC | — | KCNE1L (KCNE5) | KCNE1L | β subunit of voltage-dependent K+ channels | Gain of function of I to |
| NC | — | KCNJ8 | Kir6.1 | α subunit of I K,ATP channel | Gain of function of I K,ATP |
| NC | — | SCN1Bb | Nav β1B | β subunit of I Na channel | Loss of function of I Na and gain of function of I to |
OMIM: Online Mendelian Inheritance in Man compendium of human genes and genetic phenotypes; BrS1–BrS7: Brugada syndrome types 1–7; NC: no consensus; I Ca,L: L-type calcium current; I f: hyperpolarization-activated current; I K,ATP: ATP-sensitive potassium current; I Na: fast sodium current; I to: transient outward potassium current.
The prevalence of BrS is not well established, but may be somewhere between 1 : 2,000 and 1 : 5,000, with a higher prevalence in South-East Asia [127]. In contrast with LQTS, where ion channel-related mutations are identified in the far majority of clinically definite cases, BrS can be less well genotyped. Crotti et al. [128] recently assessed the presence of putative pathogenic mutations in each of the BrS-related genes SCN5A, GPD1-L, CACNA1C, CACNB2b, SCN1B, KCNE3, SCN3B, HCN4, CACNA2D1, MOG1, KCND3, KCNJ8, and SCN1Bb in a cohort of 129 unrelated patients referred for BrS genetic testing. These are all of the 14 genes listed in Table 2 except KCNE1L. Variants of the latter gene, which is also known as KCNE5, have been reported as BrS modulators by Ohno et al. [129]. Crotti et al. [128] identified mutations in 27 patients (21%). Most (21, or 16%) had a mutation in BrS1 gene SCN5A, in line with the study by Kapplinger et al. [130], who found an overall yield of 21% BrS1-associated SCN5A mutations derived from over 2,100 unrelated patients referred for BrS genetic testing, with the yield of the nine centers involved ranging between 11% and 28%. Crotti et al. [128] identified six patients (4.6%) with a mutation in one of the 12 other genes tested: two with a mutation in CACNB2b and one with a mutation in each of HCN4, KCND3, KCNJ8, and SCN1Bb. The low <2% yield of mutations involving the L-type calcium channel genes CACNA1C, CACNB2b, and CACNA2D1 is in contrast with previous findings of 8.5% for CACNA1C and CACNB2b only [131] and 12.3% for CACNA1C, CACNB2b, and CACNA2D1 all three [132] and may be related to the presence of a concomitant short QT interval [128], as in the study by Burashnikov et al. [132] and also reported by Hong et al. [133].
Overall, ion channel-related mutations are found in a minority of BrS patients, and if such mutation is found, it is almost always a mutation in SCN5A (BrS1). Mutations in other genes are rare and have thus far only been identified in single patients or in small families, so that one should be cautious with inferring these genes in the pathogenesis of BrS, as illustrated by the recent finding by Holst et al. [134] that support the association of SCN1Bb with BrS, but challenge that of SCN3B, MOG1, and KCND3. Many of the BrS-associated mutations listed in Table 2 have only recently been identified. This holds in particular for the mutations in CACNA2D1 [132], MOG1 [135], KCND3 [136], KCNE1L (KCNE5) [129], KCNJ8 [137], and SCN1Bb [138] and explains why there is no consensus in allocating BrS types 8 and higher to specific genes.
3.3. Short QT Syndrome
In contrast with LQTS, the short QT syndrome (SQTS) is characterized by a shortening of the QT interval on the ECG and, at the cellular level, a shortening of the action potential [139–141]. This shortening may be due to gain-of-function mutations in genes related to outward currents that flow during the repolarization phase of the action potential, for example, I Kr, I Ks, and I K1 (SQTS types 1–3; Table 3), or loss-of-function mutations in genes related to inward currents flowing during the repolarization phase, in particular the L-type calcium current, I Ca,L (SQTS types 4–6; Table 3). SQTS is a relatively recent addition to the family of inheritable arrhythmogenic diseases. After reports on familial SQTS by Gussak et al. in 2000 [142] and by Gaita et al. in 2003 [143], disease-causing mutations in KCNH2 (SQTS type 1, STQ1) [144], KCNQ1 (STQ2) [145], KCNJ2 (STQ3) [146], and CACNA1C andCACNB2b (STQ4 and STQ5) [131] were reported in 2004–2007. Recently, a sixth SQTS entity, associated with mutations in CACNA2D1, was reported by Templin et al. [147].
Table 3.
Genes linked to short QT syndrome.
| Type | OMIM | Gene | Protein | Functional role in cardiomyocytes | Effect of mutation |
|---|---|---|---|---|---|
| Autosomal dominant inheritance | |||||
|
| |||||
| SQT1 | #609620 | KCNH2 (HERG) | Kv11.1 | α subunit of I Kr channel | Gain of function of I Kr |
| SQT2 | #609621 | KCNQ1 (KVLQT1) | Kv7.1 (KvLQT1) | α subunit of I Ks channel | Gain of function of I Ks |
| SQT3 | #609622 | KCNJ2 | Kir2.1 | α subunit of I K1 channel | Gain of function of I K1 |
| SQT4 | — | CACNA1C | Cav1.2 | α 1C subunit of I Ca,L channel | Loss of function of I Ca,L |
| SQT5 | — | CACNB2b | Cav β2b | β 2b subunit of I Ca,L channel | Loss of function of I Ca,L |
| SQT6 | — | CACNA2D1 | Cav α 2 δ-1 | α 2 δ subunit of I Ca,L channel | Loss of function of I Ca,L |
OMIM: Online Mendelian Inheritance in Man compendium of human genes and genetic phenotypes; SQT1–SQT6: short QT syndrome types 1–6; I Ca, L: L-type calcium current; I K1: inward rectifier potassium current; I Kr: rapid delayed rectifier potassium current; I Ks: slow delayed rectifier potassium current.
Although data on the prevalence of SQTS are limited [148–150], it is clear that the prevalence of SQTS is much lower than that of LQTS and BrS. However, SQTS carries a high risk of sudden death and may be a cause of death in early infancy [139], thus possibly underlying cases of SIDS.
3.4. Cathecholaminergic Polymorphic Ventricular Tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a relatively rare but highly malignant inheritable arrhythmogenic disease. It is characterized by adrenergically mediated polymorphic ventricular tachyarrhythmias in the absence of electrocardiographic markers and structural heart disease and is induced by catecholamines released during physical exercise or emotional stress [151–155]. Thus far, mutations in three genes have been linked to CPVT, autosomal dominant mutations in the RYR2 gene encoding for the cardiac ryanodine receptor isoform 2 (CPVT type 1 or CPVT1) and rare autosomal recessive mutations in the CASQ2 and TRDN genes, encoding for the cardiac calsequestrin isoform 2 (CPVT2) and the triadin protein, respectively. All three proteins are involved in the intracellular cardiac calcium homeostasis (Table 4). The associations with the RYR2 and CASQ2 genes were both reported in 2001 [156, 157], whereas that with the TRDN gene was only recently reported [158]. In 2007, another autosomal recessive form of CPVT had been reported by Bhuiyan et al. [159], which mapped to a 25 Mb interval on chromosome 7p14–p22, but at present the causal gene at this locus is unknown. The TRDN variant is not listed as “CPVT3” in Table 4, because CPVT3 has already been used in relation to the study by Bhuiyan et al. [159] (OMIM entry %614021).
Table 4.
Genes linked to catecholaminergic polymorphic ventricular tachycardia.
| Type | OMIM | Gene | Protein | Functional role in cardiomyocytes | Effect of mutation |
|---|---|---|---|---|---|
| Autosomal dominant inheritance | |||||
|
| |||||
| CPVT1 | #604772 | RYR2 | RyR2 | Ryanodine receptor in SR membrane | Calcium leak from SR |
|
| |||||
| Autosomal recessive inheritance | |||||
|
| |||||
| CPVT2 | #611938 | CASQ2 | Calsequestrin-2 | Calcium buffering in SR | Disrupted SR calcium buffering capacity |
| NC | — | TRDN | Triadin | Links calsequestrin-2 and RyR2 | Impaired SR calcium release |
OMIM: Online Mendelian Inheritance in Man compendium of human genes and genetic phenotypes; CPVT1 and CPVT2: catecholaminergic polymorphic ventricular tachycardia types 1 and 2; NC: no consensus; SR: sarcoplasmic reticulum.
The calcium leak from the sarcoplasmic reticulum associated with the RYR2 mutations increases the probability of aberrant RyR2 channel opening during diastole [160]. This may result in delayed afterdepolarizations or spontaneous depolarization during diastole, which are both triggers for arrhythmias, due to the activation of the sodium-calcium exchange current I NaCa, as illustrated in Figure 6(a). As a paradigm to understand the mechanisms of arrhythmias associated to impaired Ca2+ regulation [153], CPVT has been widely studied in mouse models [160–163], identifying Purkinje cells as critical contributors to arrhythmic triggers in CPVT and suggesting a broader role for the Purkinje fiber network in the genesis of ventricular arrhythmias [163].
The prevalence of CPVT in the population is not known, but it has been estimated around 1 : 10,000 [151], which is probably an underestimation, as set out by de la Fuente et al. [164]. The potentially lethal arrhythmias associated with CPVT mostly occur in children and teenagers and are typically triggered by stress or exercise. Because sudden cardiac death may be the first manifestation of CPVT, it may well underlie cases of SIDS.
3.5. Caveat
In the above, the set of “primary electrical cardiac diseases” has been limited to LQTS, BrS, SQTS, and CPVT, as in many other papers [165–170] in which inheritable arrhythmogenic diseases are categorized and/or opposed to inheritable cardiomyopathies due to mutations in genes encoding sarcomeric or cytoskeletal proteins. There are, however, more electrical diseases, like the aforementioned inherited cardiac conduction disease [71] and the early repolarization and J wave syndromes [132, 137]. The electrical diseases are not separate entities, but may show overlap, not only in clinical symptoms but also in the underlying mutations [72, 171]. The latter is perhaps best illustrated by the 1795insD mutation in SCN5A, which has been associated with sinus node dysfunction, bradycardia, conduction disease, BrS, and LQTS, either in isolation or in combinations thereof [172–174]. Similar overlap syndromes have been reported for other mutations, in particular mutations in SCN5A, like E161K [175]. In addition, incomplete penetrance and variable expressivity may be observed [176], with family members with the same mutation demonstrating widely different clinical symptoms.
Furthermore, arrhythmogenic diseases may have some characteristics of cardiomyopathies and vice versa. As mentioned before, the pathophysiological mechanism of BrS may involve structural abnormalities that are not easily detectable [126], which would make BrS not purely an electrical disease. On the other hand, mutations in “structural genes,” as they occur in inheritable cardiomyopathies, may lead to a reduction in sodium current and intercellular coupling, thereby creating an arrhythmogenic substrate that is largely of an electrical nature. This may in particular hold for the early phase of arrhythmogenic right ventricular cardiomyopathy (ARVC; see [177] and primary references cited therein), which was initially termed arrhythmogenic right ventricular dysplasia (ARVD) or arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) and is currently being termed “arrhythmogenic cardiomyopathy” or “arrhythmogenic ventricular cardiomyopathy,” taking into account that the disease may also affect the left ventricle.
Overall, ion channel-related mutations associated with primary electrical cardiac diseases, as listed in Tables 1–4, should be taken into account in relation to SIDS. In addition, there may be early phases of genetically determined cardiomyopathies that cannot be identified macroscopically by autopsy, but add to the vulnerability of the infant and contribute to SIDS, as recently reported for hypertrophic cardiomyopathy (HCM) and HCM-associated genes [168, 178]. Disruption of sarcomeric activity might in turn disrupt intracellular calcium homeostasis and thus be responsible for arrhythmogenesis [178].
4. Cardiac Ion Channelopathies in Molecular Autopsy
In 2000 and 2001, a number of case reports were published on mutations in LQTS genes in SIDS or near SIDS [179–181], starting with the description by Schwartz et al. [179] of an infant who nearly died of SIDS and in whom LQTS was diagnosed and a spontaneous mutation in SCN5A was identified, providing a “proof of concept” of cardiac ion channelopathies as a cause of SIDS. Shortly thereafter, Ackerman et al. [182] reported the results of the first population-based “molecular autopsy” study, screening for mutations in LQTS genes as the possible cause of SIDS. A series of such population-based cohort studies of SIDS cases followed. These postmortem genetic analyses, screening for mutations in LQTS, SQTS, BrS, and CPVT genes, have revealed associations of mutations in these genes with SIDS. In the present section, I will first review the proof-of-concept findings and next the outcome of the various genetic analyses of population-based SIDS cohorts.
4.1. Proof-of-Concept Findings
In 2000, a “proof of concept” of cardiac ion channel mutations as a possible cause of SIDS was provided with the aforementioned description by Schwartz et al. [179] of an infant who was resuscitated from ventricular fibrillation and in whom LQTS was diagnosed and a de novo missense mutation in SCN5A (S941N) was identified. In another two case reports, mutations were identified in the KCNQ1 and SCN5A genes [180, 181]. The P117L mutation in KCNQ1 was identified in an SIDS victim upon genetic analysis and appeared a de novo missense mutation that had already been identified in an unrelated family that was affected by LQTS [180]. The A1330P mutation in SCN5A also appeared a de novo missense mutation, resulting in a functionally disturbed hyperactive I Na channel, as demonstrated in voltage clamp recordings from HEK-293 cells expressing the mutant channels, which showed a positive shift in voltage dependence of inactivation, a slowing of the time course of inactivation, and faster recovery from inactivation [181]. Effects of the mutation on the ventricular action potential, including a significant prolongation, were later directly demonstrated in “dynamic action potential clamp” experiments [183]. It is likely that the mutation caused the prolonged QTc interval of 600 ms on the ECG taken on day 5 as well as the lethal tachyarrhythmia in week 9 that occurred despite propranolol therapy. Sadly, cardiopulmonary resuscitation was not successful.
Several other case reports on ion channel mutations in SIDS or near SIDS have appeared in the literature since the reports by Schwartz et al. [179, 180] and Wedekind et al. [181]. A Scandinavian group found a novel mutation in KCNH2 (K101E) in a seven-week-old SIDS victim [184]. Because of a positive family history for clinical LQTS and documented Torsade de Pointes, it was assumed that this mutation may well have caused a lethal arrhythmia. Nof et al. [185] presented a family in which an inherited common polymorphism in KCNH2 (K897T) combined with a loss-of-function mutation (P926AfsX14) on separate alleles of the same gene led to sudden infant death and spontaneous abortion. Family members with only the polymorphism or only the mutation did not have any events of syncope or sudden cardiac death. Coexpression studies demonstrated a much greater loss of the function of KCNH2 current in the case of P926AfsX14/K897T than for P926AfsX14 or K897T alone.
Further case reports have all dealt with mutations in SCN5A. Skinner et al. [186] reported a near-miss SIDS case of a 19-day-old infant with documented marginal QTc interval prolongation and ventricular fibrillation. Genetic analysis revealed a missense mutation in SCN5A (R1193Q) that had been associated with BrS in a previous study [187] and had been characterized as an LQT3 mutation by Wang et al. [188]. Turillazzi et al. [189] linked the heterozygous W822X nonsense mutation in SCN5A to the death of both members of a set of apparently healthy monozygotic twins (“simultaneous sudden infant death syndrome,” SSIDS). This mutation had been linked to BrS in a previous study [190]. Huang et al. [191] described the case of a de novo mutation in SCN5A (S1333Y) identified in an SIDS victim who also carried a T20I mutation in KCNE1. Priori et al. [192] reported on two cases of SIDS in a family with BrS and the L567Q mutation in SCN5A, which was later functionally characterized as a loss-of-function mutation by Wan et al. [193]. Although direct genetic data of the two victims are not available, it is likely that they carried the familial L567Q mutation. Chockalingam et al. [194] described how an apparently healthy 4-month-old girl almost became an SIDS victim when immunization and/or the subsequent fever triggered life-threatening arrhythmias. Molecular analysis revealed a novel splice-site mutation in SCN5A that was then also identified in her father and in her 3-year-old brother. On the basis of the combination of findings in different family members, a diagnosis of BrS could be made, associated in this case with significant conduction defects.
4.2. Molecular Autopsy of Population-Based SIDS Cohorts
Starting with the study by Ackerman et al. [182] a series of population-based cohort studies have been published in which postmortem genetic analysis of SIDS cases, screening for mutations in LQTS genes and related genes (“molecular autopsy”), has revealed associations of mutations or rare variants in these genes with SIDS. The results of these studies are briefly discussed in order of publication below and summarized in Table 5.
Table 5.
Mutations or rare variants in cardiac ion channel-related genes in SIDS cohorts.
| Study‡ | Size of cohort | Genes tested | Reported mutations or rare variants† |
|---|---|---|---|
| Ackerman et al. [182] | 93 | SCN5A | A997S, R1826H |
|
| |||
| Tester and Ackerman [195] | 93 | KCNQ1 | T600M |
| KCNH2 | |||
| KCNE1 | |||
| KCNE2 | |||
|
| |||
| Wedekind et al. [196] | 41 | SCN5A | — |
| KCNQ1 | |||
| KCNH2 | |||
| KCNE1 | |||
| KCNE2 | |||
|
| |||
| Plant et al. [197] | 133 | SCN5A | S524Y (2 cases), R689H, homozygous S1103Y (3 cases), E1107K |
|
| |||
| Cronk et al. [198] | 134 | CAV3 | V14L, T78M, L79R |
|
| |||
| Arnestad et al. [199] | 201 | SCN5A | S216L, delAL586-587, R680H, R1193Q (2 cases), T1304M, F1486L, V1951L, F2004L (3 cases), P2006A (2 cases) |
| KCNQ1 | |||
| KCNH2 | |||
| KCNE1 | |||
| KCNE2 | |||
| KCNJ2 | |||
| CAV3 | |||
|
| |||
| Tester et al. [200] | 134 | RYR2 | R2267H, S4565R |
|
| |||
| Van Norstrand et al. [201] | 221 | GPD1-L | I124V, R273C |
|
| |||
| Otagiri et al. [202] | 42 | SCN5A | F532C, G1084S, F1705S |
| KCNQ1 | |||
| KCNH2 | |||
|
| |||
| Millat et al. [203] | 32 | SCN5A | Q692K, R975W, S1333Y |
| KCNQ1 | |||
| KCNH2 | |||
| KCNE1 | |||
| KCNE2 | |||
|
| |||
| Cheng et al. [204] | 292 | SNTA1 | G54R, P56S (3 cases), T262P, S287R, T372M, G460S |
|
| |||
| Tan et al. [205] | 292 | SCN1B | — |
| SCN2B | |||
| SCN3B | |||
| SCN4B | |||
|
| |||
| Tester et al. [206] | 292 | KCNJ8 | E332del, V346I |
|
| |||
| Hu et al. [207] | 292 | SCN1Bb | R214Q |
|
| |||
| Van Norstrand et al. [208] | 292 | GJA1 | E42K, S272P |
|
| |||
| Giudicessi et al. [209] | 292 | KCND3 | S530P |
‡Studies in order of (online) publication.
†Functionally significant mutations or rare variants listed in bold.
Ackerman et al. [182] genetically analyzed postmortem cardiac tissue from 45 SIDS and 48 possible SIDS cases, obtained between September 1997 and August 1999 in the State of Arkansas, USA, for mutations in SCN5A. In two of these 93 cases, a missense mutation in SCN5A was found (A997S and R1826H). In either case, the mutant channels were investigated in an expression system, where the mutant sodium current showed a slower decay and a two- to threefold increase in late current, compatible with LQTS type 3 (LQT3). The two mutations are therefore listed in bold in Table 5, indicating that they are functionally of a malignant nature. The same cohort was used by Tester and Ackerman [195] when screening for mutations in KCNQ1, KCNH2, KCNE1, and KCNE2. They found novel potassium channel variants in KCNQ1 (T600M), KCNH2 (G294V), and KCNE2 (V14I), as listed in Table 5. In addition, they discovered a novel mutation in KCNH2 (P1157L) following postmortem genetic testing in a separate case of an infant who was found dead in the prone sleep position. None of these mutations was seen in nearly 1,500 reference alleles from healthy controls. However, there is no functional evidence, for example, from in vitro expression studies, that any of these four mutations leads to potassium channel dysfunction. The KCNQ1-T600M, KCNH2-G294V, and KCNE2-V14I are therefore not classified as “functionally significant” in Table 5. Furthermore, the KCNQ1-T600M and KCNH2-G294V variants occurred in the same black victim, together with the sodium channel variant SCN5A-S1103Y, which has later been associated with sudden infant death in African Americans by Plant et al. [197] (see below).
As listed in Table 5, another 14 population-based cohort studies have been published since the studies by Ackerman et al. [182] and Tester and Ackerman [195]. If data on the functional significance of a mutation were not provided, the literature was searched for such data, in particular from linkage analysis and/or from expression studies. Part of the search results has been published in a previous study [210]. All of the 18 different genes tested, and therefore listed in Table 5, have previously been associated with LQTS, SQTS, BrS, or CPVT, and thus appear in Tables 1–4, except the SCN2B and GJA1 genes, which encode for the sodium channel β 2 subunit and the cardiac gap junction protein connexin43, respectively.
Wedekind et al. [196] performed a postmortem examination in a total of 41 SIDS cases of sudden and unexpected infant death, which occurred in 1991/1992 and 1995/1996 in the northwestern area of Germany. Apart from a number of common and rare polymorphisms, the results of screening for mutations in the LQTS genes KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2, associated with the LQTS types 1–3, 5, and 6, respectively, were limited to a single missense mutation in KCNQ1 (H105L). However, the mutant channel failed to display significant electrophysiological disturbances in vitro.
Plant et al. [197] studied the prevalence of SCN5A variants among 133 African American SIDS cases and found homozygous and rare heterozygous SCN5A variants in seven cases (5%). Three cases were homozygous for S1103Y, a variant that had previously been associated with an increased risk for arrhythmia in adults [211]. Comparison to controls gave an approximately 24-fold increase in risk of SIDS with the homozygous S1103Y genotype. In vitro, the variant Y1103 channels operated normally under control conditions, but showed abnormal function when subjected to lowered intracellular pH, which may indicate a predisposition to acidosis-induced arrhythmia. Three mutations in SCN5A at sites other than 1103 were identified in a total of four cases—S524Y (two cases), R689H, and E1107K—and characterized as gain-of-function mutations.
The cardiac fast sodium channel is localized in caveolae, membrane microdomains involved in vesicular trafficking, whose major component in cardiomyocytes is CAV3-encoded caveolin-3 [212]. Vatta et al. [212] reported a novel type of LQTS (LQT9) associated with mutations in CAV3 that result in a two- to threefold increase in late sodium current compared with wild-type caveolin-3, similar to the functional effect of LQT3-associated SCN5A mutations. Cronk et al. [198] investigated the prevalence of LQT9 in 134 cases of SIDS and identified three distinct CAV3 mutations (V14L, T78M, and L79R). At functional characterization, all of these showed a significant ≈5-fold increase in late sodium current, consistent with the LQT3-like phenotype described by Vatta et al. [212].
Between 1988 and 2004, Arnestad et al. [199] genetically analyzed 201 Norwegian cases of SIDS, screening for mutations in KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, KCNJ2, and CAV3, associated with LQTS types 1–3, 5–7, and 9, respectively. Mutations and rare variants in LQTS genes were identified in 26 cases. On the basis of the available functional data, 8 mutations and 7 rare variants found in 19 of 201 cases (9.5%) were considered as likely contributors to sudden death. Nine SCN5A mutations or rare genetic variants were found in a total of 13 cases (6.5%). Biophysical characterization of the R1193Q variant [188] and the eight other mutations or rare variants [213] revealed an increased persistent sodium current in all cases, either under control conditions or only under conditions of internal acidosis (R680H) or when expressed in the context of the common splice variant delQ1077 (delAL586-587 and V1951L). In 11 cases, nine different mutations or rare variants in the potassium channel genes KCNQ1 (5 cases), KCNH2 (5 cases), and KCNE2 (one case) were found (Table 5). No mutations were found in KCNE1 or KCNJ2. However, one rare KCNQ1 variant (P448R, three cases) appeared a common, ethnic-specific polymorphism [214], whereas the mutations V279M, R885C, and S1040G in KCNH2 (three cases in total) exhibited biophysical properties indistinguishable from wild type [215] and are probably benign variants. For the remaining five cases clear functional effects have been found [199, 215]. The functional characterization of the KCNH2-R273Q, KCNH2-R954C/K897T, and KCNE2-Q9E mutations revealed a loss of function of the associated current (I Kr), which would result in a long QT phenotype. The functional effect of the KCNQ1 mutations is a loss of function (G460S) or gain of function (I274V) of the associated current (I Ks), which would result in a long QT or short QT phenotype, respectively. Arnestad et al. [199] also identified two CAV3 variants (C72W and T78M). Since T78M had earlier been characterized as a functionally significant mutation [198], this variant was considered pathogenic, although one of the two victims with this variant also carried the functionally significant delAL586-587 mutation in SCN5A. With regard to C72W, no functional data are available. Of note, the C72W variant had actually been designated as a common polymorphism in the original study identifying CAV3 as the LQT9 locus [212].
A 2004 study from the Mayo Clinic represented the first molecular autopsy of RYR2 in cases of sudden unexplained death [216]. In the cohort of 49 cases, six distinct RYR2 missense mutations were found in seven cases (14%). Next, Tester et al. [200] performed a study with the aim to determine the spectrum and prevalence of RYR2 mutations in a cohort of 134 SIDS cases. Overall, two distinct and novel RYR2 mutations (R2267H and S4565R) were identified in two cases of SIDS that were subsequently established to be mutation negative for all known LQTS susceptibility genes. Both amino acid substitutions were absent in 400 reference alleles. Functional characterization showed that the two mutant channels were prone to display a significant gain-of-function “leaky” phenotype, especially under conditions that simulated stress during diastole. These findings are supported by the study by Mathur et al. [217], who created a knock-in mouse model of SIDS and reported that young mice with the equivalent of the gain-of-function mutation R176Q in RYR2 [218] show an increased propensity to calcium leak-induced cardiac arrhythmias and sudden death. The nocturnal occurrence of cardiac arrhythmias in SIDS can be explained by sudden increases in sympathetic activity, for example, following hypoxia or possibly even during REM sleep [200].
London et al. [219] reported that the A280V mutation in the GPD1-L gene, which encodes the glycerol-3-phosphate dehydrogenase 1-like protein (G3PD1L), reduced inward sodium current and thus caused BrS. Given this finding, Van Norstrand et al. [201] analyzed 83 cases of sudden unexplained death, including 7 SIDS cases, and identified a GPD1-L mutation in a 3-month-old boy (E83K). Further analysis of a cohort of 221 SIDS cases revealed two additional mutations (I124V and R273C). All three mutations were absent in 600 reference alleles. Compared with wild type, coexpression of mutant GPD1-L with SCN5A resulted in a significantly reduced sodium current, consistent with a loss-of-function, BrS-like phenotype. London et al. [219] found a reduced cell surface expression of SCN5A with mutant GPD1-L and hypothesized that the mutation caused impaired trafficking of the cardiac sodium channel to the cell surface. Valdivia et al. [220] later provided evidence that the effect of the mutation in GPD1-L is through a reduced enzymatic function of G3PD1L, which regulates SCN5A through direct phosphorylation, whereas another study [221] emphasizes the role of a mutation-induced increase in the intracellular concentration of NADH.
In 2008, Otagiri et al. [202] reported the results of an investigation of 42 Japanese SIDS cases between 1995 and 2004, in which they studied the LQTS genes KCNQ1,KCNH2, andSCN5A. They identified two potassium channel mutations, one in KCNQ1 (K598R) and one in KCNH2 (T895M), and three mutations in SCN5A (F532C, G1084S and F1705S). In expression studies, the latter two SCN5A mutations showed hyperpolarizing shifts in inactivation (G1084S and F1705S) and a delayed recovery from inactivation (F1705S), loss-of-function features commonly seen in BrS mutations in SCN5A. As for SCN5A-F532C, Otagiri et al. [202] found no evidence indicating a functionally perturbed channel in their expression experiments, but this mutation has more recently been associated with BrS [222]. The KCNQ1-K598R mutation did not noticeably alter the gating of KCNQ1 channels expressed in oocytes and was therefore considered a rare polymorphism. However, this mutation has later been identified as an LQT1-causing mutation in a clinical study [223]. Biophysical properties of KCNH2-T895M included a decrease in the amplitude of the steady-state current and a delay in deactivation. Since these alterations seem to exert opposite functional effects, it is difficult to predict the overall in vivo effect of this mutation, especially as the subject found in also carried the G1084S mutation in SCN5A.
Millat et al. [203] performed genetic screening of 32 French SIDS victims of Caucasian origin for mutations in KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2. They found three potassium channel mutations, one in each of the genes KCNQ1, KCNH2, and KCNE1 (KCNQ1-G626S, KCNH2-R148W, and KCNE1-T20I) and none in KCNE2. These mutations were interpreted by the authors as a possible cause of death. With regard to the KCNQ1 mutation, some evidence is available to support this hypothesis [224]. However, the infant with the KCNE1 mutation, who was the subject of the aforementioned case report by Huang et al. [191], was also the carrier of a spontaneous mutation in SCN5A that may provide a more likely explanation for her death, given that her father was found to be a carrier of the KCNE1-T20I mutation with no LQTS-related phenotype [191]. The KCNH2 defect is probably a benign polymorphism [225], as supported by the observation that the father, who also carried the mutation, was asymptomatic. Millat et al. [203] also found three mutations in SCN5A (Q692K, R975W, and S1333Y). The first has wild-type-like functional properties [226] and has been classified as a control variant [222], whereas the second is currently considered a rare control [222, 225]. However, SCN5A-S1333Y causes a defect in inactivation with the presence of a residual current, comparable to LQT3 [191].
In 2008, Ueda et al. [227] and Wu et al. [228] almost simultaneously reported that mutations in SNTA1, encoding the scaffolding protein α1-syntrophin, may lead to LQTS through a gain of function of the fast sodium channel, as in LQT3. Ueda et al. [227] found that the A390V mutation led to LQTS through increased direct S-nitrosylation of the cardiac sodium channel, resulting in a marked increase in late sodium current. The increase in late sodium current was accompanied by an increase in peak sodium current, an increase in sodium channel availability through a +6 mV shift in sodium channel inactivation, and a slower time course of sodium current decay. Wu et al. [228] studied the A257G mutation in SNTA1 and also found an increase in late sodium current, due to an increase in peak sodium current, an increase in sodium channel availability through a –9 mV shift in sodium channel activation, and a slower time course of sodium current decay. Subsequently, Cheng et al. [204] investigated the prevalence and functional properties of SNTA1 mutations in a cohort of 292 SIDS cases. Six mutations were found in eight cases, with one particular mutation (P56S) identified in three cases, and were absent in 800 reference alleles. In vitro, a significant increase in peak and late sodium current was observed for S287R, T372M, and G460S, which was reversed by a neuronal nitric oxide synthase inhibitor. The other three variants (G54R, P56S, and T262P) showed functionally insignificant changes in the sodium current.
Tan et al. [205] screened the Mayo Clinic cohort of 292 unrelated SIDS victims for mutations in the four sodium channel β subunit genes SCN1B to SCN4B and identified a total of three mutations in two of these genes (SCN3B-V36M, SCN3B-V54G, and SCN4B-S206L). The two SCN3B mutations are both localized in the extracellular loop of the β subunit, which is important for β3 membrane trafficking. Compared with wild-type channels, V36M channels showed both loss-of-function and gain-of-function phenotypes in expression studies, that is, decreased peak I Na and increased late I Na (at least when normalized to peak I Na), respectively, whereas V54G was purely a loss of function. SCN4B-S206L is associated with an increase in late sodium current, as demonstrated through the adenoviral transduction of adult rat ventricular myocytes [205].
Tester et al. [206] performed a mutational analysis of KCNJ8, which encodes the α subunit of the ATP-sensitive potassium channel, on genomic DNA obtained from the SIDS victims of the Mayo Clinic cohort. Two distinct and novel KCNJ8 mutations were identified (E332del and V346I). Both cases were negative for mutations in established channelopathic genes. In vitro, both mutations resulted in a significant decrease in the associated ATP-sensitive potassium current (I K,ATP). Loss of function of the KCNJ8-encoded cardiac K ATP channel may result in a long QT phenotype, predisposing to SIDS, but an isolated pro-arrhythmic mechanism remains speculative since KCNJ8 is expressed in multiple tissues, including vascular and neuronal tissue [206].
Nav β1B, encoded by SCN1Bb, is a splicing variant of the β 1 subunit of the I Na channel [229]. Mutations in SCN1Bb have been associated with BrS (Table 2). Hu et al. [207] have assessed the Mayo Clinic SIDS cohort for mutations in SCN1Bb and found a functional rare variant in a single SIDS victim who had died at the age of 4 months. In coexpression studies with wild-type SCN5A or KCND3, the R214Q variant had a loss-of-function effect on I Na as well as a gain-of-function effect on I to, in line with the reported association with BrS.
Van Norstrand et al. [208] detected two novel missense mutations (E42K and S272P) in GJA1, which encodes the gap junction channel protein connexin43 (Cx43), in the Mayo Clinic cohort of 292 SIDS cases. Functional studies were performed using dual whole cell patch clamp and revealed a strongly reduced gap junctional conductance for E42K compared to wild type. Such strongly reduced intercellular coupling may result in lethal ventricular arrhythmias, as demonstrated in several conditional Cx43 knockout models [230].
When screening the Mayo Clinic cohort for mutations in KCND3, which encodes the α subunit of the I to channel, Giudicessi et al. [209] found a rare missense mutation in a single SIDS victim who had died at the age of 2 months. However, when the S530P mutation was heterologously expressed in HEK293 cells, the electrophysiological phenotype did not differ from wild type.
5. Prevalence of Cardiac Ion Channelopathies in SIDS
From the data in Table 5, one can compute the prevalence of mutations in each of the genes tested thus far. Mutations in the sodium channel gene SCN5A, for example, have been assessed in the studies by Ackerman et al. [182], Wedekind et al. [196], Plant et al. [197], Arnestad et al. [199], Otagiri et al. [202], and Millat et al. [203], yielding a total of 542 SIDS cases, in which 28 mutations in SCN5A were detected (5.2%), almost all with functional significance (26, or 4.8%). Similarly, numbers for the other 17 genes can be obtained. All results appear in Table 6 and are illustrated in Figures 7 and 8. It should be noted that, when compiling Tables 5 and 6, it has been ignored that a few SIDS victims carried two ion channel-related mutations at the same time and thus they appear in these tables twice. This holds, as mentioned before, for an infant in the study by Tester and Ackerman [195] with mutations in both KCNQ1 and KCNH2, for an infant in the study by Arnestad et al. [199] with CAV3-T78M and SCN5A-delAL586-587 mutations, and for an infant in the study by Millat et al. [203] with KCNE1-T20I and SCN5A-S1333Y mutations. Furthermore, it has been ignored that there are differences in the populations between studies, which are mostly Northern American, but also African American [197], French [203], German [196], Norwegian [199], and Japanese [202].
Table 6.
Prevalence of cardiac ion channel-related mutations in SIDS cohorts‡.
| Number of SIDS cases | |||
|---|---|---|---|
| Gene | Total | with mutation | Functionally significant |
| I Na related genes | |||
| SCN5A | 542 | 28 (5.2%) | 26 (4.8%) |
| SCN1B | 292 | 0 | 0 |
| SCN1Bb | 292 | 1 (0.3%) | 1 (0.3%) |
| SCN2B | 292 | 0 | 0 |
| SCN3B | 292 | 2 (0.7%) | 2 (0.7%) |
| SCN4B | 292 | 1 (0.3%) | 1 (0.3%) |
| CAV3 | 335 | 6 (1.8%) | 5 (1.5%) |
| GPD1-L | 221 | 2 (0.9%) | 2 (0.9%) |
| SNTA1 | 292 | 8 (2.7%) | 3 (1.0%) |
| I to related genes | |||
| KCND3 | 292 | 1 (0.3%) | 0 |
| I Ks related genes | |||
| KCNQ1 | 409 | 9 (2.2%) | 4 (1.0%) |
| KCNE1 | 367 | 1 (0.3%) | 0 |
| I Kr related genes | |||
| KCNH2 | 409 | 8 (2.0%) | 2 (0.5%) |
| KCNE2 | 367 | 2 (0.5%) | 1 (0.3%) |
| I K1 related genes | |||
| KCNJ2 | 201 | 0 | 0 |
| I K,ATP related genes | |||
| KCNJ8 | 292 | 2 (0.7%) | 2 (0.7%) |
| Intracellular calcium cycling related genes | |||
| RYR2 | 134 | 2 (1.5%) | 2 (1.5%) |
| Gap junctional current related genes | |||
| GJA1 | 292 | 2 (0.7%) | 1 (0.3%) |
I K,ATP: ATP-sensitive potassium current; I K1: inward rectifier potassium current; I Kr: rapid delayed rectifier potassium current; I Ks: slow delayed rectifier potassium current; I Na: fast sodium current; I to: transient outward potassium current.
‡Based on data listed in Table 5.
Figure 7.
Prevalence of cardiac ion channelopathies in SIDS victims, based on mutations found in population-based cohort studies (Tables 5 and 6). Left: overall prevalence (blue bar) and prevalence of mutations with a known malignant phenotype (red bar). Right: prevalence of fast sodium current (I Na) related mutations (blue bar) and those of a known malignant phenotype (red bar).
Figure 8.
Prevalence of cardiac ion channelopathies in SIDS victims, based on data from population-based cohort studies (Tables 5 and 6). Blue bars indicate the prevalence of mutations and red bars that of mutations with a known malignant phenotype. (a) Mutations in genes affecting the fast sodium current I Na. (b) Mutations in genes affecting the transient outward potassium current I to (KCND3), the slow delayed rectifier potassium current I Ks(KCNQ1 and KCNE1), the rapid delayed rectifier potassium current I Kr (KCNH2 and KCNE2), the inward rectifier potassium current I K1 (KCNJ2), the ATP-sensitive potassium current I K,ATP (KCNJ8), the RyR2 calcium release channel (RYR2), and the gap junctional current I j (GJA1).
According to the data in Table 6, 20.2% of the SIDS victims carried a mutation in any of the 18 cardiac ion channel-related genes studied (Figure 7, leftmost bar). In the majority of cases (13.9%), there is evidence for malignancy of these mutations, as detailed in Section 4.2, with functional effects related to LQTS types 1–3, 5, 6, 9, 10, and 12, BrS types 1, 2, and 7, and SQTS type 2 and CPVT type 1. In the remaining cases (6.3%), there are either no data on the functional effects of the mutation or no functionally significant effects have been observed, for example, in expression studies. In the latter case, there may still be latent functional defects that are not evident under the conditions of the functional test. For example, there may be defects that only show their malignancy in the case of acidosis or in the context of a specific splice variant, as for some of the SIDS-associated SCN5A variants studied by Wang et al. [213] (see Section 4.2). Another example is the SIDS associated KCNQ1-K598R mutation, which seemed benign when expressed in oocytes [202], but was later identified as an LQT1-causing mutation in a clinical study [223]. Similarly, the D1275N mutation in SCN5A showed a clear clinical phenotype and an impaired sodium current in a mouse model, but did not generate major changes in sodium channel function in heterologous expression studies [231].
Another factor that may turn apparently benign mutations into malignant ones is high temperature. The malignant properties of a mutation may become apparent or more severe with an increase in temperature, as demonstrated for several sodium channel mutations associated with BrS, for example, in the studies by Dumaine et al. [232] and Keller et al. [233], explaining fever-induced arrhythmias observed in BrS patients. More recently, it has become clear that fever may also induce life-threatening arrhythmias in the carriers of an LQT2 mutation [234, 235]. The aforementioned case report by Chockalingam et al. [194] provides evidence that arrhythmogenic events triggered by fever, in this case vaccination related, may also play a role in the etiology of SIDS. A second case has recently been presented, as part of a study on the effects of loss-of-function sodium channelopathies in children [236]. In addition, Kanter et al. [237] reported a case of rapid ventricular tachycardia in a 5-month-old boy on the day of receiving his 6-month immunizations.
If we assume that at least some of the mutations for which functional data are lacking are of a malignant phenotype, the prevalence of malignant cardiac ion channel-related mutations among SIDS victims may actually be close to 20%. It may even be higher, given that conventional genetic analysis may fail to uncover severe mutations [238–242] and that the determination of associations between SIDS and mutations in ion channel-related genes is still an emerging field, with several associations that have only recently been reported [206–208] and others that remain to be assessed, for example, a possible association between SIDS and mutations in the L-type calcium channel-related genes CACNA1C, CACNB2b, and CACNA2D1, which have been associated with LQTS, BrS, and SQTS (Table 1–3). Of note, the prevalence of LQTS, and BrS-associated mutations among SIDS victims is much higher than the estimated prevalence of 1 : 2,000 for LQTS and 1 : 2,000 to 1 : 5,000 for BrS in the general population [97, 127].
Furthermore, we can conclude that mutations in I Na channel-related genes are the most malignant ones. Mutations in the α subunit encoding gene, SCN5A, are found in 5.2% of the SIDS victims (Table 6, Figure 8(a)), in accordance with the severe symptoms in children with LQT3 [118] and the observation in adults that LQT3 has the most severe clinical events [117] and that these events tend to occur during sleep [243], as in SIDS. In total, more than half of the reported mutations are related to the I Na channel (Figure 7). In addition to the mutations in SCN5A, there are mutations in the β subunit encoding genes SCN1Bb, SCN3B, and SCN4B (1.4%) and in “regulatory genes” (CAV3, GPD1-L, and SNTA1; 5.4%), adding up to a prevalence of 12.0% for mutations affecting I Na, with functional evidence available for 9.6% (Table 6, Figures 7 and 8(a)). Similar percentages for the “cardiac sodium Nav1.5 channelsome” have been reported in an abstract form by Van Norstrand et al. [244]. With a total prevalence of 6.0%, with functional evidence in the case of 2.4%, mutations in potassium channel genes (KCND3, KCNQ1, KCNE1, KCNH2, KCNE2, KCNJ2, and KCNJ8) seem less frequent and less malignant (Figure 8(b)).
It should be noted that mutations in ion channel-related genes as putative causes of death are not limited to cases of SIDS, but have also been reported in cohorts of sudden unexplained death cases in which the victims were >1 year of age [113, 245–247]. Also, there are reports, for example, by Cuneo et al. [248] and Miller et al. [249], that mutations in ion channel-related genes may lead to cardiac arrhythmias and death in utero.
6. Conclusion
In addition to clinical associations such as prone sleeping and exposure to cigarette smoke, several genetic factors have been identified with regard to SIDS, including polymorphisms in genes related to the immune system and the autonomic nervous system. With large prospective ECG studies, several “proof-of-concept” cases, a series of population-based cohort studies, and the ability to assess the functional effects of a mutation in vitro, the role of cardiac ion channel-related mutations as a genetic contributor to SIDS is currently the most well established. Combining data from population-based cohort studies, it can be concluded that one out of five SIDS victims carries a mutation in a cardiac ion channel-related gene and that the majority of these mutations are of a known malignant phenotype.
7. Further Reading
The intriguing issue of the role of mutations in ion channel-related genes in cases of sudden unexplained deaths has been addressed in a number of recent reviews. For further reading the reader is referred to the reviews by Tester and Ackerman [250], Van Norstrand and Ackerman [251, 252], Tfelt-Hansen et al. [253], Cerrone and Priori [254], Chopra and Knollmann [255], Insolia et al. [256], Opdal and Rognum [257], Martin et al. [258] and Perrin and Gollob [259].
References
- 1.Beckwith JB. Discussion of terminology and definition of sudden infant death syndrome. In: Bergman AB, Beckwith JB, Ray CG, editors. Proceedings of the 2nd International Conference on Causes of Sudden Death in Infants; Seattle, Wash, USA. University of Washington Press; 1970. pp. 14–22. [Google Scholar]
- 2.Willinger M, James LS, Catz C. Defining the sudden infant death syndrome (SIDS): deliberations of an expert panel convened by the National Institute of Child Health and Human Development. Pediatric Pathology. 1991;11(5):677–684. doi: 10.3109/15513819109065465. [DOI] [PubMed] [Google Scholar]
- 3.Krous HF, Beckwith JB, Byard RW, et al. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics. 2004;114(1):234–238. doi: 10.1542/peds.114.1.234. [DOI] [PubMed] [Google Scholar]
- 4.Krous HF. Sudden unexpected death in infancy and the dilemma of defining the sudden infant death syndrome. Current Pediatric Reviews. 2010;6(1):5–12. [Google Scholar]
- 5.Byard RW, Krous HF. Sudden infant death syndrome: overview and update. Pediatric and Developmental Pathology. 2003;6(2):112–127. doi: 10.1007/s10024-002-0205-8. [DOI] [PubMed] [Google Scholar]
- 6.Byard RW, Marshall D. An audit of the use of definitions of sudden infant death syndrome (SIDS) Journal of Forensic and Legal Medicine. 2007;14(8):453–455. doi: 10.1016/j.jflm.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Limerick SR, Bacon CJ. Terminology used by pathologists in reporting on sudden infant deaths. Journal of Clinical Pathology. 2004;57(3):309–311. doi: 10.1136/jcp.2003.013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gould SJ, Weber MA, Sebire NJ. Variation and uncertainties in the classification of sudden unexpected infant deaths among paediatric pathologists in the UK: findings of a National Delphi Study. Journal of Clinical Pathology. 2010;63(9):796–799. doi: 10.1136/jcp.2010.079715. [DOI] [PubMed] [Google Scholar]
- 9.Kim SY, Shapiro-Mendoza CK, Chu SY, Camperlengo LT, Anderson RN. Differentiating cause-of-death terminology for deaths coded as sudden infant death syndrome, accidental suffocation, and unknown cause: an investigation using US death certificates, 2003-2004. Journal of Forensic Sciences. 2012;57(2):364–369. doi: 10.1111/j.1556-4029.2011.01937.x. [DOI] [PubMed] [Google Scholar]
- 10.Byard RW. Sudden unexpected death in infancy (SUDI)—the role of the pathologist. Current Pediatric Reviews. 2010;6(1):21–26. [Google Scholar]
- 11.Byard RW. SUDI or “undetermined”: does it matter? Forensic Science, Medicine, and Pathology. 2009;5(4):252–253. doi: 10.1007/s12024-009-9129-1. [DOI] [PubMed] [Google Scholar]
- 12.Blair PS, Byard RW, Fleming PJ. Sudden unexpected death in infancy (SUDI): suggested classification and applications to facilitate research activity. Forensic Science, Medicine, and Pathology. 2012;8(3):312–315. doi: 10.1007/s12024-011-9294-x. [DOI] [PubMed] [Google Scholar]
- 13.Hauck FR, Tanabe KO. International trends in sudden infant death syndrome: stabilization of rates requires further action. Pediatrics. 2008;122(3):660–666. doi: 10.1542/peds.2007-0135. [DOI] [PubMed] [Google Scholar]
- 14.Kochanek KD, Kirmeyer SE, Martin JA, Strobino DM, Guyer B. Annual summary of vital statistics: 2009. Pediatrics. 2012;129(2):338–348. doi: 10.1542/peds.2011-3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson RN, Miniño AM, Hoyert DL, Rosenberg HM. Comparability of cause of death between ICD-9 and ICD-10: preliminary estimates. National Vital Statistics Reports. 2001;49(2):1–32. [PubMed] [Google Scholar]
- 16.Willinger M, Hoffman HJ, Wu KT, et al. Factors associated with the transition to nonprone sleep positions of infants in the United States: the National Infant Sleep Position Study. Journal of the American Medical Association. 1998;280(4):329–335. doi: 10.1001/jama.280.4.329. [DOI] [PubMed] [Google Scholar]
- 17.Ottolini MC, Davis BE, Patel K, Sachs HC, Gershon NB, Moon RY. Prone infant sleeping despite the “Back to Sleep” campaign. Archives of Pediatrics and Adolescent Medicine. 1999;153(5):512–517. doi: 10.1001/archpedi.153.5.512. [DOI] [PubMed] [Google Scholar]
- 18.Gibson E, Dembofsky CA, Rubin S, Greenspan JS. Infant sleep position practices 2 years into the “Back to Sleep” campaign. Clinical Pediatrics. 2000;39(5):285–289. doi: 10.1177/000992280003900505. [DOI] [PubMed] [Google Scholar]
- 19.Pollack HA, Frohna JG. Infant sleep placement after the Back to Sleep campaign. Pediatrics. 2002;109(4):608–614. doi: 10.1542/peds.109.4.608. [DOI] [PubMed] [Google Scholar]
- 20.Hauck FR, Moore CM, Herman SM, et al. The contribution of prone sleeping position to the racial disparity in sudden infant death syndrome: the Chicago Infant Mortality Study. Pediatrics. 2002;110(4):772–780. doi: 10.1542/peds.110.4.772. [DOI] [PubMed] [Google Scholar]
- 21.Dwyer T, Ponsonby AL. Sudden infant death syndrome and prone sleeping position. Annals of Epidemiology. 2009;19(4):245–249. doi: 10.1016/j.annepidem.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 22.Dwyer T, Couper D, Walter SD. Sources of heterogeneity in the meta-analysis of observational studies: the example of SIDS and sleeping position. Journal of Clinical Epidemiology. 2001;54(5):440–447. doi: 10.1016/s0895-4356(00)00313-9. [DOI] [PubMed] [Google Scholar]
- 23.Galland BC, Taylor BJ, Bolton DPG. Prone versus supine sleep position: a review of the physiological studies in SIDS research. Journal of Paediatrics and Child Health. 2002;38(4):332–338. doi: 10.1046/j.1440-1754.2002.00002.x. [DOI] [PubMed] [Google Scholar]
- 24.Gilbert R, Salanti G, Harden M, See S. Infant sleeping position and the sudden infant death syndrome: systematic review of observational studies and historical review of recommendations from 1940 to 2002. International Journal of Epidemiology. 2005;34(4):874–887. doi: 10.1093/ije/dyi088. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell E, Krous HF, Donald T, Byard RW. Changing trends in the diagnosis of sudden infant death. American Journal of Forensic Medicine and Pathology. 2000;21(4):311–314. doi: 10.1097/00000433-200012000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Malloy MH, MacDorman M. Changes in the classification of sudden unexpected infant deaths: United States, 1992–2001. Pediatrics. 2005;115(5):1247–1253. doi: 10.1542/peds.2004-2188. [DOI] [PubMed] [Google Scholar]
- 27.Shapiro-Mendoza CK, Tomashek KM, Anderson RN, Wingo J. Recent national trends in sudden, unexpected infant deaths: more evidence supporting a change in classification or reporting. American Journal of Epidemiology. 2006;163(8):762–769. doi: 10.1093/aje/kwj117. [DOI] [PubMed] [Google Scholar]
- 28.American Academy of Pediatrics Task Force on Sudden Infant Death Syndrome. The changing concept of sudden infant death syndrome: diagnostic coding shifts, controversies regarding the sleeping environment, and new variables to consider in reducing risk. Pediatrics. 2005;116(5):1245–1255. doi: 10.1542/peds.2005-1499. [DOI] [PubMed] [Google Scholar]
- 29.Blair PS, Mitchell EA, Heckstall-Smith EMA, Fleming PJ. Head covering—a major modifiable risk factor for sudden infant death syndrome: a systematic review. Archives of Disease in Childhood. 2008;93(9):778–783. doi: 10.1136/adc.2007.136366. [DOI] [PubMed] [Google Scholar]
- 30.Fleming PJ, Blair PS, Bacon C, et al. Environment of infants during sleep and risk of the sudden infant death syndrome: results of 1993–5 case-control study for confidential inquiry into stillbirths and deaths in infancy. BMJ. 1996;313(7051):191–195. doi: 10.1136/bmj.313.7051.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lahr MB, Rosenberg KD, Lapidus JA. Bedsharing and maternal smoking in a population-based survey of new mothers. Pediatrics. 2005;116(4):e530–e542. doi: 10.1542/peds.2005-0354. [DOI] [PubMed] [Google Scholar]
- 32.Ruys JH, de Jonge GA, Brand R, Engelberts AC, Semmekrot BA. Bed-sharing in the first four months of life: a risk factor for sudden infant death. Acta Paediatrica. 2007;96(10):1399–1403. doi: 10.1111/j.1651-2227.2007.00413.x. [DOI] [PubMed] [Google Scholar]
- 33.Scragg RKR, Mitchell EA, Stewart AW, et al. Infant room-sharing and prone sleep position in sudden infant death syndrome. The Lancet. 1996;347(8993):7–12. doi: 10.1016/s0140-6736(96)91554-8. [DOI] [PubMed] [Google Scholar]
- 34.Trachtenberg FL, Haas EA, Kinney HC, Stanley C, Krous HF. Risk factor changes for sudden infant death syndrome after initiation of Back-to-Sleep campaign. Pediatrics. 2012;129(4):630–638. doi: 10.1542/peds.2011-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golding J. Sudden infant death syndrome and parental smoking—a literature review. Paediatric and Perinatal Epidemiology. 1997;11(1):67–77. doi: 10.1046/j.1365-3016.1997.d01-12.x. [DOI] [PubMed] [Google Scholar]
- 36.Carpenter RG, Irgens LM, Blair PS, et al. Sudden unexplained infant death in 20 regions in Europe: case control study. The Lancet. 2004;363(9404):185–191. doi: 10.1016/s0140-6736(03)15323-8. [DOI] [PubMed] [Google Scholar]
- 37.Anderson ME, Johnson DC, Batal HA. Sudden infant death syndrome and prenatal maternal smoking: rising attributed risk in the Back to Sleep era. BMC Medicine. 2005;3, article 4 doi: 10.1186/1741-7015-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duncan JR, Garland M, Myers MM, et al. Prenatal nicotine-exposure alters fetal autonomic activity and medullary neurotransmitter receptors: implications for sudden infant death syndrome. Journal of Applied Physiology. 2009;107(5):1579–1590. doi: 10.1152/japplphysiol.91629.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.St-Hilaire M, Duvareille C, Avoine O, et al. Effects of postnatal smoke exposure on laryngeal chemoreflexes in newborn lambs. Journal of Applied Physiology. 2010;109(6):1820–1826. doi: 10.1152/japplphysiol.01378.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blair PS, Fleming PJ, Bensley D, et al. Smoking and the sudden infant death syndrome: results from 1993–5 case-control study for confidential inquiry into stillbirths and deaths in infancy. BMJ. 1996;313(7051):195–198. doi: 10.1136/bmj.313.7051.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonnell-Naughton M, McGarvey C, O'Regan M, Matthews T. Maternal smoking and alcohol consumption during pregnancy as risk factors for sudden infant death. Irish Medical Journal. 2012;105(4):105–108. [PubMed] [Google Scholar]
- 42.Alm B, Wennergren G, Norvenius G, et al. Caffeine and alcohol as risk factors for sudden infant death syndrome. Archives of Disease in Childhood. 1999;81(2):107–111. doi: 10.1136/adc.81.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ford RPK, Schluter PJ, Mitchell EA, et al. Heavy caffeine intake in pregnancy and sudden infant death syndrome. Archives of Disease in Childhood. 1998;78(1):9–13. doi: 10.1136/adc.78.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Jonge GA, Lanting CI, Brand R, Ruys JH, Semmekrot BA, van Wouwe JP. Sudden infant death syndrome in child care settings in the Netherlands. Archives of Disease in Childhood. 2004;89(5):427–430. doi: 10.1136/adc.2003.029884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moon RY, Sprague BM, Patel KM. Stable prevalence but changing risk factors for sudden infant death syndrome in child care settings in 2001. Pediatrics. 2005;116(4):972–977. doi: 10.1542/peds.2005-0924. [DOI] [PubMed] [Google Scholar]
- 46.Kiechl-Kohlendorfer U, Moon RY. Sudden infant death syndrome (SIDS) and child care centres (CCC) Acta Paediatrica. 2008;97(7):844–845. doi: 10.1111/j.1651-2227.2008.00850.x. [DOI] [PubMed] [Google Scholar]
- 47.Simpson JM. Infant stress and sleep deprivation as an aetiological basis for the sudden infant death syndrome. Early Human Development. 2001;61(1):1–43. doi: 10.1016/s0378-3782(00)00127-4. [DOI] [PubMed] [Google Scholar]
- 48.Filiano JJ, Kinney HC. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Biology of the Neonate. 1994;65(3-4):194–197. doi: 10.1159/000244052. [DOI] [PubMed] [Google Scholar]
- 49.Guntheroth WG, Spiers PS. The triple risk hypotheses in sudden infant death syndrome. Pediatrics. 2002;110(4, article e64) doi: 10.1542/peds.110.5.e64. [DOI] [PubMed] [Google Scholar]
- 50.Courts C, Madea B. Genetics of the sudden infant death syndrome. Forensic Science International. 2010;203(1–3):25–33. doi: 10.1016/j.forsciint.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 51.Nelson EAS, Taylor BJ, Mackay SC. Child care practices and the sudden infant death syndrome. Australian Paediatric Journal. 1989;25(4):202–204. doi: 10.1111/j.1440-1754.1989.tb01455.x. [DOI] [PubMed] [Google Scholar]
- 52.Blackwell CC, Weir DM, Busuttil A. Infection, inflammation and sleep: More pieces to the puzzle of sudden infant death syndrome (SIDS) Acta Pathologica, Microbiologica, et Immunologica Scandinavica. 1999;107(5):455–473. doi: 10.1111/j.1699-0463.1999.tb01581.x. [DOI] [PubMed] [Google Scholar]
- 53.Prandota J. Possible pathomechanisms of sudden infant death syndrome: key role of chronic hypoxia, infection/inflammation states, cytokine irregularities, and metabolic trauma in genetically predisposed infants. American Journal of Therapeutics. 2004;11(6):517–546. doi: 10.1097/01.mjt.0000140648.30948.bd. [DOI] [PubMed] [Google Scholar]
- 54.Highet AR. An infectious aetiology of sudden infant death syndrome. Journal of Applied Microbiology. 2008;105(3):625–635. doi: 10.1111/j.1365-2672.2008.03747.x. [DOI] [PubMed] [Google Scholar]
- 55.Blackwell CC, Moscovis SM, Gordon AE, et al. Cytokine responses and sudden infant death syndrome: genetic, developmental, and environmental risk factors. Journal of Leukocyte Biology. 2005;78(6):1242–1254. doi: 10.1189/jlb.0505253. [DOI] [PubMed] [Google Scholar]
- 56.Dashash M, Pravica V, Hutchinson IV, Barson AJ, Drucker DB. Association of sudden infant death syndrome with VEGF and IL-6 gene polymorphisms. Human Immunology. 2006;67(8):627–633. doi: 10.1016/j.humimm.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 57.Jones KL, Krous HF, Nadeau J, Blackbourne B, Zielke HR, Gozal D. Vascular endothelial growth factor in the cerebrospinal fluid of infants who died of sudden infant death syndrome: evidence for antecedent hypoxia. Pediatrics. 2003;111(2):358–363. doi: 10.1542/peds.111.2.358. [DOI] [PubMed] [Google Scholar]
- 58.Narita N, Narita M, Takashima S, Nakayama M, Nagai T, Okado N. Serotonin transporter gene variation is a risk factor for sudden infant death syndrome in the Japanese population. Pediatrics. 2001;107(4):690–692. doi: 10.1542/peds.107.4.690. [DOI] [PubMed] [Google Scholar]
- 59.Paterson DS, Rivera KD, Broadbelt KG, et al. Lack of association of the serotonin transporter polymorphism with the sudden infant death syndrome in the San Diego Dataset. Pediatric Research. 2010;68(5):409–413. doi: 10.1203/PDR.0b013e3181f2edf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kinney HC, Filiano JJ, White WF. Medullary serotonergic network deficiency in the sudden infant death syndrome: review of a 15-year study of a single dataset. Journal of Neuropathology and Experimental Neurology. 2001;60(3):228–247. doi: 10.1093/jnen/60.3.228. [DOI] [PubMed] [Google Scholar]
- 61.Paterson DS, Hilaire G, Weese-Mayer DE. Medullary serotonin defects and respiratory dysfunction in sudden infant death syndrome. Respiratory Physiology and Neurobiology. 2009;168(1-2):133–143. doi: 10.1016/j.resp.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kinney HC, Richerson GB, Dymecki SM, Darnall RA, Nattie EE. The brainstem and serotonin in the sudden infant death syndrome. Annual Review of Pathology. 2009;4:517–550. doi: 10.1146/annurev.pathol.4.110807.092322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weese-Mayer DE, Berry-Kravis EM, Zhou L, et al. Sudden infant death syndrome: case-control frequency differences at genes pertinent to early autonomic nervous system embryologic development. Pediatric Research. 2004;56(3):391–395. doi: 10.1203/01.PDR.0000136285.91048.4A. [DOI] [PubMed] [Google Scholar]
- 64.Livolsi A, Niederhoffer N, Dali-Youcef N, et al. Cardiac muscarinic receptor overexpression in sudden infant death syndrome. PLoS ONE. 2010;5(3) doi: 10.1371/journal.pone.0009464.e9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Livolsi A, Niederhoffer N, Dali-Youcef N, et al. Constitutive overexpression of muscarinic receptors leads to vagal hyperreactivity. PLoS ONE. 2010;5(12) doi: 10.1371/journal.pone.0015618.e15618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burchell A, Bell JE, Busuttil A, Hume R. Hepatic microsomal glucose-6-phosphatase system and sudden infant death syndrome. The Lancet. 1989;2(8658):291–294. doi: 10.1016/s0140-6736(89)90484-4. [DOI] [PubMed] [Google Scholar]
- 67.Forsyth L, Hume R, Howatson A, Busuttil A, Burchell A. Identification of novel polymorphisms in the glucokinase and glucose-6-phosphatase genes in infants who died suddenly and unexpectedly. Journal of Molecular Medicine. 2005;83(8):610–618. doi: 10.1007/s00109-005-0666-0. [DOI] [PubMed] [Google Scholar]
- 68.Forsyth L, Scott HM, Howatson A, Busuttil A, Hume R, Burchell A. Genetic variation in hepatic glucose-6-phosphatase system genes in cases of sudden infant death syndrome. Journal of Pathology. 2007;212(1):112–120. doi: 10.1002/path.2147. [DOI] [PubMed] [Google Scholar]
- 69.Fraser GR, Froggatt P. Unexpected cot deaths. The Lancet. 1966;288(7453):56–57. [Google Scholar]
- 70.Keeton BR, Southall E, Rutter N, Anderson RH, Shinebourne EA, Southall DP. Cardiac conduction disorders in six infants with “near-miss” sudden infant deaths. BMJ. 1977;2(6087):600–601. doi: 10.1136/bmj.2.6087.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smits JPP, Veldkamp MW, Wilde AAM. Mechanisms of inherited cardiac conduction disease. Europace. 2005;7(2):122–137. doi: 10.1016/j.eupc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 72.Remme CA, Wilde AAM, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends in Cardiovascular Medicine. 2008;18(3):78–87. doi: 10.1016/j.tcm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 73.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nature Reviews Cardiology. 2009;6(5):337–348. doi: 10.1038/nrcardio.2009.44. [DOI] [PubMed] [Google Scholar]
- 74.Schwartz PJ. Cardiac sympathetic innervation and the sudden infant death syndrome. a possible pathogenetic link. American Journal of Medicine. 1976;60(2):167–172. doi: 10.1016/0002-9343(76)90425-3. [DOI] [PubMed] [Google Scholar]
- 75.Montague TJ, Finley JP, Mukelabai K, et al. Cardiac rhythm, rate and ventricular repolarization properties in infants at risk for sudden infant death syndrome: comparison with age- and sex-matched control infants. American Journal of Cardiology. 1984;54(3):301–307. doi: 10.1016/0002-9149(84)90187-5. [DOI] [PubMed] [Google Scholar]
- 76.Haddad GG, Epstein MAF, Epstein RA. The QT interval in aborted sudden infant death syndrome infants. Pediatric Research. 1979;13(2):136–138. doi: 10.1203/00006450-197902000-00010. [DOI] [PubMed] [Google Scholar]
- 77.Leistner HL, Haddad GG, Epstein RA, Lai TL, Epstein MAF, Mellins RB. Heart rate and heart rate variability during sleep in aborted sudden infant death syndrome. Journal of Pediatrics. 1980;97(1):51–55. doi: 10.1016/s0022-3476(80)80129-6. [DOI] [PubMed] [Google Scholar]
- 78.Moss AJ, Kass RS. Long QT syndrome: from channels to cardiac arrhythmias. The Journal of Clinical Investigation. 2005;115(8):2018–2024. doi: 10.1172/JCI25537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roden DM. Long-QT syndrome. The New England Journal of Medicine. 2008;358(2):169–176. doi: 10.1056/NEJMcp0706513. [DOI] [PubMed] [Google Scholar]
- 80.Maron BJ, Clark CE, Goldstein RE, Epstein SE. Potential role of QT interval prolongation in sudden infant death syndrome. Circulation. 1976;54(3):423–430. doi: 10.1161/01.cir.54.3.423. [DOI] [PubMed] [Google Scholar]
- 81.Maron BJ, Barbour DJ, Marraccini JV, Roberts WC. Sudden unexpected death 12 years after “near-miss” sudden infant death syndrome in infancy. American Journal of Cardiology. 1986;58(11):1104–1105. doi: 10.1016/0002-9149(86)90122-0. [DOI] [PubMed] [Google Scholar]
- 82.Froggatt P, James TN. Sudden unexpected death in infants. Evidence on a lethal cardiac arrhythmia. Ulster Medical Journal. 1973;42(2):136–152. [PMC free article] [PubMed] [Google Scholar]
- 83.Kelly DH, Shannon DC, Liberthson RR. The role of the QT interval in the sudden infant death syndrome. Circulation. 1977;55(4):633–635. doi: 10.1161/01.cir.55.4.633. [DOI] [PubMed] [Google Scholar]
- 84.Kukolich MK, Telsey A, Ott J, Motulsky AG. Sudden infant death syndrome: normal QT interval on ECGs of relatives. Pediatrics. 1977;60(1):51–54. [PubMed] [Google Scholar]
- 85.Steinschneider A. Sudden infant death syndrome and prolongation of the QT interval. American Journal of Diseases of Children. 1978;132(7):688–691. doi: 10.1001/archpedi.1978.02120320048011. [DOI] [PubMed] [Google Scholar]
- 86.Weinstein SL, Steinschneider A. QTc and R-R intervals in victims of the sudden infant death syndrome. American Journal of Diseases of Children. 1985;139(10):987–990. doi: 10.1001/archpedi.1985.02140120033023. [DOI] [PubMed] [Google Scholar]
- 87.Southall DP, Arrowsmith WA, Stebbens V, Alexander JR. QT interval measurements before sudden infant death syndrome. Archives of Disease in Childhood. 1986;61(4):327–333. doi: 10.1136/adc.61.4.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Southall DP, Richards JM, Rhoden KJ, et al. Prolonged apnea and cardiac arrhythmias in infants discharged from neonatal intensive care units: failure to predict an increased risk for sudden infant death syndrome. Pediatrics. 1982;70(6):844–851. [PubMed] [Google Scholar]
- 89.Southall DP, Richards JM, de Swiet M, et al. Identification of infants destined to die unexpectedly during infancy: evaluation of predictive importance of prolonged apnoea and disorders of cardiac rhythm or conduction. BMJ. 1983;286(6371):1092–1096. doi: 10.1136/bmj.286.6371.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Southall DP, Arrowsmith WA, Oakley JR, McEnery G, Anderson RH, Shinebourne EA. Prolonged QT interval and cardiac arrhythmias in two neonates: sudden infant death syndrome in one case. Archives of Disease in Childhood. 1979;54(10):776–779. doi: 10.1136/adc.54.10.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schwartz PJ, Montemerlo M, Facchini M, et al. The QT interval throughout the first 6 months of life: a prospective study. Circulation. 1982;66(3):496–501. doi: 10.1161/01.cir.66.3.496. [DOI] [PubMed] [Google Scholar]
- 92.Sadeh D, Shannon DC, Abboud S, Saul JP, Akselrod S, Cohen RJ. Altered cardiac repolarization in some victims of sudden infant death syndrome. The New England Journal of Medicine. 1987;317(24):1501–1505. doi: 10.1056/NEJM198712103172404. [DOI] [PubMed] [Google Scholar]
- 93.Guntheroth WG. Theories of cardiovascular causes in sudden infant death syndrome. Journal of the American College of Cardiology. 1989;14(2):443–447. doi: 10.1016/0735-1097(89)90200-3. [DOI] [PubMed] [Google Scholar]
- 94.Schwartz PJ, Stramba-Badiale M, Segantini A, et al. Prolongation of the QT interval and the sudden infant death syndrome. The New England Journal of Medicine. 1998;338(24):1709–1714. doi: 10.1056/NEJM199806113382401. [DOI] [PubMed] [Google Scholar]
- 95.Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120(18):1761–1767. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schulze-Bahr E. Long QT syndromes: genetic basis. Cardiac Electrophysiology Clinics. 2012;4(1):1–16. [Google Scholar]
- 97.Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circulation Arrhythmia and Electrophysiology. 2012;5(4):868–877. doi: 10.1161/CIRCEP.111.962019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval, and sudden death. American Heart Journal. 1957;54(1):59–68. doi: 10.1016/0002-8703(57)90079-0. [DOI] [PubMed] [Google Scholar]
- 99.Mönnig G, Schulze-Bahr E, Wedekind H, et al. Klinik und Molekulargenetik des Jervell- und Lange-Nielsen-Syndroms. Zeitschrift für Kardiologie. 2002;91(5):380–388. doi: 10.1007/s00392-002-0789-z. [DOI] [PubMed] [Google Scholar]
- 100.Splawski I, Timothy KW, Michael Vincent G, Atkinson DL, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. The New England Journal of Medicine. 1997;336(22):1562–1567. doi: 10.1056/NEJM199705293362204. [DOI] [PubMed] [Google Scholar]
- 101.Schulze-Bahr E, Wang Q, Wedekind H, et al. KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nature Genetics. 1997;17(3):267–268. doi: 10.1038/ng1197-267. [DOI] [PubMed] [Google Scholar]
- 102.Lang F, Vallon V, Knipper M, Wangemann P. Functional significance of channels and transporters expressed in the inner ear and kidney. American Journal of Physiology–Cell Physiology. 2007;293(4):C1187–C1208. doi: 10.1152/ajpcell.00024.2007. [DOI] [PubMed] [Google Scholar]
- 103.Andersen ED, Krasilnikoff PA, Overvad H. Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. A new syndrome? Acta Paediatrica Scandinavica. 1971;60(5):559–564. doi: 10.1111/j.1651-2227.1971.tb06990.x. [DOI] [PubMed] [Google Scholar]
- 104.Tawil R, Ptacek LJ, Pavlakis SG, et al. Andersen’s syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Annals of Neurology. 1994;35(3):326–330. doi: 10.1002/ana.410350313. [DOI] [PubMed] [Google Scholar]
- 105.Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105(4):511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 106.Jongsma HJ, Wilders R. Channelopathies: Kir2.1 mutations jeopardize many cell functions. Current Biology. 2001;11(18):R747–R750. doi: 10.1016/s0960-9822(01)00437-7. [DOI] [PubMed] [Google Scholar]
- 107.Tristani-Firouzi M, Jensen JL, Donaldson MR, et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome) The Journal of Clinical Investigation. 2002;110(3):381–388. doi: 10.1172/JCI15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Splawski I, Timothy KW, Sharpe LM, et al. Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 109.Splawski I, Timothy KW, Decher N, et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(23):8089–8096. doi: 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bidaud I, Lory P. Hallmarks of the channelopathies associated with L-type calcium channels: a focus on the Timothy mutations in Cav1.2 channels. Biochimie. 2011;93(12):2080–2086. doi: 10.1016/j.biochi.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 111.Liao P, Soong TW. Cav1.2 channelopathies: from arrhythmias to autism, bipolar disorder, and immunodeficiency. Pflügers Archiv European Journal of Physiology. 2010;460(2):353–359. doi: 10.1007/s00424-009-0753-0. [DOI] [PubMed] [Google Scholar]
- 112.Zaklyazminskaya EV, Abriel H. Prevalence of significant genetic variants in congenital long QT syndrome is largely underestimated. Frontiers in Pharmacology. 2012;3, article 73 doi: 10.3389/fphar.2012.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Winkel BG, Larsen MK, Berge KE, et al. The prevalence of mutations in KCNQ1, KCNH2, and SCN5A in an unselected national cohort of young sudden unexplained death cases. Journal of Cardiovascular Electrophysiology. 2012;23(10):1092–1098. doi: 10.1111/j.1540-8167.2012.02371.x. [DOI] [PubMed] [Google Scholar]
- 114.Kapplinger JD, Tester DJ, Salisbury BA, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6(9):1297–1303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT Syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2 . Circulation. 2000;102(10):1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 116.Napolitano C, Priori SG, Schwartz PJ, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. Journal of the American Medical Association. 2005;294(23):2975–2980. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 117.Zareba W, Moss AJ, Schwartz PJ, et al. Influence of the genotype on the clinical course of the long-QT syndrome. The New England Journal of Medicine. 1998;339(14):960–965. doi: 10.1056/NEJM199810013391404. [DOI] [PubMed] [Google Scholar]
- 118.Blaufox AD, Tristani-Firouzi M, Seslar S, et al. Congenital long QT 3 in the pediatric population. American Journal of Cardiology. 2012;109(10):1459–1465. doi: 10.1016/j.amjcard.2012.01.361. [DOI] [PubMed] [Google Scholar]
- 119.Hedley PL, Jørgensen P, Schlamowitz S, et al. The genetic basis of Brugada syndrome: a mutation update. Human Mutation. 2009;30(9):1256–1266. doi: 10.1002/humu.21066. [DOI] [PubMed] [Google Scholar]
- 120.Campuzano O, Brugada R, Iglesias A. Genetics of Brugada syndrome. Current Opinion in Cardiology. 2010;25(3):210–215. doi: 10.1097/HCO.0b013e32833846ee. [DOI] [PubMed] [Google Scholar]
- 121.Berne P, Brugada J. Brugada syndrome 2012. Circulation Journal. 2012;76(7):1563–1571. doi: 10.1253/circj.cj-12-0717. [DOI] [PubMed] [Google Scholar]
- 122.Mizusawa Y, Wilde AAM. Brugada syndrome. Circulation Arrhythmia and Electrophysiology. 2012;5(3):606–616. doi: 10.1161/CIRCEP.111.964577. [DOI] [PubMed] [Google Scholar]
- 123.Antzelevitch C. The Brugada syndrome: ionic basis and arrhythmia mechanisms. Journal of Cardiovascular Electrophysiology. 2001;12(2):268–272. doi: 10.1046/j.1540-8167.2001.00268.x. [DOI] [PubMed] [Google Scholar]
- 124.Meregalli PG, Wilde AAM, Tan HL. Pathophysiological mechanisms of Brugada syndrome: depolarization disorder, repolarization disorder, or more? Cardiovascular Research. 2005;67(3):367–378. doi: 10.1016/j.cardiores.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 125.Wilde AAM, Postema PG, Di Diego JM, et al. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. Journal of Molecular and Cellular Cardiology. 2010;49(4):543–553. doi: 10.1016/j.yjmcc.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hoogendijk MG, Opthof T, Postema PG, Wilde AAM, de Bakker JMT, Coronel R. The Brugada ECG pattern a marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the Brugada syndrome. Circulation Arrhythmia and Electrophysiology. 2010;3(3):283–290. doi: 10.1161/CIRCEP.110.937029. [DOI] [PubMed] [Google Scholar]
- 127.Postema PG. About Brugada syndrome and its prevalence. Europace. 2012;14(7):925–928. doi: 10.1093/europace/eus042. [DOI] [PubMed] [Google Scholar]
- 128.Crotti L, Marcou CA, Tester DJ, et al. Spectrum and prevalence of mutations involving BrS1- through BrS12-susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: implications for genetic testing. Journal of the American College of Cardiology. 2012;60(15):1410–1418. doi: 10.1016/j.jacc.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ohno S, Zankov DP, Ding WG, et al. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. American Journal of Physiology–Heart and Circulatory Physiology. 2011;4(3):352–361. doi: 10.1161/CIRCEP.110.959619. [DOI] [PubMed] [Google Scholar]
- 130.Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7(1):33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Burashnikov E, Pfeiffer R, Barajas-Martinez H, et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7(12):1872–1882. doi: 10.1016/j.hrthm.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hong K, Hu J, Yu J, Brugada R. Concomitant Brugada-like and short QT electrocardiogram linked to SCN5A mutation. European Journal of Human Genetics. 2012;20(11):1189–1192. doi: 10.1038/ejhg.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Holst AG, Saber S, Houshmand M, et al. Sodium current and potassium transient outward current genes in Brugada syndrome: screening and bioinformatics. Canadian Journal of Cardiology. 2012;28(2):196–200. doi: 10.1016/j.cjca.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 135.Kattygnarath D, Maugenre S, Neyroud N, et al. MOG1: a new susceptibility gene for Brugada syndrome. Circulation Cardiovascular Genetics. 2011;4(3):261–268. doi: 10.1161/CIRCGENETICS.110.959130. [DOI] [PubMed] [Google Scholar]
- 136.Giudicessi JR, Ye D, Tester DJ, et al. Transient outward current (Ito) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. 2011;8(7):1024–1032. doi: 10.1016/j.hrthm.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Barajas-Martínez H, Hu D, Ferrer T, et al. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8 . Heart Rhythm. 2012;9(4):548–555. doi: 10.1016/j.hrthm.2011.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Olesen MS, Holst AG, Svendsen JH, Haunsø S, Tfelt-Hansen J. SCN1Bb R214Q found in 3 patients: 1 with Brugada syndrome and 2 with lone atrial fibrillation. Heart Rhythm. 2012;9(5):770–773. doi: 10.1016/j.hrthm.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 139.Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. European Heart Journal. 2006;27(20):2440–2447. doi: 10.1093/eurheartj/ehl185. [DOI] [PubMed] [Google Scholar]
- 140.Hedley PL, Jørgensen P, Schlamowitz S, et al. The genetic basis of long QT and short QT syndromes: a mutation update. Human Mutation. 2009;30(11):1486–1511. doi: 10.1002/humu.21106. [DOI] [PubMed] [Google Scholar]
- 141.Patel C, Yan GX, Antzelevitch C. Short QT syndrome: from bench to bedside. Circulation Arrhythmia and Electrophysiology. 2010;3(4):401–408. doi: 10.1161/CIRCEP.109.921056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94(2):99–102. doi: 10.1159/000047299. [DOI] [PubMed] [Google Scholar]
- 143.Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: a familial cause of sudden death. Circulation. 2003;108(8):965–970. doi: 10.1161/01.CIR.0000085071.28695.C4. [DOI] [PubMed] [Google Scholar]
- 144.Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109(1):30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- 145.Bellocq C, van Ginneken ACG, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109(20):2394–2397. doi: 10.1161/01.CIR.0000130409.72142.FE. [DOI] [PubMed] [Google Scholar]
- 146.Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circulation Research. 2005;96(7):800–807. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 147.Templin C, Ghadri JR, Rougier JS, et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6) Euopean Heart Journal. 2011;32(9):1077–1088. doi: 10.1093/eurheartj/ehr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation. 2007;116(7):714–720. doi: 10.1161/CIRCULATIONAHA.106.676551. [DOI] [PubMed] [Google Scholar]
- 149.Funada A, Hayashi K, Ino H, et al. Assessment of QT intervals and prevalence of short QT syndrome in Japan. Clinical Cardiology. 2008;31(6):270–274. doi: 10.1002/clc.20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm. 2009;6(5):652–657. doi: 10.1016/j.hrthm.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 151.Liu N, Ruan Y, Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Progress in Cardiovascular Diseases. 2008;51(1):23–30. doi: 10.1016/j.pcad.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 152.Katz G, Arad M, Eldar M. Catecholaminergic polymorphic ventricular tachycardia from bedside to bench and beyond. Current Problems in Cardiology. 2009;34(1):9–43. doi: 10.1016/j.cpcardiol.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 153.Cerrone M, Napolitano C, Priori SG. Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated to impaired Ca2+ regulation. Heart Rhythm. 2009;6(11):1652–1659. doi: 10.1016/j.hrthm.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 154.Ylänen K, Poutanen T, Hiippala A, Swan H, Korppi M. Catecholaminergic polymorphic ventricular tachycardia. European Journal of Pediatrics. 2010;169(5):535–542. doi: 10.1007/s00431-010-1154-2. [DOI] [PubMed] [Google Scholar]
- 155.Leenhardt A, Denjoy I, Guicheney P. Catecholaminergic polymorphic ventricular tachycardia. Circulation Arrhythmia and Electrophysiology. 2012;5(5):1044–1052. doi: 10.1161/CIRCEP.111.962027. [DOI] [PubMed] [Google Scholar]
- 156.Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103(2):196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 157.Lahat H, Pras E, Olender T, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. American Journal of Human Genetics. 2001;69(6):1378–1384. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Human Molecular Genetics. 2012;21(12):2759–2767. doi: 10.1093/hmg/dds104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bhuiyan ZA, Hamdan MA, Shamsi ETA, et al. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. Journal of Cardiovascular Electrophysiology. 2007;18(10):1060–1066. doi: 10.1111/j.1540-8167.2007.00913.x. [DOI] [PubMed] [Google Scholar]
- 160.Wehrens XHT, Lehnart SE, Huang F, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113(7):829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 161.Cerrone M, Colombi B, Santoro M, et al. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circulation Research. 2005;96(10):e77–e82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 162.Cerrone M, Noujaim SF, Tolkacheva EG, et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circulation Research. 2007;101(10):1039–1048. doi: 10.1161/CIRCRESAHA.107.148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kang G, Giovannone SF, Liu N, et al. Purkinje cells from RyR2 mutant mice are highly arrhythmogenic but responsive to targeted therapy. Circulation Research. 2010;107(4):512–519. doi: 10.1161/CIRCRESAHA.110.221481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.de la Fuente S, van Langen IM, Postma AV, Bikker H, Meijer A. A case of catecholaminergic polymorphic ventricular tachycardia caused by two calsequestrin 2 mutations. Pacing and Clinical Electrophysiology. 2008;31(7):916–919. doi: 10.1111/j.1540-8159.2008.01111.x. [DOI] [PubMed] [Google Scholar]
- 165.Priori SG. Inherited arrhythmogenic diseases: the complexity beyond monogenic disorders. Circulation Research. 2004;94(2):140–145. doi: 10.1161/01.RES.0000115750.12807.7E. [DOI] [PubMed] [Google Scholar]
- 166.Rodríguez-Calvo MS, Brion M, Allegue C, Concheiro L, Carracedo A. Molecular genetics of sudden cardiac death. Forensic Science International. 2008;182(1–3):1–12. doi: 10.1016/j.forsciint.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 167.Kaufman ES. Mechanisms and clinical management of inherited channelopathies: long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, and short QT syndrome. Heart Rhythm. 2009;6(8, supplement):S51–S55. doi: 10.1016/j.hrthm.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 168.Dettmeyer RB, Kandolf R. Cardiomyopathies—misdiagnosed as Sudden Infant Death Syndrome (SIDS) Forensic Science International. 2010;194(1–3):e21–e24. doi: 10.1016/j.forsciint.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 169.Schimpf R, Veltmann C, Wolpert C, Borggrefe M. Arrhythmogenic hereditary syndromes: Brugada syndrome, long QT syndrome, short QT syndrome and CPVT. Minerva Cardioangiologica. 2010;58(6):623–636. [PubMed] [Google Scholar]
- 170.Cerrone M, Cummings S, Alansari T, Alansari T, Priori SG. A clinical approach to inherited arrhythmias. Circulation Cardiovascular Genetics. 2012;5(5):581–590. doi: 10.1161/CIRCGENETICS.110.959429. [DOI] [PubMed] [Google Scholar]
- 171.Chockalingam P, Wilde A. The multifaceted cardiac sodium channel and its clinical implications. Heart. 2012;98(17):1318–1324. doi: 10.1136/heartjnl-2012-301784. [DOI] [PubMed] [Google Scholar]
- 172.Bezzina C, Veldkamp MW, van den Berg MP, et al. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circulation Research. 1999;85(12):1206–1213. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 173.Veldkamp MW, Wilders R, Baartscheer A, Zegers JG, Bezzina CR, Wilde AAM. Contribution of sodium channel mutations to bradycardia and sinus node dysfunction in LQT3 families. Circulation Research. 2003;92(9):976–983. doi: 10.1161/01.RES.0000069689.09869.A8. [DOI] [PubMed] [Google Scholar]
- 174.Postema PG, van den Berg MP, van Tintelen JP, et al. Founder mutations in the Netherlands: SCN5A 1795insD, the first described arrhythmia overlap syndrome and one of the largest and best characterised families worldwide. Netherlands Heart Journal. 2009;17(11):422–428. doi: 10.1007/BF03086296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Smits JPP, Koopmann TT, Wilders R, et al. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. Journal of Molecular and Cellular Cardiology. 2005;38(6):969–981. doi: 10.1016/j.yjmcc.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 176.Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. doi: 10.1016/j.trsl.2012.08.005. Translational Research. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wilders R. Arrhythmogenic right ventricular cardiomyopathy: considerations from in silico experiments. Frontiers in Physiology. 2012;3, article 168 doi: 10.3389/fphys.2012.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Brion M, Allegue C, Santori M, et al. Sarcomeric gene mutations in sudden infant death syndrome (SIDS) Forensic Science International. 2012;219(1–3):278–281. doi: 10.1016/j.forsciint.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 179.Schwartz PJ, Priori SG, Dumaine R, et al. A molecular link between the sudden infant death syndrome and the long-QT syndrome. The New England Journal of Medicine. 2000;343(4):262–267. doi: 10.1056/NEJM200007273430405. [DOI] [PubMed] [Google Scholar]
- 180.Schwartz PJ, Priori SG, Bloise R, et al. Molecular diagnosis in a child with sudden infant death syndrome. The Lancet. 2001;358(9290):1342–1343. doi: 10.1016/S0140-6736(01)06450-9. [DOI] [PubMed] [Google Scholar]
- 181.Wedekind H, Smits JPP, Schulze-Bahr E, et al. De novo mutation in the SCN5A gene associated with early onset of sudden infant death. Circulation. 2001;104(10):1158–1164. doi: 10.1161/hc3501.095361. [DOI] [PubMed] [Google Scholar]
- 182.Ackerman MJ, Siu BL, Sturner WQ, et al. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. Journal of the American Medical Association. 2001;286(18):2264–2269. doi: 10.1001/jama.286.18.2264. [DOI] [PubMed] [Google Scholar]
- 183.Berecki G, Zegers JG, Bhuiyan ZA, Verkerk AO, Wilders R, van Ginneken ACG. Long-QT syndrome-related sodium channel mutations probed by the dynamic action potential clamp technique. Journal of Physiology. 2006;570, part 2:237–250. doi: 10.1113/jphysiol.2005.096578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Christiansen M, Tønder N, Larsen LA, et al. Mutations in the HERG K+-ion channel: a novel link between long QT syndrome and sudden infant death syndrome. American Journal of Cardiology. 2005;95(3):433–434. doi: 10.1016/j.amjcard.2004.09.054. [DOI] [PubMed] [Google Scholar]
- 185.Nof E, Cordeiro JM, Pérez GJ, et al. A common single nucleotide polymorphism can exacerbate long-QT type 2 syndrome leading to sudden infant death. Circulation Cardiovascular Genetics. 2010;3(2):199–206. doi: 10.1161/CIRCGENETICS.109.898569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Skinner JR, Chung SK, Montgomery D, et al. Near-miss SIDS due to Brugada syndrome. Archives of Disease in Childhood. 2005;90(5):528–529. doi: 10.1136/adc.2004.058115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Vatta M, Dumaine R, Varghese G, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Human Molecular Genetics. 2002;11(3):337–345. doi: 10.1093/hmg/11.3.337. [DOI] [PubMed] [Google Scholar]
- 188.Wang Q, Chen S, Chen Q, et al. The common SCN5A mutation R1193Q causes LQTS-type electrophysiological alterations of the cardiac sodium channel. Journal of Medical Genetics. 2004;41(5, article e66) doi: 10.1136/jmg.2003.013300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Turillazzi E, La Rocca G, Anzalone R, et al. Heterozygous nonsense SCN5A mutation W822X explains a simultaneous sudden infant death syndrome. Virchows Archiv. 2008;453(2):209–216. doi: 10.1007/s00428-008-0632-7. [DOI] [PubMed] [Google Scholar]
- 190.Keller DI, Barrane FZ, Gouas L, et al. A novel nonsense mutation in the SCN5A gene leads to Brugada syndrome and a silent gene mutation carrier state. Canadian Journal of Cardiology. 2005;21(11):925–931. [PubMed] [Google Scholar]
- 191.Huang H, Millat G, Rodriguez-Lafrasse C, et al. Biophysical characterization of a new SCN5A mutation S1333Y in a SIDS infant linked to long QT syndrome. FEBS Letters. 2009;583(5):890–896. doi: 10.1016/j.febslet.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 192.Priori SG, Napolitano C, Giordano U, Collisani G, Memmi M. Brugada syndrome and sudden cardiac death in children. The Lancet. 2000;355(9206):808–809. doi: 10.1016/S0140-6736(99)05277-0. [DOI] [PubMed] [Google Scholar]
- 193.Wan X, Chen S, Sadeghpour A, Wang Q, Kirsch GE. Accelerated inactivation in a mutant Na+ channel associated with idiopathic ventricular fibrillation. American Journal of Physiology–Heart and Circulatory Physiology. 2001;280(1):H354–H360. doi: 10.1152/ajpheart.2001.280.1.H354. [DOI] [PubMed] [Google Scholar]
- 194.Chockalingam P, Rammeloo LA, Postema PG, et al. Fever-induced life-threatening arrhythmias in children harboring an SCN5A mutation. Pediatrics. 2011;127(1):e239–e244. doi: 10.1542/peds.2010-1688. [DOI] [PubMed] [Google Scholar]
- 195.Tester DJ, Ackerman MJ. Sudden infant death syndrome: how significant are the cardiac channelopathies? Cardiovascular Research. 2005;67(3):388–396. doi: 10.1016/j.cardiores.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 196.Wedekind H, Bajanowski T, Friederich P, et al. Sudden infant death syndrome and long QT syndrome: an epidemiological and genetic study. International Journal of Legal Medicine. 2006;120(3):129–137. doi: 10.1007/s00414-005-0019-0. [DOI] [PubMed] [Google Scholar]
- 197.Plant LD, Bowers PN, Liu Q, et al. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. The Journal of Clinical Investigation. 2006;116(2):430–435. doi: 10.1172/JCI25618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Cronk LB, Ye B, Kaku T, et al. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007;4(2):161–166. doi: 10.1016/j.hrthm.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Arnestad M, Crotti L, Rognum TO, et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115(3):361–367. doi: 10.1161/CIRCULATIONAHA.106.658021. [DOI] [PubMed] [Google Scholar]
- 200.Tester DJ, Dura M, Carturan E, et al. A mechanism for sudden infant death syndrome (SIDS): stress-induced leak via ryanodine receptors. Heart Rhythm. 2007;4(6):733–739. doi: 10.1016/j.hrthm.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.van Norstrand DW, Valdivia CR, Tester DJ, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1-like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. 2007;116(20):2253–2259. doi: 10.1161/CIRCULATIONAHA.107.704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Otagiri T, Kijima K, Osawa M, et al. Cardiac ion channel gene mutations in sudden infant death syndrome. Pediatric Research. 2008;64(5):482–487. doi: 10.1203/PDR.0b013e3181841eca. [DOI] [PubMed] [Google Scholar]
- 203.Millat G, Kugener B, Chevalier P, et al. Contribution of long-QT syndrome genetic variants in sudden infant death syndrome. Pediatric Cardiology. 2009;30(4):502–509. doi: 10.1007/s00246-009-9417-2. [DOI] [PubMed] [Google Scholar]
- 204.Cheng J, van Norstrand DW, Medeiros-Domingo A, et al. α1-syntrophin mutations identified in sudden infant death syndrome cause an increase in late cardiac sodium current. Circulation Arrhythmia and Electrophysiology. 2009;2(6):667–676. doi: 10.1161/CIRCEP.109.891440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tan BH, Pundi KN, van Norstrand DW, et al. Sudden infant death syndrome-associated mutations in the sodium channel beta subunits. Heart Rhythm. 2010;7(6):771–778. doi: 10.1016/j.hrthm.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Tester DJ, Tan BH, Medeiros-Domingo A, Song C, Makielski JC, Ackerman MJ. Loss-of-function mutations in the KCNJ8-encoded Kir6.1 KATP channel and sudden infant death syndrome. Circulation Cardiovascular Genetics. 2011;4(5):510–515. doi: 10.1161/CIRCGENETICS.111.960195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hu D, Barajas-Martínez H, Medeiros-Domingo A, et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Nav1.5 and Kv4.3 channel currents. Heart Rhythm. 2012;9(5):760–769. doi: 10.1016/j.hrthm.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.van Norstrand DW, Asimaki A, Rubinos C, et al. Connexin43 mutation causes heterogeneous gap junction loss and sudden infant death. Circulation Arrhythmia and Electrophysiology. 2012;125(3):474–481. doi: 10.1161/CIRCULATIONAHA.111.057224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Giudicessi JR, Ye D, Kritzberger CJ, et al. Novel mutations in the KCND3-encoded Kv4.3 K+ channel associated with autopsy-negative sudden unexplained death. Human Mutation. 2012;33(6):989–997. doi: 10.1002/humu.22058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Klaver EC, Versluijs GM, Wilders R. Cardiac ion channel mutations in the sudden infant death syndrome. International Journal of Cardiology. 2011;152(2):162–170. doi: 10.1016/j.ijcard.2010.12.051. [DOI] [PubMed] [Google Scholar]
- 211.Splawski I, Timothy KW, Tateyama M, et al. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297(5585):1333–1336. doi: 10.1126/science.1073569. [DOI] [PubMed] [Google Scholar]
- 212.Vatta M, Ackerman MJ, Ye B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114(20):2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 213.Wang DW, Desai RR, Crotti L, et al. Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation. 2007;115(3):368–376. doi: 10.1161/CIRCULATIONAHA.106.646513. [DOI] [PubMed] [Google Scholar]
- 214.Sharma D, Glatter KA, Timofeyev V, et al. Characterization of a KCNQ1/KVLQT1 polymorphism in Asian families with LQT2: implications for genetic testing. Journal of Molecular and Cellular Cardiology. 2004;37(1):79–89. doi: 10.1016/j.yjmcc.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 215.Rhodes TE, Abraham RL, Welch RC, et al. Cardiac potassium channel dysfunction in sudden infant death syndrome. Journal of Molecular and Cellular Cardiology. 2008;44(3):571–581. doi: 10.1016/j.yjmcc.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Tester DJ, Spoon DB, Valdivia HH, Makielski JC, Ackerman MJ. Targeted mutational analysis of the RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: a molecular autopsy of 49 medical examiner/coroner's cases. Mayo Clinic Proceedings. 2004;79(11):1380–1384. doi: 10.4065/79.11.1380. [DOI] [PubMed] [Google Scholar]
- 217.Mathur N, Sood S, Wang S, et al. Sudden infant death syndrome in mice with an inherited mutation in RyR2 . Circulation Arrhythmia and Electrophysiology. 2009;2(6):677–685. doi: 10.1161/CIRCEP.109.894683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tester DJ, Kopplin LJ, Will ML, Ackerman MJ. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm. 2005;2(10):1099–1105. doi: 10.1016/j.hrthm.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 219.London B, Michalec M, Mehdi H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1-like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116(20):2260–2268. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Valdivia CR, Ueda K, Ackerman MJ, Makielski JC. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A . American Journal of Physiology–Heart and Circulatory Physiology. 2009;297(4):H1446–H1452. doi: 10.1152/ajpheart.00513.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Liu M, Sanyal S, Gao G, et al. Cardiac Na+ current regulation by pyridine nucleotides. Circulation Research. 2009;105(8):737–745. doi: 10.1161/CIRCRESAHA.109.197277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7(1):33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Itoh H, Shimizu W, Hayashi K, et al. Long QT syndrome with compound mutations is associated with a more severe phenotype: a Japanese multicenter study. Heart Rhythm. 2010;7(10):1411–1418. doi: 10.1016/j.hrthm.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 224.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2(5):507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 225.Kapa S, Tester DJ, Salisbury BA, et al. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120(18):1752–1760. doi: 10.1161/CIRCULATIONAHA.109.863076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Ye D, Ackerman MJ. Role of heterologous expression studies in distinguishing pathogenic SCN5A mutations from background genetic noise. Heart Rhythm. 2009;6(supplement 1):S229–S230. [Google Scholar]
- 227.Ueda K, Valdivia C, Medeiros-Domingo A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(27):9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wu G, Ai T, Kim JJ, et al. α-1-syntrophin mutation and the long-QT syndrome: a disease of sodium channel disruption. Circulation Arrhythmia and Electrophysiology. 2008;1(3):193–201. doi: 10.1161/CIRCEP.108.769224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Qin N, D’Andrea MR, Lubin ML, Shafaee N, Codd EE, Correa AM. Molecular cloning and functional expression of the human sodium channel β 1B subunit, a novel splicing variant of the β 1 subunit. European Journal of Biochemistry. 2003;270(23):4762–4770. doi: 10.1046/j.1432-1033.2003.03878.x. [DOI] [PubMed] [Google Scholar]
- 230.Jansen JA, van Veen TAB, de Bakker JMT, van Rijen HVM. Cardiac connexins and impulse propagation. Journal of Molecular and Cellular Cardiology. 2010;48(1):76–82. doi: 10.1016/j.yjmcc.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 231.Watanabe H, Yang T, Stroud DM, et al. Striking in vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation. 2011;124(9):1001–1011. doi: 10.1161/CIRCULATIONAHA.110.987248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circulation Research. 1999;85(9):803–809. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- 233.Keller DI, Huang H, Zhao J, et al. A novel SCN5A mutation, F1344S, identified in a patient with Brugada syndrome and fever-induced ventricular fibrillation. Cardiovascular Research. 2006;70(3):521–529. doi: 10.1016/j.cardiores.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 234.Amin AS, Herfst LJ, Delisle BP, et al. Fever-induced QTc prolongation and ventricular arrhythmias in individuals with type 2 congenital long QT syndrome. The Journal of Clinical Investigation. 2008;118(7):2552–2561. doi: 10.1172/JCI35337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lim SM, Pak HN, Lee MH, et al. Fever-induced QTc prolongation and ventricular fibrillation in a healthy young man. Yonsei Medical Journal. 2011;52(6):1025–1027. doi: 10.3349/ymj.2011.52.6.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Chockalingam P, Clur SAB, Breur JMPJ, et al. The diagnostic and therapeutic aspects of loss-of-function cardiac sodium channelopathies in children. Heart Rhythm. 2012;9(12):1986–1992. doi: 10.1016/j.hrthm.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 237.Kanter RJ, Pfeiffer R, Hu D, Barajas-Martinez H, Carboni MP, Antzelevitch C. Brugada-like syndrome in infancy presenting with rapid ventricular tachycardia and intraventricular conduction delay. Circulation. 2012;125(1):14–22. doi: 10.1161/CIRCULATIONAHA.111.054007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Koopmann TT, Alders M, Jongbloed RJ, et al. Long QT syndrome caused by a large duplication in the KCNH2 (HERG) gene undetectable by current polymerase chain reaction-based exon-scanning methodologies. Heart Rhythm. 2006;3(1):52–55. doi: 10.1016/j.hrthm.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 239.Eddy CA, MacCormick JM, Chung SK, et al. Identification of large gene deletions and duplications in KCNQ1 and KCNH2 in patients with long QT syndrome. Heart Rhythm. 2008;5(9):1275–1281. doi: 10.1016/j.hrthm.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 240.Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. Journal of the American College of Cardiology. 2009;54(22):2065–2074. doi: 10.1016/j.jacc.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Tester DJ, Benton AJ, Train L, Deal B, Baudhuin LM, Ackerman MJ. Prevalence and spectrum of large deletions or duplications in the major long QT syndrome-susceptibility genes and implications for long QT syndrome genetic testing. American Journal of Cardiology. 2010;106(8):1124–1128. doi: 10.1016/j.amjcard.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Medlock MM, Tester DJ, Will ML, Bos JM, Ackerman MJ. Repeat long QT syndrome genetic testing of phenotype-positive cases: prevalence and etiology of detection misses. Heart Rhythm. 2012;9(12):1977–1982. doi: 10.1016/j.hrthm.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 243.Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103(1):89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 244.van Norstrand DW, Tester DJ, Medeiros-Domingo A. The cardiac sodium Nav1.5 channelsome and sudden infant death syndrome. Circulation. 2010;122, supplementA13448 [Google Scholar]
- 245.Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. Journal of the American College of Cardiology. 2007;49(2):240–246. doi: 10.1016/j.jacc.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 246.Gladding PA, Evans CA, Crawford J, et al. Posthumous diagnosis of long QT syndrome from neonatal screening cards. Heart Rhythm. 2010;7(4):481–486. doi: 10.1016/j.hrthm.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 247.Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM, Ackerman MJ. Cardiac channel molecular autopsy: insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clinic Proceedings. 2012;87(6):524–539. doi: 10.1016/j.mayocp.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Cuneo BF, Ovadia M, Strasburger JF, et al. Prenatal diagnosis and in utero treatment of torsades de pointes associated with congenital long QT syndrome. American Journal of Cardiology. 2003;91(11):1395–1398. doi: 10.1016/s0002-9149(03)00343-6. [DOI] [PubMed] [Google Scholar]
- 249.Miller TE, Estrella E, Myerburg RJ, et al. Recurrent third-trimester fetal loss and maternal mosaicism for long-QT syndrome. Circulation. 2004;109(24):3029–3034. doi: 10.1161/01.CIR.0000130666.81539.9E. [DOI] [PubMed] [Google Scholar]
- 250.Tester DJ, Ackerman MJ. Cardiomyopathic and channelopathic causes of sudden unexplained death in infants and children. Annual Review of Medicine. 2009;60:69–84. doi: 10.1146/annurev.med.60.052907.103838. [DOI] [PubMed] [Google Scholar]
- 251.van Norstrand DW, Ackerman MJ. Sudden infant death syndrome: do ion channels play a role? Heart Rhythm. 2009;6(2):272–278. doi: 10.1016/j.hrthm.2008.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.van Norstrand DW, Ackerman MJ. Genomic risk factors in sudden infant death syndrome. Genome Medicine. 2010;2(11, article 86) doi: 10.1186/gm207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Tfelt-Hansen J, Winkel BG, Grunnet M, Jespersen T. Cardiac channelopathies and sudden infant death syndrome. Cardiology. 2011;119(1):21–33. doi: 10.1159/000329047. [DOI] [PubMed] [Google Scholar]
- 254.Cerrone M, Priori SG. Genetics of sudden death: focus on inherited channelopathies. European Heart Journal. 2011;32(17):2109–2118. doi: 10.1093/eurheartj/ehr082. [DOI] [PubMed] [Google Scholar]
- 255.Chopra N, Knollmann BC. Genetics of sudden cardiac death syndromes. Current Opinion in Cardiology. 2011;26(3):196–203. doi: 10.1097/HCO.0b013e3283459893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Insolia R, Ghidoni A, Dossena C, Mastantuono E, Schwartz PJ. Sudden infant death syndrome and cardiac channelopathies: from mechanisms to prevention of avoidable tragedies. Cardiogenetics. 2011;1(supplement 1, article e6) [Google Scholar]
- 257.Opdal SH, Rognum TO. Gene variants predisposing to SIDS: current knowledge. Forensic Science, Medicine, and Pathology. 2011;7(1):26–36. doi: 10.1007/s12024-010-9182-9. [DOI] [PubMed] [Google Scholar]
- 258.Martin CA, Matthews GD, Huang CL. Sudden cardiac death and inherited channelopathy: the basic electrophysiology of the myocyte and myocardium in ion channel disease. Heart. 2012;98(7):536–543. doi: 10.1136/heartjnl-2011-300953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Perrin MJ, Gollob MH. Genetics of cardiac electrical disease. doi: 10.1016/j.cjca.2012.07.847. Canadian Journal of Cardiology. In press. [DOI] [PubMed] [Google Scholar]