

Abstract
Although it is well understood that selection shapes the polymorphism pattern in Drosophila, signatures of classic selective sweeps are scarce. Here, we focus on Drosophila mauritiana, an island endemic, which is closely related to Drosophila melanogaster. Based on a new, annotated genome sequence, we characterized the genome-wide polymorphism by sequencing pooled individuals (Pool-seq). We show that the interplay between selection and recombination results in a genome-wide polymorphism pattern characteristic for D. mauritiana. Two large genomic regions (>500 kb) showed the signature of almost complete selective sweeps. We propose that the absence of population structure and limited geographic distribution could explain why such pronounced sweep patterns are restricted to D. mauritiana. Further evidence for strong adaptive evolution was detected for several nucleoporin genes, some of which were not previously identified as genes involved in genomic conflict. Since this adaptive evolution is continuing after the split of D. mauritiana and Drosophila simulans, we conclude that genomic conflict is not restricted to short episodes, but rather an ongoing process in Drosophila.
Intragenomic conflict describes the phenomenon that within an organism some genetic elements (e.g., segregation distorters) increase their transmission at the expense of others (Werren 2011). Due to the preferential transmission, such elements spread in the population and can leave a characteristic trace of strongly reduced variability in the genome that resembles a selective sweep (Derome et al. 2004). Population genetic analyses of segregation distortion systems in Drosophila did not find a molecular signature similar to a classic selective sweep (Derome et al. 2004, 2008; Presgraves et al. 2009; Kingan et al. 2010; Bastide et al. 2011). The patterns of variability instead resembled partial selective sweeps, suggesting that the genetic element increased in frequency but did not reach fixation. This observation is consistent with the fact that elements of intragenomic conflict are frequently deleterious when homozygous (Wallace 1948; Curtsinger and Feldman 1980) or that suppressors of the intragenomic conflict have evolved (Hamilton 1967).
In the Drosophila melanogaster complex, only a small number of genes involved in intragenomic conflict have been identified within natural populations (e.g., Sandler et al. 1959; Mercot et al. 1995). While this may suggest that intragenomic conflict is a relatively rare event, it needs to be considered that there is a strong ascertainment bias: The rapid spread of driver alleles is either prevented by a quick fixation of suppressor alleles, or, in case of sex chromosome–linked segregation distorters, populations with an advanced intragenomic conflict become extinct (Gershenson 1928; Hamilton 1967; Lyttle 1977). In both cases, past episodes of genomic conflict cannot be recognized in an intraspecific polymorphism analysis.
Indeed, consistent with the idea that genomic conflict is a common phenomenon, detailed analysis of hybrids showed that “speciation” genes tend to be involved in intragenomic conflict, but their effect could be only detected in hybrids (Perez et al. 1993; Dermitzakis et al. 2000; Tao et al. 2001; Presgraves et al. 2003; Phadnis and Orr 2009; Tang and Presgraves 2009).
Several genes involved in intragenomic conflict in Drosophila were discovered in the Drosophila simulans clade that consists of three recently diverged species, the cosmopolitan D. simulans and the island endemics Drosophila mauritiana and Drosophila sechellia. D. mauritiana was the first species for which a “speciation” gene could be characterized at the molecular level: In hybrid crosses with D. simulans, the Odysseus (OdsH) allele of D. mauritiana together with additional tightly linked factors causes hybrid male sterility in the F1 generation (Perez and Wu 1995; Ting et al. 1998) and has been later identified as a gene involved in genomic conflict (Bayes and Malik 2009). Another D. mauritiana gene, too much yin (tmy), causes both, hybrid male sterility and segregation distortion in crosses between D. mauritiana and D. simulans (Tao et al. 2001), whereas the heterochromatic hlx locus causes hybrid lethality between D. mauritiana and both of its sister species (Cattani and Presgraves 2009).
Additional elements of intragenomic conflict have been identified between the more distantly related D. melanogaster and D. simulans, in which the interaction between the genes Hmr and Lhr contributes to hybrid male lethality in crosses between D. melanogaster and D. simulans (Brideau et al. 2006; Maheshwari and Barbash 2012).
The D. simulans alleles of two nucleoporin genes, Nup962 and Nup160, cause recessive male lethality when crossed to a D. melanogaster X chromosome (Presgraves et al. 2003; Tang and Presgraves 2009), a phenomenon that has been also linked to genomic conflict (Presgraves 2007; Presgraves and Stephan 2007). While the NUP96 protein is highly conserved between D. simulans and D. mauritiana, the D. mauritiana allele of Nup96 has no hybrid-lethal effect, which suggests more complex genetic interactions leading to Nup96-dependent incompatibility (Barbash 2007).
Despite the importance of D. mauritiana as a model for understanding the genetic basis of speciation, an annotated genome sequence is not yet available. Using de novo assembly, we generated a draft genome of D. mauritiana and estimated genome-wide polymorphism patterns from Pool-seq data. Our data show the impact of genes involved in genomic conflict on the evolution of the D. mauritiana lineage. Nucleoporin genes, implicated in hybrid incompatibilities that have evolved between D. simulans and D. melanogaster, are possible targets of recurrent positive selection due to ongoing genomic conflict (Presgraves and Stephan 2007). Unlike previous genome-wide polymorphism surveys of D. simulans and D. melanogaster (Begun et al. 2007; Langley et al. 2012), we find that in the D. mauritiana lineage, nucleoporins are among the genes showing the strongest evidence of recurrent adaptive evolution. Furthermore, the presence of a pair of meiotic drive genes and a “speciation” gene at the center of two valleys of strongly reduced variability suggests that these sweeps have been caused by genes involved in genomic conflict.
Results
The recent advances in sequencing technology provide the opportunity to perform population genetic analyses on a genome scale. Even for species with no available reference genome, it has become feasible to generate draft genomes that can be used for population genomic analysis. Here we pursue this strategy for D. mauritiana, for which no annotated reference genome is available yet. We sequenced the D. mauritiana strain MS17 using a mixture of single-end and paired-end Illumina reads (Supplemental Table S1), and assembled and annotated the draft genome (for further details, see Supplemental Results). To study the impact of selection on the polymorphism pattern in D. mauritiana, we sequenced a pool of 107 isofemale lines (Supplemental Table S2).
Faster rate of evolution on the X chromosome
Since the X chromosome is hemizygous in males, rates of sequence evolution can be contrasted between the X chromosome and the autosomes to shed some light on the operating selective forces. Under the assumption that new mutations are recessive, population genetics theory predicts a higher rate of evolution on the X chromosome than on the autosomes (Maynard Smith and Haigh 1974; Charlesworth et al. 1987).
Mean pairwise divergence (Dxy) between D. melanogaster and D. mauritiana is significantly higher on the X chromosome (mean Dxy = 0.0548) than on the major autosomes (mean Dxy of the major autosomal arms = 0.0491, two-tailed Wilcoxon rank-sum test based on nonoverlapping 10-kb windows, P < 2.2 × 10−16). The same pattern is observed for the species pair D. melanogaster and D. simulans (mean Dxy on the X chromosome = 0.0540, mean Dxy on the major autosomes = 0.0483) (Fig. 1; Table 1), which is consistent with the genome-wide data of Begun et al. (2007).
Figure 1.

Mean pairwise divergence (Dxy) along each major chromosomal arm between species of the D. melanogaster complex. The following species pairs are shown: D. mauritiana–D. simulans (yellow), D. sechellia–D. simulans (orange), D. mauritiana–D. sechellia (green), D. simulans–D. melanogaster (gray), and D. mauritiana–D. melanogaster (red). The sliding window analysis was performed using a window size of 500 kb and a step size of 100 kb; chromosomal coordinates are those of D. mauritiana.
Table 1.
Mean pairwise divergence (Dxy) between D. mauritiana, D. simulans, and D. melanogaster based on alignments of repeat-masked genomes
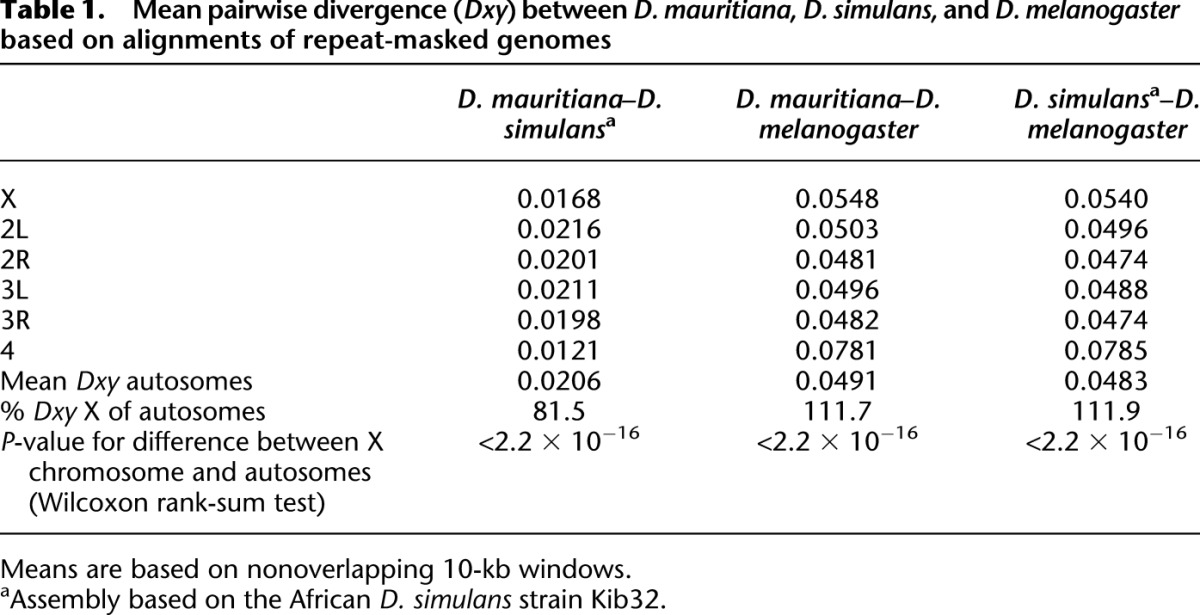
Interestingly, comparisons within the D. simulans clade show the opposite pattern: Mean pairwise divergence between D. mauritiana and D. simulans, for example, is higher on the major autosomal arms (Dxy = 0.0206) than on the X chromosome (Dxy = 0.0168, two-tailed Wilcoxon rank-sum test based on nonoverlapping 10-kb windows, P < 2.2 × 10−16). This pattern of a higher divergence on the autosomes holds for all comparisons among species of the D. simulans clade (Fig. 1; Table 1; Supplemental Table S3) and has been noted previously in the D. simulans–D. sechellia comparison (Singh et al. 2008). Reduced divergence on the X chromosome compared with the autosomes could be explained by hybridization between species of the D. simulans clade (Ballard 2000; Morton et al. 2004; Nunes et al. 2010). If the X chromosome experiences more interspecific gene flow than the autosomes, this would result in a higher divergence on the autosomes. Nevertheless, since Garrigan et al. (2012) found twice as many fragments with a putative introgession signal on the autosomes than on the X chromosome (Garrigan et al. 2012), we consider this scenario not very likely.
Another cause of lower divergence on the X chromosome could be less ancestral polymorphism on the X chromosome than on the autosomes (Singh et al. 2008). Alternatively, selection on the short time scale could be mainly operating on standing variation rather than on new mutations (Orr and Betancourt 2001). Hence, assuming that in the D. simulans clade selection acts mainly on shared standing variation, the time scale may be too short to notice a higher substitution rate on the X chromosome. In contrast, comparisons involving D. melanogaster encompass longer time intervals allowing for more novel mutations and fewer shared mutations, which makes the higher substitution rate on the X chromosome visible.
Impact of the recombination landscape on the partitioning of variation
When comparing levels of polymorphism in D. mauritiana to those in D. melanogaster, we find that D. mauritiana is 40%–50% more variable than a cosmopolitan D. melanogaster population (Table 2; for further details, see Supplemental Results). While a higher level of overall polymorphism has been suggested previously based on a small number of loci (Hey and Kliman 1993; Moriyama and Powell 1996), our Pool-seq data allow us to address the distribution of variability along all chromosomal arms.
Table 2.
Mean nucleotide diversity (π) and mean Tajima's D per chromosomal arm in D. mauritiana compared with the D. melanogaster population from Portugal
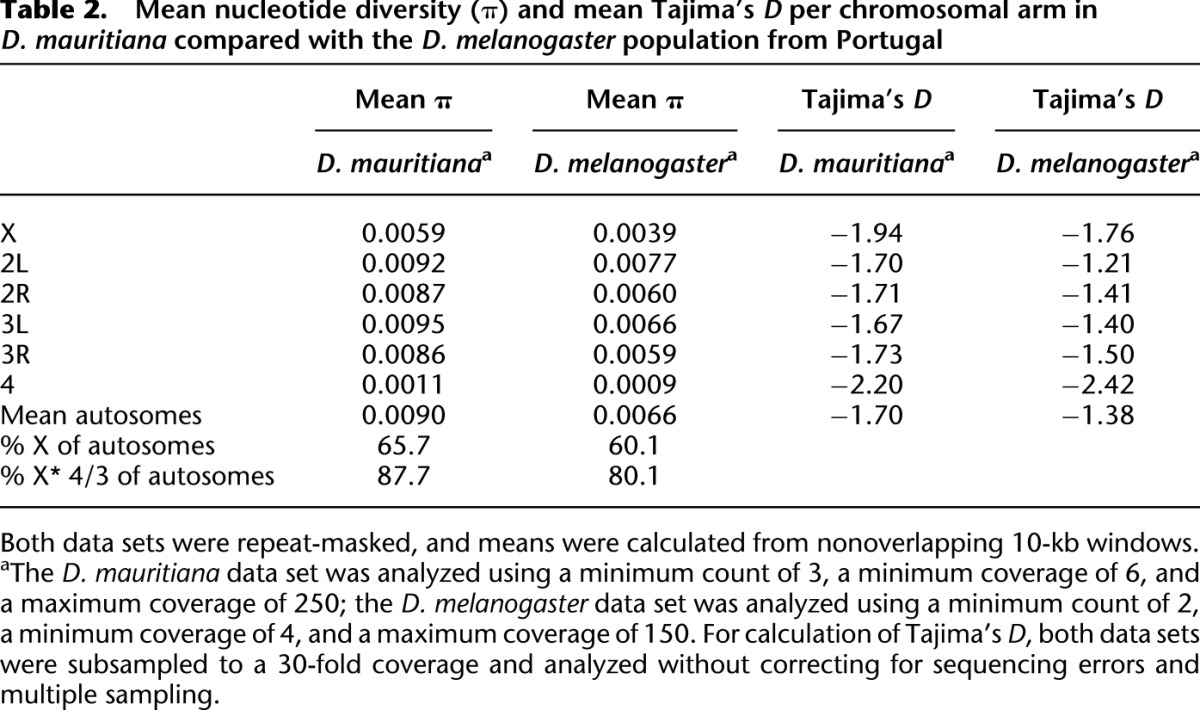
It is well understood that the recombination landscape in D. melanogaster varies along the chromosomes. Both telomeres and centromeres have a reduced recombination rate, but while the drop in recombination rate is abrupt at the telomeres, a gradual decrease in recombination rate over several megabases is observed toward the centromere on all major autosomal arms (True et al. 1996). D. mauritiana not only has a higher genome-wide recombination rate but also shows an important difference in the recombination landscape: Instead of an extended gradual decrease in recombination rate near the centromere, the suppression of recombination is restricted to a very small pericentric region (True et al. 1996).
Since the correlation between recombination rate and variability is well-studied in D. melanogaster (Begun and Aquadro 1992; Hudson 1994), we were interested if the change in recombination landscape affects the pattern of variability in genomic regions toward the centromere. Figure 2 shows that in D. melanogaster, polymorphism declines toward the centromeres, whereas in D. mauritiana, levels of variability remain almost flat throughout the entire chromosome. Moreover, not only is the level of variability reduced in low-recombining regions in D. melanogaster, but the allele frequency spectrum is affected as well. Tajima's D (Tajima 1989) is a frequently used summary statistic, which describes deviations of the allele frequency spectrum from the standard neutral model. We plotted Tajima's D along the D. melanogaster and D. mauritiana chromosomes and observed more negative Tajima's D values toward the centromere in D. melanogaster (Fig. 3). A similar trend was seen for Tajima's D of synonymous sites (Supplemental Fig. S1): Consistent with no reduced recombination rate, Tajima's D remains unaffected by proximity to the centromere for most of the D. mauritiana chromosomes. This shift toward more negative Tajima's D values in low-recombining regions of D. melanogaster is consistent with selection at linked sites affecting neutral variability, either due to recurrent sweeps of favorable mutations (hitchhiking) (Maynard Smith and Haigh 1974) or, possibly, due to background selection, caused by the removal of linked deleterious mutations (Charlesworth et al. 1993).
Figure 2.

Nucleotide diversity (π) along the major chromosomal arms in D. mauritiana (red line) and D. melanogaster (gray line). The sliding window analysis was performed using 500-kb windows with a step size of 100 kb; chromosomal coordinates have been adjusted to D. melanogaster.
Figure 3.

Tajima's D along the major chromosomal arms in D. mauritiana (red line) and D. melanogaster (gray line). The sliding window analysis was performed using 500-kb windows with a step size of 100 kb; chromosomal coordinates have been adjusted to D. melanogaster.
Because low recombination rates will decrease the efficacy of selection, we compared the ratio of nonsynonymous to synonymous polymorphisms along the chromosomes of both species (as in Presgraves 2005; Betancourt et al. 2009). In D. melanogaster, the number of nonsynonymous substitutions relative to synonymous ones increases with the decrease in recombination rate toward the centromere (Presgraves 2005). In D. mauritiana, however, almost no effect can be noticed (Fig. 4).
Figure 4.

Ratio of nonsynonymous and synonymous nucleotide diversity (πN/πS) along the major chromosomal arms in D. mauritiana (red line) and D. melanogaster (gray line). The sliding window analysis was performed using 50 genes per window and a step size of one gene. For D. mauritiana, genes from all four gene sets were included and matched with the orthologous gene in D. melanogaster. Genes with a πN/πS ratio >3 were excluded from both data sets.
The effects of elevated recombination rates in D. mauritiana are further apparent from the patterns of codon usage (for details, see Supplemental Results).
Signatures of positive selection in D. mauritiana
The neutral theory predicts a correlation between polymorphism and divergence. The McDonald–Kreitman test builds on this prediction and compares the ratio of synonymous and nonsynonymous polymorphism to the ratio of synonymous and nonsynonymous divergence; under neutrality, these quantities will be equal (McDonald and Kreitman 1991). Using a polarized McDonald–Kreitman test, we surveyed polymorphism and divergence (from D. melanogaster and Drosophila yakuba) for 10,217 genes in D. mauritiana.
We found 43 genes (FDR ≤ 0.05) that deviated significantly from the neutral expectation in the polarized test of D. mauritiana with D. melanogaster as reference and D. yakuba as outgroup (Supplemental Table S4). A detailed list of significant genes, including those identified by unpolarized versions of the McDonald–Kreitman test, are shown in Supplemental Tables S5–S7.
While several of these genes overlapped with previous studies (for further details, see Supplemental Results), we made three particularly interesting observations in D. mauritiana.
First, we find strong evidence for positive selection for a gene that has been proposed to cause morphological divergence (number of sex comb teeth) between the two sister species D. simulans and D. mauritiana. Graze et al. (2007) identified CD98hc by tissue-specific gene expression differences between both species but did not characterize its molecular evolution. Our analysis suggests that not only regulatory changes but also structural variation (Hoekstra and Coyne 2007) contribute to morphological divergence.
Second, we did not detect an accelerated rate of evolution for olfactory and gustatory receptor (Or and Gr) or Accessory gland protein (Acp) genes, some of which have been found to evolve rapidly in other Drosophila species (Begun and Lindfors 2005; Guo and Kim 2007). While gene families are more likely to be excluded from de novo assemblies, we think that species-specific selection patterns are the better explanation (for further details, see the Supplemental Discussion).
Third, the overrepresentation of nucleoporin genes among the selected genes suggests that genomic conflict may be one major driver of adaptive evolution in D. mauritiana. The most significant Gene Ontology (GO) term for genes significant at FDR 0.05 (43 genes) in the polarized MK test is “SMAD protein import into the nucleus” (Supplemental Table S8; for the GO enrichment analysis of the unpolarized McDonald–Kreitman tests, see Supplemental Tables S9, S10). This pointed to nucleoporin genes, some of which have been previously described to be rapidly evolving and to cause hybrid incompatibility (Presgraves 2003; Presgraves and Stephan 2007; Tang and Presgraves 2009).
Further manual inspection of the top candidates from the polarized test identified three nucleoporin genes. The gene associated with the most significant MK test, CG8771, is a homolog of the human nucleoporin gene Nup188, which is involved in controlling membrane protein traffic and maintenance of nuclear membrane homeostasis (Theerthagiri et al. 2010). The yeast homolog Nup188 plays a role in structural organization of the nuclear pore (Nehrbass et al. 1996; Miller et al. 2000). As expected for a member of a nucleopore complex, CG8771 interacts with several other nucleopore proteins (as indicated in the protein–protein interaction database STRING 9.0) (Jensen et al. 2009). Interestingly, two of the interacting partners, Nup107 and CG11943, a homolog of the human NUP205 gene, were also found among the top 43 candidates. One of them, CG11943, has been previously described as a rapidly evolving gene in a comparison of D. simulans and D. melanogaster (Jagadeeshan and Singh 2005). Both Nup107, part of the Nup107–160 complex (Vasu and Forbes 2001), and CG11943, a member of the Nup53–93 complex (Chen and Xu 2010), appear not only to interact with CG8771 but also with each other (Jensen et al. 2009) (but see Theerthagiri et al. [2010] for evidence against an interaction between human NUP188 and NUP205). Since several Nups have not yet been identified as nucleoporins in the D. melanogaster annotation r.5.32 and are thus missing in the GO databases (e.g., CG8771), the GO term analysis does not adequately address whether or not Nups are overrepresented among our candidate genes. We thus tested further for an overrepresentation of nucleoporins among our candidate genes by assuming that about 30 nucleoporins exist in the D. mauritiana genome (Wente and Rout 2010) and find that Nups are highly significantly overrepresented (two-tailed P < 0.0001, χ2-test with Yates correction).
Given this overrepresentation of Nups, we searched for further evidence of positive selection operating on additional Nups by relaxing our search criteria. Nup154 is significant in the unpolarized test with D. melanogaster at FDR 0.1 and at FDR 0.001 with D. yakuba as outgroup. Nup154 is a conserved nucleoporin essential for viability (Kiger et al. 1999) and crucial for normal oogenesis and spermatogenesis (Gigliotti et al. 1998; Colozza et al. 2011), and interacts with CG8771 and CG11943 (Jensen et al. 2009), homologs of the human NUP188 and NUP205 genes, respectively. Nup160, a hybrid lethality gene between D. simulans and D. melanogaster (Tang and Presgraves 2009), is significant at FDR 0.01 (FDR 0.001) in the unpolarized test with D. melanogaster (D. yakuba) as reference, and Nup133 at FDR 0.1 in the polarized test (Supplemental Tables S5–S7).
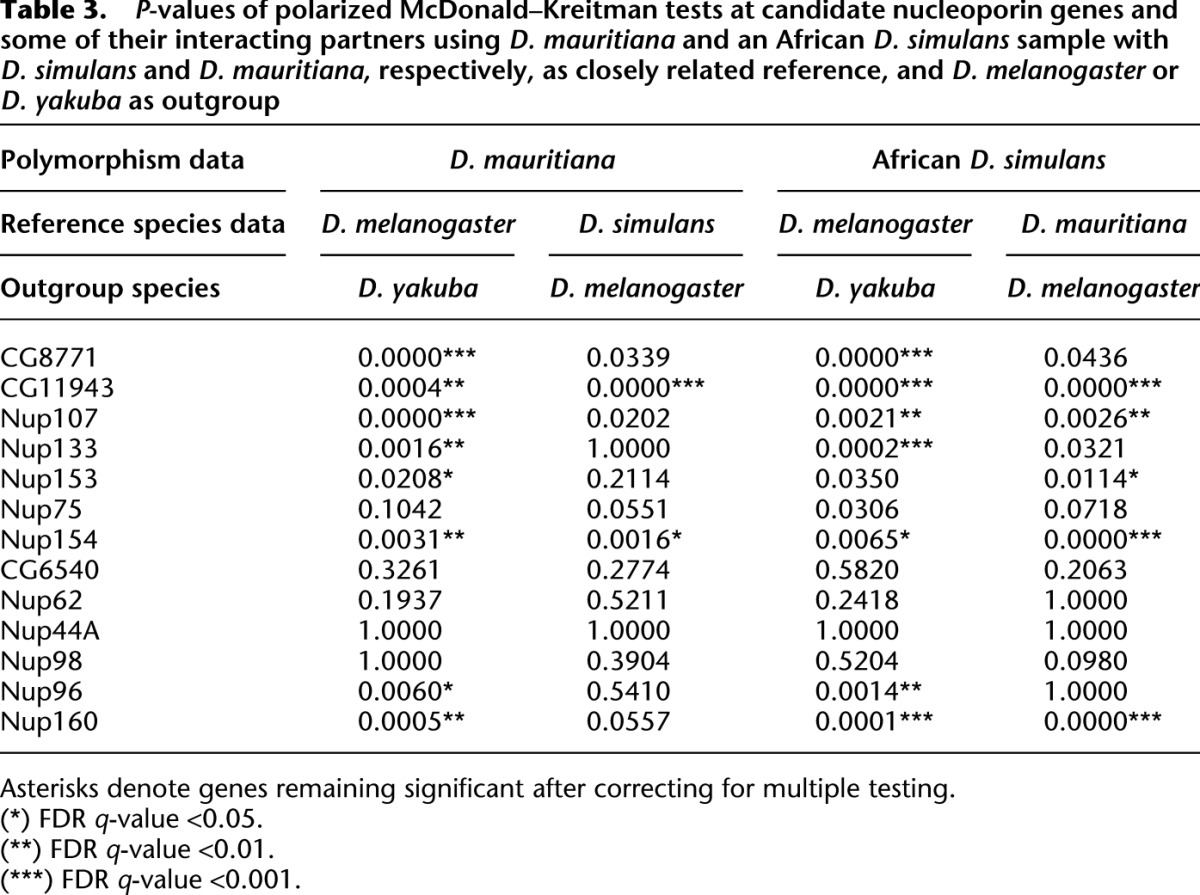
Polarized tests based on D. melanogaster as reference are not suited to determine whether positive selection predates the split of D. simulans and D. mauritiana or is still ongoing after the split of the two species. Given the strong evidence for positive selection operating on Nups, we reasoned that we should have enough power to identify ongoing positive selection after the species split and repeated the polarized tests using D. simulans as reference and D. melanogaster as outgroup. We analyzed the three candidate Nups CG8771, CG11943, and Nup107, their interaction partners as listed in the STRING interaction database v. 9.0 (Jensen et al. 2009), as well as Nup160, and found strong evidence for ongoing positive selection after the split of D. mauritiana and D. simulans for several Nups (Table 3). Analyzing Pool-seq data from African D. simulans (V Nolte and C Schlötterer, unpubl.) with D. mauritiana as reference, we also find evidence for ongoing positive selection in D. simulans. The X-linked gene, CG11943, a homolog of the human NUP205 gene, shows one of the strongest signatures of recent positive selection in both species. Overall, the evidence for positive selection was stronger in D. simulans than in D. mauritiana: Most nucleoporins with signatures of ongoing rapid evolution in D. mauritiana show an even more significant test result in D. simulans, and two nucleoporins (Nup133 and Nup153) appear to evolve rapidly in D. simulans only, but not in D. mauritiana.
Table 3.
P-values of polarized McDonald–Kreitman tests at candidate nucleoporin genes and some of their interacting partners using D. mauritiana and an African D. simulans sample with D. simulans and D. mauritiana, respectively, as closely related reference, and D. melanogaster or D. yakuba as outgroup
Given the striking evidence for rapid evolution of Nups, which is possibly driven by intragenomic conflict, we turned our attention to RNAi genes, which are also thought to evolve rapidly due to genomic conflict (Obbard et al. 2006, 2009a,b). Only 16 out of 23 RNAi genes studied by Obbard et al. (2006, 2009a) and Kolaczkowski et al. (2011) were included in the initial D. mauritiana annotation. Hence, we manually curated the annotation of the seven missing RNAi genes (AGO2, armi, krimp, AGO3, mael, rhi, and squ). Consistent with positive selection, we found two RNAi genes (aub and AGO2) to show a significant polarized MK test with D. melanogaster as reference and D. yakuba as outgroup (Supplemental Table S11). Three other genes (armi, Fmr1, and Dcr-2) were only significant (P < 0.05) when no correction for multiple testing was applied. Two genes in the D. mauritiana (aub and dcr-2) and one gene in the African D. simulans data set (armi) showed significant evidence for ongoing positive selection after the species split (Supplemental Table S12).
A polymorphism trough around two loci involved in genomic conflict
Classic selective sweeps, in which the favorable allele starts at a very low frequency and increases until it (almost) reaches fixation, cause a characteristic imprint on the polymorphism pattern in the genome (Smith and Haigh 1974; Kaplan et al. 1989). The variability in the genomic region flanking the target of selection is strongly reduced and increases gradually with distance from the selected site. The shape of such a trough depends on various parameters, such as the initial frequency of the selected allele, the selection coefficient, and the recombination rate. Figure 5 shows the partitioning of variation along the D. mauritiana X chromosome. Two very pronounced troughs in variability can be recognized that could not be attributed to alignment artifacts (see Supplemental Results). In both regions with reduced variability, we noticed a high differentiation (FST) (data not shown) from African D. simulans, but no increase in sequence divergence (Dxy) (Fig. 1).
Figure 5.

Nucleotide diversity (π) along the D. mauritiana X chromosome. The location of genes potentially causing the two selective sweeps are indicated: (large red diamond) MDox/Dox; (large blue diamond) OdsH; (small red diamond) E(Dox). Nucleotide diversity (π) is plotted in nonoverlapping 10-kb windows.
The first region encompasses ∼600 kb with a threefold reduction in variability relative to the average X-chromosomal diversity (mean π for the region = 0.002 vs. mean X-linked π = 0.0059; two-tailed Wilcoxon rank-sum test, nonoverlapping 10-kb windows, P < 2.2 × 10−16). In the central position of the trough, the variability is even further reduced (mean π = 0.001, coordinates 8.75–9.05 Mb). In addition, we observed a pronounced reduction in Tajima's D values compared with the remainder of the X chromosome (two-tailed Wilcoxon rank-sum test, nonoverlapping 10-kb windows, P < 2.2 × 10−16) (Fig. 3).
The width of the trough suggests an exceptionally strong selective sweep, since it is located in a genomic region of normal to high recombination (True et al. 1996), and no common inversion polymorphism has been described in D. mauritiana (for review, see Aulard et al. 2004). A close inspection of the function of the about 37 genes in the genomic region of reduced variability did not show any gene for which adaptive evolution was previously suggested. The only gene that could tentatively be associated with positive selection is Ser7, since it seems to be involved in immune response (Irving et al. 2001; Hill-Burns and Clark 2009). We thus turned our attention to other possible causes of selective sweeps. In addition to beneficial alleles that provide some fitness benefit to the organism, alleles involved in genomic conflict can also have very strong selective advantages (Presgraves et al. 2009) and thus the potential to drive selective sweeps. We note that a pair of genes causing sex-ratio distortion in D. simulans is located within the region with the most extreme reduction in variability (Fig. 5; Supplemental Fig. S2): Alleles of the paralogous genes Mother of Dox (MDox) and Dox function as drivers in a well-characterized sex-ratio meiotic drive system in D. simulans (Tao et al. 2007a; Kingan et al. 2010). The estimated selection coefficient s ranges from 0.12 to 0.39, depending on the parameter values used (Supplemental Table S13), which could be consistent with strong selection during genomic conflict (Curtsinger 1984).
The second trough in variability on the X chromosome extends over an even larger region but shows a less pronounced reduction in variability. A genomic region of ∼1000 kb between coordinates 16–17 Mb of the D. mauritiana reference genome shows an approximately twofold reduction in variability (mean π = 0.0030 vs. mean X-linked π = 0.0059, two-tailed Wilcoxon rank-sum test, nonoverlapping 10-kb windows, P < 2.2 × 10−16) (Fig. 5). Similar to the first sweep region, Tajima's D is also lower than in the remainder of the X chromosome (two-tailed Wilcoxon rank-sum test of Tajima's D values for 10-kb windows in each sweep region vs. 10-kb windows in the remainder of the X chromosome, P < 2.2 × 10−16) (Fig. 3). Estimates for the selection coefficient range from 0.04 to 0.46 (Supplemental Table S13).
The region of reduced variability contains more than 60 genes with many of them having no known function. The strongest reduction in variability is observed at the proximal border of the sweep window and harbors the haplolethal 16F gene cluster described in D. melanogaster (Prado et al. 1999).
Surprisingly, at the center of the window of reduced variability, we find another gene with a well-documented role in speciation and, possibly, genomic conflict, Odysseus (OdsH) (Ting et al. 1998; Bayes and Malik 2009). In addition, the sweep around the OdsH gene extends to the region in which the enhancer of Dox (E[Dox]) has been located, a not yet precisely mapped factor proximal to the gene forked that enhances the sex-ratio distorting effect of Dox (Fig. 5; Tao et al. 2007a).
Discussion
Quality of draft genomes based on paired-end Illumina sequencing
Here, we have built a high-quality draft genome of a Drosophila species using only short paired-end reads. Using a conservative, D. melanogaster–centric annotation, we recovered a similar number of genes as a previous genome project did for D. simulans and D. sechellia (Clark et al. 2007). The analyses in this study demonstrate that a draft genome facilitates addressing several important evolutionary questions. Nevertheless, we caution that it has some shortcomings. First, we are not able to provide a correct annotation of transposable elements in the D. mauritiana reference genome, since repetitive structures cannot be reliably assembled with short reads (Phillippy et al. 2008). Furthermore, heterochromatic sequences are poorly represented. Since the traces of genomic conflict are so apparent in D. mauritiana, and heterochromatic sequences may be important players in genomic conflict and speciation (Brideau et al. 2006; Cattani and Presgraves 2009; Ferree and Barbash 2009; Meiklejohn et al. 2011), we caution that the genomic signatures of ongoing genomic conflict are probably incomplete without the corresponding heterochromatic regions. Finally, gene families composed of closely related paralogs tend to be collapsed into a single copy during de novo assembly (e.g., the Hsp70 gene cluster).
Nevertheless, our assembly recovered a large fraction of gene families that are frequently identified as targets of positive selection (Supplemental Table S14). Accessory gland protein (Acp) and seminal fluid protein (Sfp) genes, which belong to recently duplicated gene families, frequently evolve under positive selection (for review, see Ram and Wolfner 2007), but we did not find genes in these categories among the top candidates for adaptively evolving genes in the polarized McDonald–Kreitman test. Similarly, olfactory (Or) and gustatory receptor (Gr) genes are frequently involved into ecological adaptation and speciation (Guo and Kim 2007; McBride 2007; Tunstall et al. 2007; Gardiner et al. 2008, 2009), but we also failed to identify such genes among the top candidates. Since our annotation recovered a large fraction of the adaptively evolving genes described in D. melanogaster, we consider it unlikely that the absence of a molecular signature of adaptation in D. mauritiana is an annotation artifact. Rather, we speculate that the selective forces driving an adaptive response of these genes in other Drosophila species are less prominent in D. mauritiana.
Nucleoporins as a preferential target for positive selection
Despite the fact that the function and composition of nuclear pore complexes are highly conserved, recent work showed that some of their components, the nucleoporins, evolve rapidly, and two of them cause hybrid lethality in Drosophila (Presgraves et al. 2003; Presgraves and Stephan 2007; Tang and Presgraves 2009). Presgraves and Stephan (2007) suggest three forms of genetic conflict that could drive the rapid evolution of nucleoporins: (1) host–parasite conflict due to viruses that need to enter via the gatekeeper nuclear pore complexes, which function to exclude invading viruses; (2) intragenomic conflict due to centromeric drive, since some Nups are associated with kinetochores; (3) intragenomic conflict due to other forms of segregation distortion, since nuclear pore complexes may potentially suppress them. Our analyses cannot distinguish between these hypotheses, but they provide additional evidence that many Nups evolve unusually rapidly due to positive selection. Furthermore, we show that the rapid evolution is not restricted to some time during the divergence between D. melanogaster and D. simulans (Presgraves et al. 2003; Presgraves and Stephan 2007; Tang and Presgraves 2009), but that positive selection is an ongoing process that continues after the split between D. mauritiana and D. simulans.
Our evolutionary analyses provide some insights into how nucleoporin genes may be involved in genomic conflict. CG11943, a homolog of the human NUP205 gene, shows strong evidence of ongoing selection in D. mauritiana and D. simulans. Presgraves et al. (2003) and Tang and Presgraves (2009) previously identified Nup96 and Nup160 as one cause of hybrid lethality between D. simulans and D. melanogaster, due to an interaction with an as-yet-unidentified X-linked factor. Given that CG11943 is located on the X chromosome, we speculate that it may be an alternative interaction partner of Nup160 and/or Nup96 instead of (or in addition to) their suggested Nup153.
While the McDonald–Kreitman test with D. simulans as a reference indicates that the high rate of sequence evolution is ongoing in D. mauritiana, there was no clear signature of a selective sweep at nucleoporin genes in the polymorphism data (Fig. 6). Since this observation is consistent with the analyses of Presgraves and Stephan (2007) and Tang and Presgraves (2009), we hypothesize that positively selected mutations in interacting proteins may lead to complex sweep dynamics, which could retard the spread of a beneficial mutation. As a consequence, beneficial mutations at Nups may result in a signature that resembles more a soft sweep (Hermisson and Pennings 2005) rather than a hard sweep.
Figure 6.

Nucleotide diversity (π) and Tajima's D at selected nucleoporin genes in D. mauritiana. (A) Nucleotide diversity (π) at selected nucleoporin genes in comparison to the average chromosome-wide nucleotide diversity (π) of the corresponding chromosome (dashed line). (B) Tajima's D at selected nucleoporin genes in comparison to the average chromosome-wide Tajima's D of the corresponding chromosome (dashed line).
Two recent selective sweeps potentially associated with genomic conflict
One of our most striking findings is that the D. mauritiana genome harbors two large regions—0.6 and 1 Mb—of profoundly reduced diversity, suggesting that two exceptionally strong selective sweeps have occurred in this species. In comparison, genomic signatures of previously described sweeps in Drosophila are much narrower with estimated selection coefficients being about an order of magnitude lower: The broadest valley of reduced variability described to date is caused by the insertion of a transposable element next to the gene Cyp6g1, which confers resistance to DDT—the sweep extends over ∼100 kb and has an associated selection coefficient of 0.022 (Schlenke and Begun 2004).
The gene that could most parsimoniously be assumed to drive one of the sweeps on the D. mauritiana X chromosome is OdsH. This gene was initially described as a “speciation gene” that causes hybrid male sterility between D. mauritiana and D. simulans and shows a strongly accelerated rate of evolution in D. mauritiana (Ting et al. 1998). Later it was recognized that the OdsH gene product interacts with Y-linked heterochromatin in hybrids between D. mauritiana and D. simulans but not in pure species, suggesting that it could be involved in genomic conflict (Bayes and Malik 2009; Meiklejohn et al. 2011). Consistent with previous reports, we observe a much higher number of nonsynonymous fixations along the D. mauritiana than the D. simulans lineage, but in both species neither the homeodomain nor the entire gene showed evidence of positive selection in a polarized McDonald–Kreitman test.
The second selective sweep might be caused by the Winters meiotic drive system, which is well-characterized in D. simulans (Tao et al. 2007a,b; Kingan et al. 2010). This system consists of at least three components: the drivers MDox and Dox, the autosomal dominant suppressor Nmy, and the enhancer of Dox [E(Dox)]. The sequences of these genes are highly similar, partially derived from one another by tandem duplication and retrotransposition, and contain tandem repeat structures (Tao et al. 2007a,b). As a result of the sequence similarity and repetitive structure of these loci, reliable sequence analysis of this region is difficult, even with targeted PCR approaches (Tao et al. 2007a,b), and essentially impossible with genome-wide short read sequencing.
Our D. mauritiana strains did not show obvious signs of sex-ratio distortion, but theoretical models predict that ongoing intragenomic conflict results in rapid cycles during which competing alleles rise and fall in frequency (Charlesworth and Hartl 1978; Carvalho and Vaz 1999; Hall 2004). Driver alleles, such as sex-ratio distorters, will increase in frequency until a suppressor allele arrives, which spreads, and the genomic conflict ultimately disappears (i.e., the population reaches a balanced sex ratio). While an almost complete sweep of a strongly distorting allele appears unlikely, theoretical models have described situations under which such a pattern is predicted (Charlesworth and Hartl 1978; Carvalho and Vaz 1999; Hall 2004).
In some D. simulans populations, the Dox gene shows evidence of a partial selective sweep (Kingan et al. 2010), but it is difficult to distinguish highly localized selective sweeps from random fluctuations in variability due to the bottleneck associated with the out of Africa expansion (Jensen et al. 2005). We further scrutinized the genomic region around Dox using Pool-seq data from African D. simulans (V Nolte and C Schlötterer, unpubl.) and did not note any pronounced trough in variability around the Dox region, suggesting that, at least in the African population sample, no evidence for a selective sweep comparable to the one in D. mauritiana could be detected (Supplemental Fig. S3).
The Dox system was initially discovered in D. simulans (Dermitzakis et al. 2000), and the driver loci MDox and Dox have not yet been functionally analyzed in D. mauritiana. While an analysis of the D. mauritiana alleles present at MDox, Dox, and Nmy is not possible from the Pool-seq data (see above), Tao et al. (2007b) inferred from sequence comparison that a functional suppressor allele at Nmy is present in D. mauritiana, suggesting the existence of a functional distorter (Tao et al. 2007a,b). No selective sweep could be detected in the Nmy gene region (data not shown), suggesting that no new allele at Nmy has swept through D. mauritiana.
Given the dynamic evolution of repetitive structures in the Winters sex-ratio genes, it is possible that in D. mauritiana, a new driver allele at Dox evolved and caused the pronounced sweep signature, while in D. simulans, the signature of an older sweep has already been erased. Alternatively, we could speculate that it may be easier for an allele to sweep in D. mauritiana since D. mauritiana has almost no population structure (Nunes et al. 2010), while the cosmopolitan species D. simulans shows a higher level of population differentiation (Hamblin and Veuille 1999).
Theoretical studies predict that a beneficial allele will spread much faster in panmictic populations, whereas population subdivision and low migration rates lead to a delay in the fixation of a beneficial mutation (Barton 2000; Santiago and Caballero 2005; Kim and Maruki 2011).
The importance of population structure for the detection of meiotic drive dynamics has also been highlighted in recent theoretical work. Hall (2004) suggested that the hitherto absence of documented cycling behavior in natural Drosophila populations may be the result of migration between subdivided populations with different drive parameters. Instead, in isolated populations that share the same drive dynamics due to panmixia large fluctuations in driver and suppressor can be seen. Since D. mauritiana shows no population differentiation on Mauritius, we think that this could explain the difference with the other Drosophila species.
It is apparent that more work is needed to characterize the driver, responder, and suppressor alleles in both species to shed further light onto the differences in evolutionary signatures observed between the two species.
Methods
D. mauritiana strains and Illumina sequencing
We used the D. mauritiana isofemale strain MS17 (http://kyotofly.kit.ac.jp/cgi-bin/ehime/index.cgi, stock number E-18912) to generate a D. mauritiana reference genome. Pool-seq data were obtained from 107 D. mauritiana lines collected at different time points and locations in Mauritius (Supplemental Table S2). Illumina libraries were generated following the instructions of the Illumina Paired-End Sample Preparation Kit and sequenced on a GAIIx.
De novo assembly and annotation of a D. mauritiana reference genome
To generate the D. mauritiana reference genome sequence, we initially performed a de novo assembly of Illumina reads using the software CLC Assembly Cell v. 3.1.0 beta2 (CLC Bio). In the second phase of the assembly procedure, we anchored de novo contigs on the reference genome of D. melanogaster r. 5.22 using the nucmer module in the MUMmer package v. 3.0 (Kurtz et al. 2004). The D. mauritiana chromosomes were built by overlapping or concatenating contigs. The longest isoform of each D. melanogaster protein from FlyBase release 5.32 served as template for annotation. Each protein sequence was aligned to the D. mauritiana reference genome using exonerate v. 2.0 (Slater and Birney 2005). We generated four sets of gene annotations, using varying degrees of filtering criteria that are described in detail in Supplemental Methods.
Divergence estimates and codon usage analysis
We performed multiple alignments of the D. mauritiana, a D. simulans, the D. sechellia r.1.3, and the D. melanogaster r. 5.32 genome sequences using MAUVE (Darling et al. 2010) and calculated pairwise divergence Dxy between them using the PoPoolation package (Kofler et al. 2011). We used CAIcal v. 1.4 (Puigbo et al. 2008) to determine the Codon Adaptation Index (CAI), originally developed by Sharp and Li (1987).
Reference mapping and variability estimates in D. mauritiana Pool-seq data
Paired-end reads of the pooled D. mauritiana sample were aligned to the MS17 draft genome (or D. melanogaster genome) using bwa v. 0.5.8 (Li and Durbin 2009). Alignments were filtered for a minimum mapping quality of 20 and for properly paired reads using SAMtools v. 0.1.9 (http://samtools.sourceforge.net/). Minimum requirements for coverage and allele count used in SNP calling are detailed in Supplemental Methods. Analyses of π and Tajima's D were performed with the PoPoolation package (Kofler et al. 2011). To test for recurrent positive selection in the D. mauritiana lineage, we performed McDonald–Kreitman tests. Multiple alignments of the coding sequence of each D. mauritiana gene with the orthologs of D. melanogaster r. 5.32 and D. yakuba r. 1.3 were generated using PRANK v. 100701 (Loytynoja and Goldman 2005). We combined the interspecific with the intraspecific alignments using custom Perl scripts and performed McDonald–Kreitman tests using the MK.pl script obtained from http://www.dpgp.org/aholloway/Software.html (Holloway et al. 2007). We calculated false discovery rates (FDR) using the LBE package (Dalmasso et al. 2005) and performed an analysis of Gene Ontology enrichment with GOrilla (http://cbl-gorilla.cs.technion.ac.il/) (Eden et al. 2009). Details of all analyses are provided in Supplemental Methods.
Statistical tests
Statistical tests were performed using R version 2.11.1 (The R Core Team 2010) unless stated otherwise.
Data access
All Illumina short reads used in this study are available from the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under the following accession numbers: the single D. mauritiana reference strain MS17 under SRA058420, the D. mauritiana Pool-seq data under SRA058664, the D. simulans reference strain Kib32 under SRA059282, and the African D. simulans Pool-seq data under SRA059292. The D. mauritiana strain MS17 reference genome and annotation are available at http://www.popoolation.at/mauritiana_genome/index.html. A BAM file containing D. mauritiana Pool-seq data is available at http://www.popoolation.at/mauritiana_genome/index.html. A searchable, user-friendly version of the D. mauritiana Pool-seq and the African D. simulans Pool-seq data is available at http://www.popoolation.at/pgt/dmau_browse.html and http://www.popoolation.at/pgt/dsim_browse.html.
Acknowledgments
We thank all members of the Institute of Population Genetics for discussion and support throughout the entire project. We are grateful to A.J. Betancourt for drawing our attention to the MDox/Dox locus and to N. de Maio for mathematical support. A.J. Betancourt, B.F. McAllister, C. Willett, R. Tobler, and three anonymous reviewers provided helpful comments on earlier versions of the manuscript. We are grateful to the Ehime-Fly Drosophila Stock center for providing us with fly stocks. This work was supported by the Austrian Science Funds (FWF) grant P19467 to C.S.
Author contributions: V.N. and C.S. conceived and designed the experiments; V.N. performed the experiments; V.N. analyzed the data; R.V.P. and R.K. contributed analysis tools; and V.N. and C.S. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.139873.112.
Freely available online through the Genome Research Open Access option.
Throughout the manuscript, we refer to “Nup96 gene” as the part of the Nup98-96 gene that corresponds to amino acid residues 1029–1961 in the resulting protein; this part of the protein frequently referred to as “NUP96.” Similarly, we refer to “Nup98 gene” as the part of the Nup98-96 gene that corresponds to amino acid residues 1–1028 in the resulting protein; this part of the protein is frequently referred to as “NUP98,” e.g., in Presgraves et al. (2003).
References
- Aulard S, Monti L, Chaminade N, Lemeunier F 2004. Mitotic and polytene chromosomes: Comparisons between Drosophila melanogaster and Drosophila simulans. Genetica 120: 137–150 [DOI] [PubMed] [Google Scholar]
- Ballard JW 2000. When one is not enough: Introgression of mitochondrial DNA in Drosophila. Mol Biol Evol 17: 1126–1130 [DOI] [PubMed] [Google Scholar]
- Barbash DA 2007. Nup96-dependent hybrid lethality occurs in a subset of species from the simulans clade of Drosophila. Genetics 176: 543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NH 2000. Genetic hitchhiking. Philos Trans R Soc Lond B Biol Sci 355: 1553–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastide H, Cazemajor M, Ogereau D, Derome N, Hospital F, Montchamp-Moreau C 2011. Rapid rise and fall of selfish sex-ratio X chromosomes in Drosophila simulans: Spatio-temporal analysis of phenotypic and molecular data. Mol Biol Evol 28: 2461–2470 [DOI] [PubMed] [Google Scholar]
- Bayes JJ, Malik HS 2009. Altered heterochromatin binding by a hybrid sterility protein in Drosophila sibling species. Science 326: 1538–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Aquadro CF 1992. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356: 519–520 [DOI] [PubMed] [Google Scholar]
- Begun DJ, Lindfors HA 2005. Rapid evolution of genomic Acp complement in the melanogaster subgroup of Drosophila. Mol Biol Evol 22: 2010–202115987879 [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh YP, Hahn MW, Nista PM, Jones CD, Kern AD, Dewey CN, et al. 2007. Population genomics: Whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol 5: e310 doi: 10.1371/journal.pbio.0050310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt AJ, Welch JJ, Charlesworth B 2009. Reduced effectiveness of selection caused by a lack of recombination. Curr Biol 19: 655–660 [DOI] [PubMed] [Google Scholar]
- Brideau NJ, Flores HA, Wang J, Maheshwari S, Wang X, Barbash DA 2006. Two Dobzhansky-Muller genes interact to cause hybrid lethality in Drosophila. Science 314: 1292–1295 [DOI] [PubMed] [Google Scholar]
- Carvalho AB, Vaz SC 1999. Are Drosophila SR drive chromosomes always balanced? Heredity (Edinb) 83: 221–228 [DOI] [PubMed] [Google Scholar]
- Cattani MV, Presgraves DC 2009. Genetics and lineage-specific evolution of a lethal hybrid incompatibility between Drosophila mauritiana and its sibling species. Genetics 181: 1545–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Hartl DL 1978. Population dynamics of the segregation distorter polymorphism of Drosophila melanogaster. Genetics 89: 171–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Coyne JA, Barton NH 1987. The relative rates of evolution of sex chromosomes and autosomes. Am Nat 130: 113–146 [Google Scholar]
- Charlesworth B, Morgan MT, Charlesworth D 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu L 2010. Specific nucleoporin requirement for Smad nuclear translocation. Mol Cell Biol 30: 4022–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, Iyer VN, et al. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218 [DOI] [PubMed] [Google Scholar]
- Colozza G, Montembault E, Quenerch'du E, Riparbelli MG, D'Avino PP, Callaini G 2011. Drosophila nucleoporin Nup154 controls cell viability, proliferation and nuclear accumulation of Mad transcription factor. Tissue Cell 43: 254–261 [DOI] [PubMed] [Google Scholar]
- Curtsinger JW 1984. Components of selection in X chromosome lines of Drosophila melanogaster: Sex ratio modification by meiotic drive and viability selection. Genetics 108: 941–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtsinger JW, Feldman MW 1980. Experimental and theoretical analysis of the “sex-ratio” polymorphism in Drosophila pseudoobscura. Genetics 94: 445–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmasso C, Broet P, Moreau T 2005. A simple procedure for estimating the false discovery rate. Bioinformatics 21: 660–668 [DOI] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT 2010. progressiveMauve: Multiple genome alignment with gene gain, loss, and rearrangement. PLoS ONE 5: e11147 doi: 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET, Masly JP, Waldrip HM, Clark AG 2000. Non-Mendelian segregation of sex chromosomes in heterospecific Drosophila males. Genetics 154: 687–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derome N, Metayer K, Montchamp-Moreau C, Veuille M 2004. Signature of selective sweep associated with the evolution of sex-ratio drive in Drosophila simulans. Genetics 166: 1357–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derome N, Baudry E, Ogereau D, Veuille M, Montchamp-Moreau C 2008. Selective sweeps in a 2-locus model for sex-ratio meiotic drive in Drosophila simulans. Mol Biol Evol 25: 409–416 [DOI] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z 2009. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48 doi: 10.1186/1471-2105-10-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferree PM, Barbash DA 2009. Species-specific heterochromatin prevents mitotic chromosome segregation to cause hybrid lethality in Drosophila. PLoS Biol 7: e1000234 doi: 10.1371/journal.pbio.1000234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner A, Barker D, Butlin RK, Jordan WC, Ritchie MG 2008. Drosophila chemoreceptor gene evolution: Selection, specialization and genome size. Mol Ecol 17: 1648–1657 [DOI] [PubMed] [Google Scholar]
- Gardiner A, Butlin RK, Jordan WC, Ritchie MG 2009. Sites of evolutionary divergence differ between olfactory and gustatory receptors of Drosophila. Biol Lett 5: 244–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigan D, Kingan SB, Geneva AJ, Andolfatto P, Clark AG, Thornton KR, Presgraves DC 2012. Genome sequencing reveals complex speciation in the Drosophila simulans clade. Genome Res 22: 1499–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershenson S 1928. A new sex-ratio abnormality in Drosophila obscura. Genetics 13: 488–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigliotti S, Callaini G, Andone S, Riparbelli MG, Pernas-Alonso R, Hoffmann G, Graziani F, Malva C 1998. Nup154, a new Drosophila gene essential for male and female gametogenesis is related to the Nup155 vertebrate nucleoporin gene. J Cell Biol 142: 1195–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graze RM, Barmina O, Tufts D, Naderi E, Harmon KL, Persianinova M, Nuzhdin SV 2007. New candidate genes for sex-comb divergence between Drosophila mauritiana and Drosophila simulans. Genetics 176: 2561–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Kim J 2007. Molecular evolution of Drosophila odorant receptor genes. Mol Biol Evol 24: 1198–1207 [DOI] [PubMed] [Google Scholar]
- Hall DW 2004. Meiotic drive and sex chromosome cycling. Evolution 58: 925–931 [DOI] [PubMed] [Google Scholar]
- Hamblin MT, Veuille M 1999. Population structure among African and derived populations of Drosophila simulans: Evidence for ancient subdivision and recent admixture. Genetics 153: 305–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton WD 1967. Extraordinary sex ratios. Science 156: 477–488 [DOI] [PubMed] [Google Scholar]
- Hermisson J, Pennings PS 2005. Soft sweeps: Molecular population genetics of adaptation from standing genetic variation. Genetics 169: 2335–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Kliman RM 1993. Population genetics and phylogenetics of DNA sequence variation at multiple loci within the Drosophila melanogaster species complex. Mol Biol Evol 10: 804–822 [DOI] [PubMed] [Google Scholar]
- Hill-Burns EM, Clark AG 2009. X-linked variation in immune response in Drosophila melanogaster. Genetics 183: 1477–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra HE, Coyne JA 2007. The locus of evolution: evo devo and the genetics of adaptation. Evolution 61: 995–1016 [DOI] [PubMed] [Google Scholar]
- Holloway AK, Lawniczak MK, Mezey JG, Begun DJ, Jones CD 2007. Adaptive gene expression divergence inferred from population genomics. PLoS Genet 3: 2007–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR 1994. How can the low levels of DNA sequence variation in regions of the Drosophila genome with low recombination rates be explained? Proc Natl Acad Sci 91: 6815–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving P, Troxler L, Heuer TS, Belvin M, Kopczynski C, Reichhart JM, Hoffmann JA, Hetru C 2001. A genome-wide analysis of immune responses in Drosophila. Proc Natl Acad Sci 98: 15119–15124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadeeshan S, Singh RS 2005. Rapidly evolving genes of Drosophila: Differing levels of selective pressure in testis, ovary, and head tissues between sibling species. Mol Biol Evol 22: 1793–1801 [DOI] [PubMed] [Google Scholar]
- Jensen JD, Kim Y, Dumont VB, Aquadro CF, Bustamante CD 2005. Distinguishing between selective sweeps and demography using DNA polymorphism data. Genetics 170: 1401–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et al. 2009. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 37: D412–D416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan NL, Hudson RR, Langley CH 1989. The “hitchhiking effect” revisited. Genetics 123: 887–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiger AA, Gigliotti S, Fuller MT 1999. Developmental genetics of the essential Drosophila nucleoporin Nup154: Allelic differences due to an outward-directed promoter in the P-element 3′ end. Genetics 153: 799–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Maruki T 2011. Hitchhiking effect of a beneficial mutation spreading in a subdivided population. Genetics 189: 213–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingan SB, Garrigan D, Hartl DL 2010. Recurrent selection on the Winters sex-ratio genes in Drosophila simulans. Genetics 184: 253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler R, Orozco-terWengel P, De Maio N, Pandey RV, Nolte V, Futschik A, Kosiol C, Schlotterer C 2011. PoPoolation: A toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS ONE 6: e15925 doi: 10.1371/journal.pone.0015925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowski B, Hupalo DN, Kern AD 2011. Recurrent adaptation in RNA interference genes across the Drosophila phylogeny. Mol Biol Evol 28: 1033–1042 [DOI] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12 doi: 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley CH, Stevens K, Cardeno C, Lee YC, Schrider DR, Pool JE, Langley SA, Suarez C, Corbett-Detig RB, Kolaczkowski B, et al. 2012. Genomic variation in natural populations of Drosophila melanogaster. Genetics 192: 533–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loytynoja A, Goldman N 2005. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci 102: 10557–10562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyttle TW 1977. Experimental population genetics of meiotic drive systems I. Pseudo-Y chromosomal drive as a means of eliminating cage populations of Drosophila melanogaster. Genetics 86: 413–445 [PMC free article] [PubMed] [Google Scholar]
- Maheshwari S, Barbash DA 2012. Cis-by-trans regulatory divergence causes the asymmetric lethal effects of an ancestral hybrid incompatibility gene. PLoS Genet 8: e1002597 doi: 10.1371/journal.pgen.1002597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J, Haigh J 1974. The hitch-hiking effect of a favorable gene. Genet Res 23: 23–35 [PubMed] [Google Scholar]
- McBride CS 2007. Rapid evolution of smell and taste receptor genes during host specialization in Drosophila sechellia. Proc Natl Acad Sci 104: 4996–5001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654 [DOI] [PubMed] [Google Scholar]
- Meiklejohn CD, Landeen EL, Cook JM, Kingan SB, Presgraves DC 2011. Sex chromosome-specific regulation in the Drosophila male germline but little evidence for chromosomal dosage compensation or meiotic inactivation. PLoS Biol 9: e1001126 doi: 10.1371/journal.pbio.1001126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercot H, Atlan A, Jacques M, Montchamp-Moreau C 1995. Sex-ratio distortion in Drosophila simulans: Co-occurrence of a meiotic drive and a suppressor of drive. J Evol Biol 8: 283–300 [Google Scholar]
- Miller BR, Powers M, Park M, Fischer W, Forbes DJ 2000. Identification of a new vertebrate nucleoporin, Nup188, with the use of a novel organelle trap assay. Mol Biol Cell 11: 3381–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama EN, Powell JR 1996. Intraspecific nuclear DNA variation in Drosophila. Mol Biol Evol 13: 261–277 [DOI] [PubMed] [Google Scholar]
- Morton RA, Choudhary M, Cariou ML, Singh RS 2004. A reanalysis of protein polymorphism in Drosophila melanogaster, D. simulans, D. sechellia and D. mauritiana: Effects of population size and selection. Genetica 120: 101–114 [DOI] [PubMed] [Google Scholar]
- Nehrbass U, Rout MP, Maguire S, Blobel G, Wozniak RW 1996. The yeast nucleoporin Nup188p interacts genetically and physically with the core structures of the nuclear pore complex. J Cell Biol 133: 1153–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes MD, Wengel PO, Kreissl M, Schlötterer C 2010. Multiple hybridization events between Drosophila simulans and Drosophila mauritiana are supported by mtDNA introgression. Mol Ecol 19: 4695–4707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obbard DJ, Jiggins FM, Halligan DL, Little TJ 2006. Natural selection drives extremely rapid evolution in antiviral RNAi genes. Curr Biol 16: 580–585 [DOI] [PubMed] [Google Scholar]
- Obbard DJ, Gordon KH, Buck AH, Jiggins FM 2009a. The evolution of RNAi as a defence against viruses and transposable elements. Philos Trans R Soc Lond B Biol Sci 364: 99–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obbard DJ, Welch JJ, Kim KW, Jiggins FM 2009b. Quantifying adaptive evolution in the Drosophila immune system. PLoS Genet 5: e1000698 doi: 10.1371/journal.pgen.1000698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA, Betancourt AJ 2001. Haldane's sieve and adaptation from the standing genetic variation. Genetics 157: 875–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez DE, Wu CI 1995. Further characterization of the Odysseus locus of hybrid sterility in Drosophila: One gene is not enough. Genetics 140: 201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez DE, Wu CI, Johnson NA, Wu ML 1993. Genetics of reproductive isolation in the Drosophila simulans clade: DNA marker-assisted mapping and characterization of a hybrid-male sterility gene, Odysseus (Ods). Genetics 134: 261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phadnis N, Orr HA 2009. A single gene causes both male sterility and segregation distortion in Drosophila hybrids. Science 323: 376–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillippy AM, Schatz MC, Pop M 2008. Genome assembly forensics: Finding the elusive mis-assembly. Genome Biol 9: R55 doi: 10.1186/gb-2008-9-3-r55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado A, Canal I, Ferrus A 1999. The haplolethal region at the 16F gene cluster of Drosophila melanogaster: Structure and function. Genetics 151: 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC 2003. A fine-scale genetic analysis of hybrid incompatibilities in Drosophila. Genetics 163: 955–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC 2005. Recombination enhances protein adaptation in Drosophila melanogaster. Curr Biol 15: 1651–1656 [DOI] [PubMed] [Google Scholar]
- Presgraves DC 2007. Does genetic conflict drive rapid molecular evolution of nuclear transport genes in Drosophila? Bioessays 29: 386–391 [DOI] [PubMed] [Google Scholar]
- Presgraves DC, Stephan W 2007. Pervasive adaptive evolution among interactors of the Drosophila hybrid inviability gene, Nup96. Mol Biol Evol 24: 306–314 [DOI] [PubMed] [Google Scholar]
- Presgraves DC, Balagopalan L, Abmayr SM, Orr HA 2003. Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature 423: 715–719 [DOI] [PubMed] [Google Scholar]
- Presgraves DC, Gerard PR, Cherukuri A, Lyttle TW 2009. Large-scale selective sweep among Segregation Distorter chromosomes in African populations of Drosophila melanogaster. PLoS Genet 5: e1000463 doi: 10.1371/journal.pgen.1000463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigbo P, Bravo IG, Garcia-Vallve S 2008. CAIcal: A combined set of tools to assess codon usage adaptation. Biol Direct 3: 38 doi: 10.1186/1745-6150-3-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The R Core Team. 2010. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. [Google Scholar]
- Ram KR, Wolfner MF 2007. Seminal influences: Drosophila Acps and the molecular interplay between males and females during reproduction. Integr Comp Biol 47: 427–445 [DOI] [PubMed] [Google Scholar]
- Sandler L, Hiraizumi Y, Sandler I 1959. Meiotic drive in natural populations of Drosophila Melanogaster. I. The cytogenetic basis of segregation-distortion. Genetics 44: 233–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago E, Caballero A 2005. Variation after a selective sweep in a subdivided population. Genetics 169: 475–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenke TA, Begun DJ 2004. Strong selective sweep associated with a transposon insertion in Drosophila simulans. Proc Natl Acad Sci 101: 1626–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH 1987. The codon Adaptation Index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15: 1281–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Larracuente AM, Clark AG 2008. Contrasting the efficacy of selection on the X and autosomes in Drosophila. Mol Biol Evol 25: 454–467 [DOI] [PubMed] [Google Scholar]
- Slater GS, Birney E 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6: 31 doi: 10.1186/1471-2105-6-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JM, Haigh J 1974. The hitch-hiking effect of a favourable gene. Genet Res 23: 23–35 [PubMed] [Google Scholar]
- Tajima F 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Presgraves DC 2009. Evolution of the Drosophila nuclear pore complex results in multiple hybrid incompatibilities. Science 323: 779–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Hartl DL, Laurie CC 2001. Sex-ratio segregation distortion associated with reproductive isolation in Drosophila. Proc Natl Acad Sci 98: 13183–13188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Araripe L, Kingan SB, Ke Y, Xiao H, Hartl DL 2007a. A sex-ratio meiotic drive system in Drosophila simulans. II: An X-linked distorter. PLoS Biol 5: e293 doi: 10.1371/journal.pbio.0050293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Masly JP, Araripe L, Ke Y, Hartl DL 2007b. A sex-ratio meiotic drive system in Drosophila simulans. I: An autosomal suppressor. PLoS Biol 5: e292 doi: 10.1371/journal.pbio.0050292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theerthagiri G, Eisenhardt N, Schwarz H, Antonin W 2010. The nucleoporin Nup188 controls passage of membrane proteins across the nuclear pore complex. J Cell Biol 189: 1129–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting CT, Tsaur SC, Wu ML, Wu CI 1998. A rapidly evolving homeobox at the site of a hybrid sterility gene. Science 282: 1501–1504 [DOI] [PubMed] [Google Scholar]
- True JR, Mercer JM, Laurie CC 1996. Differences in crossover frequency and distribution among three sibling species of Drosophila. Genetics 142: 507–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunstall NE, Sirey T, Newcomb RD, Warr CG 2007. Selective pressures on Drosophila chemosensory receptor genes. J Mol Evol 64: 628–636 [DOI] [PubMed] [Google Scholar]
- Vasu SK, Forbes DJ 2001. Nuclear pores and nuclear assembly. Curr Opin Cell Biol 13: 363–375 [DOI] [PubMed] [Google Scholar]
- Wallace B 1948. Studies on sex-ratio in Drosophila pseudoobscura. I. Selection and sex-ratio. Evolution 2: 189–217 [PubMed] [Google Scholar]
- Wente SR, Rout MP 2010. The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol 2: a000562 doi: 10.1101/cshperspect.a000562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH 2011. Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc Natl Acad Sci (Suppl 2) 108: 10863–10870 [DOI] [PMC free article] [PubMed] [Google Scholar]