

Abstract
Motivation: Accurate alignment of high-throughput RNA-seq data is a challenging and yet unsolved problem because of the non-contiguous transcript structure, relatively short read lengths and constantly increasing throughput of the sequencing technologies. Currently available RNA-seq aligners suffer from high mapping error rates, low mapping speed, read length limitation and mapping biases.
Results: To align our large (>80 billon reads) ENCODE Transcriptome RNA-seq dataset, we developed the Spliced Transcripts Alignment to a Reference (STAR) software based on a previously undescribed RNA-seq alignment algorithm that uses sequential maximum mappable seed search in uncompressed suffix arrays followed by seed clustering and stitching procedure. STAR outperforms other aligners by a factor of >50 in mapping speed, aligning to the human genome 550 million 2 × 76 bp paired-end reads per hour on a modest 12-core server, while at the same time improving alignment sensitivity and precision. In addition to unbiased de novo detection of canonical junctions, STAR can discover non-canonical splices and chimeric (fusion) transcripts, and is also capable of mapping full-length RNA sequences. Using Roche 454 sequencing of reverse transcription polymerase chain reaction amplicons, we experimentally validated 1960 novel intergenic splice junctions with an 80–90% success rate, corroborating the high precision of the STAR mapping strategy.
Availability and implementation: STAR is implemented as a standalone C++ code. STAR is free open source software distributed under GPLv3 license and can be downloaded from http://code.google.com/p/rna-star/.
Contact: dobin@cshl.edu.
1 INTRODUCTION
Although genomes are composed of linearly ordered sequences of nucleic acids, eukaryotic cells generally reorganize the information in the transcriptome by splicing together non-contiguous exons to create mature transcripts (Hastings and Krainer, 2001). The detection and characterization of these spliced RNAs have been a critical focus of functional analyses of genomes in both the normal and disease cell states. Recent advances in sequencing technologies have made transcriptome analyses at the single nucleotide level almost routine. However, hundreds of millions of short (36 nt) to medium (200 nt) length sequences (reads) generated by such high-throughput sequencing experiments present unique challenges to detection and characterization of spliced transcripts. Two key tasks make these analyses computationally intensive. The first task is an accurate alignment of reads that contain mismatches, insertions and deletions caused by genomic variations and sequencing errors. The second task involves mapping sequences derived from non-contiguous genomic regions comprising spliced sequence modules that are joined together to form spliced RNAs. Although the first task is shared with DNA resequencing efforts, the second task is specific and crucial to the RNA-seq, as it provides the connectivity information needed to reconstruct the full extent of spliced RNA molecules. These alignment challenges are further compounded by the presence of multiple copies of identical or related genomic sequences that are themselves transcribed, making precise mapping difficult.
Various sequence alignment algorithms have been recently developed to tackle these challenges (Au et al., 2010; De Bona, et al., 2008; Grant et al., 2011; Han et al., 2011; Trapnell et al., 2009; Wang et al., 2010; Wu and Nacu, 2010; Zhang et al., 2012). However, application of these algorithms invokes compromises in the areas of mapping accuracy (sensitivity and precision) and computational resources (run time and disk space) (Grant et al., 2011). With current advances in sequencing technologies, the computational component is increasingly becoming a throughput bottleneck. High mapping speed is especially important for large consortia efforts, such as ENCODE (http://www.genome.gov/encode/), which continuously generate large amounts of sequencing data.
Furthermore, most of the cited algorithms were designed to deal with relatively short reads (typically ≤200 bases), and are ill-suited for aligning long read sequences generated by the emerging third-generation sequencing technologies (Flusberg et al., 2010; Rothberg et al., 2011). The longer read sequences, ideally reaching full lengths of RNA molecules, have a great potential for enhancing transcriptome studies by providing more complete RNA connectivity information.
This report describes an alignment algorithm entitled ‘Spliced Transcripts Alignment to a Reference (STAR)’, which was designed to specifically address many of the challenges of RNA-seq data mapping, and uses a novel strategy for spliced alignments. We performed high-throughput validation experiments that corroborated STAR’s precision for detection of novel splice junctions. STAR’s high mapping speed and accuracy were crucial for analyzing the large ENCODE transcriptome (Djebali et al., 2012) dataset (>80 billion Illumina reads). We also demonstrated that STAR has a potential for accurately aligning long (several kilobases) reads that are emerging from the third-generation sequencing technologies.
2 ALGORITHM
Many previously described RNA-seq aligners were developed as extensions of contiguous (DNA) short read mappers, which were used to either align short reads to a database of splice junctions or align split-read portions contiguously to a reference genome, or a combination thereof. In contrast to these approaches, STAR was designed to align the non-contiguous sequences directly to the reference genome. STAR algorithm consists of two major steps: seed searching step and clustering/stitching/scoring step.
2.1 Seed search
The central idea of the STAR seed finding phase is the sequential search for a Maximal Mappable Prefix (MMP). MMP is similar to the Maximal Exact (Unique) Match concept used by the large-scale genome alignment tools Mummer (Delcher et al., 1999, 2002; Kurtz et al.) and MAUVE (Darling et al., 2004, 2010). Given a read sequence R, read location i and a reference genome sequence G, the MMP(R,i,G) is defined as the longest substring (Ri, Ri+ 1, … , Ri+MML− 1) that matches exactly one or more substrings of G, where MML is the maximum mappable length. We will explain this concept using a simple example of a read that contains a single splice junction and no mismatches (Fig. 1a). In the first step, the algorithm finds the MMP starting from the first base of the read. Because the read in this example comprises a splice junction, it cannot be mapped contiguously to the genome, and thus the first seed will be mapped to a donor splice site. Next, the MMP search is repeated for the unmapped portion of the read, which, in this case, will be mapped to an acceptor splice site. Note that this sequential application of MMP search only to the unmapped portions of the read makes the STAR algorithm extremely fast and distinguishes it from Mummer and MAUVE, which find all possible Maximal Exact Matches. This approach represents a natural way of finding precise locations of splice junctions in a read sequence and is advantageous over an arbitrary splitting of read sequences used in the split-read methods. The splice junctions are detected in a single alignment pass without any a priori knowledge of splice junctions’ loci or properties, and without a preliminary contiguous alignment pass needed by the junction database approaches. The MMP in STAR search is implemented through uncompressed suffix arrays (SAs) (Manber and Myers, 1993). Notably, finding MMP is an inherent outcome of the standard binary string search in uncompressed SAs, and does not require any additional computational effort compared with the full-length exact match searches. The binary nature of the SA search results in a favorable logarithmic scaling of the search time with the reference genome length, allowing fast searching even against large genomes. Advantageously, for each MMP the SA search can find all distinct exact genomic matches with little computational overhead, which facilitates an accurate alignment of the reads that map to multiple genomic loci (“multimapping” reads).
Fig. 1.
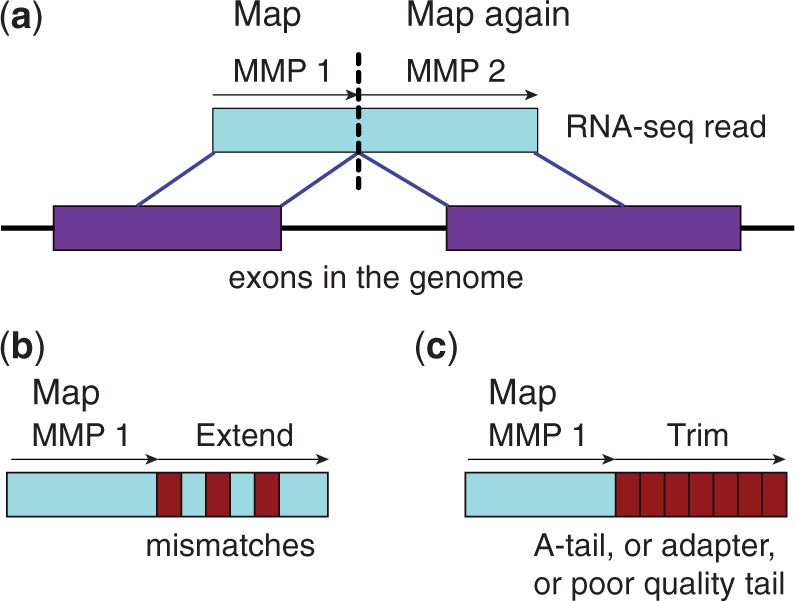
Schematic representation of the Maximum Mappable Prefix search in the STAR algorithm for detecting (a) splice junctions, (b) mismatches and (c) tails
In addition to detecting splice junctions, the MMP search, implemented in STAR, enables finding multiple mismatches and indels, as illustrated in Figure 1b. If the MMP search does not reach the end of a read because of the presence of one or more mismatches, the MMPs will serve as anchors in the genome that can be extended, allowing for alignments with mismatches. In some cases, the extension procedure does not yield a good genomic alignment, which allows identification of poly-A tails, library adapter sequences or poor sequencing quality tails (Fig. 1c). The MMP search is performed in both forward and reverse directions of the read sequence and can be started from user-defined search start points throughout the read sequence, which facilitates finding anchors for reads with errors near the ends and improves mapping sensitivity for high sequencing error rate conditions.
Besides the efficient MMP search algorithm, uncompressed SAs also demonstrate a significant speed advantage over the compressed SAs implemented in many popular short read aligners (Supplementary Section 1.8). This speed advantage is traded off against the increased memory usage by uncompressed arrays, which is assessed further in Section 3.3.
2.2 Clustering, stitching and scoring
In the second phase of the algorithm, STAR builds alignments of the entire read sequence by stitching together all the seeds that were aligned to the genome in the first phase. First, the seeds are clustered together by proximity to a selected set of ‘anchor’ seeds. We found that an optimal procedure for anchor selection is through limiting the number of genomic loci the anchors align to. All the seeds that map within user-defined genomic windows around the anchors are stitched together assuming a local linear transcription model. The size of the genomic windows determines the maximum intron size for the spliced alignments. A frugal dynamic programming algorithm (see Supplementary Section 1.5 for details) is used to stitch each pair of seeds, allowing for any number of mismatches but only one insertion or deletion (gap).
Importantly, the seeds from the mates of paired-end RNA-seq reads are clustered and stitched concurrently, with each paired-end read represented as a single sequence, allowing for a possible genomic gap or overlap between the inner ends of the mates. This is a principled way to use the paired-end information, as it reflects better the nature of the paired-end reads, namely, the fact that the mates are pieces (ends) of the same sequence. This approach increases the sensitivity of the algorithm, as only one correct anchor from one of the mates is sufficient to accurately align the entire read.
If an alignment within one genomic window does not cover the entire read sequence, STAR will try to find two or more windows that cover the entire read, resulting in a chimeric alignment, with different parts of the read mapping to distal genomic loci, or different chromosomes, or different strands (Supplementary Fig. S1). STAR can find chimeric alignments in which the mates are chimeric to each other, with a chimeric junction located in the unsequenced portion of the RNA molecule between two mates. STAR can also find chimeric alignments in which one or both mates are internally chimerically aligned, thus pinpointing the precise location of the chimeric junction in the genome. An example of the BCR-ABL fusion transcript detection from the K562 erythroleukemia cell line is given in the Supplementary Section 1.7 (Supplementary Fig. S2).
The stitching is guided by a local alignment scoring scheme, with user-defined scores (penalties) for matches, mismatches, insertions, deletions and splice junction gaps, allowing for a quantitative assessment of the alignment qualities and ranks (see Supplementary Section 1.4 for details). The stitched combination with the highest score is chosen as the best alignment of a read. For multimapping reads, all alignments with scores within a certain user-defined range below the highest score are reported.
Although the sequential MMP search only finds the seeds exactly matching the genome, the subsequent stitching procedure is capable of aligning reads with a large number of mismatches, indels and splice junctions, scalable with the read length. This characteristic has become ever more important with the emergence of the third-generation sequencing technologies (such as Pacific Biosciences or Ion Torrent) that produce longer reads with elevated error rates.
3 RESULTS
3.1 Performance on simulated RNA-seq data
First, we used simulated data to evaluate performance of STAR and compare it with other RNA-seq mappers. Simulations allow for a precise calculation of false-positive and -negative rates, although artificial error models, used to generate simulated reads, may not adequately represent experimental errors. We used a simulated dataset from a recent study (Grant et al., 2011), in which 10 million of 2 × 100 nt Illumina-like read sequences with a reasonably high error rate were generated from the mouse transcriptome, including annotated transcripts and artificial ones. Various types of genomic variations and sequencing errors were introduced to mimic real RNA-seq data.
The latest available versions of STAR 2.1.3, TopHat2 2.0.0 (Trapnell et al., 2009), GSNAP 2012-07-03 (Wu and Nacu, 2010), RUM 1.11 (Grant et al., 2011) and MapSplice 1.15.2 (Wang et al., 2010) were run on the simulated dataset labeled as ‘SIM1-TEST2’ in (Grant et al., 2011). Because the TopHat2 2.0.0 release represents a major new development of the TopHat aligner, which has not been peer reviewed yet, we also made the comparisons with the previous TopHat version 1.4. We found that the new version yields a slightly better accuracy and faster mapping speed (Supplementary Section 2.1 and Fig. S3). All aligners were run in the de novo mode, i.e. without using gene/transcript annotations. The maximum number of mismatches was set at 10 per paired-end read, and the minimum/maximum intron sizes were set at 20 b/500 kb (Supplementary Section 2 for additional information). Note that running comparison between mappers with their default parameters is a reasonable and commonly accepted practice, as all considered aligners were, by default, optimized for mammalian genomes and recent RNA-seq data.
The resulting alignments were compared with the true genomic origin of the simulated reads, and true-/false-positive rates of splice junction detection were calculated using procedures and scripts developed by Grant et al. (2011). ROC curves (Fig. 2) were computed with the detection (discrimination) threshold given by the number of reads mapped across each junction, i.e. for each aligner, only junctions supported by at least N reads were selected for each point along the ROC curves, with N varied from 1 (lowest threshold) to 100 (high threshold). All aligners exhibit desirable steep ROC curves at high values of detection threshold. At the lowest detection threshold of 1 read per junction, STAR exhibits the lowest false-positive rate while achieving high sensitivity. Supplementary Figure S5 shows the same analysis for a low error rate-simulated dataset, which yields similar conclusions.
Fig. 2.
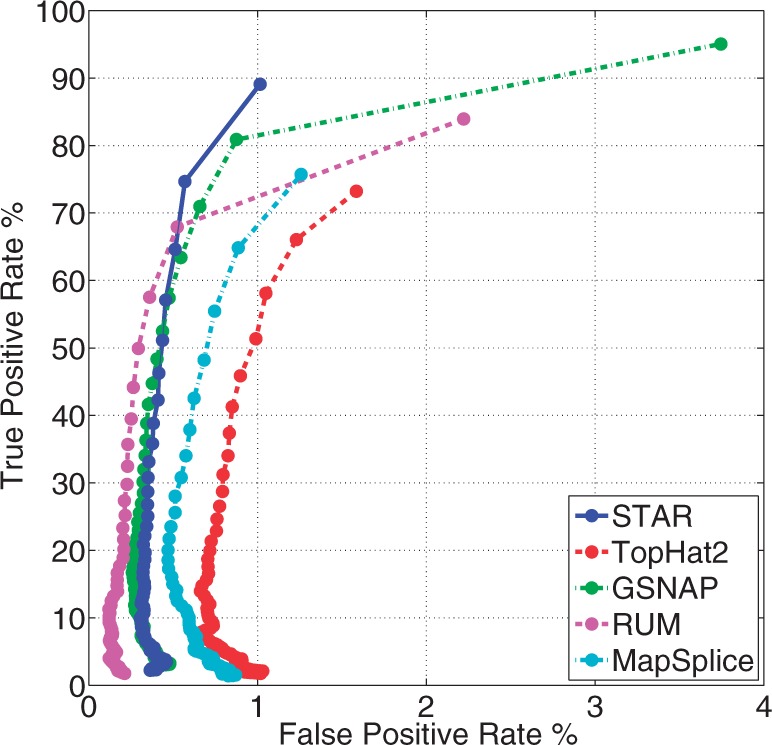
True-positive rate versus false-positive rate (ROC-curve) for simulated RNA-seq data for STAR, TopHat2, GSNAP, RUM and MapSplice
3.2 Performance on experimental RNA-seq data
For evaluation of the RNA-seq mappers’ performance on experimental RNA-seq data STAR, TopHat2, GSNAP, RUM and MapSplice were run (see Supplementary Section 2 for additional information) on an ENCODE long RNA-seq dataset (K562 whole cell A + sample, 1 Illumina GAIIx lane of 40 million 2 × 76 reads). STAR and GSNAP aligned the largest percentage of reads (94% both), followed by RUM (86%), MapSplice (85%) and TopHat2 (71%).
Different accuracy metrics for splice junction detection with respect to the Gencode 7 (Harrow et al., 2012) annotations are plotted in Figure 3a–c as a function of the detection threshold, defined as the minimum number of RNA-seq reads per junction. Although all aligners detect a similar number of annotated junctions (Fig. 3a, solid lines), there are noticeable differences between mappers in the number of detected unannotated junctions (Fig. 3a, dashed lines). The percentage of the unannotated among all detected junctions is plotted in Figure 3b as a function of detection threshold. Because all aligners show similar sensitivities to annotated junctions, the proportion of annotated among all detected junctions may serve as a surrogate of precision. STAR, RUM and TopHat2 perform similarly, while GSNAP exhibits lower precision at a lower detection threshold, and MapSplice shows unusual non-monotonic and non-saturating behavior, which was also noted in Zhang et al. (2012). Pseudo-ROC curve, i.e. the proportion of annotated junctions that are detected (pseudo-sensitivity) versus the proportion of detected junctions that are unannotated (pseudo-false-positive rate), is plotted in Figure 3c. All aligners (except MapSplice) perform similarly at high values of the detection threshold.
Fig. 3.

Various accuracy metrics for splice junction detection in the experimental RNA-seq data. The color-coding scheme for mappers is the same in all plots. X-axis in plots (a), (b), (d) and (e) is the detection threshold defined as the number of reads mapped across each junction, i.e. each point with the X-value of N represents all junctions that are supported by at least N reads mapped by a given aligner. (a) Total number of detected junctions, annotated (solid lines) and unannotated (dashed lines); (b) percentage of detected junctions that are annotated; (c) pseudo-ROC curve: percentage of all annotated junctions that are detected versus percentage of detected junctions that are unannotated; (d) number of unannotated junctions detected by at least two mappers (solid lines) and number of unannotated junctions detected exclusively by only one mapper (dashed lines); (e) percentage of detected unannotated junctions that are detected exclusively by only one mapper and (f) pseudo-ROC curve: percentage of unannotated junctions that are detected by at least two mappers versus percentage of detected unannotated junctions that are detected exclusively by only one mapper
Because many unannotated junctions represent true novel splicing events and are not false positives, the percentage of unannotated among all detected junctions is not an accurate proxy for the false-positive rate. To obtain a more accurate estimate of the false-positive rate, we followed another frequently used approach (Zhang et al., 2012) and plotted (Fig. 3d) the number of junctions detected by at least two mappers (pseudo-true positive) and the number of junctions detected exclusively by each mapper (pseudo-false positive). STAR alignments yield the lowest pseudo-false-positive rate, i.e. the lowest proportion of exclusively detected junctions (Fig. 3e), while at the same time achieving the second in class pseudo-sensitivity (Fig. 3f). GSNAP exhibits the highest pseudo-sensitivity at the cost of a high pseudo-false-positive rate. These results qualitatively agree with the aligners’ performance on the simulated data, whereas the quantitative differences may be attributed to disparities between real and simulated errors. Supplementary Figure S6 shows the same analysis for a shorter RNA-seq dataset (2 × 50 b), which indicates that STAR retains high sensitivity and precision even for short reads.
Note that the pseudo-true-/false-positive definitions are based on the assumption that junctions detected by only one aligner are more likely to be false positive than the junctions detected by two or more aligners; however, these definitions are not rigorous because the true/false assessments cannot be made for experimental data. We would also like to stress that these comparisons were done for current versions of each tool, with the default parameters and for the present state of Illumina sequencing technology. As both sequencing technologies and tools improve, these rankings may change and have to be reevaluated.
Similarly to other RNA-seq aligners, STAR’s default parameters are optimized for mammalian genomes. Other species may require significant modifications of some alignment parameters; in particular, the maximum and minimum intron sizes have to be reduced for organisms with smaller introns.
3.3 Speed benchmarks
Speed benchmarks were performed on a server equipped with two 6-core Intel Xeon CPUs X5680@ 3.33 GHz and 148 GB of RAM (random-access memory). Six or 12 threads were requested for each run, using half or full capacity of the server. All mappers were run with their default parameters on the ∼40 million 2 × 76 Illumina human RNA-seq dataset described in the previous section.
The ‘wall’ time (i.e. the total run time required to complete the mapping) and RAM usage are presented in Table 1. STAR achieves a speed of 550 million 2 × 76 Illumina paired-end reads per hour using 12 threads (full capacity of the server), i.e. 45 million paired reads per hour per processor, outperforming the second fastest mapper (TopHat2) by a factor >50. STAR exhibits close to linear scaling of the throughput rate with the number of threads, losing ∼10% of per thread mapping speed when the number of threads is increased from 6 to 12.
Table 1.
Mapping speed and RAM benchmarks on the experimental RNA-seq dataset
| Aligner | Mapping speed: million read pairs/hour |
Peak physical RAM, GB |
||
|---|---|---|---|---|
| 6 threads | 12 threads | 6 threads | 12 threads | |
| STAR | 309.2 | 549.9 | 27.0 | 28.4 |
| STAR sparse | 227.6 | 423.1 | 15.6 | 16.0 |
| TopHat2 | 8.0 | 10.1 | 4.1 | 11.3 |
| RUM | 5.1 | 7.6 | 26.9 | 53.8 |
| MapSplice | 3.0 | 3.1 | 3.3 | 3.3 |
| GSNAP | 1.8 | 2.8 | 25.9 | 27.0 |
STAR’s high mapping speed is traded off against RAM usage: STAR requires ∼27 GB of RAM for aligning to the human genome. Like all other aligners, with the exception of RUM, the amount of RAM used by STAR does not increase significantly with the number of threads, as the SA is shared among all threads. Although STAR’s RAM requirements would have been prohibitively expensive several years ago, at the time when the first short read aligners were developed, recent progress in semiconductor technologies resulted in a substantial drop of RAM prices, and modern high performance servers are commonly equipped with RAM >32 GB. STAR has an option to use sparse SAs, reducing the RAM consumption to <16 GB for the human genome at the cost of ∼25% decrease in the mapping speed, while maintaining the alignment accuracy.
3.4 Experimental validation
As part of the characterization of human transcriptome by the ENCODE (Djebali et al., 2012), STAR was used to map polyadenylated (poly A+) long (>200 nt) transcripts isolated from whole cell extracts of primary human H1ES (embryonic stem cells) and HUVEC (umbilical vein endothelial cells) cell lines. These RNAs were sequenced using a duplex-specific nuclease protocol (Parkhomchuk et al., 2009) that generated 2 × 76 bp strand-specific reads.
Not surprisingly, unannotated (novel) splice sites show lower abundance levels than the annotated junctions, as indicated by the significant drop in the number of unannotated junctions with the number of supporting reads (Supplementary Fig. S7). Because each of the cell lines was sequenced in biological duplicates, a collection of high confidence splice sites could be identified based on their reproducibility between replicas. To assess the reproducibility of the detected splice junctions, we developed a non-parametric irreproducible discovery rate (npIDR) approach, specifically suitable for the discrete nature of the RNA-seq data (see Supplementary Materials for the detailed description). This approach is similar to the npIDR concept extensively used in the analysis of the ENCODE ChIP-seq experiments (Landt et al., 2012). Supplementary Figure S8 shows the dependence of npIDR = 0.1 on the read count per junction, providing a principled method for selecting the read count threshold with a desired level of reproducibility. For example, five staggered reads per junction are required to achieve an npIDR of 0.1, i.e. the 90% likelihood that these junctions will be observed again in another experiment on the same cell line with the same sequencing depth.
Experimental validation was carried out on 1920 novel splice junctions in a wide range of RNA-seq reads support, both below and above the npIDR threshold. Only splice junctions mapped to intergenic or antisense loci to Gencode 7 genes (Harrow et al., 2012) were chosen for validation, as these junctions are more likely to be false positive than the junctions that map within the annotated genes. The high-throughput validation pipeline involved reverse transcription polymerase chain reaction amplification of targeted regions followed by Roche 454 sequencing of the pooled products. The reverse transcription polymerase chain reaction primer design took advantage of the ∼250 nt insert length of the paired-end reads supporting targeted junctions, and entailed the production of long 300–600 nt amplicons. These amplicons were pooled and sequenced by a Roche 454 sequencer to provide long and more confidently mappable reads that were aligned to the genome with BLAT. Detailed description of the experimental protocols can be found in Djebali et al. (2012).
We selected 1920 intergenic and antisense splice junctions from H1ES and HUVEC cell lines, including both highly (npIDR < 0.1) and poorly (npIDR > 0.1) reproducible junctions. Of all the tested novel intergenic/antisense junctions supported by at least five RNA-seq reads (corresponding to npIDR < 0.1), ∼82–89% (H1ES) and 84–95% (HUVEC) were corroborated by at least two amplicons sequenced by 454 (Table 2). Notably, the validation rate remains at a high level of 72% (H1ES) and 74% (HUVEC) even for the candidate junctions that were supported by as few as two RNA-seq reads. These results confirm high precision of the STAR’s splicing detection algorithm even for rare novel junctions.
Table 2.
Number of selected junctions and percentage of selected junctions that were validated by at least two 454 reads, as a function of the RNA-seq read count per junction
| H1ES |
HUVEC |
||||
|---|---|---|---|---|---|
| Read count per junction from two replicates | Number of tested junctions | Proportion of junctions validated by at least two 454 reads (%) | Read count per junction from two replicates | Number of tested junctions | Proportion of junctions validated by at least two 454 reads (%) |
| 2 | 192 | 72.4 | 2 | 192 | 74.0 |
| 3 | 192 | 77.6 | 3 | 192 | 75.0 |
| 4 | 96 | 74.0 | 4 | 96 | 76.0 |
| 5 | 96 | 82.3 | 5–6 | 96 | 84.4 |
| 6–7 | 96 | 79.2 | 7–8 | 96 | 84.4 |
| 8–11 | 96 | 81.3 | 9–12 | 96 | 86.5 |
| 12–24 | 96 | 87.5 | 13–23 | 96 | 94.8 |
| ≥25 | 96 | 88.5 | ≥24 | 96 | 90.6 |
The upper bound of the false discovery rate (FDR) can be estimated from the validation rate (≡VR) as FDR ≤ 1 − VR. For low abundance junctions, the experimental FDR is lower than the npIDR predicted from the dissimilarity between the replicates: for example, although 45% of junctions, supported by just two reads, are not reproducible (Supplementary Fig. S8), > 70% of them are successfully validated (Table 2). Hence, npIDR can serve as a conservative upper bound FDR estimate in cases where validation experiments are impractical.
4 DISCUSSION
Despite several years of ongoing improvements, alignment of the non-contiguous RNA-seq reads to a reference genome is not a solved problem yet, owing both to its intrinsic complexity and rapid transformations of the sequencing technologies. Several critical problems have been found to afflict previously published approaches, such as high mapping error rate, alignment biases, low sensitivity for unannotated transcripts, poor scalability with the read length, restrictions in the number of junctions/mismatches/indels per read, inability to detect non-linear transcripts (such as chimeric RNAs), and, crucially, low mapping throughput.
In this work, we described STAR, a novel algorithm for aligning high-throughput long and short RNA-seq data to a reference genome, developed to overcome the aforementioned issues. Unlike many other RNA-seq mappers, STAR is not an extension of a short-read DNA mapper, but was developed as a stand-alone C++ code. STAR is capable of running parallel threads on multicore systems with close to linear scaling of productivity with the number of cores. STAR is fast: on a modern, but not overly expensive, 12-core server, it can align 550 million 2 × 76 nt reads per hour to the human genome, surpassing all other existing RNA-seq aligners by a factor of 50. At the same time, STAR exhibits better alignment precision and sensitivity than other RNA-seq aligners for both experimental and simulated data.
One of the main inherent problems of all de novo RNA-seq aligners is the inability to accurately detect splicing events that involve short (<5–10 nt) sequence overhangs on the donor or acceptor sides of a junction. This causes a significant underdetection of splicing events, and also increases significantly the misalignment rate, as such reads are likely to be mapped with a few mismatches to a similar contiguous genomic region. In addition, this effect also biases the alignments toward processed pseudogenes, which are abundant in the human genome. Similarly to other RNA-seq aligners, to mitigate this problem, STAR has an option to obtain information about possible splice junction loci from annotation databases (Supplementary Section 4). It is also possible to run a second mapping pass, supplying it with splice junction loci found in the first mapping pass. In this case, STAR will not discover any new junctions but will align spliced reads with short overhangs across the previously detected junctions.
To demonstrate STAR’s ability to align long reads, we have mapped the long (0.5–5 kb) human mRNA sequences from GenBank (see Supplementary Section 5 for details). The accuracy of STAR alignments is similar or higher than that of BLAT (Kent, 2002) a popular EST/mRNA aligner. At the same time, STAR outperforms BLAT by more than two orders of magnitude in the alignment speed, which is important for high-throughput sequencing applications.
The algorithm extensibility to long reads shows that STAR has a potential to serve as a universal alignment tool across a broad spectrum of emerging sequencing platforms. STAR can align reads in a continuous streaming mode, which makes it compatible with novel sequencing technologies such as the one recently announced by Oxford Nanopore Technologies. As the sequencing technologies and protocols evolve, new mapping strategies will have to be developed, and STAR core algorithm can provide a flexible framework to address arising alignment challenges.
Data access
GEO: GSE38886 (Roche 454 sequencing)
GEO: GSE30567 (Illumina long RNA-Seq)
Funding: This work was funded by NHGRI (NIH) grant U54HG004557.
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Au KF, et al. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;38:4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AC, et al. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, et al. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bona F, et al. Optimal spliced alignments of short sequence reads. Bioinformatics. 2008;24:i174–180. doi: 10.1093/bioinformatics/btn300. [DOI] [PubMed] [Google Scholar]
- Delcher AL, et al. Alignment of whole genomes. Nucleic Acids Res. 1999;27:2369–2376. doi: 10.1093/nar/27.11.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, et al. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30:2478–2483. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flusberg BA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods. 2010;7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant GR, et al. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM) Bioinformatics. 2011;27:2518–2528. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, et al. Pre-mRNA splicing: where and when in the nucleus. Trends Cell. Biol. 2011;21:336–343. doi: 10.1016/j.tcb.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, et al. GENCODE: The reference human genome annotation for the ENCODE project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings ML, Krainer AR. Pre-mRNA splicing in the new millennium. Curr. Opin. Cell. Biol. 2001;13:302–309. doi: 10.1016/s0955-0674(00)00212-x. [DOI] [PubMed] [Google Scholar]
- Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WS. BLAT–the BLAST-like alignment tool. . Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manber U, Myers G. Suffix arrays—a new method for online string searches. SIAM J. Comput. 1993;22:935–948. [Google Scholar]
- Parkhomchuk D, et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothberg JM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- Trapnell C, et al. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. PASSion: a pattern growth algorithm-based pipeline for splice junction detection in paired-end RNA-Seq data. Bioinformatics. 2012;28:479–486. doi: 10.1093/bioinformatics/btr712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.