

Abstract
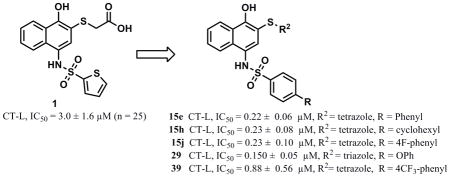
Screening efforts led to the identification of PI-8182 (1), an inhibitor of the chymotrypsin-like (CT-L) activity of the proteasome. Compound 1 contains a hydronaphthoquinone pharmacophore with a thioglycolic acid side chain at position 2 and thiophene sulfonamide at position 4. An efficient synthetic route to the hydronaphthoquinone sulfonamide scaffold was developed and compound 1 was synthesized in-house to confirm the structure and activity (IC50 = 3.0 ± 1.6 μM [n=25]). Novel hydronaphthoquinone derivatives of the hit 1 were designed, synthesized and evaluated as proteasome inhibitors. The structure activity relationship (SAR) guided synthesis of more than 170 derivatives revealed that the thioglycolic acid side chain is required and the carboxylic acid group of this side chain is critical to the CT-L inhibitory activity of compound 1. Furthermore, replacement of the carboxylic acid with carboxylic acid isosteres such as tetrazole or triazole greatly improves potency. Compounds with a thio-tetrazole or thio-triazole side chain in position 2, where the thiophene was replaced by hydrophobic aryl moieties were the most active compounds with up to 20-fold greater CT-L inhibitory than compound 1 (compounds 15e, 15f, 15h 15j, IC50 values around 200 nM and compound 29, IC50 = 150 nM). The synthetic iterations described here not only led to improving potency in vitro but also resulted in the identification of compounds that are more active such as 39 (IC50 = 0.44 to 1.01 μM) than 1 (IC50 = 3.54 to 7.22 μM) at inhibiting the proteasome CT-L activity in intact breast cancer cells. Treatment with 39 also resulted in the accumulation of ubiquitinated cellular proteins and inhibition of tumor cell proliferation of breast cancer cells. The hit 1 and its analog 39 inhibited proteasome CT-L activity irreversibly.
INTRODUCTION
Regulated protein degradation is an essential aspect of cell signaling.1 In 2004, Ciechanover2, Hershko3 and Rose4 were awarded the Nobel Prize for chemistry in elucidating the importance of proteolytic degradation inside cells and the role of ubiquitin in proteolytic pathways. Proteasomes are highly conserved compartmentalized protease complexes belonging to the family of N-terminal nucleophilic hydrolases.5 The proteasome degrades ubiquitinated proteins into small peptides6 and the ubiquitin proteasome system (UPS) is responsible for the degradation of cellular proteins (e.g. potentially toxic, oxidized, misfolded proteins, and cell cycle regulatory proteins). In cancer cells, the UPS is essential to the mechanisms underlying tumorigenesis, metastasis, angiogenesis and apoptosis.7,8, 9 Therefore, targeting the regulation of protein production and degradation that mediates proliferation and other hallmarks characteristics of malignancy has been a major focus of cancer research. Since the approval of Bortezomib (Velcade™) for multiple myeloma in 2003 by the FDA (Figure 1), the proteasome has been validated as an important target for cancer therapy.10 In addition, several studies have shown that proteasome inhibition is also important for inflammatory and autoimmune diseases.11,12 Most natural and synthetic proteasome inhibitors reported to date including Bortezomib contain a reactive moiety in the pharmacophore that forms an irreversible and/or slowly reversible covalent bond with the nucleophilic N-terminal Thr in the β5 subunit of the proteasome.13,14 Proteasome inhibitors reported to date fall into five classes15; peptide boronates, peptide aldehydes, peptide vinyl sulfones, peptide epoxyketones and β-lactones. Peptide aldehydes and vinyl sulfones also inhibit other proteases (cathepsin A, tripeptidyl peptidase II) in addition to the proteasome.16 The β-lactone salinosporamide A17,18 and tetrapeptide epoxyketone Carfilzomib19 (Figure 1) represent two classes of irreversible and/or slowly reversible proteasome inhibitors that are in clinical trials.20 The proteolytic activity of the chymotrypsin-like (CT-L) active site (β5 subunit) of the proteasome cleaves peptides that contain amino acid residues with large hydrophobic side chains.13 The trypsin-like (T-L, β2 subunit) and postglutamylpeptidase hydrolysis (PGPH, β1 subunit) proteolytic sites of the proteasome cleave peptides after acidic and basic amino acid residues respectively.13 Therefore, the selectivity of proteasome inhibition using peptide based compounds would be hard to achieve just by simply manipulating the peptide backbone. Examples of non-peptidic, small drug-like synthetic molecules which act as proteasome inhibitors are rare, and these molecules would have potential advantages over the existing inhibitors for therapeutic interventions. Recently, we disclosed identification of PI-083 (NSC-45382), a small synthetic molecule with a naphthoquinone pharmacophore as a reversible proteasome inhibitor that selectively targets cancer cells over non-transformed cells in vitro as well as in vivo.21,22 In the course of our search for new classes of inhibitors of the 20S proteasome, compound 1 (Figure 1) was recently identified and characterized as a ‘hit’ for proteasome inhibition with CT-L inhibitory activity from our in-house ChemDiv library. The synthesis of compound 1 is not reported in the literature. Herein we describe an efficient synthetic route to 1 and detailed in vitro structure activity relationship (SAR) studies of 1 via focused library synthesis as a part of our on-going efforts in the development of proteasome inhibitors.
Figure 1.

Structures of representative proteasome inhibitors; Bortezomib (clinically approved), Carfilzomib (in clinical trials) and the proteasome activity profile of the “hit” 1.
CHEMISTRY
Compound 1 was recently identified in our program as a ‘hit’ from our in-house ChemDiv 20,000 compound library that showed CT-L proteasome inhibitory activity with an IC50 value of 3.0 ± 1.6 (n=25, >95% pure by HPLC). This result prompted us to further investigate 1 as a proteasome inhibitor and establish structure and activity relationship studies (SAR) via synthetic modifications around the hydronaphthoquinone pharmacophore. The hydronaphthoquinone pharmacophore in compound 1 exhibits desirable structural diversity that was exploited for focused library synthesis and medicinal chemistry. Synthetic modifications were primarily focused on the side chain at the 2-position of the hydronaphthoquinone pharmacophore, (Figure 2) and the sulfonamide moiety of 1 to understand the structural moieties responsible for proteasome inhibition. Proposed synthetic modifications to probe binding interactions in the β5 subunit of the proteasome are described in Figure 2.
Figure 2.

Proposed synthetic modifications around compound 1 (hit); B side chain at 1-position to probe interactions.
A synthetic route to compound 1 is not reported to the best of our knowledge. Initially, halogenated hydronaphthoquinones 4a–c were synthesized starting from commercially available 4-aminonaphthol hydrochloride salt (2) and thiophene-2-sulfonyl chloride in good yields (Scheme 1). The intermediate 3 was easily obtained with high yields (Scheme 1, condition a, this reaction is reported with NaHCO3 as a base with moderate yields23) and reacted with hydrogen peroxide and 4M HCl in dioxane solution, similar to a literature reported protocol24 to obtain the compound 4a. Compounds 4b and 4c were also synthesized in a similar manner in the presence of bromine and iodine respectively with triethylamine in DMF (Scheme 1) according to a reported method25 (see 1H-NMR and HRMS of 4a, 4b and 4c in the experimental section). Our strategy was to oxidize the hydronaphthoquinone 4a to obtain the oxidized 2-chloronaphthoquinone (structure shown for oxidized 4a in Scheme 1) that would facilitate the synthesis of the final compound 1. Our attempts to oxidize compound 4a (using H2O2, Pb(OAc)4) were not successful. Furthermore, attempts to directly introduce the thioether carboxylic ester side chain in compound 1 via the intermediate 5 (Scheme 1) similar to a reported protocol with hydrazides26 and thiols27 were not successful. Syntheses of derivatives of intermediate 5 are reported from derivatives of compound 3 using lead tetraacetate22 in acetic acid, hydrogen peroxide23 or MnO2 28 in methanol. In spite of repeated attempts, we were unable to obtain the required oxidized intermediate 5 from the hydroxynaphthalene sulfonamide intermediate 3 using these reported protocols. Our reactions produced unidentified impurities by 1H NMR and TLC. Hence, this route to obtain compound 1 was abandoned.
Scheme 1.

Synthesis of hydronaphthoquinone sulfonamide scaffold
Reagents and conditions: a) thiophene-2-sulfonyl chloride, Et3N, dichloromethane, r.t., 14 h, 90%; b) for 4a (i) H2O2 (35%), CH3OH, r.t., 2 hrs, (ii) 4M HCl/dioxane, r.t., 2 h, 22%; for 4b Br2, Et3N, DMF, 0 °C to r.t., 12–14 h, 36 % and for 4c I2, Et3N, DMF, 0 °C to r.t., 12 h, 100%.
To combine the hydronaphthoquinone ring and the thioether side chain in structure 1 as required, we then validated the synthetic protocols shown in Schemes 2 and 3. Commercially available 1,4-naphthoquinone (6) (Scheme 2), 2-hydroxy-1,4-naphthoquinone (7) (Scheme 2) or 2-chloro-1,4-naphthoquinone (12) (Scheme 3) were used as starting materials. First, naphthoquinone intermediates 8a and 8b were obtained in good yield using ethyl 2-mercaptoacetate (one carbon side chain) or methyl 3-mercaptopropionate (2-carbon side chain) respectively with 2 equivalents of 1,4-naphthoquinone (6) using EtOH as the solvent. (Scheme 2, condition a). The intermediate 8c was obtained via alkylation of 2-hydroxy-1,4-naphthoquinone (7) with tert-butyl bromoacetate using silver(I) oxide as a base using a reported protocol (Scheme 2, condition b). 29 The sulfonamide building blocks that were not commercially available were synthesized according to a reported protocol.30 The sulfonamide building blocks either commercially available or in-house synthesized were then regioselectively coupled to naphthoquinone intermediates 8a–c according to a procedure that has been widely used in the formation of sulfonimide building blocks 31 using titanium(IV) chloride and triethylamine with conventional heating. We modified the conditions of this reaction (i.e. synthesis of intermediate 9 in Scheme 2, condition c) using microwave assisted heating (60 °C, 5–20 min. 50–60% yield) to accommodate parallel synthesis. This reaction was a key reaction in building the sulfonimide naphthoquinone scaffold 9 in our synthetic route to final compounds and microwave assisted heating was convenient for library synthesis. Coupling intermediates 8 with various sulfonamides proceeded regioselectively at the 4-carbonyl of the naphthoquinone ring. We observed the change of the chemical shift of hydrogen at 3-position of 8a from 6.7 ppm to 7.9 ppm in 9a. (see NMR spectra in supplementary information). However, the coupling reactions of 3-methyl substituted naphthoquinone (i.e. 8 with a methyl group at 3-position, Scheme 2) with arylsulfonamides were not successful using this protocol possibly due to the steric hindrance of the adjacent methyl group.
Scheme 2.

Synthetic route to compound 1 (hit) and derivatives of the hit
Reagents and conditions: a) HS(CH2)nCOOR1 (R1 = ethyl, n =1 for 8a, 89 % and R1 = methyl, n =2 for 8b, 94 %), ethanol (2.5 mL/mmol), r.t. 0.5 h; b) BrCH2COOtBu, Ag2O, CHCl3, cat. KI, reflux under Ar, 36 h, 33%; c) R2SO2NH2, TiCl4·2THF, Et3N, DCM, microwave, 60 °C, 5~20 min, 26~62%; d) Na2S2O4, THF, 1 h, r.t. or EtOAc, H2O, r.t. 10 min., 59–93%; e) Oxone, H2O, acetone, r.t., 12–14 h, 100%; f) conc. HCl/dioxane (1:1, 16 mL/mol), r.t 3–36 h or microwave, 100 °C, 10 min. 54–97%.
Scheme 3.

Optimized synthetic route to hit and final compounds
Reagents and conditions: a) R1SO2NH2, TiCl4·2THF or 1M TiCL4 in DCM, Et3N, THF, microwave, 60 °C, 15–30 min., 30~86%; b) EtOH :DCM (1:1) for tetrazole-5-thiol, r.t., 2 h, THF for 1-methyl-tetrazole-5-thiol or 3-mercapto-1,2,4-triazole, r.t., 2 h; THF, 1 eq. pyridine for thioglycolic acid, r.t., 10–30 min.; c) Na2S2O4, EtOAc and H2O (mixing in a separating funnel), 54~100 %; d). NaN3, water, reflux 2 h, 67%; e) TFA/anisole (5:1, 2 mL/mmole), microwave 100 °C, 2 h, 93%.
In the coupling reactions with intermediate 8, formation of several impurities were observed by TLC, and one of the major by-products formed was shown to be the reduced structure hydronaphthoquinone (i.e. compound 10). We were able to isolate this impurity using SiO2 chromatography and confirmed it to be, by NMR and LCMS, the hydronaphthoquinone derivative 10. The key intermediates 9 were reduced in situ using sodium hydrosulfite32 to hydronaphthoquinone carboxylic esters 10 (Scheme 2, condition d,). Hydrolysis of 10 in a 1:1 mixture of concentrated HCl and dioxane gave the final hydronaphthoquinone acid library 11 in moderate yield (Scheme 2, condition f). Compound 1 (Figure 1) was synthesized via this route to confirm the structure and CT-L proteasome inhibitory activity. The sulfone moiety in compound 11e was formed by oxidation of compound 10a with oxone33 followed by acid hydrolysis (Scheme 2, conditions e and f). The by-product formation in the sulfonamide coupling reaction (i.e. step c in Scheme 2) made the isolation of intermediates 9 laborious and inconvenient for library synthesis. Therefore, further optimization of the synthetic route was carried out with commercially available 2-chloro-1,4-naphthoquinone (12)as shown in the Scheme 3.
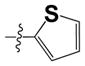
The hydronaphthoquinone focused library 15 (Scheme 3) was synthesized via coupling various sulfonamides to commercially available 2-chloro-1,4-naphthoquinone (12) using the same synthetic protocol validated for naphthoquinone intermediate 9 (Scheme 2, condition c). The yield of this reaction was significantly improved from 56% to 83% by replacing DCM with THF (Scheme 3, condition a). The key intermediates 13 (Scheme 3) were then easily purified by either re-crystallization or triturating from appropriate solvents (see experimental section), was more convenient for library synthesis. Substitution of the 2-chlorine intermediates 13 with various nucleophiles was carried out in the presence of pyridine when thioglycolic acid was used and without a base when tetrazole-5-thiol (Scheme 3, condition b), 1-methyl-tetrazole-5-thiol or 3-mercapto-1,2,4-triazole (commercially available building blocks) was used to generate the library 14 with the appropriate thioether moieties.34 Synthesis of the tetrazole-5-thiol building block (Scheme 3, condition d) was carried out from para-methoxybenzylisothiocyanate and sodium azide. Deprotection of the para-methylbenzyltetrazole intermediate was achieved with trifluoroacetic acid to synthesize the final tetrazole-5-thiol building block (Caution!!! This reaction can be explosive, isolation and drying large quantities of the thiotetrazole building block should be avoided. Once prepared, the thio-tetrazole building block should be stored in a solvent until further use). The final step in the Scheme 3 involved in situ reduction of the library 14 with thioether side chains (XR2 = thioglycolic acid, thiolactic acid, thiotetrazoles, thiotriazoles, thiomethyltetrazoles, thioaromatic groups, thioalkyl and thioamide side chains [not reported here]) with high yields to afford the required final library 15 with > 95% purity by NMR and LCMS. The purity of the most potent biologically active thioether analogs was > 95% as determined by HPLC (see experimental). Few of the intermediates of the library 14 leading to library 15 were characterized and confirmed by NMR, LCMS and formula guided mass spectroscopy. The library members 15 with thioether side chains appeared more stable at room temperature compared to the compound bearing ether moiety (i.e. 11b). The synthesis of the final library 15 with thioether side chains via 14 using the key naphthoquinone intermediate 13 proved to be more convenient (3 steps) and we used this optimized synthetic route to generate >170 compounds (varying the sulfonamide moiety and the side chain) including the original hit 1.
Moving the carboxylic acid side chain from the 2-position to 1-position of the naphthalene ring in compound 1 (Figure 1) is outlined in the Scheme 4. Our aim was to synthesize a small number of derivatives of the hit with O-substituted and S-substituted carboxylic acid side chains at 1-position of the naphthoquinone ring to probe interactions in the CT-L subunit of the proteasome. First, compounds 16a and 16b were obtained by directly alkylating the compound 3 (synthesis of 3 is shown in Scheme 1) with 2-bromoacetate or 4-bromobutyrate using DBU (Scheme 4, condition a). Compound 3 was less reactive and 15 equivalents of 2-bromoacetate or 4-bromobutyrate were required to push the reactions. Acidic hydrolysis of the intermediate esters 16a and 16b gave 17a and 17b respectively in good yields. The synthesis of the intermediates 16a and 16b and the final compounds 17a (one carbon side chain) and 17b (3-carbon side chain) were confirmed by NMR, LCMS and formula guided mass spectroscopy.
Scheme 4.

Synthesis of the thioglycolic acid side chain at 1-position
Reagents and conditions: a)15 eq. Br(CH2)nCOOR, 2 eq. DBU, DMF, microwave, 90 °C, 50–60 min, 51%; b) conc. HCl/dioxane (1:1, 20 mL/mmol), r.t., 1 ~ 12 h, 50–80%. c) Me2NCSCl, K2CO3, NMP, 50 °C, 2 h, 90%; d) NMP, microwave, 180 °C, 20 min., 72%; e) (i) 4 eq. KOH, CH3OH, inert conditions, r.t., 12 h; (ii) BrCH2COOC(CH3)3, r.t., 12 h, 86%; f) H-Cube: H2 (40 bar, 25 °C), Pd/C (10%), CH3OH, flow rate: 1 ml/min, 100%; g) (i) Pyridine, THF/H2O (4:1), 0 °C, (ii) thiophene-2-sulfonyl chloride, 0 °C → r.t., 4 h, 58%.
Reaction of commercially available 4-nitronaphthol with N,N-dimethylthiocarbamoyl chloride (Me2NCSCl) produced thiocarbamate 19 in good yield. A Newman-Kwart rearrangement was employed to build the thioether scaffold (intermediate 20 in Scheme 4, condition d) in the 1-position of the naphthalene ring. Hydrolysis of N,N-dimethylthioamide 20 in KOH solution (1 M, 4eq.) under inert conditions, followed by alkylation with tert-butyl 2-bromoacetate directly gave the acid intermediate 21. Hydrogenation of the 4-nitro group using an H-cube reactor (Scheme 4, condition f) gave intermediate 22 in quantitative yield. The intermediate 22 was used in the final step that involved sulfonamide formation with thiophene-2-sulfonyl chloride and pyridine in an aqueous solution to afford the desired compound thioether carboxylic acid 23 in good yield. Formation of compound 23 was confirmed by NMR, LCMS and formula guided mass spectroscopy and the purity of 23 was shown to be > 95% as determined by HPLC.
RESULTS AND DISCUSSION
The in-house synthesis (via both Schemes 2 and 3) of compound 1 confirmed the structure and CT-L proteasome inhibitory activity (IC50 = 3.0 ± 1.6 μM [n=25]), which in turn allowed us to develop novel compounds with improved activity and selectivity. In an effort to determine the important moieties of the pharmacophore that are critical to its CT-L inhibitory activity and to improve its in vitro and whole cell activities, we have synthesized over 170 derivatives and performed SAR studies using a fluorogenic assay as previously described.21, 22 In the initial screen the % of CT-L inhibitory activity was determined, and on the basis of these results a dose response (IC50 values) was obtained for compounds that displayed > 70% inhibition at 10 μM. The compounds that displayed potent CT-L inhibitory activity were also tested for in vitro trypsin-like (T-L) inhibitory activity (Table 1).21,22
Table 1.
SAR around the side chain S-R2, sulfonamide moiety R1 of compound 1.
| Compound ID | S-R2 | R1 | IC50a (μM) Proteasome Activity (In Vitro) | |
|---|---|---|---|---|
| CT-L | T-L | |||
| 1 (15a and 11a) | S-CH2COOH |
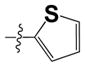
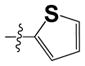
|
3.0 ± 1.6 | ND |
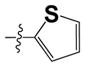
| 15b |
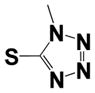
|
|
4.2 ± 2.1 | ND |
| 15c |
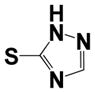
|
|
4.2 ± 2.7 | ND |
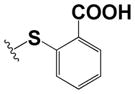
| 24 |
|
|
4.05 ± 2.64 | ND |
| 25 |
|
|
0.28± 0.8 | 4.22 ± 0.30 |
| 26 |
|
|
0.73 ± 0.03 | 2.35 ± 0.05 |
| 27 |
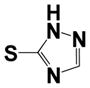
|
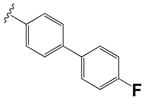
|
0.18 ± 0.08 | 2.37 ± 0.05 |
| 28 |
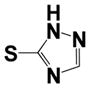
|
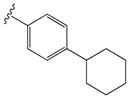
|
0.3 ± 0.07 | 7 ± 0.8 |
| 29 |
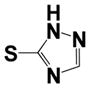
|
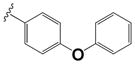
|
0.150 ± 0.05 | 4.2 ± 1.8 |
| 30 |
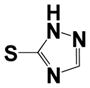
|
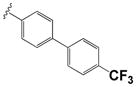
|
2.47 ± 0.52 | 7.2 ± 0.9 |
| 31 |
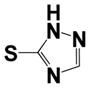
|
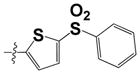
|
1.97 ± 0.08 | ND |
| 32 |
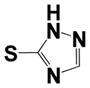
|
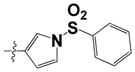
|
1.61 ± 0.06 | ND |
| 33 |
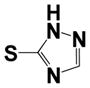
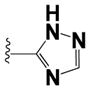
|
|
0.30± 0.01 | 2.0 ± 0.2 |
| 34 |
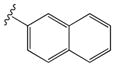
|
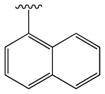
|
0.65 ± 0.09 | ND |
| 15d |
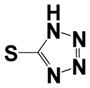
|
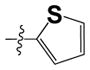
|
0.51 ± 0.12 | 0.20 ± 0.10 |
| 15e |
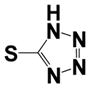
|
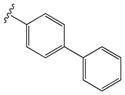
|
0.22 ± 0.06 | 4.80 ± 0.54 |
| 15f |
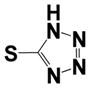
|
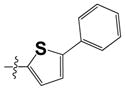
|
0.23 ± 0.02 | 0.55 ± 0.32 |
| 15g |
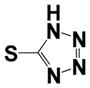
|
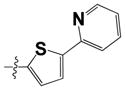
|
0.480 ± 0.09 | 1.55 ± 0.45 |
| 15h |
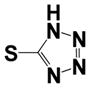
|
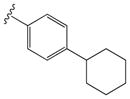
|
0.23 ± 0.08 | 1.85 ± 0.67 |
| 15i |
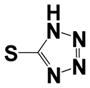
|
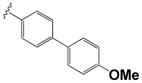
|
0.40 ± 0.12 | 2.80 ± 0.12 |
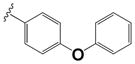
| 15j |
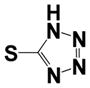
|
|
0.23 ± 0.1 | 2.0 ± 0.60 |
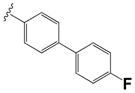
| 15k |
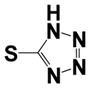
|
|
0.22 ± 0.01 | 1.3 ± 0.4 |
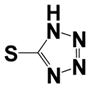
| 15l |
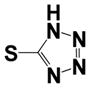
|
|
0.40 ± 0.14 | 1.35 ± 0.56 |
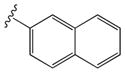
| 15m |
|
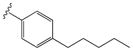
|
0.47 ± 0.18 | 1.45 ± 0.14 |
| 35 |
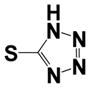
|
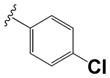
|
0.570 ± 0.06 | 0.75 ± 0.13 |
| 36 |
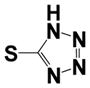
|
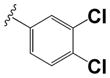
|
0.54 ± 0.08 | 0.57 ± 0.31 |
| 37 |
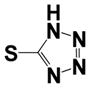
|
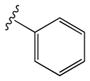
|
0.6 ± 0.1 | 1.38 ± 0.60 |
| 38 |
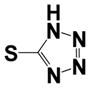
|
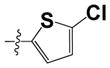
|
0.63 ± 0.1 | 1.10 ± 0.16 |
| 39 |
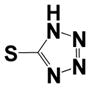
|
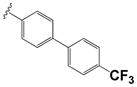
|
0.88 ± 0.56 | 1.14 ± 0.16 |
Each IC50 is at least the mean of 3 determinations.
ND-IC50 not determined, these compounds showed less than 70% T-L inhibition at 10 μM.
In the hit-to-lead optimization chemistry, we first investigated in detail the importance of the thioglycolic acid side chain while maintaining the thiophene sulfonamide moiety in structure 1 (Figure 2 A). Removal of the side chain from 1 as in the 2-unsubstituted counterpart 3 (Scheme 1, IC50 > 30 μM) was not tolerated indicating that the thioglycolic acid side chain is required for CT-L inhibitory activity. Hydronaphthoquinone analogs bearing halogens Cl, Br and I at the 2-position 4a, 4b and 4c respectively (Scheme 1) resulted in loss of CT-L inhibitory activity (IC50 > 30 μM) further confirming the importance of the thioglycolic acid side chain. Furthermore, direct replacement of ‘sulfur’ by ‘oxygen’ (ether side chain, 11b in Scheme 2) or ‘sulfone’ (11e in Scheme 2) was also detrimental to CT-L inhibitory activity (IC50 > 100 μM) indicating that the thio-ether moiety in compound 1 is important. The replacement of the thioglycolic acid with groups such as S-ethyl, O-ethyl, S-ethanol, thio-N,N-dimethylpropionamide (S(CH2)2CO-N-Me2) and thio-N-methylpropionamide (S(CH2)2CO-NH-CH3), showed weaker CT-L proteasome activity (IC50 > 5 μM, compounds not reported here), suggesting that the carboxylic acid moiety was essential for proteasome activity. The compound 10a (Scheme 2) with ethyl ester was shown to be less potent (IC50 > 10 μM) than the corresponding acid further confirming the importance of the H-bond donor/acceptor features of the carboxylic group for CT-L activity. The length of the side chain is also important as demonstrated by the 2-fold decreased potency in the extended thioether carboxylic acid side chain as in 11d (Scheme 2, IC50 = 5.7 μM). The position of the thioglycolic acid side chain is critical as demonstrated by the loss of CT-L inhibitory activity (IC50 >100 uM) in compound 23 (Scheme 4) where the thioglycolic acid side chain was moved from the 2 to the 1-position. Our attempts to substitute the thioglycolic acid chain of 1 with amines (not reported here) were not successful due to oxidized impurities formed in the final products. Finally, the naphthoquinone analogs of library 14 (Schemes 3) showed 2–3 fold weaker activity compared to the corresponding active analogs of hydronaphthoquinone library 15, indicating that the H-bond donor properties of the hydronaphthoquinone may be important for binding interactions with the β5 subunit of the protein. The SAR studies described above demonstrate that the thioglycolic acid side chain is required for the CT-L inhibitory activity of compound 1 and that the carboxylic acid moiety in this side chain is critical. Subsequently our chemistry efforts for side chain modifications were focused on substituting the carboxylic acid moiety with carboxylic acid isosteres such as tetrazoles or triazoles with a thioether moiety. By incorporating the tetrazole or triazole moieties we were aiming to improve the potency, solubility, and cell permeability of this class of compounds. Replacement of the carboxylic acid moiety with tetrazole is well known and has increasingly been used with success in drug discovery.35 Comparison of carboxylic acid and tetrazole groups at physiological pH reveals that tetrazole group is almost 10 times more lipophilic while having similar acidity pKa = 4.9, to that observed for carboxylic acids (pKa = 4.2–4.4).36
The replacement of the thioglycoloic acid side chain in structure 1 (Figure 1) with thio-tetrazole (compound 15d in Scheme 3) showed improved CT-L inhibitory activity (IC50 = 0.51 ± 0.12 μM, Table 1). Furthermore, replacement of the thioglycoloic acid side chain with thio-methyltetrazole (15b, Scheme 3), thio-triazole (15c, Scheme 3), and thiobenzoic acid (24, Table 1) resulted in retention of the CT-L proteasome activity (IC50 = 4.2, 4.2 and 4 respectively, Table 1). We were encouraged by these results and our next generation of compounds (Scheme 3) were synthesized with thio-tetrazole, -triazole or -methyl tetrazole to mimic the H-bond donor/acceptor features of the carboxylic acid moiety in our original hit. The compound 24 (Table 1) with thiobenzoic acid side chain was not further pursued for library synthesis due to poor solubility. In the next generation of analogs, we varied the sulfonamide moiety while keeping the thio-tetrazole, -triazole or -methyltetrazole moieties as side chains at the 2-position of the hydronaphthoquinone pharmacophore. Compounds with thio-tetrazole and thio-triazole side chains containing biaryl sulfonamide moieties (Table 1) displayed the best CT-L activities. For example, compounds 15e, 15f, 15h, 15j 15k, 27 and 29, (Table 1) displayed up to 20 fold (IC50 values around 150–250 nM) improved CT-L inhibitory activity. These results indicate that aromatic hydrophobic sulfonamide groups are tolerated in the binding region. Interestingly, compounds bearing 5-methyl-thio-tetrazole side chain with biaryl sulfonamide moieties (analogs of 15b in Table 1, not reported here) failed to improve the CT-L activity indicating that the tetrazole anion is contributing to potency (tetrazole is deprotonated at pH 7 similar to a carboxylic acid group). Overall, compounds with methyl-thiotetrazole side chain did not improve the CT-L proteasome activity.
The stability of some of the potent hydronaphthoquinone analogs (15f and 25, Table 1) with thioether side chain (thioglycolic acid, thio-tetrazole, thio-triazole) including the ‘hit’ were examined by 1H NMR in deuterated DMSO up to 3 weeks at room temperature. The HPLC and LCMS were also determined under the assay conditions (in tris buffer at room temperature). Our data concluded the hydronaphthoquinone class of compounds showed > 95% purity by LCMS, HPLC and 1H NMR and were stable at room temperature for up to two weeks. Approximately 3–5% impurities (oxidized material) were shown after 2 weeks. Further analysis of stability of the compounds 1, 15k and 39 was carried out using the 2 biological media we used for our in vivo studies (Dulbecco’s Modified Eagle Medium [DMEM] and Roswell Park Memorial Institute-1640 [RPMI1640]) without serum. Stock solutions of compounds 1, 15k and 39 (1 mM solutions) were prepared using DMSO and 10 μl aliquot of the each of the stock solution was diluted to 1 mL using the biological medium. The final concentration of each of the samples was 10 μM with 1% DMSO. These samples were left in an incubator at 37 °C and analyzed by HPLC to determine the amount of respective compound present in each sample vial at 1, 3, 5, and 7 day time intervals. The HPLC results, peak area against time were plotted as shown in the Figure 3, which shows the peak area of each of the compounds in either DMEM or RPMI did not significantly change or deviate with the time. From these data we concluded compounds 1, 15k and 39 were stable up to 7 days at 37 °C in the biological media we used.
Figure 3.

Stability assessment of compounds 1, 15k and 39 in biological medium (DMEM and RPMI-1640) up to 7 days
One of the important aims in this study was to obtain a compound highly selective for CT-L activity. Therefore, we next determined if those compounds that are potent proteasome inhibitors are selective for CT-L over the T-L activity of the proteasome. Table 1 show that most compounds were more selective for CT-L over T-L activity of the proteasome. The most potent and selective CT-L activity (28-fold, in-vitro) was displayed by compound 29 (Table 1). Interestingly, compounds 35 and 36 were equipotent against CT-L and T-L activities while compound 15d was more selective for T-L over CT-L.
Our initial goal was to identify compounds that were more potent in the primary in vitro assay as well as the cell culture assay. Our in vitro study showed several tetrazole containing analogues to be more potent than the initial hit (Table 1). We, therefore, determined whether some of these compounds were cell permeable and could inhibit the CT-L activity of the proteasome in intact cells. Treatment of the human breast cancer cell lines MDA-MB-468 and MDA-MB-231 with various doses of 1 for 48 hours resulted in inhibition of the CT-L activity in these cells with an IC50 value of 7.22 μM and 3.54 μM, respectively. Out of several compounds tested for intact cell CT-L inhibitory activity in MDA-MB-468 and MDA-MB-231 cells, we identified 39 that potently inhibited CT-L activity with IC50 values of 1.01 and 0.44 μM respectively. To determine if the most potent compound 39 could inhibit CT-L activity at earlier time points and if this results in the accumulation of ubiquitinated cellular proteins, we treated MDA-MB-468 cells with various concentrations of 39 for 11 hours and processed the cells for CT-L assays and western blotting as described in Methods. After 11 hours of treatment, 39 inhibited CT-L activity in MDA-MB-468 cells with IC50 value of 15.6 μM (Figure 4A). Figure 4B shows that treatment of MDA-MB-468 cells with 39 resulted in a concentration-dependent increase in ubiquitination. Finally, to determine the effects of 39 on MDA-MB-468 tumor cell proliferation, we treated the cells as described above and analyzed cell proliferation by MTT assay after 11 hours. Figure 4C shows that 39 inhibited tumor cell proliferation with IC50 value of 17.0 μM.
Figure 4.

Effects of compound 39 on proteasomal CT-L activity, ubiquitination and tumor cell viability in human breast cancer MDA-MB-468 cells. Exponentially growing human breast cancer cells were treated with different concentrations of compound 39 for 11 hr, followed by measurement of CT-L activity in whole cell extracts (A), determination of ubiquitination by Western blot analysis (B) and cell viability as measured by MTT assay (C).
To investigate whether compound 1 and 39 mediated proteasome inhibition is reversible or irreversible; we performed a dialysis experiment with compounds 1, 39 and Bortezomib, a covalent slowly reversible proteasome inhibitor that was used as an internal control. Figure 5 shows that in the absence of dialysis, 1, 39 and Bortezomib were able to inhibit the CT-L activity of the 20S proteasome by 68%, 73% and 87%, respectively. During dialysis, the CT-L activity did not show any recovery in the compounds 1 and 39 treated samples. By contrast, the Bortezomib treated samples, CT-L activity recovery started within 4 hrs. These results suggest that both compounds 1 and 39 behave differently than Bortezomib. It is likely that both 1 and 39 behave as irreversible CT-L proteasome inhibitors.
Figure 5.

Recovery of CT-L activity upon dialysis of the 20S proteasome-compound complexes after pre-incubation with Bortezomib (▲), compound 39 (■) and compound 1 (●).
CONCLUSIONS
In this report we describe the design and synthesis of novel class of compounds as proteasome inhibitors. The hit 1 was identified from screening of an in-house ChemDiv 20,000 compound library. The synthetic route to 1-hydroxy-4-thiophenesulfonamido naphthalene-2-thioacetic acid (1) class of compounds was established and analogs of 1 were shown to be potent inhibitors of CT-L activity of protesome both in vitro and in vivo. The SAR- guided lead optimization showed incorporation of the tetrazole moiety, a carboxylic acid isostere and triazole at the 2-position of the hydronaphthoquinone moieties was crucial to achieving improved potency. The most potent compounds 25, 27, 29, 15e, 15f, 15h, 15j and 15k (Table 1) with IC50 values 150–300 nM were obtained with hydrophobic sulfonamide moieties. Compounds 1 and 39 (that showed improved whole cell activity) were further tested to understand the binding mode with the proteasome. It was concluded compounds that contain 1-hydroxy-4-thiophenesulfonamidonaphthalene scaffold with 2-thoacetic acid or thio-tetrazole moieties as side chains are irreversible proteasome inhibitors. Furthermore, compound 39 was much more potent than 1 at inhibiting CT-L activity in intact human breast cancer cells. Compound 39 also induced the accumulation of ubiquitinated cellular proteins and inhibited breast tumor cell proliferation.
EXPERIMENTAL SECTION
All reagents were purchased from commercial suppliers and used without further purification. Melting points were determined using a Barnstead international melting point apparatus and remain uncorrected. 1H NMR spectra were recorded on a Varian Mercury 400 MHz spectrometer with CD2Cl2, CDCl3, CD3CN or DMSO-d6 as the solvent. 13C NMR spectra are recorded at 100 MHz. All coupling constants are measured in Hertz (Hz) and the chemical shifts (δH and δC) are quoted in parts per million (ppm) relative to TMS (δ 0), which was used as the internal standard. The definition apparent is used as app to describe 1H NMR signals. Liquid chromatography mass spectroscopy (LCMS) and high resolution mass spectroscopy (HRMS) were carried out on an Agilent 6210 LC/MS (ESI-TOF). For LCMS and HRMS the compounds were eluted between 2–5 minutes using Rapid Resolution Cartridge (2.1 × 30 mm, particle size 3.5 μm) from Agilent Technologies. LCMS was used to detect ions of mass 100–1000 Da, and single peak was observed in the chromatogram after purification. HPLC was carried out using Jasco UV-2075 plus uv-vis detector (column: ultra C18, 5μm, 150 mm × 4.6 mm). H-Cube® (ThalesNano) continuous-flow hydrogenation reactor was used for hydrogenation reactions. Microwave reactions were performed in CEM Discover 908005 model and Biotage initiator 8 machines. Thin layer chromatography was performed using silica gel 60 F254 plates (Fisher), with observation under UV when necessary. Anhydrous solvents were used as purchased: dichloromethane (DCM) (anhydrous, 99.8% contains 50–150 ppm hydrocarbon as stabilizer from Aldrich), dimethyl formamide (DMF) (anhydrous, 99.9% from Aldrich), tetrahydrofuran (THF) (anhydrous, 99.9%, inhibitor free, Aldrich), acetonitrile (anhydrous, 99.8%, Aldrich), toluene (anhydrous, 99.8%, Aldrich), methanol (MeOH) (anhydrous, 99.8%, Aldrich), ethanol (EtOH) (absolute, 99.5%, Aldrich). N-methyl-2-pyrolidinone was purchased from Acros (99%) and used as a solvent. All biologically characterized compounds were > 95% pure as determined by HPLC and LCMS except 4b, 10a, 10c, 11b, 11c, 15b, 15c,15e, 15i, 24, 37 that showed purity between 89–94% by HPLC. All biologically characterized compounds were analyzed by 1H NMR, 13C NMR, and HRMS (formula guided mass spectroscopy). Most of the intermediates of the library 14 were not isolated since the reaction produced a mixture of naphthoquinone sulfonimides (14) and hydronathoquinone sulfonamides (15). This mixture was treated with Na2S2O4 to get the final library 15 with high purity. However, the 14a, 14b, and 14c were isolated and reported here.
Analysis of stability of compound 1, 15 and 39 using HPLC
Using fresh powder, 1mM stock solutions were made (in DMSO) for each compound 1, 15k and 39. 10 μl aliquot from each stock solution was taken and diluted to 1 ml with biological media (DMEM and RMPI). We prepared 5 replicates per sample per media and the final concentration of each solution was 10 μM with 1% DMSO. These sample solutions were incubated at 37 °C for 1, 3, 5 and 7 days respectively. The vials containing the sample solution were taken out from the incubator at day-1, day-3, day-5 and day-7 and cooled to 0 °C in an ice bath. The sample solutions were analyzed immediately with HPLC. Peak areas were plotted against time (day).
HPLC conditions
Angilent Eclipse XDB-C18, 5 μm, 4.6×150mm, 35% CH3CN, 65% H2O (with 0.1% TFA), 1 ml/min, 30 min for compound 1; 50% CH3CN, 50% H2O (with 0.1% TFA), 1 ml/min, 30 min for compound 15k and 39.
Cell culture, and cell lysate preparation and determination of proteolytic activity in cell lysates
Human MDA-MB-468 breast cancer cells were cultured in DMEM medium containing 10% fetal calf serum (FCS) and 100 units/mL of penicillin and 100 μg/mL of streptomycin. Cells were maintained at 37 °C in a humidified incubator in an atmosphere of 5% CO2. Cells were treated with different concentrations of compounds 1, 39 or vehicle control (DMSO) for 48 h. Cells were then harvested, washed with PBS twice, and homogenized in lysis buffer (50 mM Tris-HCl, pH = 8.0, 5 mM EDTA, 150 mM NaCl, 0.5% NP-40) for 30 min at 4 °C. Cell lysates were centrifuged at 12,000 g for 15 min, and the supernatants were collected as cell lysates. To determine the proteasome CT-L activity in whole cell extracts from cultured cells, we used the same method 21, 22 described for in vitro CT-L activity assay, except instead of using 20S rabbit proteasome. We used 5 μg of cell lysates.
Western blot analysis
Cell lysates (30 μg) were separated by SDS-PAGE and transferred to a nitrocellulose membrane, probed with ubiquitin antibody (Santa Cruz Biotechnnology Inc. (Santa Cruz, CA), and signals were visualized by enhanced chemoluminescence (ECL, Amersham, Piscataway, NJ) according to the manufacturer’s protocol.
MTT (3-(4,5-dimethythiazol-2-yl)-2,5-diphenyltetrazolium bromide) metabolism assay
Cells were plated in 96-well plates in 100 μl medium and allowed to attach overnight. Cells were then incubated for 11 hr with varying concentrations of compound 39, and appropriate control. Media was aspirated and replaced with 100 μl complete media containing 1 mg/ml MTT and incubated for three hours at 37°C in 5% CO2 humidified incubator. Media was then aspirated and DMSO was added. Cells were incubated for 10 min at room temperature while shaking, and the absorbance was determined at 540 nm using a μQuant spectrophotometric plate reader (Bio-TEK, Winooski, VT).
Dialysis using purified rabbit 20S proteasome
To measure the effect of dialysis on CT-L activity, compounds 1, 39 (10 μM) or vehicle (0.1 % DMSO) were added to rabbit 20S proteasome at a final concentration of 1.5 nM in proteasome assay buffer (50 mM Tris-HCl, pH = 7.6) and incubated at room temperature for 30 min. After 30 min of incubation, proteasome-compound mixtures were added to 3,500 MWCO Thermo Scientific Slide-A-Lyzer MINI Dialysis Unit (Rockford, IL) and dialyzed against proteasome assay buffer. Immediately (t = 0) and 0.5 h, 1 h, 4 h, 8h, and 18 h of dialysis at 4 °C, samples were removed from the dialysis unit and the CT-L 20S proteasome activity was determined as described previously. 21, 22 Proteasome activity was normalized against proteasome activity of DMSO control.
N-(4-hydroxynaphthalen-1-yl)thiophene-2-sulfonamide (3)
The 4-aminonaphthol hydrochloride salt (2) (1.957 g, 0.01 mol) was suspended in DCM (80 mL) and added triethylamine (3.0 mL, 22 mmol) at 0 °C (the suspension became a dark brown solution). Thiophene-2-sulfonyl chloride (1.827 g, 0.01 mol) was added and the reaction mixture was stirred at r.t. overnight (14 h). The reaction mixture was diluted with DCM (200 mL) washed with aqueous HCl (1 N, 30 mL×3), water (30 mL×3) and brine (30 mL). The organic layer was separated, dried (MgSO4) and concentrated to obtain a dark brown solid. The crude solid was suspended in methanol/water (1:1, ~ 50 mL), filtered and washed with methanol/H2O (1:1, ~15 mL × 2) to obtain pure compound 3 (2.7 g, 90%) as a brown solid. Rf = 0.23 (TLC, EtOAc: Hexane [1:2]); m.p.: 146–148 °C; HPLC 96% (Rt = 3.51 min, 60% CH3CN in 0.1% TFA water, 20 min); 1H-NMR (400 MHz, CD2Cl2) δ 7.83-1.78 (m, 2H), 7.70 (dd, J = 5.8, 1.2 Hz, 1H), 7.60 (dd, J = 3.6, 1.2 Hz, 1H), 7.49-7.42 (m, 2H), 7.07 (d, J = 8.0 Hz, 1H), 7.06 (dd, J = 5.2, 4.0 Hz 1H), 6.66 (d, J = 8.4 Hz, 1H), 4.30 (brs, 2H, disappeared on D2O shake); 13C NMR (100 MHz, CDCl3) δ 141.83, 138.30, 135.52, 135.19, 134.61, 128.06, 127.69, 126.93, 125.81, 124.11, 122.19, 121.18, 119.63, 108.26; LC-MS (ESI+) 306.03 (M+H)+; HRMS (ESI+) m/z calculated for C14H12NO3S2 (M+H)+ 306.0253, found 306.0266.
N-(3-chloro-4-hydroxynaphthalen-1-yl)thiophene-2-sulfonamide (4a)
The compound 3 (987 mg, 3.2 mmol) was suspended in MeOH (10 mL), added hydrogen peroxide (35% in water, 3 mL) and stirred at r.t. for 2 h. Additional hydrogen peroxide (3 mL) was added followed by HCl (4 M in dioxane, 1 mL) and continued stirring at r.t. for another 2 h. The organic phase was evaporated and the residue was dissolved in ethyl acetate (100 mL), washed with water (20 mL×3) and brine (20 mL×2). The organic phase was dried (Mg2SO4), filtered and concentrated to obtain a crude solid. The crude solid was purified using SiO2 chromatography (Hexane/EtOAc gradient elution) to obtain pure compound 4a (244 mg, 22.2%) as a brown solid. Rf = 0.46 (TLC, EtOAc: Hexane [1:2]); m.p.: 113–115 °C; HPLC 98% (Rt = 6.79 min, 60% CH3CN in 0.1% TFA water, 20 min); 1H-NMR (400 MHz, CDCl3) δ 7.79 (d, J = 8.4 Hz, 1H), 7.69 (d, J = 8.4 Hz, 1H), 7.64 (dd, J = 4.8, 1.1 Hz, 1H), 7.60 (dd, J = 4.0, 1.2 Hz, 1H), 7.44-7.35 (m, 2H), 7.19 (s, 1H), 7.02 (dd, J = 5.2, 4.0, Hz, 1H), 4.46 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 138.07, 137.40, 135.81, 134.98, 134.78, 127.84, 126.96, 126.92, 126.82, 123.74, 122.44, 121.08, 120.54, 111.99; LC-MS (ESI+) 339.98 (M+H)+; HRMS (ESI+) m/z calculated for C14H11ClO3S2 (M+H)+ 339.9863, found 339.9856.
N-(3-bromo-4-hydroxynaphthalen-1-yl)thiophene-2-sulfonamide (4b)
The compound 3 (306 mg, 1 mmol) was dissolved in DMF (1 mL) at 0 °C, Br2 (320 mg in 1 mL of DCM solution, 2 mmol) was added and stirred at r.t. for 1 h. Triethylamine (0.558 mL, 4 mmol) was added at 0 °C and continued stirring at r.t. overnight (12–14 h). The reaction mixture was diluted with ethyl acetate (50 mL), washed with water (10 mL), brine (10 mL), dried (Na2SO4) and concentrated to obtain a brown crude solid. The crude product was purified using SiO2 chromatography (Hexane/EtOAc gradient elution) to obtain 4b (140 mg, 36.4%) as an orange-red solid. Rf = 0.38 (TLC, EtOAc: Hexane [1:2]); m.p.: 128–130 °C; HPLC 88% (Rt = 7.33 min, 60% CH3CN in 0.1% TFA water 20 min); 1H-NMR (400 MHz, CDCl3) δ 7.80 (d, J = 7.6 Hz, 1H), 7.70 (d, J = 7.6 Hz, 1H), 7.66 (appdd, J = 4.8, 1.2 Hz, 1H), 7.61 (appdd, J = 4.0, 1.2 Hz, 1H), 7.47-7.39 (m, 2H), 7.32 (s, 1H), 7.04 (dd, J = 4.8, 3.6 Hz, 1H), 4.75 (brs, 2H); LC-MS (ESI+) 383.93 (M+H)+; HRMS (ESI+) m/z calculated for C14H11BrO3S2 (M+H)+ 383.9358, found 383.9348.
N-(3-iodo-4-hydroxynaphthalen-1-yl)thiophene-2-sulfonamide (4c)
This compound was prepared using the procedure described for compound 4b, except using I2 (152 mg, 0.6 mmol). The pure product 4c (129.4 mg, 100%) was obtained as a brown solid. Rf = 0.45 (TLC, EtOAc: Hexane [1:2]); m.p.: 134–136 °C; HPLC 97% (Rt = 8.19 min, 60% CH3CN in 0.1% TFA water 20 min); 1H-NMR (400 MHz, CDCl3) δ 7.83-7.81 (m, 1H), 7.75-7.72 (m, 1H), 7.68 (dd, J = 4.8, 1.2 Hz, 1H), 7.62 (dd, J = 4.0, 1.2 Hz, 1H), 7.46-7.41 (m, 3H), 7.06 (dd, J = 5.0, 3.8 Hz, 1H), 4.7 (brs, 1H); 13C NMR (100 MHz, CDCl3) δ 142.59, 137.62, 135.83, 134.95, 134.82, 128.37, 127.89, 127.83, 127.28, 126.82, 122.64, 122.62, 121.56, 75.24; LC-MS (ESI+) 431.92 (M+H)+; HRMS (ESI+) m/z calculated for C14H11IO3S2 (M+H)+ 431.9220, found 431.9216.
Ethyl 2-(1,4-dioxo-1,4-dihydronaphthalen-2-ylthio)acetate (8a)
The 1,4-naphthoquinone (791 mg, 5 mmol) was added portion wise to EtOH (10 mL) containing ethyl mercaptoacetate (0.27 mL, 25 mmol) at room temperature. The reaction mixture was stirred at room temperature for 30 min. The yellow solid obtained was filtered, washed with ethanol and dried under vacuum to obtain naphthoquinone intermediate 8a (491 mg, 88.9%) as a yellow solid. Rf = 0.60 (TLC, EtOAc: Hexane = 1:2); m.p.: 150–152 °C; 1H-NMR (400 MHz, CDCl3) δ 8.11-8.07 (m, J = 7.4 Hz, 2H), 7.78-7.69 (m, 2H), 6.70 (s, 1H), 4.24 (q, J = 7.1 Hz, 2H), 3.66 (s, 2H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 182.15, 181.73, 167.68, 153.42, 134.69, 133.67, 132.27, 131.88, 128.12, 127.08, 126.84, 62.60, 33.14, 14.33; LC-MS (ESI+) 299.01 (M+Na)+; HRMS (ESI+) m/z calculated for C14H13O4S (M+H)+ 277.0529, found 277.0529.
Methyl 3-(1,4-dioxo-1,4-dihydronaphthalen-2-ylthio)propanoate (8b)
This compound was prepared using the procedure described for compound 8a except using methyl 3-mercaptopropionate (1.582 g, 0.01 mol). The pure product 8b (1.298 g, 93.9%) was obtained as a yellow solid. Rf = 0.40 (TLC, EtOAc: Hexane [1:2]); m.p.: 108–110 °C; 1H-NMR (400 MHz, CDCl3) δ 8.09 (t, J = 7.6 Hz, 2H), 7.76 (td, J = 7.6, 1.6 Hz, 1H), 7.71 (td, J = 7.6, 1.2 Hz, 1H), 6.64 (s, 1H), 3.74 (s, 3H), 3.13 (t, J = 7.4 Hz, 2H), 2.78 (t, J = 7.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 182.16, 181.66, 171.43, 154.29, 134.63, 133.59, 132.31, 132.02, 127.36, 127.10, 126.81, 52.42, 32.29, 25.46; LC-MS (ESI+) 299.04 (M+Na)+, 277.05 (M+H)+; HRMS (ESI+) m/z calculated for C14H13O4S (M+H)+ 277.0529, found 277.0539.
tert-Butyl 2-(1,4-dioxo-1,4-dihydronaphthalen-2-yloxy)acetate (8c)
A mixture of 2-hydroxynaphthalene-1,4-dione 6 (174.2 mg, 1.0 mmol), tert-butyl 2-bromoacetate (0.2 mL, 1.33 mmol), silver oxide (308 mg, 1.33 mmol) and potassium iodide (16.6 mg, 0.1 mmol) in chloroform (5 mL) was refluxed under argon atmosphere for 12 hours. Additional tert-butyl 2-bromoacetate (1.5 mL, 10 mmol) was added and the reaction was continued under reflux for further 24 hours. The reaction mixture was filtered through a pad of celite, washed with DCM (3 × 10 mL) and the filtrate was concentrated to obtain a grey yellow solid. The crude product was purified using SiO2 chromatography (5g silica gel, EtOAc/Hexane gradient elution) to give pure compound 8c (93.2 mg, 32%) as a light yellow solid. Rf = 0.50 (TLC, EtOAc: Hexane [1:2]); m.p.: 120–122 °C; 1H NMR (400 MHz, CDCl3) δ 8.15 (m, 1H), 8.08 (m, 1H), 7.77-7.71 (m, 2H), 6.04 (s, 1H), 4,61 (s, 2H), 1.41 (s, 9H); LC-MS (ESI+) 233.03 (M+H-tBu)+, 289.10 (M+H)+; HRMS (ESI+) m/z calculated for C16H17O5 (M+H)+ 289.1071, found 289.1090.
Ethyl 2-(1-oxo-4-(thiophen-2-ylsulfonylimino)-1,4-dihydronaphthalen-2-ylthio)acetate (9a)
To a mixture of 8a (63 mg, 0.228 mmol) and thiophene-2-sulfonamide (37 mg, 0.228 mmol) in DCM (2.5 mL), at 0 °C was added TiCl4·2THF complex (76 mg, 0.228 mmol) followed by triethylamine (70 μL, 0.5 mmol). The mixture was heated at 60 °C using the microwave reactor for 20 min. The reaction mixture was diluted with DCM (60 mL), washed with water (~15 mL) and brine (~15 mL). The organic phase was separated, dried (Na2SO4) and concentrated to obtain a brown crude semi-solid. The crude mixture was purified using SiO2 chromatography (EtOAc/Hexane gradient elution) to obtain the required pure compound 9a (58 mg, 60 %) as an orange oil which solidified on standing at room temperature. Rf = 0.30 (TLC, EtOAc: Hexane [1:2]); m.p.: 110–112 °C; HPLC 95% (Rt = 4.63 min, 70% CH3CN in 0.1% TFA water, 20 min); 1H-NMR (400 MHz, CDCl3) δ 8.22 (dd, J = 6.0, 3.2 Hz, 1H), 8.15 (dd, J = 5.6, 3.2 Hz, 1H), 7.96 (s, 1H), 7.83 (dd, J = 4.0, 1.6 Hz, 1H), 7.71-7.68 (m, 3H), 7.15 (dd, J = 5.0, 4.0 Hz, 1H), 4.31 (q, J = 7.2 Hz, 2H), 3.80 (s, 2H), 1.35 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 180.80, 167.34, 160.01, 153.40, 142.08, 134.42, 133.61, 133.59, 133.27, 133.00, 131.81, 127.56, 127.23, 120.91, 62.95, 33.56, 14.29; LC-MS (ESI+) 422.03 (M+H)+; HRMS (ESI+) m/z calculated for C18H16NO5S3 (M+H)+ 422.0185, found 422.0192.
tert-Butyl-2-(1-oxo-4-(thiophen-2-ylsulfonylimino)-1,4-dihydronaphthalen-2-yloxy)acetate (9b)
Triethylamine (0.12 mL, 0.88 mmol) was added to a mixture of tert-butyl 2-(1,4-dioxo-1,4-dihydronaphthalen-2-yloxy)acetate 8c (115 mg, 0.4 mmol) and thiophene-2-sulfonamide (78 mg, 0.48 mmol) in anhydrous DCM (4 mL). TiCl4.2THF (134 mg, 0.4 mmol) was added to the reaction mixture and heated at 60 °C using the microwave reactor for 20 min. The crude mixture was poured into EtOAC (20 mL), filtered using a celite pad and the filtrate was concentrated and purified (SiO2 chromatography, EtOAc/Hexane gradient elution) to give pure compound 9b (0.068 g, 39%) as a yellow solid. m.p.: 153–155 °C; HPLC 94% (Rt = 4.86 min, 70% CH3CN in 0.1% TFA water 20 min); 1H NMR (400 MHz, CDCl3) δ 8.08-8.06 (m, 1H), 8.02-8.00 (m, 1H), 7.64 (appdd, J = 3.7, 1.2 Hz, 1H), 7.55-7.53 (m, 3H), 7.10 (s, 1H), 6.98 (t, J = 4.0 Hz, 1H), 4.58 (s, 2H), 1.39 (s, 9H); LC-MS (ESI+) 334.02 (M+H)+.
Ethyl 2-(1-oxo-4-(phenylsulfonylimino)-1,4-dihydronaphthalen-2-ylthio)acetate (9c)
This compound was prepared using the procedure described for compound 9a except using phenylsulfonamide (157 mg, 1 mmol) to give pure product 9c (132 mg, 31.7%) as a yellow solid. Rf = 0.50 (TLC, EtOAc: Hexane [1:2]); m.p.: 93–95 °C; HPLC 95% (Rt = 5.11 min, 70% CH3CN in 0.1% TFA water 20 min); 1H-NMR (400 MHz, CDCl3) δ 8.11 (appdd, J = 7.2, 2.4 Hz, 2H), 8.07 ( appd, J = 7.2 Hz, 2H), 7.99 (s, 1H), 7.65-7.62 (m, 3H), 7.58 (appt, J = 8.0 Hz, 2H), 4.32 (q, J = 7.2 Hz, 2H), 3.81 (s, 2H), 1.34 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 180.89, 167.41, 160.31, 152.98, 141.25, 134.32, 133.74, 133.45, 133.37, 131.78, 129.26, 127.49, 127.46, 127.09, 121.29, 62.94, 33.56, 14.30; LC-MS (ESI+) 416.06 (M+H)+; HRMS (ESI+) m/z calculated for C20H18NO5S2 (M+H)+ 416.0621, found 416.0621.
Methyl 3-(1-oxo-4-(thiophen-2-ylsulfonylimino)-1,4-dihydronaphthalen-2-ylthio) propanoate (9d)
This compound was prepared according to the procedure described for compound 9a except using 8b (276 mg, 1 mmol). The title compound 9d (110 mg, 26.1%) was obtained as an orange color solid. Rf = 0.50 (TLC, EtOAc: Hexane [1:2]); m.p.: 140–142 °C; HPLC 99% (Rt = 4.47 min, 70% CH3CN in 0.1% TFA water 20 min); 1H-NMR (400 MHz, CDCl3) δ 8.24-8.22 (m, 1H), 8.15-8.13 (m, 1H), 7.91 (s, 1H), 7.83-7.82 (m, 1H), 7.71-7.68 (m, 3H), 7.16-7.14 (m, 1H), 3.75 (s, 3H), , 3.26 (t, J = 6.9 Hz, 2H), 2.85 (t, J = 6.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 180.79, 171.35, 160.09, 154.37, 142.10, 134.39, 133.63, 133.53, 133.23, 132.95, 131.99, 127.59, 127.57, 127.23, 120.19, 52.46, 32.07, 25.88; LC-MS (ESI+) 422.03 (M+H)+; HRMS (ESI+) m/z calculated for C18H16NO5S3 (M+H)+ 422.0185, found 422.0181.
Ethyl 2-(1-hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-ylthio)acetate (10a)
The (E)-ethyl 2-(1-oxo-4-(thiophen-2-ylsulfonylimino)-1,4-dihydronaphthalen-2-ylthio)acetate (9a) (52 mg, 0.123 mmol) was dissolved in THF (2 mL), and added Na2S2O4 (107 mg, 0.62 mmol in 2 mL of water), The biphasic mixture obtained was stirred at r.t. for 1 h until it turned pale yellow. The mixture was diluted with ethyl acetate (~30 mL), washed with water (~10 mL) and brine (~10 mL). The organic phase was dried (Na2SO4), filtered and the filtrate was concentrated. The crude product obtained was purified (SiO2 chromatography, EtOAc/Hexane gradient elution) to obtain pure 10a (0.025 g, 58.7 %) as an off-white solid. Rf = 0.25 (TLC, EtOAc: Hexane [1:2]); m.p.: 116–118 °C; HPLC 93% (Rt = 4.72 min, 60% CH3CN in 0.1% TFA water 20 min); 1H-NMR (400 MHz, CDCl3) δ 8.42 (s, 1H), 8.29 (dd, J = 6.3, 3.2 Hz, 1H), 7.80 (dd, J = 6.3, 3.1 Hz, 1H), 7.52 (d, J = 4.0 Hz, 1H), 7.48 (dd, J = 6.4, 3.2 Hz, 2H), 7.38 (d, J = 2.4 Hz, 1H), 7.34 (s, 1H), 6.95 (t, J = 4.8 Hz, 1H), 6.7 (brs, 1H), 4.18 (q, J = 7.1 Hz, 2H), 3.48 (s, 2H), 1.21 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.46, 155.83, 139.71, 133.32, 132.69, 131.87, 128.70, 127.59, 126.46, 124.78, 124.05, 123.41, 122.18, 110.19, 62.75, 39.73, 14.24; LC-MS (ESI+) 441.05 (M+NH4)+; HRMS (ESI+) m/z calculated for C18H18NO5S3 (M+H)+ 424.0342, found 424.0335.
tert-Butyl-2-(1-hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-yloxy)acetate (10b)
The tert-butyl-2-(1-oxo-4-(thiophen-2-ylsulfonylimino)-1,4-dihydronaphthalen-2-yloxy)acetate 9b (70 mg, 0.16 mmol) was dissolved in THF (3 mL) and stirred at r.t. for 5 min. The Na2S2O4 (139 mg, 0.8 mmol) and water (1 mL) was added to the mixture and stirred vigorously until Na2S2O4 was completely dissolved. The reaction mixture was further stirred at r.t. for 10 min. The color changed from orange to light yellow. EtOAc (40 mL) was added into the mixture and washed with water (40 mL) followed by brine. The organic phase was dried (MgSO4), filtered and the filtrate was concentrated. The required product 10b was obtained by crystallization with EtOAc/Hexane as a light yellow solid (50 mg, 71%). m.p.= 155–157 °C; HPLC 96% (Rt = 3.39 min, 70% CH3CN in 0.1% TFA water 20 min); 1H NMR (400 MHz, CDCl3) δ 8.52 (brs, 1H, disappeared on D2O shake), 8.24 (d, J = 8.4 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.46 (dd, J = 4.8, 1.3 Hz, 1H), 7.44-7.40 (m, 1H), 7.35-7.29 (m, 2H), 7.23 (s, 1H), 6.90 (dd, J = 5.2, 4.0 Hz, 1H), 6.68 (s, 1H, disappeared on D2O shake), 4.53 (s, 2H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 171.34, 143.83, 140.29, 139.79, 133.09, 132.62, 127.98, 127.54, 126.44, 125.97, 125.64, 123.13, 122.78, 121.04, 119.32, 84.20, 71.12, 28.29; LC-MS (ESI−) 434.07 (M−H)−; HRMS (ESI−) m/z calculated for C20H20NO6S2 (M−H)− 434.0738, found 434.0755.
Ethyl 2-(1-hydroxy-4-(phenylsulfonamido)naphthalen-2-ylthio)acetate (10c)
This compound was prepared using the procedure described for 10a except using the starting material 9c (83 mg, 0.2 mmol). The title compound 10c was obtained as a pale yellow solid (68 mg, 81.5%). Rf = 0.22 (TLC, EtOAc: Hexane [1:2]); m.p.: 118–120 °C. HPLC 92% (Rt = 5.04 min, 60% CH3CN in 0.1% TFA water 20 min); 1H-NMR (400 MHz, CD2Cl2) δ 8.51 (s, 1H), 8.27-8.24 (m, 1H), 7.87-7.85 (m, 1H), 7.69 (d, J = 7.4 Hz, 2H), 7.56 (t, J = 7.2 Hz, 1H), 7.51-7.47 (m, 2H), 7.43 (t, J = 7.9 Hz, 2H), 7.17 (s, 1H), 6.59 (brs, 1H), 4.17 (q, J = 7.1 Hz, 2H), 3.43 (s, 2H), 1.21 (t, J = 7.2 Hz, 3H); LC-MS (ESI+) 440.07 (M+Na)+; HRMS (ESI+) m/z calculated for C20H19NNaO5S2 (M+Na)+ 440.0597, found 440.0596.
Methyl 3-(1-hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-ylthio)propanoate (10d)
This compound was prepared using the procedure described for 10a except using starting material 9d (30 mg, 0.071 mmol). The title compound 10d was obtained without any purification as a pale yellow solid (28 mg, 93%). Rf = 0.20 (TLC, EtOAc: Hexane [1:2]); m.p.: 117–119 °C; HPLC 97% (Rt = 4.09 min, 60% CH3CN in 0.1% TFA water 20 min); 1H-NMR (400 MHz, DMSO-d6) δ 8.25 (dd, J = 6.4, 3.2 Hz, 1H), 7.87 (dd, J = 6.4, 3.2 Hz, 1H), 7.60 (brs, 1H), 7.52-7.48 (m, 3H), 7.41 (dd, J = 3.6 1.2 Hz, 1H), 7.32 (s, 1H), 6.95 (dd, J = 4.8, 3.6 Hz, 1H), 6.91 (brs, 1H), 3.70 (s, 3H), 2.93 (t, J = 7.1 Hz, 2H), 2.51 (t, J = 7.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 172.41, 154.89, 139.74, 133.29, 132.77, 132.48, 131.35, 128.54, 127.60, 126.54, 124.24, 123.76, 123.51, 122.35, 109.96, 52.31, 34.09, 31.79; LC-MS (ESI+) 446.01 (M+Na)+; HRMS (ESI+) m/z calculated for C18H17NNaO5S3 (M+Na)+ 446.0161, found 446.0157.
Ethyl 2-(1-hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-ylsulfonyl)acetate (10e)
Ethyl 2-(1-hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-ylthio)acetate (10a) (42.4 mg, 0.1 mmol) was dissolved in acetone (2 mL), to which was added oxone (307.4 mg in 2 mL of aqueous solution). The resulting mixture was stirred at r.t. overnight (12–14 h). The organic solvent was evaporated and the aqueous phase was extracted with ethyl acetate. The combined organic extracts were washed with water, brine and dried over Na2SO4. The organic phase was filtered and the filtrate was concentrated to obtain 10e (47 mg, 100%) as a pale yellow solid, m.p.: 154–156 °C. 1H-NMR (400 MHz, CDCl3), δ 10.02 (s, 1H), 8.43 (d, J = 8.4 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.70 (t, J = 8.0 Hz, 1H), 7.62 (td, J = 8.0, 0.8 Hz, 1H), 7.58 (appdd, J = 5.0, 1.2 Hz, 1H), 7.43 (appdd, J = 4.0, 1.6 Hz, 1H), 7.29 (s, 1H), 7.00 (dd, J = 4.8, 3.6 Hz, 1H), 6.58 (s, 1H), 4.16 (q, J = 7.2 Hz, 2H), 4.12 (s, 2H), 1.15 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CD2Cl2) δ 162.16, 155.75, 139.23, 134.91, 133.62, 133.21, 131.25, 127.86, 127.66, 125.75, 124.53, 124.26, 122.85, 122.26, 112.67, 63.05, 61.22, 13.77. LC-MS (ESI−) 454.01 (M−H)−; HRMS (ESI−) m/z calculated for C18H17NO7S3 (M−H)− 454.0094, found 454.0112.
2-(1-Hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-ylthio)acetic acid 11a (compound 1)
The ethyl 2-(1-hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-ylthio)acetate (10a) (8 mg, 0.019 mmol) was dissolved in dioxane, (0.5 mL) and added HCl (0.5 mL, 4 N). The reaction mixture was heated at 100 °C using the microwave reactor for 10 min and diluted with ethyl acetate (20 mL) and washed with water and brine. The organic phase was dried (Na2SO4), filtered and the filtrate was concentrated to afford the title compound as a pale yellow solid (6 mg, 80%). Rf = 0.25 (TLC, CH3OH: DCM [1:10]); m.p.: 175–177 °C; HPLC 97% (Rt = 15.6 min, 35% CH3CN in 0.1% TFA water, 30 min); 1H-NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H, disappeared on D2O shake), 9.87 (brs, 1H, disappeared on D2O shake), 8.13 (d, J = 8.0 Hz, 1H), 7.85-7.83 (m, 1H, 2H), 7.48-7.39 (m, 2H), 7.33 (dd, J = 4.0, 1.6 Hz, 1H), 7.06-7.03 (m, 2H), 3.52 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 171.11, 152.44, 140.21, 133.13, 132.38, 131.31, 129.38, 127.61, 126.78, 125.82, 125.05, 123.95, 123.21, 122.41, 112.95, 36.83.; LC-MS (ESI−) 393.99 (M−H)−; HRMS (ESI−) m/z calculated for C16H12NO5S3 (M−H)− 393.9883, found 393.9885.
2-(1-Hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-yloxy)acetic acid (11b)
The tert-butyl-2-(1-hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-yloxy)acetate (10b) (0.025 g, 0.057 mmol) was dissolved in dioxane and concentrated hydrochloric acid (dioxane: HCl, 1:1, 4 mL) and stirred at r.t. for 3 h (The solution changed from clear to white cloudy). The solvent was evaporated and the solid obtained was washed with DCM and hexane separately to get pure product as a gray solid (0.021 g, 97%). m.p.= 155–157 °C; HPLC 90% (Rt = 6.48 min, 40% CH3CN in 0.1% TFA water 20 min); 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H, disappeared on D2O shake), 9.68 (brs, 1H, disappeared on D2O shake), 8.04 (d, J = 8.8 Hz, 1H), 7.82 (d, J = 4.0 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.38 (t, J = 7.2 Hz, 1H), 7.33 (appd, J = 2.4 Hz, 1H), 7.27 (t, J = 7.2 Hz, 1H), 7.03 (appt, J = 4.0 Hz, 1H), 6.89 (s, 1H), 4.55 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 171.66, 141.36, 140.97, 140.60, 133.76, 132.95, 128.26, 127.96, 125.98, 125.94, 125.26, 123.74, 123.56, 122.26, 117.23, 68.16; LC-MS (ESI−) 378.01 (M−H)−; HRMS (ESI−) m/z calculated for C16H12NO6S2 (M−H)− 378.0112, found 378.0117.
2-(1-Hydroxy-4-(phenylsulfonamido)naphthalen-2-ylthio)acetic acid (11c)
This compound was prepared using the procedure described for 11a except using 10c (40 mg, 0.096 mmol) at r.t. overnight (12–14 h). The title compound was obtained as an off white solid (20 mg, 53.6%). m.p.: 100–102 °C; HPLC 94% (Rt = 9.3 min, 40% CH3CN in 0.1% TFA water, 40 min); 1H-NMR (400 MHz, DMSO-d6) δ 12.75 (brs, 1H), 9.91 (s, 1H), 9.80 (brs, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.83 (d, J = 8.3 Hz, 1H), 7.60 (d, J = 7.7 Hz, 2H), 7.56 (d, J = 7.3 Hz, 1H), 7.46 (t, J = 7.6 Hz, 2H), 7.39-7.36 (m, 2H), 6.96 (s, 1H), 3.47 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 171.74, 152.82, 140.39, 133.31, 131.82, 129.79, 129.72, 127.51, 127.33, 126.45, 125.71, 124.82, 124.02, 123.02, 113.60, 37.40; LC-MS (ESI−) 388.03 (M−H)−; HRMS (ESI−) m/z calculated for C18H14NO5S2 (M−H)− 388.0319, found 388.0330.
3-(1-Hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-ylthio)propanoic acid (11d)
This compound was prepared using the procedure described for 11a except using 10d (43 mg, 0.1 mmol) at r.t. overnight (12–14 h). The title compound was obtained as a white solid (38 mg, 90.4%). m.p.: 185 °C (dec.); HPLC 96% (Rt = 8.8 min, 40% CH3CN in 0.1% TFA water, 40 min); 1H-NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 10.07 (s, 1H), 9.70 (brs, 1H), 8.13 (d, J = 7.6 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), , 7.85 (td, J = 4.8, 0.5 Hz, 1H), 7.49-7.42 (m, 2H), 7.34 (td, J = 4.0, 1.6 Hz, 1H), 7.07-7.05 (m, 1H), 6.96 (s, 1H), 2.84 (t, J = 7.6 Hz, 2H), 2.37 (t, J = 6.8 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 173.40, 153.02, 140.76, 133.87, 133.07, 131.86, 129.69, 128.30, 127.31, 126.49, 125.77, 124.67, 124.06, 123.05, 113.58, 34.61, 29.92; LC-MS (ESI−) 408.00 (M−H)−; HRMS (ESI−) m/z calculated for C17H14NO5S3 (M−H)− 408.0040, found 408.0054.
2-(1-Hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-ylsulfonyl)acetic acid (11e)
Ethyl 2-(1-hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-ylsulfonyl)-acetate (10e) (20 mg, 0.044 mmol) was dissolved in dioxane (2 mL) and added conc. HCl (2 mL). The mixture was stirred at r.t. for 36 h. The solvent was removed using the rotary evaporator and the solid obtained was washed with dichloromethane affording title compound 11e (18.3 mg, 97.3%) as an off-white solid, m.p.: 210–212 °C; HPLC 97% (Rt = 7.45 min, 35% CH3CN in 0.1% TFA water, 20 min); 1H-NMR (400 MHz, DMSO-d6) δ 11.62 (brs, 1H), 10.24 (s, 1H), 8.37 (d, J = 7.6 Hz, 1H), 7.97 (d, J = 7.6 Hz, 1H), 7.86 (dd, J = 5.2, 0.8 Hz, 1H), 7.64-7.58 (m, 2H), 7.35 (s, 1H), 7.33 (dd, J = 4.0, 0.8 Hz, 1H), 7.05 (dd, J = 4.9, 3.9 Hz, 1H), 4.52 (s, 2H). 13C NMR (100 MHz, CD3OD) δ 164.35, 154.42, 139.92, 135.05, 132.84, 132.62, 129.98, 127.39, 126.83, 125.81, 124.99, 123.52, 123.48, 122.58, 115.59, 59.78; LC-MS (ESI−) 426.00 (M−H)−; HRMS (ESI−) m/z calculated for C16H13NO7S3 (M−H)− 425.9781, found 425.9788.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)thiophene-2-sulfonamide (13a)
The 2-chloro-1,4-naphthoquinone 12 (385 mg, 2 mmol) was added to thiophene-2-sulfonamide (326 mg, 2 mmol) in DCM (15 mL) at 0 °C followed by TiCl4 (2mL, 1M solution in DCM) and TEA (0.613 mL, 4.4 mmol). The mixture was heated at 60 °C using the microwave reactor for 15 min and the black mixture obtained was poured into ethyl acetate (100 mL). The insoluble particles were removed by filtering through a pad of celite. The filtrate was concentrated and the residue obtained was suspended in DCM (100 mL). The brown insoluble particles were removed by filtration and the filtrate was again concentrated to dryness. The residue was suspended in ethyl acetate/hexane (1:1 ~20 mL,) and the yellow solid obtained was filtered. The solid was washed with ethyl acetate/hexane (1:1, 5 mL×2) and dried under vacuum to afford the title compound as a yellow solid (378 mg, 55.9%). When TiCl4·2THF (668 mg, 2 mol), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) were used instead of TiCl4 in DCM, the reaction afforded the title compound with better yields also as a yellow solid (522 mg 77.3 %). Rf = 0.53 (TLC, EtOAc: Hexane [1:2]); m.p.: 167–169 °C; 1H-NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 8.24-8.20 (m, 2H), 7.86 (dd, J = 3.6, 1.2 Hz, 1H), 7.78-7.70 (m, 3H), 7.18 (dd, J = 4.8, 3.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 177.11, 160.88, 145.59, 141.08, 134.45, 134.28, 134.04, 133.81, 132.98, 131.61, 129.60, 128.16, 127.73, 127.24; LC-MS (ESI+) 355.00 (M+NH4)+; HRMS (ESI+) m/z calculated for C14H9ClNO3S2 (M+H)+ 337.9707, found 337.9708.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)biphenyl-4-sulfonamide (13b)
This compound was prepared using the procedure described for 13a except using 4-biphenylsulfonamide (233 mg, 1 mmol). The title compound 13b was obtained as a yellow solid (154 mg, 37.8%). When TiCl4·2THF (668 mg, 2 mmol), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) were used in the reaction, the title compound 13b was obtained also as a yellow solid with improved yield (586.7 mg, 71.9%). Rf = 0.60 (TLC, EtOAc: Hexane [1:2]); m.p.: 190–192 °C; 1H-NMR (400 MHz, CDCl3) δ 8.71 (s, 1H), 8.24 (appt, J = 8.8 Hz, 2H), 8.14 (d, J = 8.6 Hz, 2H), 7.81 (d, J = 8.6 Hz, 2H), 7.74 (td, J = 7.2, 1.6 Hz, 1H), 7.69 (td, J = 8.0, 2.0 Hz, 1H), 7.64 (d, J = 8.0 Hz, 2H), 7.51 (t, J = 6.8 Hz, 2H), 7.46-7.43 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 177.21, 161.14, 146.82, 145.33, 139.40, 138.95, 134.37, 134.14, 133.12, 131.59, 129.91, 129.35, 128.93, 128.26, 128.11, 128.04, 127.64, 127.14; LC-MS (ESI+) 425.06 (M+NH4)+; HRMS (ESI+) m/z calculated for C22H15ClNO3S (M+H)+ 408.0456, found 408.0460.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-5-phenylthiophene-2-sulfonamide (13c)
This compound was prepared using the procedure described for 13a except using 5-phenylthiophene-2-sulfonamide (477 mg, 2 mmol), TiCl4 (2 mL, 1M solution in DCM), TEA (0.615 mL, 4.4 mol) and THF (15 mL) as the solvent. The title compound was obtained as an orange solid (520 mg, 63.0%). m.p.: 112.4 °C (Dec.). 1H NMR (400 MHz, CDCl3) δ 8.65 (s, 1H), 8.27-8.25 (m, 1H), 8.22-8.19 (m, 1H), 7.81 (d, J = 4.0 Hz, 1H), 7.77-7.70 (m, 2H), 7.65 (dd, J = 8.4, 1.6 Hz, 2H), 7.47-7.41 (m, 3H), 7.32 (d, J = 3.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 177.14, 160.72, 153.69, 145.55, 138.88, 134.73, 134.45, 134.25, 133.03, 132.73, 131.62, 129.73, 129.54, 129.52, 128.15, 127.28, 126.65, 123.29; LC-MS (ESI+) 414.00 (M+H)+; HRMS (ESI+) m/z calculated for C20H13ClNO3S2 (M+H)+ 414.0020, found 414.0018.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-4-phenoxybenzenesulfonamide (13d)
This compound was prepared using the procedure described for 13a except using 4-phenoxyphenylesulfonamide (434 mg, 2 mmol), TiCl4 (2 mL, 1M solution in DCM), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) as the solvent. The title compound was obtained as a yellow solid, (526 mg, 67.2%) m.p.: 124.8 °C (Dec.). 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 8.19 (appdd, J = 7.6, 1.6 Hz, 1H), 8.16 (appdd, J = 7.6, 1.6 Hz, 1H), 8.00 (d, J = 9.2 Hz, 2H), 7.73 (td, J = 7.6, 1.6 Hz, 1H), 7.67 (td, J = 7.6, 1.6 Hz, 1H), 7.44-7.40 (m, 2H), 7.26-7.22 (m, 1H), 7.15-7.08 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 177.22, 162.67, 160.78, 155.11, 145.21, 134.33, 134.08, 133.81, 133.15, 131.58, 130.48, 130.09, 129.78, 128.08, 127.07, 125.42, 120.69, 117.76; LC-MS (ESI+) 424.03 (M+H)+; HRMS (ESI+) m/z calculated for C22H15ClNO4S (M+H)+ 424.0405, found 424.0403.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)naphthalene-2-sulfonamide (13e)
This compound was prepared using the procedure described for 13a except using naphthalene-2-sulfonamide (415 mg, 2 mmol), TiCl4·2THF (668 mg, 2 mmol), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) as the solvent. The title compound was obtained as a yellow solid (0. 595 g, 77.9%). Rf = 0.55 (TLC, EtOAc: Hexane [1:2]); m.p.: 200–202 °C; 1H-NMR (400 MHz, CDCl3) δ 8.73 (s, 1H), 8.64 (s, 1H), 8.20 (appd, 7.6 Hz, 1H), 8.14 (appd, J = 8.0 Hz, 1H), 8.05-8.03 (m, 3H), 7.96 (d, 8.4 Hz, 1H), 7.72 (appq, J = 7.4 Hz, 2H), 7.66 (appt, J = 7.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 177.05, 160.98, 145.15, 137.09, 135.34, 134.38, 134.14, 132.92, 132.11, 131.42, 129.77, 129.64, 129.57, 129.00, 128.15, 127.95, 127.04, 122.61; LC-MS (ESI+) 382.03 (M+H)+; HRMS (ESI+) m/z calculated for C20H13ClNO3S (M+H)+ 382.0299, found 382.0301.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-4′-fluorobiphenyl-4-sulfonamide (13f)
This compound was prepared using the procedure described for 13a except using 4′-fluorobiphenyl-4-sulfonamide (251 mg, 1 mmol), TiCl4 (1 mL,1M solution in DCM), TEA (308 mL, 2.2 mmol) and THF (10 mL) as the solvent. The title compound was obtained as a yellow solid (348 mg, 81.6%). m.p.: 172.1 – 172.8 °C. 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.20 (dd, J = 7.6, 1.2 Hz, 1H), 8.17 (dd, J = 8.0, 1.2 Hz, 1H), 8.13 (d, J = 8.4 Hz, 2H), 7.77-7.75 (m, 2H), 7.72-7.66 (m, 2H), 7.60 (dd, J = 8.8, 5.2 Hz, 2H), 7.19 (t, J = 8.8 Hz, 2H); 19F NMR (376 MHz, CDCl3) δ −113.53 (m); 13C NMR (100 MHz, CDCl3) δ 177.17, 163.46, (d, J = 245 Hz), 161.20, 145.74, 145.38, 139.04, 135.53 (d, J = 3.5 Hz), 134.36, 134.16, 133.09, 131.59, 129.35 (d, J = 8 Hz), 129.31, 128.33, 128.12, 127.87, 127.10, 116.36 (d, J = 21.5 Hz); LC-MS (ESI+) 426 (M+H)+, 443.05 (M+NH4)+; HRMS (ESI+) m/z calculated for C22H14ClFNO3S (M+H)+ 426.0362, found 426.0356.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-5-(pyridin-2-yl)thiophene-2-sulfonamide (13g)
This compound was prepared using the procedure described for 13a except using 5-(pyridin-2-yl)thiophene-2-sulfonamide (481 mg, 2 mmol), TiCl4 (2 mL, 1M solution in DCM), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) as the solvent. The title compound was obtained as an orange solid (511 mg, 61.5%). m.p.: 155.3 °C (Dec.). 1H NMR (400 MHz, CDCl3) δ 8.66 (d, J = 5.2 Hz, 1H), 8.60 (s, 1H), 8.28-8.26 (m, 1H), 8.21-8.18 (m, 1H), 7.87 (td, J = 7.8, 1.5 Hz, 1H), 7.84 (d, J = 4.0 Hz, 1H), 7.79 (s, 1H), 7.77 (s, 1H), 7.75-7.71 (m, 2H), 7.37 (dd, J = 6.7, 5.1 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 176.99, 162.05, 153.71, 150.64, 150.45, 145.71, 138.40, 135.74, 135.18, 135.13, 132.96, 132.22, 128.59, 128.00, 127.09, 125.94, 125.01, 120.42, 104.99; LC-MS (ESI+) 414.99 (M+H)+; HRMS (ESI+) m/z calculated for C19H12ClN2O3S2 (M+H)+ 414.9972, found 414.9969.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-4-cyclohexylbenzenesulfonamide (13h)
This compound was prepared using the procedure described for 13a except using 4-cyclohexylphenylsulfonamide (290 mg, 1.2 mmol), TiCl4 (1.2 mL, 1M solution in DCM), TEA (0.372 mL, 2.7 mmol) and THF (10 mL) as the solvent. The title compound was obtained as a yellow solid (213 mg, 42.4 %). 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.19-8.15 (m, 2H), 7.97 (d, J = 8.4 Hz, 2H), 7.72 (td, J = 7.5, 1.5 Hz, 1H), 7.67 (td, J = 7.6, 1.6 Hz, 1H), 7.42 (d, J = 8.4 Hz, 2H), 2.64-2.58 (m, 1H), 1.91-1.85 (m, 4H), 1.79-1.75 (m, 1H), 1.50-1.34 (m, 4H), 1.31-1.23 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 177.24, 160.85, 154.67, 145.12, 137.66, 134.32, 134.03, 133.18, 131.56, 129.85, 128.03, 127.90, 127.83, 127.12, 44.93, 34.32, 26.87, 26.17; LC-MS (ESI+) 414.09 (M+H)+; HRMS (ESI+) m/z calculated for C22H21ClNO3S (M+H)+ 414.0925, found 414.0924.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-4′-methoxybiphenyl-4-sulfonamide (13i)
This compound was prepared using the procedure described for 13a except using 4′-methoxybiphenyl-4-sulfonamide (527 mg, 2 mmol), TiCl4 (2 mL, 1M solution in DCM), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) as solvent. The title compound was obtained as a yellow solid (650 mg, 74.2 %). m.p.: 168.1 °C (Dec.). 1H NMR (400 MHz, CDCl3) δ 8.70 (s, 1H), 8.19 (apptd, J = 8.8, 1.6 Hz, 2H), 8.10 (d, J = 8.6 Hz, 2H), 7.76 (d, J = 8.6 Hz, 2H, partially overlapped with the adjacent multiplet), 7.76-7.71 (m, 1H, partially overlapped with the adjacent doublet), 7.68 (td, J = 7.6, 1.6 Hz, 1H), 7.59 (d, J = 8.8 Hz, 2H), 7.02 (d, J = 8.8 Hz, 2H), 3.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 177.21, 160.99, 160.48, 146.39, 145.26, 138.18, 134.34, 134.08, 133.15, 131.70, 131.58, 129.87, 128.77, 128.27, 128.07, 127.38, 127.12, 114.79, 55.65; LC-MS (ESI+) 438.04 (M+H)+; HRMS (ESI+) m/z calculated for C23H17ClNO4S (M+H)+ 438.0561, found 438.0562.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-4-pentylbenzenesulfonamide (13j)
This compound was prepared using the procedure described for 13a except using 4-pentylphenylsulfonamide (455 mg, 2 mmoL), TiCl4 (2 mL, 1M solution in DCM), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) as solvent and the product was purified using SiO2 chromatography (Hexane/EtOAc gradient elution) to afford the title compound as a yellow solid (452 mg, 56.2 %), m.p.: 111.7 – 112.8 °C. 1H NMR (400 MHz, CDCl3) δ 8.69 (s, 1H), 8.20 (dd, J = 7.6, 1.2 Hz, 1H), 8.16 (dd, J = 7.2, 1.6 Hz, 1H), 7.97 (d, J = 8.4 Hz, 2H), 7.73 (td, J = 7.5, 1.5 Hz, 1H), 7.68 (td, J = 7.6, 1.6 Hz, 1H), 7.40 (d, J = 8.4 Hz, 2H), 2.72 (t, J = 7.7 Hz, 2H), 1.66 (q, J = 7.5 Hz, 2H), 1.42-1.30 (m, 4H), 0.90 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 177.24, 160.86, 149.79, 145.15, 137.64, 134.30, 134.02, 133.19, 131.57, 129.86, 129.39, 128.04, 127.77, 127.10, 36.19, 31.62, 30.99, 22.70, 14.22; LC-MS (ESI+) 402 (M+H)+, 419.11 (M+NH4)+; HRMS (ESI+) m/z calculated for C21H21ClNO3S (M+H)+ 402.0925, found 402.0926.
5-Chloro-N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)thiophene-2-sulfonamide (13k)
This compound was prepared using the procedure described for 13a except using 5-chlorothiophene-2-sulfonamide (395 mg, 2 mmol), TiCl4 (2 mL, 1M solution in DCM), TEA (0.615 mL, 4.4 mol) and DCM (15 mL) as solvent. The title compound was obtained as a yellow solid (320 mg, 43.0%). Rf = 0.50 (TLC, EtOAc:Hexane [1:2]); m.p.: 149–151 °C; 1H NMR, (400 MHz, CDCl3) δ 8.55 (s, 1H), 8.23-8.20 (m, 2H), 7.79-7.72 (m, 2H), 7.64 (d, J = 4.1 Hz, 1H), 7.01 (d, J = 4.1 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 176.99, 161.21, 145.85, 139.77, 134.53, 134.47, 133.10, 132.78, 131.60, 129.58, 128.25, 127.24, 127.00, 104.99; LC-MS (ESI+) 761.89 (2M+NH4)+; HRMS (ESI+) m/z calculated for C14H8Cl2NO3S2 (M+H)+ 371.9317, found 371.9324.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-4′-trifluoromethylbiphenyl-4-sulfonamide (13l)
This compound was prepared using the procedure described for 13a except using 4′-trifluoromethylbiphenyl-4-sulfonamide (603 mg, 2 mmol), TiCl4 (2 mL, 1M solution in DCM), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) as the solvent. The title compound was obtained as a yellow solid (794 mg, 83.4%), m.p.: 190.4–191.3 °C; 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 8.21 (dd, J = 7.6, 1.2 Hz, 1H), 8.18-8.16 (m, 2H), 8.17 (d, J = 8.6 Hz, 1H), 7.81 (d, J = 8.6 Hz, 2H), 7.78-7.72 (m, 5H), 7.68 (td, J = 7.6, 1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 177.12, 161.41, 145.50, 145.20, 142.93, 140.04, 134.36, 134.22, 133.05, 131.61, 130.95 (q, J = 32.5 Hz), 129.92, 128.42, 128.26, 128.16, 128.01, 127.09, 126.30 (q, J = 3.7 Hz), 124.23 (d, J = 270.7 Hz); 19F NMR (376 MHz, CDCl3) δ −63.01 (s); LC-MS (ESI+) 493.05 (M+NH4)+; HRMS (ESI+) m/z calculated for C23H14ClF3NO3S (M+H)+ 476.0330, found 476.0325.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-5-(phenylsulfonyl)thiophene-2-sulfonamide (13m)
This compound was prepared using the procedure described for 13a except using 5-(phenylsulfonyl)thiophene-2-sulfonamide (341 mg, 1.2 mmol), TiCl4 (1.1 mL, 1M solution in DCM), TEA (0.345 mL, 2.5 mmol) and THF (10 mL) as the solvent. The title compound was obtained as a yellow solid (272 mg, 50.7%), m.p.: 165.3 °C (Dec.); 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 1H), 8.22-8.19 (m, 2H), 8.06-8.01(m, 2H), 7.79-7.76 (m, 2H), 7.73 (d, J = 4.0 Hz, 1H), 7.68-7.64 (m, 2H), 7.60-7.56 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.41, 150.49, 148.23, 146.44, 140.75, 134.85, 134.73, 134.52, 132.52, 132.46, 132.34, 131.59, 129.98, 129.70, 128.40, 128.00, 127.40, 104.99; elemental analysis for 13m: Calculated: C, 50.26; H, 2.53; N, 2.93; Found: C, 50.53; H, 2.85; N, 2.68.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-1-(phenylsulfonyl)-1H-pyrrole-3-sulfonamide (13n)
This compound was prepared using the procedure described for 13a except using 1-(phenylsulfonyl)-1H-pyrrole-3-sulfonamide (390 mg, 1.4 mmol), TiCl4 (1.4 mL, 1M solution in DCM), TEA (0.42 mL, 3 mmol) and THF (10 mL) as the solvent. The title compound was obtained as a yellow solid (444 mg, 70.7%), m.p.: 127.5–131.3 °C; 1H NMR (400 MHz, CDCl3) δ 8.55 (s, 1H), 8.20-8.16 (m, 2H), 7.97-7.95 (m, 2H), 7.85 (dd, J = 2.4, 1.6 Hz, 1H), 7.76-7.68 (m, 3H), 7.60 (t, J = 7.6 Hz, 2H), 7.27 (dd, J = 3.4, 2.4 Hz, 1H), 6.74 (dd, J = 3.4, 1.7 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 177.11, 161.01, 145.34, 137.75, 135.29, 134.41, 134.21, 132.97, 131.58, 130.18, 129.76, 128.88, 128.13, 127.71, 127.11, 124.19, 122.43, 111.92; LC-MS (ESI+) 461.00 (M+H)+; HRMS (ESI+) m/z calculated for C20H14ClN2O5S2 (M+H)+ 461.0027, found 461.0024.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)naphthalene-1-sulfonamide (13o, intermediate for compound compound 34)
This compound was prepared using the procedure described for 13a except using naphthalene-1-sulfonamide (415 mg, 2 mmol), TiCl4 (2 mL, 1M solution in DCM), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) as the solvent. The title compound was obtained as an orange-red solid (0.653 mg, 85.5%) Rf = 0.67 (TLC, EtOAc: Hexane [1:2]); m.p.: 185.6 °C (Dec.); 1H NMR (400 MHz, CDCl3) δ 8.75 (s, 1H), 8.67 (dd, J = 8.8, 0.8 Hz, 1H), 8.44 (dd, J = 7.4, 1.6 Hz, 1H), 8.21-8.17 (m, 2H), 8.00 (appdt, J = 7.9, 1.9 Hz, 2H), 7.72-7.62 (m, 4H), 7.59 (td, J = 8.0, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 177.20, 161.53, 145.34, 135.61, 135.51, 134.47, 134.41, 134.08, 133.21, 131.61, 130.28, 129.32, 129.22, 128.78, 128.64, 128.08, 127.29, 127.14, 125.41, 124.44; LC-MS (ESI+) 382 (M+H)+, 399.06 (M+NH4)+; HRMS (ESI+) m/z calculated for C20H13ClNO3S (M+H)+ 382.0299, found 382.0300.
4-Chloro-N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)benzenesulfonamide (13p)
This compound was prepared using the procedure described for 13a except using 4-chlorobenzenesulfonamide (383 mg, 2 mmol), TiCl4 (2 mL, 1M solution in DCM), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) as the solvent. The title compound was obtained as a yellow solid (399 mg, 54.5%). Rf = 0.60 (TLC, EtOAc: Hexane [1:2]); m.p.: 138–140 °C; 1H-NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 8.21 (dd, J = 7.6, 1.6 Hz, 1H), 8.12 (dd, J = 7.6, 1.2 Hz, 1H), 8.01 (d, J = 8.7 Hz, 2H), 7.75 (td, J = 8.8,1.2, Hz, 1H), 7.68 (td, J = 7.6, 1.6 Hz, 1H), 7.58 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 177.07, 161.51, 145.59, 140.47, 138.99, 134.41, 134.30, 132.93, 131.58, 129.88, 129.72, 129.17, 128.18, 127.07; LC-MS (ESI+) 383.00 (M+NH4)+; HRMS (ESI+) m/z calculated for C16H10Cl2NO3S (M+H)+ 365.9753, found 365.9763.
3,4-Dichloro-N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)benzenesulfonamide (13q)
This compound was prepared using the procedure described for 13a except using 3, 4-dichlorobenzenesulfonamide (452 mg, 2 mmol), TiCl4 (2 mL, 1M solution in DCM), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) as the solvent. The title compound was obtained as a yellow solid (622 mg, 77.6%). Rf = 0.41 (TLC, EtOAc: Hexane [1:2]); m.p.: 140.2 – 141.7 °C; 1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H), 8.22 (dd, J = 7.7, 1.4 Hz, 1H), 8.16 (d, J = 2.2 Hz, 1H), 8.12 (dd, J = 8.0, 1.6 Hz, 1H), 7.90 (dd, J = 8.4, 2.2 Hz, 1H), 7.77 (td, J = 7.2, 1.2 Hz, 1H), 7.72-7.68 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 176.97, 162.00, 145.89, 140.15, 138.81, 134.49, 134.10, 132.75, 131.57, 131.47, 129.90, 129.68, 128.27, 127.14, 126.73; LC-MS (ESI+) 817.90 (2M+NH4)+; HRMS (ESI+) m/z calculated for C16H9Cl3NO3S (M+H)+ 399.9363, found 399.9375.
N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)benzenesulfonamide (13r)
This compound was prepared using the procedure described for 13a except using phenylsulfonamide (314 mg, 2 mmol), TiCl4 (2 mL, 1M solution in DCM), TEA (0.615 mL, 4.4 mmol) and THF (15 mL) as the solvent. The title compound was obtained as a yellow solid (508 mg, 76.6%). Rf = 0.31 (TLC, EtOAc: Hexane [1:4]); m.p.: 184.8–186.4 °C; 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 8.20 (dd, J = 7.6, 1.2 Hz, 1H), 8.15 (d, J = 7.6 Hz, 1H), 8.09 (d, J = 7.2 Hz, 2H), 7.74 (td, J = 7.2, 1.2 Hz, 1H), 7.71-7.66 (m, 2H), 7.62 (t, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 177.18, 161.22, 145.33, 140.49, 134.36, 134.14, 133.81, 133.08, 131.57, 129.93, 129.40, 128.09, 127.68, 127.09; LC-MS (ESI+) 349.03 (M+NH4)+; HRMS (ESI+) m/z calculated for C16H11ClNO3S (M+H)+ 332.0143, found 332.0145.
2-(1-Oxo-4-(thiophen-2-ylsulfonylimino)-1,4-dihydronaphthalen-2-ylthio)acetic acid (14a)
The 13a (188 mg, 0.56 mmol) was dissolved in THF (5 mL), and added THF solution containing pyridine (0.56 mL, 0.5 M) followed by THF solution containing 2-mercaptoglycolic acid (0.56 mL, 0.5 M). Additional pyridine (0.56 mL, 0.5 M in THF) and 2-mercaptoglycolic acid (0.56 mL, 0.5 M in THF) were added to the mixture. The mixture was stirred at room temperature for 30 min., 2,3-dichloro-5,6-dicyanobenzoquinone (114 mg, 0.5 mmol) was added and stirred at room temperature overnight (12 h). The insoluble solid obtained was filtered and the filtrate was concentrated. The crude residue was first purified via preparative TLC (2.5% CH3OH in DCM) and then using SiO2 column chromatography (CH3OH/CH3COOH/DCM [1.5: 0.5: 100]) to afford a yellow solid as the desired pure product 14a (20 mg, 9.2%) and a mixture of desired product and reduced product (80.9 mg, 36.9%). Rf = 0.33 (TLC, CH3OH:DCM [1:20]); m.p.: 131.7 °C (Dec.); 1H NMR (400 MHz, CD3CN) δ 8.21-8.19 (m, 1H), 8.12-8.10 (m, 1H), 7.92-7.90 (m, 2H), 7.87 (dd, J = 3.8, 1.3 Hz, 1H), 7.80-7.78 (m, 2H), 7.24 (dd, J = 4.8, 3.6 Hz, 1H), 3.88 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 180.77, 168.99, 160.11, 153.94, 141.97, 134.37, 133.54, 133.34, 133.22 132.94, 131.78, 127.59, 127.43, 127.17, 120.53, 33.90; LC-MS (ESI−) 391.97 (M−H)−; HRMS (ESI−) m/z calculated for C16H10NO5S3 (M−H)− 391.9727, found 391.9729.
N-(3-(1-methyl-1H-tetrazol-5-ylthio)-4-oxonaphthalen-1(4H)-ylidene)thiophene-2-sulfonamide (14b)
The 13a (36 mg, 0.1 mmol) was dissolved in THF (3 mL), and added 1-methyl-tetrazole-5-thiol (11.7 mg, 0.1 mol). The reaction mixture was stirred at r.t. for 2 hrs and concentrated. The residue was dissolved in DCM and triturated with hexane. The precipitate obtained was filtered and washed with DCM/hexane (1:1, 2 mL×3) to afford the title compound as an orange-red solid (29 mg, 68.9%). Rf = 0.80 (TLC, EtOAc: Hexane [7:3]); m.p.: 172 °C (dec.); 1H-NMR (400 MHz, CDCl3) δ 8.26-8.24 (m, 1H), 8.17-8.15 (m, 1H), 8.01 (s, 1H), 7.78 (d, J = 3.8 Hz, 1H), 7.76-7.72 (m, 3H), 7.15 (dd, J = 5.2, 4.4 Hz, 1H), 4.21 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 180.28, 160.98, 149.29, 147.61, 140.87, 136.15, 135.46, 134.90, 134.26, 133.33, 132.00, 128.82, 127.64, 127.15, 122.73, 35.38; LC-MS (ESI+) 418.01 (M+H)+; HRMS (ESI+) m/z calculated for C16H12N5O3S3 (M+H)+ 418.0097, found 418.0099.
N-(3-(1H-1,2,4-triazol-5-ylthio)-4-oxonaphthalen-1(4H)-ylidene)thiophene-2-sulfonamide (14c)
This compound was prepared using the procedure described for 14b except using 3-mercapto-1,2,4-triazole (10 mg, 0.1 mmol). The title compound 14c was obtained as an orange-red solid (39 mg, 96.5%). Rf = 0.33 (TLC, EtOAc: Hexane [1:1]); m.p.: 197 °C (dec.); HPLC 95% (Rt = 4.08 min, 55% CH3CN in 0.1% TFA water 20 min); 1H-NMR (400 MHz, CD3CN), δ 8.63 (s, 1H), 8.19-8.17 (m, 1H), 8.15-8.14 (m, 1H), 7.90-7.88 (m, 1H), 7.81-7.79 (m, 3H), 7.75-7.74 (m, 1H), 7.22-7.19 (m,1H); 13C NMR (100 MHz, DMSO-d6) δ 179.55, 160.03, 152.17, 150.60, 146.29, 134.58, 134.15, 133.49, 132.73, 132.23, 130.92, 127.58, 126.46, 125.92, 120.99, 117.56; LC-MS (ESI+) 402.99 (M+H)+; HRMS (ESI+) m/z calculated for C16H11N4O3S3 (M+H)+ 402.9988, found 402.9989.
2-(1-Hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-ylthio)acetic acid (15a)
The (E)-N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)thiophene-2-sulfonamide (13 a) (34 mg, 0.1 mmol) was dissolved in THF (2 mL) and added a THF solution containing thioglycolic acid (0.1 mL, 1M in THF) followed by pyridine (1 eq, 0.2 mL, 0.5 M in THF). The mixture was stirred at r.t. for 10 min and the solvent was removed using a rotary evaporator. The orange-red residue obtained was re-dissolved in ethyl acetate (50 mL). The mixture was transferred to a separation funnel and washed with NaHSO4 (0.5 M solution, 10 mL×2) to remove pyridine. The sodium hydrosulfite solid (5 eq, 87 mg, 0.5 mmol) was added followed by water (10 mL). The mixture was shaken until the organic phase turned colorless. The organic phase was separated and washed with water (10 mL×2) and brine (10 mL), dried (Na2SO4) and filtered. The filtrate was concentrated. The solid obtained was suspended in DCM/hexane (1:1, ~5 mL) filtered and the product was washed with DCM (1 mL×3) to afford title compound as an off-white solid (25 mg, 63.6%). The analytical data obtained for compound 15a are identical to that obtained for 11a.
N-(4-hydroxy-3-(1-methyl-1H-tetrazol-5-ylthio)naphthalen-1-yl)thiophene-2-sulfonamide (15b)
The 14b (13 mg, 0.031 mmol) was dissolved in ethyl acetate (50 mL), and added sodium hydrosulfite (27 mg, 0.156 mmol) followed by water (10 mL). The mixture was shaken in a separating funnel until the yellow organic phase turned colorless. The organic phase was washed with water (10 mL×3), brine (10 mL), dried (Na2SO4) and concentrated. The residue obtained was dissolved in DCM (3 mL) and triturated with hexane (3 mL). The precipitate was washed with DCM/hexane (1:1, 1 mL×3) and dried under vacuum to afford the title compound 15b as a white solid (13 mg, 99%). Rf = 0.44 (TLC, EtOAc: Hexane [1:1]); m.p.: 130 °C (dec.); HPLC 93% (Rt = 8.1 min, 40% CH3CN in 0.1% TFA water 40 min); 1H-NMR (400 MHz, CD3CN) δ 8.28 (dd, J = 6.4, 2.8 Hz, 1H), 8.14 (brs, 1H, disappeared on D2O shake), 7.98 (dd, J = 6.8, 3.5 Hz, 1H), 7.94 (brs, 1H, disappeared on D2O shake), 7.65 (dd, J = 4.8, 1.2 Hz, 1H), 7.61 (dd, J = 6.5, 3.4 Hz, 2H), 7.38 (dd, J = 4.0, 1.2 Hz, 1H), 7.16 (s, 1H), 6.99 (dd, J = 4.8, 3.6 Hz, 1H), 4.00 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 154.04, 152.73, 140.64, 133.86, 133.07, 132.82, 129.49, 128.42, 128.23, 127.00, 126.30, 125.42, 124.16, 123.42, 107.44, 34.73; LC-MS (ESI+) 420.02 (M+H)+, 442.00 (M+Na)+; HRMS (ESI+) m/z calculated for C16H14N5O3S3 (M+H)+ 420.0253, found 420.0240.
N-(3-(1H-1,2,4-triazol-5-ylthio)-4-hydroxynaphthalen-1-yl)thiophene-2-sulfonamide (15c)
This compound was prepared using the procedure described for 15b using 14c (20 mg, 0.05 mmol) and sodium hydrosulfite (43 mg, 0.25 mmol) to afford the title compound as a white solid (20 mg, 100%). Rf = 0.40 (TLC, EtOAc: Hexane [1:1]); m.p.: 180 °C (dec.); HPLC 92% (Rt = 5.4 min, 40% CH3CN in 0.1% TFA water 40 min); 1H-NMR (400 MHz, DMSO-d6) δ 10.19 (brs, 1H), 10.05 (s, 1H), 8.61 (brs, 1H), 8.16 (d, J = 7.6 Hz, 1H), 7.89 (d, J = 7.4 Hz, 1H), 7.81 (d, J = 4.2 Hz, 1H), 7.48 (brt, J = 7.2 Hz, 2H), 7.28 (d, J = 2.5 Hz, 1H), 6.99 (brt, J = 4.2 Hz, 1H), 6.90 (brs, 1H); 13C NMR (100 MHz, DMSO-d6) δ 140.86, 133.68, 133.00, 132.93, 129.47, 128.16, 126.66, 126.60, 126.28, 124.90, 124.04, 123.14, 104.99. LC-MS (ESI−) 403.00 (M−H)−; HRMS (ESI−) m/z calculated for C16H11N4O3S3 (M−H)− 402.9999, found 402.9989.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)thiophene-2-sulfonamide (15d)
The 13a (34 mg, 0.1 mmole) and 1H-tetrazole-5-thiol (10 mg, 0.1 mmol) were dissolved in EtOH/DCM (2 mL, 1:1) and stirred at r.t. for 2 hrs. The reaction mixture was diluted with EtOAc (20 mL) and Na2S2O4 (0.5 mmol in 5 mL of water) was added. The mixture was shaken in a separation funnel until the color of the organic phase turned colorless (from bright yellow). The organic phase was separated and washed with water (5 mL), brine (5 mL) and dried (Na2SO4). The organic phase was filtered, concentrated and the residue was slurred in DCM (5 mL). The solid obtained was washed with DCM (1 mL×2) and dried to afford the title compound 15d (28 mg, 69.9%, via 2 steps) as an off-white solid. m.p.: 186.6 °C (dec.); HPLC 99% (Rt = 9.5 min, 35% CH3CN in 0.1% TFA water 30 min); 1H NMR (400 MHz, DMSO-d6) δ 10.63 (brs, 1H), 10.14 (s, 1H), 8.25-8.23 (m, 1H), 7.96-7.94 (m, 1H), 7.82 (d, J = 4.8 Hz, 1H), 7.56-7.54 (m, 2H), 7.32 (d, J = 3.6 Hz, 1H), 6.97 (t, J = 4.0 Hz, 1H), 6.92 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 154.80, 140.56, 133.81, 133.17, 133.13, 130.21, 128.41, 128.13, 126.87, 126.47, 125.29, 124.16, 123.57; LC-MS (ESI+) 406.01 (M+H)+; HRMS (ESI+) m/z calculated for C15H12N5O3S3 (M+H)+ 406.0097, found 406.0075.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)biphenyl-4-sulfonamide (15e)
This compound was prepared using the procedure described for 15d except using 13b (41 mg, 0.1 mmol) to afford the title compound 15e (32 mg, 67.4%, via 2 steps) as a white solid. m.p.: 158.8 °C (dec.); HPLC 94% (Rt = 7.8 min, 50% CH3CN in 0.1% TFA water 30 min); 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, 1H), 8.24-8.22 (m, 1H), 8.02-8.00 (m, 1H), 7.72-7.63 (m, 6H), 7.55-7.53 (m, 2H), 7.51-7.47 (m, 2H), 7.45-7.40 (m, 1H), 6.87 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 154.63, 144.81, 139.08, 138.97, 133.08, 129.95, 129.77, 129.20, 128.34, 128.20, 127.78, 127.70, 126.86, 126.51, 125.58, 124.39, 123.55; LC-MS (ESI+) 476.09 (M+H)+; HRMS (ESI+) m/z calculated for C23H18N5O3S2 (M+H)+ 476.0846, found 476.0842.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)-5-phenylthiophene-2-sulfonamide (15f)
This compound was prepared using the procedure described for 15d except using 13c (41 mg, 0.1 mmol), to afford the title compound 15f (34 mg, 70.1%, via 2 steps) as an off-white solid. m.p.: 167.9 °C (dec.); HPLC 98% (Rt = 7.7 min, 50% CH3CN in 0.1% TFA water, 30 min); 1H NMR (400 MHz, DMSO-d6) δ 10.65 (brs, 1H), 10.23 (s, 1H), 8.26-8.24 (m, 1H), 8.02-7.90 (m, 1H), 7.60 (dd, J = 8.1, 2.4 Hz, 2H), 7.56-7.54 (m, 2H), 7.46-7.39 (m, 3H), 7.33 (d, J = 3.9 Hz, 1H), 7.29 (d, J = 3.9 Hz, 1H), 7.04 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 154.95, 150.37, 139.01, 134.22, 133.17, 132.73, 130.38, 130.01, 129.84, 128.47, 126.90, 126.59, 126.49, 125.21, 124.41, 124.17, 123.61; LC-MS (ESI+) 482.04 (M+H)+; HRMS (ESI+) m/z calculated for C21H16N5O3S3 (M+H)+ 482.0410, found 482.0415
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)-5-(pyridin-2-yl)thiophene-2-sulfonamide (15g)
This compound was prepared using the procedure described for 15d except using 13g (42 mg, 0.1 mmol), to afford the title compound 15g (40 mg, 84.8%, via 2 steps) as an off-white solid. m.p.: 163.2 °C (dec.); HPLC 96% (Rt = 7.9 min, 40% CH3CN in 0.1% TFA water, 30 min); 1H NMR (400 MHz, , DMSO-d6) δ 10.63 (br s, 1H), 10.29 (s, 1H), 8.53 (app. d, J = 4.6 Hz, 1H), 8.23 (dd, J = 6.8, 3.6 Hz, 1H), 7.98 (dd, J = 6.5, 3.2 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.87 (td, J = 7.8, 1.6 Hz, 1H), 7.60 (d, J = 4.0 Hz, 1H), 7.54-7.50 (m, 2H), 7.37-7.34 (m, 1H), 7.28 (d, J = 4.0 Hz, 1H), 7.05 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 154.87, 151.23, 150.88, 150.31, 141.44, 138.20, 134.06, 133.03, 130.29, 128.48, 126.90, 126.47, 125.26, 125.17, 124.51, 124.09, 123.62, 120.02, 106.90; LC-MS (ESI+) 483.04 (M+H)+; HRMS (ESI+) m/z calculated for C20H15N6O3S3 (M+H)+ 483.0362, found 483.0348.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)-4-cyclohexylbenzenesulfonamide (15h)
This compound was prepared using the procedure described for 15d except using 13h (41 mg, 0.1 mmol) to afford the title compound 15h (22 mg, 46.3%, via 2 steps) as an off-white solid. m.p.: 169.2 °C (dec.); HPLC 99% (Rt = 14.5 min, 50% CH3CN in 0.1% TFA water, 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.64 (brs, 1H), 9.79 (s, 1H), 8.20 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.54-7.49 (m, 2H), 7.46 (d, J = 8.3 Hz, 2H), 7.20 (d, J = 8.0 Hz, 2H), 6.71 (s, 1H), 1.76-1.64 (m, 5H), 1.34-1.17 (m, 5H); 13C NMR (100 MHz, DMSO-d6) δ 154.83, 153.32, 137.35, 133.29, 130.33, 128.27, 127.80, 127.72, 126.77, 126.46, 125.63, 124.42, 123.53, 44.19, 34.12, 26.82, 26.11; LC-MS (ESI−) 480.11 (M−H)−; HRMS (ESI+) m/z calculated for C23H24N5O3S2 (M+H)+ 482.1315, found 482.1318
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)-4′-methoxybiphenyl-4-sulfonamide (15i)
This compound was prepared using the procedure described for 15d except using 13i (44 mg, 0.1 mmol) to afford the title compound 15i (42 mg, 82.0%, via 2 steps) as a white solid. m.p.: 156.8 °C (dec.); HPLC 93% (Rt = 12.0 min, 45% CH3CN in 0.1% TFA water, 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.61 (brs, 1H), 9.95 (s, 1H), 8.24-8.21 (m, 1H), 8.02-7.99 (m, 1H), 7.68-7.59 (m, 4H), 7.55-7.52 (m, 2H), 7.03 (d, J = 9.2 Hz, 2H), 6.87 (s, 1H), 3.79 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 160.39, 154.61, 144.43, 138.15, 133.08, 131.26, 129.91, 128.90, 128.34, 128.18, 127.04, 126.86, 126.50, 125.64, 124.40, 123.55, 115.20, 106.77, 55.95; LC-MS (ESI+) 506.10 (M+H)+; HRMS (ESI+) m/z calculated for C24H20N5O4S2 (M+H)+ 506.0951, found 506.0936.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)-4′-fluorobiphenyl-4-sulfonamide (15j)
This compound was prepared using the procedure described for 15d except using 13f (43 mg, 0.1 mmol) to afford the title compound 15j (40 mg, 81.8%, via 2 steps) as a white solid. m.p.: 168.1 °C (dec.); HPLC 95% (Rt = 13.7 min, 45% CH3CN in 0.1% TFA water 30 min); 1H NMR (400 MHz, DMSO-d6) δ 10.56 (brs, 1H), 9.99 (s, 1H), 8.24-8.22 (m, 1H), 8.02-8.00 (m, 1H), 7.71-7.64 (m, 6H), 7.55-7.52 (m, 2H), 7.32 (t, J = 8.9 Hz, 2H), 6.85(s, 1H); 19F NMR (376 MHz, DMSO-d6) δ −114.25 (m); 13C NMR (100 MHz, DMSO-d6) δ 163.13 (d, J = 245 Hz), 154.57, 143.72, 138.96, 135.57, 133.05, 129.85, 129.84 (d, J = 8 Hz), 128.35, 128.21, 127.74, 126.88, 126.51, 125.57, 124.39, 123.54, 116.63 (d, J = 21 Hz), 106.90; LC-MS (ESI+) 494.07 (M+H)+; HRMS (ESI+) m/z calculated for C23H17FN5O3S2 (M+H)+ 494.0751, found 494.0739.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)-4-phenoxybenzenesulfonamide (15k)
This compound was prepared using the procedure described for 15d except using 13d (42 mg, 0.1 mmol) to afford the title compound 15k (22 mg, 45.1%, via 2 steps) as a white solid. m.p.: 172.5 °C (dec.); HPLC 99% (Rt = 8.2 min, 50% CH3CN in 0.1% TFA water 30 min); 1H NMR (400 MHz, DMSO-d6) δ 10.58 (brs, 1H, disappeared on D2O shake), 9.88 (s, 1H, disappeared on D2O shake), 8.22-8.20 (m, 1H), 7.94-7.92 (m, 1H), 7.53 (d, J = 8.8 Hz, 4H), 7.41 (t, J = 7.6 Hz, 2H), 7.21 (t, J = 7.2 Hz, 1H), 6.98 (d, J = 7.6 Hz, 2H), 6.92 (d, J = 8.8 Hz, 2H), 6.83 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 161.03, 155.79, 154.58, 134.15, 133.06, 130.99, 130.21, 130.09, 128.30, 126.81, 126.48, 125.57, 125.36, 124.35, 123.54, 120.31, 118.45, 106.87; LC-MS (ESI+) 492.07 (M+H)+; HRMS (ESI+) m/z calculated for C23H18N5O4S2 (M+H)+ 492.0795, found 492.0805
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)naphthalene-2-sulfonamide (15l)
This compound was prepared using the procedure described for 15d except 13e (38 mg, 0.1 mmol) to afford the title compound 15l (29 mg, 64.9%, via 2 steps) as a grey solid. m.p.: 173.6 °C (dec.); HPLC 98% (Rt = 5.4 min, 50% CH3CN in 0.1% TFA water 30 min); 1H NMR (400 MHz, DMSO-d6) δ 10.54 (brs, 1H), 10.06 (s, 1H), 8.19-8.16 (m, 2H), 7.99-7.93 (m, 4H), 7.70 (dd, J = 8.6, 1.5 Hz, 1H), 7.63 (t, J = 7.2 Hz, 1H), 7.55 (t, J = 7.5 Hz, 1H), 7.49-7.43 (m, 2H), 6.88 (s, 1H); LC-MS (ESI+) 450.07 (M+H)+; 13C NMR (100 MHz, DMSO-d6) δ 154.69, 137.30, 134.78, 133.03, 132.10, 130.08, 129.79, 129.77, 129.46, 128.54, 128.43, 128.32, 128.16, 126.82, 126.44, 125.49, 124.29, 123.55, 123.10, 104.99; LC-MS (ESI+) 450.07 (M+H)+; HRMS (ESI+) m/z calculated for C21H16N5O3S2 (M+H)+ 450.0689, found 450.0702.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)-4-pentylbenzenesulfonamide (15m)
This compound was prepared using the procedure described for 15d except using 13j (40 mg, 0.1 mmol) to afford the title compound 15m (39 mg, 83.6%, via 2 steps) as a grey solid. m.p.: 155.6 °C (dec.); HPLC 99% (Rt = 15.3 min, 50% CH3CN in 0.1% TFA water 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.53 (brs, 1H), 9.80 (s, 1H), 8.21-8.19 (m, 1H), 7.96-7.94 (m, 1H), 7.56-7.49 (m, 2H), 7.48 (d, J = 8.3 Hz, 2H), 7.16 (d, J = 8.3 Hz, 2H), 6.76 (s, 1H), 2.54 (t, J = 7.5 Hz, 2H), 1.50 (p, J = 7.5 Hz, 2H), 1.30-1.23 (m, 2H), 1.18-1.11 (m, 2H), 0.83 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 154.75, 148.33, 137.34, 133.23, 130.26, 129.40, 128.27, 127.62, 126.76, 126.47, 125.63, 124.42, 123.53, 106.51, 35.43, 31.24, 30.77, 22.55, 14.56; LC-MS (ESI+) 470.13 (M+H)+; HRMS (ESI+) m/z calculated for C22H24N5O3S2 (M+H)+ 470.1315, found 470.1310.
2-(4-(5-Chlorothiophene-2-sulfonamido)-1-hydroxynaphthalen-2-ylthio)acetic acid (15n)
This compound was prepared using the procedure described for 15a using 13k (52 mg, 0.14 mmol) and a solution of thioglycolic acid (0.14 mL, 1M solution in THF). The title compound 15n (32 mg, 54%) was obtained as a white solid. m.p.: 143–145 °C; HPLC 95% (Rt = 17.1 min, 40% CH3CN in 0.1% TFA water, 40 min); 1H-NMR (400 MHz, DMSO-d6) δ 12.77 (brs, 1H), 10.30 (s, 1H), 9.96 (brs, 1H), 8.14 (d, J = 7.7 Hz, 1H), 7.85 (d, J = 8.8 Hz, 1H), 7.50-7.43 (m, 2H), 7.22 (d, J = 4.1 Hz, 1H), 7.12-7.11 (m, 2H), 3.55 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 171.78, 153.29, 139.27, 135.68, 132.92, 131.87, 130.16, 128.59, 127.59, 126.59, 125.76, 124.15, 123.69, 123.18, 113.74, 37.42; LC-MS (ESI−) 427.94 (M−H)−; HRMS (ESI+) m/z calculated for C16H12ClNO5S3Na (M+Na)+ 451.9458, found 451.9468.
2-(4-(Biphenyl-4-ylsulfonamido)-1-hydroxynaphthalen-2-ylthio)acetic acid (15o)
This compound was prepared using the procedure described for 15a using 13b (50 mg, 0.12 mmol) and a solution of thioglycolic acid (0.12 mL, 1M solution in THF). The title compound 15o (19 mg, 40 %) was obtained as a grey solid. m.p.: 170 °C (dec.); HPLC 97% (Rt = 10.44 min, 50% CH3CN in 0.1% TFA water 20 min); 1H-NMR (400 MHz, DMSO-d6) δ 12.78 (brs, 1H), 9.95 (s, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.76 (d, J = 8.4 Hz, 2H), 7.68-7.65 (m, 4H), 7.548-7.39 (m, 5H), 7.00 (s, 1H), 5.73 (brs, 1H), 3.46 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 171.73, 152.91, 144.78, 139.23, 139.14, 131.89, 129.77, 129.17, 128.25, 127.87, 127.71, 127.28, 126.41, 125.79, 124.79, 124.13, 123.09, 113.72, 37.44; LC-MS (ESI+) 464.07 (M+H)+; HRMS (ESI−) m/z calculated for C24H18NO5S2 (M−H)− 464.0632, found 464.0638.
tert-Butyl 2-(4-(thiophene-2-sulfonamido)naphthalen-1-yloxy)acetate (16a)
The compound 3 (61 mg, 0.2 mmol) was dissolved in DMF (1 mL), and added tert-butyl bromoacetate (0.060 mL, 0.4 mmol) followed by DBU (0.4 mL, 1 M solution in NMP) and catalytic amount of KI. The reaction mixture was heated using a microwave reactor at 90 °C for 20 min. The TLC analysis (EtOAc: Hexane [1:2]) showed the presence of starting material. To drive the reaction to completion, additional tert-butyl bromoacetate (13.5 eq., 0.4 mL, 2.7 mmol) was added and the reaction was continued at 90 °C for further 30 min. The resulting mixture was diluted with ethyl acetate (40 mL) and washed with NaHSO4 (10 mL×2, 0.5 M), water (10 mL×6) and brine (10 mL). The organic phase was dried (Na2SO4) and concentrated. The solid obtained was purified via SiO2 chromatography (Hexane/EtOAc, gradient elution) to afford 16a (43 mg, 51.3%) as a gray solid. Rf = 0.27 (TLC, EtOAc: Hexane [1:2]); m.p.: 128.6 °C (dec.). 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 7.6 Hz, 1H), 7.80 (dd, J = 7.6, 1.2 Hz, 1H), 7.61 (dd, J = 4.8, 1.2 Hz, 1H), 7.56 (dd, J = 3.6, 1.2 Hz, 1H), 7.46-7.36 (m, 2H), 7.16 (d, J = 8.4 Hz, 1H), 6.99 (dd, J = 5.0, 3.8 Hz, 1H), 6.31 (d, J = 8.4 Hz, 1H), 3.92 (s, 2H), 1.52 (s, 9H).; LC-MS (ESI+) 420.09 (M+H)+; HRMS (ESI+) m/z calculated for C20H22NO5S2 (M+H)+ 420.0934, found 420.0932.
Methyl 4-(4-(thiophene-2-sulfonamido)naphthalen-1-yloxy)butanoate (16b)
This compound was synthesized according to the procedure described for 16a except using 15 eq. of ethyl bromobutanoate (0.44 mL, 3 mmol), and the reaction mixture was heated using a microwave reactor at 90 °C for 30 min. The TLC analysis indicated presence of starting material and additional 30 min. at 90 °C was required to complete the reaction. The crude solid 16b was purified (SiO2 chromatography, Hexane/EtOAc gradient elution) to afford 16b (43 mg, 51.3%) as a brown oil. The base line impurities were still observed after SiO2 purification. This product was used in the next step without further purification. Rf = 0.36 (TLC, EtOAc: Hexane=1:2); 1H NMR (400 MHz, CDCl3) δ 7.82-7.80 (m, 1H), 7.78-7.75 (m, 1H), 7.62 (dd, J = 5.2, 1.6 Hz, 1H), 7.58 (dd, J = 4.0, 1.2 Hz, 1H), 7.42-7.36 (m, 2H), 7.13 (d, J = 8.4 Hz, 1H), 7.00 (dd, J = 4.8, 4.0 Hz, 1H), 6.44 (d, J = 8.4 Hz, 1H), 4.15 (q, J = 7.1 Hz, 2H), 3.31 (t, J = 6.7 Hz, 2H), 2.52 (t, J = 6.8 Hz, 2H), 2.11 (p, J = 6.7 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H).
2-(4-(Thiophene-2-sulfonamido)naphthalen-1-yloxy)acetic acid (17a)
The 16a was dissolved in dioxane (1 mL) and added conc. HCl (1mL, 32%). The mixture was stirred at room temperature for 1 hour (TLC analysis EtOAc: Hexane [1:2] indicated completion of the reaction) and concentrated. The residue was suspended in DCM/Hexane (1:1, 2 mL) and left in a refrigerator (overnight). The precipitate was collected by filtration and washed with DCM/Hexane (1:1, 3 mL) affording 17a (21.4 mg, 82.3%) as a gray solid. Rf = 0.33 (TLC, CH3OH: DCM [1:20]); m.p.: 148.9 °C (dec.). HPLC 98% (Rt = 6.19 min, 50% CH3CN in 0.1% TFA water 20 min); 1H NMR (400 MHz, DMSO-d6) δ 12.64 (brs, 1H), 8.14-8.13 (m, 1H), 8.11 (dd, J = 5.2, 1.4 Hz, 1H), 7.78 (dd, J = 3.8, 1.4 Hz, 1H), 7.66-7.63 (m, 1H), 7.46-7.43 (m, 2H), 7.19 (dd, J = 4.8, 3.6 Hz, 1H), 7.01 (d, J = 8.5 Hz, 1H), 6.80 (brs, 1H), 6.23 (d, J = 8.4 Hz, 1H), 3.96 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 172.94, 143.97, 137.63, 136.87, 135.95, 134.32, 129.07, 127.88, 127.43, 125.75, 123.72, 122.55, 121.54, 120.25, 101.97, 45.39; LC-MS (ESI−) 362.01 (M−H)−; HRMS (ESI−) m/z calculated for C16H12NO5S2 (M−H)− 362.0162, found 362.0174.
4-(4-(Thiophene-2-sulfonamido)naphthalen-1-yloxy)butanoic acid (17b)
This compound was synthesized using the procedure described for 17a except using 16b (43 mg, 0.1 mmol), dioxane (2 mL), conc. HCl (4 mL, 32%). The required product 17b (23 mg, 56.6%) was obtained as a brown greasy solid. Rf = 0.09 (TLC, CH3OH: DCM [1:20]); HPLC 97% (Rt = 7.43 min, 50% CH3CN in 0.1% TFA water 20 min); 1H NMR (400 MHz, DMSO-d6) δ 12.11 (brs, 1H), 8.18-8.15 (m, 1H), 8.10 (dd, J = 4.8, 1.2 Hz, 1H), 7.77 (dd, J = 2.8, 0.8 Hz, 1H), 7.64–7.62 (m, 1H), 7.42–7.40 (m, 2H), 7.18 (t, J = 4.0 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 6.42-6.39 (m, 2H), 3.20-3.17 (m, 2H), 2.36 (t, J = 7.2 Hz, 2H), 1.92–1.85 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 175.19, 144.40, 137.55, 136.84, 135.46, 134.32, 129.02, 127.94, 127.30, 125.42, 123.75, 122.75, 121.45, 120.37, 101.52, 43.20, 32.04, 24.05; LC-MS (ESI−) 390.05 (M−H)−; HRMS (ESI−) m/z calculated for C18H16NO5S2 (M−H)− 390.0475, found 390.0476.
O-4-nitronaphthalen-1-yl dimethylcarbamothioate (19)
The 4-nitro-1-naphthol (378 mg, 2 mmol) was dissolved in NMP (2 mL) and added K2CO3 (276 mg, 2 mmol). The mixture was heated to 50 °C and added a solution of N,N -dimethylthiocarbamoyl chloride (260 mg, 2.1 mmol) in NMP (1 mL) drop-wise over 5 min. The resulting solution was stirred at 50 °C for 2 hours and quenched with water (30 mL) at 50 °C. The brown precipitate obtained was filtered and washed with water and dried to obtain intermediate 19 (496 mg, 89.8%) as a dark brown solid. Rf = 0.40 (TLC, EtOAc: Hexane [1:2]); m.p.: 110.4 °C (dec.). 1H NMR (400 MHz, CDCl3) δ 8.67 (d, J = 8.8 Hz, 1H), 8.33 (d, J = 8.4 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.77-7.72 (m, 1H), 7.66-7.62 (m, 1H), 7.30 (d, J = 8.4 Hz, 1H), 3.53 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 186.71, 154.44, 144.13, 130.11, 128.49, 127.95, 126.84, 124.77, 123.93, 122.62, 118.66, 43.77, 39.35; LC-MS (ESI+) 277.07 (M+H)+; HRMS (ESI+) m/z calculated for C13H13N2O3S (M+H)+ 277.0641, found 277.0637.
S-4-nitronaphthalen-1-yl dimethylcarbamothioate (20)
The intermediate 19 (276 mg, 1 mmol) was dissolved in NMP (2 mL) and the solution was heated to 180 °C in a microwave reactor for 20 min. The reaction mixture was diluted with ethyl acetate (50 mL) and washed with water and brine. The organic phase was dried (Na2SO4), concentrated and the crude product obtained was purified using chromatography (10g silica gel, Hexane/EtOAc gradient elution) to afford compound 20 as a brown solid (198 mg, 71.5%). Rf = 0.38 (TLC, EtOAc:Hexane [1:2]); m.p.: 91.2 °C (dec.). 1H NMR (400 MHz, CDCl3) δ 8.50-8.46 (m, 2H), 8.13 (d, J = 7.9 Hz, 1H), 7.87 (d, J = 7.9 Hz, 2H), 7.77-7.64 (m, 1H), 3.24 (s, 3H), 3.03 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 165.00, 148.27, 136.12, 134.33, 134.22, 129.61, 128.46, 126.75, 125.69, 123.66, 122.83, 37.41; LC-MS (ESI+) 277.07 (M+H)+; HRMS (ESI+) m/z calculated for C13H13N2O3S (M+H)+ 277.0641, found 277.0642.
2-(4-Nitronaphthalen-1-ylthio)acetic acid (21)
The intermediate 20 (97 mg, 0.35 mmol) was suspended in methanol (1 mL) under argon atmosphere and added KOH (1.4 mL, 1 M solution in CH3OH). The reaction mixture was stirred at room temperature for 12 hours affording a homogenous solution. The tert-butyl bromoacetate (0.103 mL, 0.7 mmol) was added and stirring was continued for 12 hours. The reaction mixture was concentrated and the residue was dissolved in water and acidified (pH=1–2) using solid NaHSO4. The yellow precipitate obtained was filtered, washed with water and air dried to obtain a yellow solid, (79 mg, 85.6%, via 2 steps). Rf = 0.18 (TLC, CH3OH: DCM: HOAc [1:20: 0.1]); m.p.: 144.9 °C (dec.). 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J = 8.8 Hz, 1H), 8.40 (d, J = 8.8 Hz, 1H), 8.18 (d, J = 8.1 Hz, 1H), 7.77-7.73 (m, 1H), 7.69-7.65 (m, 1H), 7.52 (d, J = 8.4 Hz, 1H), 3.91 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 170.63, 144.42, 143.51, 131.71, 129.92, 127.67, 125.31, 124.74, 124.16, 124.00, 121.35, 35.19. LC-MS (ESI−) 218.03 (M-H-CO2)−; HRMS (ESI−) m/z calculated for C12H8NO4S (M−H)− 262.0179, found 262.0179.
2-(4-Aminonaphthalen-1-ylthio)acetic acid (22)
The intermediate 21 (70 mg, 0.27 mmol) was dissolved in methanol (50 mL) and the solution was passed through the H-cube hydrogenator (10% Pd-C cat. cartridge, 1.0 mL/min., 25 °C, 40 bar) to obtain a slightly brown color solution. The solution was concentrated to dryness to afford the required product 22 (70 mg, 100%) as a gray solid. Rf = 0.40 (TLC, CH3OH: DCM [1:10]); m.p.: 138.9 °C (dec.). 1H NMR (400 MHz, CD3OD) δ 8.48 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.59 (d, J = 8.0, 1H), 7.55-7.51 (m, 1H), 7.46-7.42 (m, 1H), 6.71 (d, J = 7.6 Hz, 1H), 3.42 (s, 2H); 13C NMR (100 MHz, CD3OD) δ 172.73, 146.04, 135.53, 135.20, 126.58, 125.77, 124.47, 124.40, 122.33, 117.42, 108.43, 38.58; LC-MS (ESI−) 188.05 (M-H-CO2)−; HRMS (ESI−) m/z calculated for C12H10NO2S (M−H)− 232.0438, found 232.0435.
2-(4-(Thiophene-2-sulfonamido)naphthalen-1-ylthio)acetic acid (23)
The compound 22 (33 mg, 0.14 mmol) was dissolved in THF (1 mL), added pyridine (0.28 mL, 0.5 M solution in THF) at 0 °C, followed by water (0.5 mL). Thiophene-2-sulfonyl chloride (26 mg in 1 mL THF) was added drop-wise followed by additional pyridine (0.28 mL 0.5M solution in THF). The resulting solution was stirred vigorously at 0 °C and slowly warmed to room temperature. After 4 hours, the organic phase was removed and the aqueous portion was diluted with water (10 mL) and acidified (pH= 2–3) at 0 °C using NaHSO4 solution (0.5 M). The mixture was extracted with ethyl acetate. The organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The crude product obtained was purified using chromatography (SiO2, DCM/CH3OH [20:1]) to afford 23 (31 mg, 58.4%) as a gray solid. Rf = 0.17 (TLC, CH3OH: DCM: HOAc [1:20: 0.1]); m.p.: 159.8 °C (dec.). HPLC 99% (Rt = 4.43 min, 50% CH3CN in 0.1% TFA water 20 min); 1H NMR (400 MHz, CD3CN) δ 8.36 (d, J = 8.4 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.68 (dd, J = 4.8, 1.3 Hz, 1H), 7.64-7.60 (m, 1H), 7.57-7.51 (m, 2H), 7.43 (dd, J = 3.6, 1.2 Hz, 1H), 7.28 (d, J = 7.6 Hz, 1H), 7.04 (dd, J = 5.2, 4.0 Hz, 1H), 3.78 (s, 2H); LC-MS (ESI−) 378.00 (M−H)−; HRMS (ESI−) m/z calculated for C16H12NO4S3 (M−H)− 377.9934, found 377.9937.
2-(1-Hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-ylthio)benzoic acid (24)
This compound was prepared using the procedure described for 15a except using thiosalicylic acid (15 mg, 0.1 mmol). The title compound 24 (19 mg, 99%) was obtained as a white solid. m.p.: 215 °C (dec.); HPLC 91% (Rt = 18.9 min, 40% CH3CN in 0.1% TFA water, 40 min); 1H-NMR (400 MHz, DMSO-d6) δ 13.15 (brs, 1H), 10.09 (s, 1H), 8.19 (d, J = 8.0 Hz, 1H), 8.01 (d, J = 7.6 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.76 (d, J = 4.8 Hz, 1H), 7.58-7.50 (m, 2H), 7.34-7.31 (m, 2H), 7.17 (t, 7.2 Hz, 1H), 6.91 (s, 2H), 6.47 (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 168.15, 156.08, 141.87, 140.39, 133.88, 133.34, 133.21, 133.17, 132.65, 131.78, 128.38, 128.17, 126.70, 126.58, 126.18, 125.15, 125.06, 124.21, 123.69, 109.66, 104.98; LC-MS (ESI+) 475.05 (M+NH4)+; HRMS (ESI+) m/z calculated for C21H15NO5S3Na (M+Na)+ 480.0005, found 479.9992.
N-(3-(1H-1,2,4-triazol-3-ylthio)-4-hydroxynaphthalen-1-yl)-3,4-dichlorobenzenesulfonamide (25)
This compound was prepared using the procedure described for 15d except using 13b (82 mg, 0.2 mmol) and 1H-1,2,4-triazole-3-thiol (24 mg, 0.24 mmol). The title compound 25 (23 mg, 91.3%, via 2 steps) was obtained as a white solid. HPLC 98% (Rt = 21.0 min, 40% CH3CN in 0.1% TFA water, 40 min); m.p.: 154.8 °C (Dec.); 1H NMR (400 MHz, DMSO-d6) δ 10.17 (brs, 1H), 9.95 (s, 1H), 8.55 (brs, 1H), 8.145 (d, J = 7.6 Hz, 1H), 7.91 (d, J = 7.6 Hz, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.68-7.58 (m, 4H), 7.50-7.39 (m, 5H), 6.94 (brs, 1H); 13C NMR (100 MHz, DMSO-d6) δ 152.68, 144.69, 139.25, 139.08, 132.05, 129.79, 129.39, 129.18, 128.11, 127.77, 127.70, 127.59, 126.58, 126.30, 125.15, 124.15, 123.19; LC-MS (ESI+) 475.09 (M+H)+; HRMS (ESI+) m/z calculated for C24H19N4O3S2 (M+H)+ 475.0893, found 475.0898.
N-(3-(1H-1,2,4-triazol-3-ylthio)-4-hydroxynaphthalen-1-yl)-5-phenylthiophene-2-sulfonamide (26)
This compound was prepared using the procedure described for 15d except using 13c (83 mg, 0.2 mmol) and 1H-1,2,4-triazole-3-thiol (20 mg, 0.2 mmol). The title compound 26 (12 mg, 43.0%, via 2 steps) was obtained as a grey solid. m.p.: 172.9 °C (dec.); HPLC 95% (Rt = 20.1 min, 40% CH3CN in 0.1% TFA water, 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.17 (br s, 2H), 8.52 (brs, 1H), 8.17 (d, J = 8.8 Hz, 1H), 7.94 (d, J = 8.8 Hz, 1H), 7.62 (appd, J = 7.2 Hz, 2H), 7.50-7.39 (m, 6H), 7.29 (d, J = 4.0 Hz, 1H), 7.04 (brs, 1H); 13C NMR (100 MHz, DMSO-d6) δ 152.94, 150.22, 139.29, 134.02, 132.78, 132.25, 130.04, 129.82, 129.73, 127.73, 126.66, 126.58, 126.31, 124.83, 124.48, 124.03, 123.23, 111.15; LC-MS (ESI+) 481.05 (M+H)+; HRMS (ESI+) m/z calculated for C22H17N4O3S3 (M+H)+ 481.0457, found 481.0456.
N-(3-(1H-1,2,4-triazol-3-ylthio)-4-hydroxynaphthalen-1-yl)-4′-fluorobiphenyl-4-sulfonamide (27)
This compound was prepared using the procedure described for 15d except using 13f (85 mg, 0.2 mmol) and 1H-1,2,4-triazole-3-thiol (20 mg, 0.2 mmol). The title compound 27 (29 mg, 38.1%, via 2 steps) was obtained as an off-white solid. m.p.: 194.1 °C (dec.); HPLC 97% (Rt = 24.2 min, 40% CH3CN in 0.1% TFA water, 40 min); 1H NMR (400 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.51 (brs, 1H), 8.13 (d, J = 7.6 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.70-7.67 (m, 4H), 7.61 (d, J = 8.6 Hz, 2H), 7.45-7.42 (m, 2H), 7.30 (t, J = 8.8 Hz, 2H), 6.88 (brs, 1H); 13C NMR (100 MHz, DMSO-d6) δ 163.12 (d, J = 244.3 Hz), 145.75, 143.61, 139.26, 135.56, 129.84 (d, J = 8.3 Hz), 128.12, 127.72, 127.54, 126.56, 126.29, 125.04, 124.14, 123.18, 116.64 (d, J = 21.4 Hz). 19F NMR (376 MHz, CDCl3) δ −114.28 (m); LC-MS (ESI+) 493.09 (M+H)+.
N-(3-(1H-1,2,4-triazol-3-ylthio)-4-hydroxynaphthalen-1-yl)-4-cyclohexylbenzenesulfonamide (28)
This compound was prepared using the procedure described for 15d except using 13h (83 mg, 0.2 mmol) and 1H-1,2,4-triazole-3-thiol (20 mg, 0.2 mmol). The title compound 28 (40 mg, 68.0%, via 2 steps) was obtained as an off-white solid. m.p.: 154.7 °C (dec.); HPLC 95% (Rt = 14.6 min, 50% CH3CN in 0.1% TFA water 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.18 (brs, 1H), 9.77 (s, 1H), 8.59 (brs, 1H), 8.14 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 8.4 Hz, 1H), 7.48-7.39 (m, 4H), 7.24 (d, J = 8.4, 2H), 6.84 (brs, 1H), 1.76-1.67 (m, 5H), 1.35-1.30 (m, 5H), 1.25-1.20 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 153.21, 137.76, 132.17, 129.70, 127.86, 127.57, 127.48, 126.48, 126.25, 125.23, 124.13, 123.16, 44.22, 34.18, 26.83, 26.12; LC-MS (ESI+) 481.15 (M+H)+; HRMS (ESI+) m/z calculated for C24H25N4O3S2 (M+H)+ 481.1363, found 481.1371.
N-(3-(1H-1,2,4-triazol-3-ylthio)-4-hydroxynaphthalen-1-yl)-4-phenoxybenzenesulfonamide (29)
This compound was prepared using the procedure described for 15d except using 13d (78 mg, 0.2 mmol) and 1H-1,2,4-triazole-3-thiol (20 mg, 0.2 mmol). The title compound 29 (36 mg, 56.5%, via 2 steps) was obtained as an off-white solid. m.p.: 176.1 °C (dec.); HPLC 99% (Rt = 23.2 min, 40% CH3CN in 0.1% TFA water, 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.12 (brs, 1H, disappeared on D2O shake), 9.78 (s, 1H, disappeared on D2O shake), 8.53 (brs, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.49 (d, J = 8.8 Hz, 2H), 7.46-7.35 (m, 4H), 7.18-7.14 (m, 1H), 6.95 (appd, J = 7.7 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 6.82 (brs, 1H); 13C NMR (100 MHz, DMSO-d6) δ 160.92, 155.92, 152.67, 134.42, 132.11, 130.99, 130.01, 129.74, 127.56, 126.53, 126.29, 125.28, 125.13, 124.15, 123.18, 120.23, 118.60, 104.99, LC-MS (ESI+) 491.09 (M+H)+; HRMS (ESI+) m/z calculated for C24H19N4O4S2 (M+H)+ 491.0842, found 491.0847.
N-(3-(1H-1,2,4-triazol-3-ylthio)-4-hydroxynaphthalen-1-yl)-4′-(trifluoromethyl)biphenyl-4-sulfonamide (30)
This compound was prepared using the procedure described for 15d except using 13l (95 mg, 0.2 mmol) and 1H-1,2,4-triazole-3-thiol (20 mg, 0.2 mmol). The title compound 30 (31 mg, 79.2%, via 2 steps) was obtained as an off-white solid. m.p.: 193.8 °C (dec.); HPLC 95% (Rt = 13.7 min, 50% CH3CN in 0.1% TFA water, 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.18 (brs, 1H), 9.97 (s, 1H), 8.48 (brs, 1H), 8.12 (d, J = 7.9 Hz, 1H), 7.90-7.79 (m, 5H), 7.76 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8 Hz, 2H), 7.44-7.41 (m 2H), 6.86 (s, 1H);. 19F NMR (376 MHz, DMSO-d6) δ -61.44 (s); LC-MS (ESI+) 543.08 (M+H)+; HRMS (ESI+ve) m/z calculated for C25H18F3N4O3S2 (M+H)+ 543.0767, found 543.0781.
N-(3-(1H-1,2,4-triazol-3-ylthio)-4-hydroxynaphthalen-1-yl)-5-(phenylsulfonyl)thiophene-2-sulfonamide (31)
This compound was prepared using the procedure described for 15d except using N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-5-(phenylsulfonyl)thiophene-2-sulfonamide (13m) (96 mg, 0.2 mmol) and 1H-1,2,4-triazole-3-thiol (20 mg, 0.2 mmol). The title compound 31 (36 mg, 68.9%, via 2 steps) was obtained as an off-white solid. m.p.: 153.3 °C (dec.); HPLC 98% (Rt = 11.5 min, 40% CH3CN in 0.1% TFA water 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H), 10.33 (brs, 1H), 8.46 (brs, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.89 (d, J = 7.6 Hz, 2H), 7.74 (d, J = 7.6 Hz, 1H), 7.69 (d, J = 4.0 Hz, 1H), 7.64 (t, J = 7.6 Hz, 3H), 7.40 (t, J = 7.2 Hz, 1H), 7.29 (d, J = 4.4 Hz, 1H), 7.17 (t, J = 7.6 Hz, 1H), 6.99 (s, 1H), approximately 5% base line impurity was observed; 13C NMR (100 MHz, DMSO-d6) δ 153.26, 148.25, 147.60, 145.27, 140.93, 140.66, 135.23, 134.43, 133.19, 131.81, 130.76, 130.38, 127.87, 127.70, 126.71, 126.21, 123.81, 123.32, 123.24; LC-MS (ESI+) 545.00 (M+H)+; HRMS (ESI+) m/z calculated for C22H17N4O5S4 (M+H)+ 545.0076, found 545.0096.
N-(3-(1H-1,2,4-triazol-3-ylthio)-4-hydroxynaphthalen-1-yl)-1-(phenylsulfonyl)-1H-pyrrole-3-sulfonamide (32)
This compound was prepared using the procedure described for 15d except using N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)-1-(phenylsulfonyl)-1H-pyrrole-3-sulfonamide (13n) (92 mg, 0.2 mmol) and 1H-1,2,4-triazole-3-thiol (20 mg, 0.2 mmol). The title compound 32 (34 mg, 75.2%, via 2 steps) was obtained as an off-white solid. m.p.: 157.1 °C (dec.); HPLC 99.8% (Rt = 9.8 min, 50% CH3CN in 0.1% TFA water 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.19 (brs, 1H), 9.81 (s, 1H), 8.47 (brs, 1H), 8.11 (d, J = 8.3 Hz, 1H), 7.92 (d, J = 7.5 Hz, 2H), 7.81 (d, J = 8.5 Hz, 1H), 7.75 (t, J = 7.5 Hz, 1H), 7.59 (t, J = 7.9 Hz, 2H), 7.53-7.52 (m, 1H), 7.44-7.36 (m, 3H), 7.30 (t, J = 7.3 Hz, 1H), 6.79 (s, 1H), 6.36 (dd, J = 3.3, 1.6 Hz, 1H), approximately 5% base line impurity was observed.; 13C NMR (100 MHz, DMSO-d6) δ 171.01, 152.65, 137.54, 136.00, 131.87, 130.77, 129.37, 128.83, 127.84, 127.80, 127.44, 126.53, 126.21, 124.98, 123.99, 123.43, 123.21, 123.17, 112.26; LC-MS (ESI+) 528.05 (M+H)+; HRMS (ESI+) m/z calculated for C22H18N5O5S3 (M+H)+ 528.0465, found 528.0476.
N-(3-(1H-1,2,4-triazol-3-ylthio)-4-hydroxynaphthalen-1-yl)-3,4-dichlorobenzenesulfonamide(33)
This compound was prepared using the procedure described for 15d except using 13e (76 mg, 0.2 mmol) and 1H-1,2,4-triazole-3-thiol (20 mg, 0.2 mmol). The title compound 33 (71 mg, 78.9%, 2 steps) was obtained as a white solid. m.p: 183.2 °C (dec.); HPLC 99.9% (Rt = 21.9 min, 35% CH3CN in 0.1% TFA water 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.02 (brs, 1H), 8.30 (brs, 1H), 8.17 (appd, J = 1.3 Hz, 1H), 8.12 (d, J = 8.0 Hz, 1H), 8.00-7.93 (m, 5H), 7.70-7.63 (m, 2H), 7.59 (appt, J = 7.6 Hz, 1H), 7.45-7.37 (m, 2H), 6.87 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 137.61, 134.79, 132.15, 132.03, 129.85, 129.79, 129.41, 129.23, 128.45, 128.37, 128.15, 127.50, 126.55, 126.28, 125.13, 124.18, 123.12, 123.06; LC-MS (ESI+) 449.08 (M+H)+; HRMS (ESI+) m/z calculated for C22H17N4O3S2 (M+H)+ 449.0737, found 449.0746.
N-(3-(1H-1,2,4-triazol-3-ylthio)-4-hydroxynaphthalen-1-yl)naphthalene-1-sulfonamide (34)
This compound was prepared according to the procedure for 15d using N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)naphthalene-1-sulfonamide (13o) (76 mg, 0.2 mmol) and 1H-1,2,4-triazole-3-thiol (20 mg, 0.2 mmol). The title compound 34 (29 mg, 74.5%, via 2 steps) was obtained as a white solid. m.p.: 158.1 °C (dec.); HPLC 98% (Rt = 10.6 min, 40% CH3CN in 0.1% TFA water, 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.24 (s, 1H), 10.10 (brs, 1H), 8.73 (d, J = 8.4 Hz, 1H), 8.50 (brs, 1H), 8.11 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 7.6 Hz, 1H), 7.83-7.782 (m, 2H), 7.69-7.61 (m, 2H), 7.44-7.38 (m, 2H), 7.26 (t, J = 7.2 Hz, 1H), 6.74 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 152.14, 135.53, 134.70, 134.34, 131.79, 130.06, 129.64, 128.88, 128.49, 128.24, 127.46, 127.27, 126.50, 126.16, 125.33, 124.96, 123.88, 123.04; LC-MS (ESI+) 449.08 (M+H)+; HRMS (ESI+) m/z calculated for C22H17N4O3S2 (M+H)+ 449.0737, found 449.0741.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)-4-chlorobenzenesulfonamide (35)
This compound was prepared using the procedure described for 15d using 4-chloro-N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)benzenesulfonamide (13p) (37 mg, 0.1 mmol) and 1H-terazole-5-thiol ( 10 mg, 0.1 mmol). The title compound 35 (35 mg, 81.6%, via 2 steps) was obtained as an off-white solid. m.p.: 180.9 °C (dec.); HPLC 98% (Rt = 11.0 min, 40% CH3CN in 0.1% TFA water, 30 min); 1H NMR (400 MHz, DMSO-d6) δ 10.61 (brs, 1H), 10.06 (s, 1H), 8.323–8.20 (m, 1H), 7.94-7.91 (m, 1H), 7.57 (d, J = 8.8 Hz, 2H), 7.55-7.52 (m, 2H), 7.45 (d, J = 8.8 Hz, 2H), 6.83 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 154.72, 138.99, 138.27, 132.96, 130.12, 129.78, 129.44, 128.41, 126.93, 126.50, 125.23, 124.24, 123.60, 106.88; LC-MS (ESI+) 434.01 (M+H)+; HRMS (ESI+) m/z calculated for C17H13ClN5O3S2 (M+H)+ 434.0143, found 434.0129.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)-3,4-dichlorobenzenesulfonamide (36)
This compound was prepared using the procedure for 15d using 3,4-dichloro-N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)benzenesulfonamide (13q) (40 mg, 0.1 mmol) and 1H-terazole-5-thiol (10 mg, 0.1 mmol). The title compound 36 (40 mg, 84.8%, via 2 steps) was obtained as a white solid. m.p.: 174.4 °C (dec.); HPLC 97% (Rt = 10.1 min, 45% CH3CN in 0.1% TFA water, 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.64 (brs, 1H), 10.18 (s, 1H), 8.24-8.22 (m, 1H), 7.94-7.92 (m, 1H), 7.75 (appd, J = 2.0 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.56-7.54 (m, 2H), 7.51 (dd, J = 8.4, 1.6 Hz, 1H), 6.85 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 154.80, 140.35, 136.54, 132.95, 132.65, 132.07, 130.17, 129.21, 128.46, 127.63, 127.00, 126.55, 124.86, 124.15, 123.63, 107.07; LC-MS (ESI+) 467.98 (M+H)+; HRMS (ESI+) m/z calculated for C17H12Cl2N5O3S2 (M+H)+ 467.9753, found 467.9752.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)benzenesulfonamide (37)
This compound was prepared using the procedure described for 15d using N-(3-chloro-4-oxonaphthalen-1(4H)-ylidene)benzenesulfonamide (13r) (32 mg, 0.1 mmol) and 1H-terazole-5-thiol ( 10 mg, 0.1 mmol). The title compound 37 (32 mg, 78.9%, via 2 steps) was obtained as a white solid. m.p.: 154.8 °C (dec.); HPLC 92% (Rt = 6.5 min, 40% CH3CN in 0.1% TFA water 30 min); 1H NMR (400 MHz, DMSO-d6) δ 10.57 (brs, 1H), 9.94 (s, 1H), 8.21-8.19 (m, 1H), 7.95-7.92 (m, 1H), 7.59-7.56 (m, 2H), 7.54-7.48 (m, 3H), 7.39 (t, J = 8.0 Hz, 2H), 6.79 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 154.57, 140.16, 133.33, 133.03, 129.97, 129.60, 128.31, 127.48, 126.83, 126.47, 125.53, 124.32, 123.52, 106.87; LC-MS (ESI+) 400.06 (M+H)+; HRMS (ESI+) m/z calculated for C17H14N5O3S2 (M+H)+ 400.0526, found 400.0529.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)-5-chlorothiophene-2-sulfonamide (38)
This compound was prepared using the procedure described for 15d using 13k (37 mg, 0.1 mmol) and 1H-terazole-5-thiol ( 10 mg, 0.1 mmol). The title compound 38 (36 mg, 81.6%, via 2 steps) was obtained as a gray solid. m.p.: 159.4 °C (dec.); HPLC 96% (Rt = 6.9 min, 45% CH3CN in 0.1% TFA water, 40 min); 1H NMR (400 MHz, DMSO-d6) δ 10.68 (brs, 1H), 10.32 (s, 1H), 8.25-8.23 (m, 1H), 7.95-7.93 (m, 1H), 7.57-7.55 (m, 2H), 7.20 (d, J = 4.0 Hz, 1H), 7.04 (d, J = 4.0 Hz, 1H), 6.98 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 154.92, 138.97, 135.76, 133.03, 133.00, 130.29, 128.54, 128.45, 126.97, 126.50, 124.88, 124.02, 123.65, 107.13; LC-MS (ESI+) 439.96 (M+H)+; HRMS (ESI+) m/z calculated for C15H11ClN5O3S3 (M+H)+ 439.9707, found 439.9707.
N-(3-(1H-tetrazol-5-ylthio)-4-hydroxynaphthalen-1-yl)-4′-(trifluoromethyl)biphenyl-4-sulfonamide (39)
This compound was prepared using the procedure described for 15d using 13l (48 mg, 0.1 mmol) and 1H-terazole-5-thiol ( 10 mg, 0.1 mmol). The title compound 39 (41 mg, 76.1%, via 2 steps) was obtained as a white solid. m.p.: 168 °C (dec.); HPLC 99% (Rt = 14.6 min, 50% CH3CN in 0.1% TFA water 30 min); 1H NMR (400 MHz, DMSO-d6) δ 10.59 (brs, 1H), 10.04 (s, 1H), 8.23-8.20 (m, 1H), 8.00-7.97 (m, 1H), 7.88-7.82 (m, 4H), 7.77 (d, J = 8.6 Hz, 2H), 7.69 (d, J = 8.6 Hz, 2H), 7.54-7.51 (m, 2H), 6.83 (s, 1H); LC-MS (ESI+) 544.08 (M+H)+; HRMS (ESI+) m/z calculated for C24H17F3N5O3S2 (M+H)+ 544.0719, found 544.0704.
Supplementary Material
Acknowledgments
We acknowledge the NIH for funding (1PO1 CA118210-03), PI- Dr. Saïd M. Sebti. We also acknowledge Experimental Lab, Chemical Biology Core for high throughput screening.
ABBREVIATIONS USED
- CT-L
chymotrypsin like
- T-L
trypsin like
- SAR
structure activity relationship
- PGPH
postglutamylpeptidase hydrolysis
- DCM
dichloromethane
- THF
terahydrofuran
- DMF
dimethylformamide
- DBU
diazabicycloundecene
- DMSO
dimethylsulfoxide
- TFA
trifluoroacetic acid
- DMEM
Dulbecco’s Modified Eagle Medium
- RPMI1640
Roswell Park Memorial Institute-1640
Footnotes
Supporting Information Available: Supporting analytical data; 1H NMR, 13C NMR, LCMS and HPLC for key compounds. This information is availablefree of charge via the internet at http://pubs.acs.org
References
- 1.Pickart CM. Back to the future with ubiquitin. Cell (Cambridge, MA, U S) 2004;116:181–190. doi: 10.1016/s0092-8674(03)01074-2. [DOI] [PubMed] [Google Scholar]
- 2.Ciechanover A. Intracellular protein degradation: From a vague idea, through the lysosome and the ubiquitin proteasome system, and onto human diseases and drug targeting (Nobel lecture) Angew Chem Int Ed. 2005;44:5944–5967. doi: 10.1002/anie.200501428. [DOI] [PubMed] [Google Scholar]
- 3.Hershko A. The ubiquitin system for protein degradation and some of its roles in the control of the cell-division cycle (Nobel lecture) Angew Chem Int Ed Eng. 2005;44:5932–5943. doi: 10.1002/anie.200501724. [DOI] [PubMed] [Google Scholar]
- 4.Rose I. Ubiquitin at fox chase. Prix Nobel. 2005:218–225. [Google Scholar]
- 5.Verdoes M, Florea BI, van der Marel GA, Overkleeft HS. Chemical Tools To Study the Proteasome. Eur J Org Chem. 2009:3301–3313. [Google Scholar]
- 6.Ciechanover A, Schwartz AL. The ubiquitin-proteasome pathway: the complexity and myriad functions of proteins death. Proc Natl Acad SciUSA. 1998;95:2727–2730. doi: 10.1073/pnas.95.6.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ciechanover A, Schwartz Alan L. Ubiquitin-mediated degradation of cellular proteins in health and disease. Hepatology. 2002;35:3–6. doi: 10.1053/jhep.2002.30316. [DOI] [PubMed] [Google Scholar]
- 7.King RW, Deshaies RJ, Peters JM, Kirschner MW. How proteolysis drives the cell cycle. Science (Washington, D C) 1996;274:1652–1658. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]; Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–421. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 8.Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol. 2005;23:630–639. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 9.Chauhan D, Hideshima T, Anderson KC. Proteasome inhibition in multiple myeloma: therapeutic implication. Annu Rev Pharmacol Toxicol. 2005;45:465–476. doi: 10.1146/annurev.pharmtox.45.120403.100037. 2 plates. [DOI] [PubMed] [Google Scholar]
- 10.Papandreou CN, Daliani D, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, Kim J, Adams J, Elliott P, Esseltine D, Petrusich A, Dieringer P, Perez C, Logothetis CJ. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol. 2004;22:2108–2121. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 11.Palombella VJ, Conner EM, Fuseler JW, Destree A, Davis JM, Laroux FS, Wolf RE, Huang J, Brand S, Elliott PJ, Lazarus D, McCormack T, Parent L, Stein R, Adams J, Grisham MB. Role of the proteasome and NF-kB in streptococcal cell wall-induced polyarthritis. Proc Natl Acad SciUS A. 1998;95:15671–15676. doi: 10.1073/pnas.95.26.15671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Maldonado MA. The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol Immunol. 2006;3:255–261. [PubMed] [Google Scholar]; Meng L, Kwok BH, Sin N, Crews CM. Eponemycin exerts its antitumor effect through the inhibition of proteasome function. Cancer Res. 1999;59:2798–2801. [PubMed] [Google Scholar]
- 13.Groll M, Kim KB, Kairies N, Huber R, Crews CM. Crystal structure of epoxomicin:20S proteasome reveals a molecular basis for selectivity of a′,b′-epoxyketone proteasome inhibitors. J Am Chem Soc. 2000;122:1237–1238. [Google Scholar]
- 14.Groll M, McArthur Katherine A, Macherla Venkat R, Manam Rama R, Potts Barbara C. Snapshots of the fluorosalinosporamide/20S complex offer mechanistic insights for fine tuning proteasome inhibition. J Med Chem. 2009;52:5420–5428. doi: 10.1021/jm900559x. [DOI] [PubMed] [Google Scholar]
- 15.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 16.Meiners S, Ludwig A, Stangl V, Stangl K. Proteasome inhibitors: Poisons and remedies. Med Res Rev. 2008;28:309–327. doi: 10.1002/med.20111. [DOI] [PubMed] [Google Scholar]
- 17.Fuchs O. Proteasome inhibition as a therapeutic strategy in patients with multiple myeloma. Mult Myeloma. 2009:101–125. [Google Scholar]
- 18.Lam KS, Lloyd GK, Neuteboom STC, Palladino MA, Sethna KM, Spear MA, Potts BC. From natural products to clinical trials: NPI-0052 (salinosporamide A), a marine actinomycete-derived anticancer agent. Nat Prod Chem Drug Discovery. 2010:355–373. [Google Scholar]
- 19.Zhou HJ, Aujay Monette A, Bennett Mark K, Dajee M, Demo Susan D, Fang Y, Ho Mark N, Jiang J, Kirk Christopher J, Laidig Guy J, Lewis Evan R, Lu Y, Muchamuel T, Parlati F, Ring E, Shenk Kevin D, Shields J, Shwonek Peter J, Stanton T, Sun Congcong M, Sylvain C, Woo Tina M, Yang J. Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047) J Med Chem. 2009;52:3028–3038. doi: 10.1021/jm801329v. [DOI] [PubMed] [Google Scholar]
- 20.Sterz J, von Metzler I, Hahne JC, Lamottke B, Rademacher J, Heider U, Terpos E, Sezer O. The potential of proteasome inhibitors in cancer therapy. Expert Opin Inv Drug. 2008;17:879–895. doi: 10.1517/13543784.17.6.879. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence HR, Kazi A, Luo Y, Kendig R, Ge Y, Jain S, Daniel K, Santiago D, Guida WC, Sebti SM. Synthesis and biological evaluation of naphthoquinone analogs as a novel class of proteasome inhibitors. Bioorg Med Chem. 2010;18:5576–5592. doi: 10.1016/j.bmc.2010.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kazi A, Lawrence H, Guida Wayne C, McLaughlin Mark L, Springett Gregory M, Berndt N, Yip Richard ML, Sebti Said M. Discovery of a novel proteasome inhibitor selective for cancer cells over non-transformed cells. Cell Cycle. 2009;8:1940–1951. doi: 10.4161/cc.8.12.8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avdeenko AP, Velichko NV, Romanenko EA, Pirozhenko VV. Chlorination of N-arylsulfonyl-1,4-aminonaphthols and N-arylsulfonyl-1,4-naphthoquinone-1,4-imines. Zh Org Khim. 1991;27:1747–1757. [Google Scholar]
- 24.Altland HW, Briffa BF., Jr 1, 4-Addition of triazolium thiolates to quinones. J Org Chem. 1985;50:433–437. [Google Scholar]
- 25.Avdeenko AP, Evgrafova NI, Ryazantsev VP, Luk’yanenko LV, Titov EV, Velichko NV. U.S.S. R patent 1558901. Preparation of N-arylsulfonyl-2-chloro-1,4-naphthoquinone imines 86-4156772, 1558901, 19861204, 1990. 1990
- 26.Avdeenko AP, Evgrafova NI. Reaction of N-(arylsulfonyl)-p-naphthoquinonimines with acylhydrazines. Zh Org Khim. 1987;23:1060–1063. [Google Scholar]
- 27.Thiols. Coj E, Afanas’eva GB, Chupakhin ON, Sidorov EO. Reactions of benzo analogs of N-phenyl-1,4-benzoquinone monoimine with thiols. Zh Org Khim. 1989;25:2409–2416. [Google Scholar]
- 28.Fujita S, Sano K. Reduction of quinones and quinonemonosulfonimides with N,N-diethylhydroxylamine. J Org Chem. 1979;44:2647–2651. [Google Scholar]
- 29.Lien JC, Huang LJ, Teng CM, Wang JP, Kuo SC. Synthesis of 2-alkoxy 1,4-naphthoquinone derivatives as antiplatelet, antiinflammatory, and antiallergic agents. Chem Pharm Bull. 2002;50:672–674. doi: 10.1248/cpb.50.672. [DOI] [PubMed] [Google Scholar]
- 30.Obafemi CA. Studies in the heterocyclic compounds. II. The mass spectra of some thiophene-sulfonyl derivatives. Phosphorus Sulfur. 1980;8:201–204. [Google Scholar]
- 31.Matsubara R, Doko T, Uetake R, Kobayashi S. Enesulfonamides as nucleophiles in catalytic asymmetric reactions. Angew Chem, Int Ed. 2007;46:3047–3050. doi: 10.1002/anie.200605054. [DOI] [PubMed] [Google Scholar]
- 32.Adams R, Whitaker L. Quinone imides. XXXIX. Adducts of quinone monoimides and conversion of active methylene adducts to benzofurans. J Am Chem Soc. 1956;78:658–663. [Google Scholar]
- 33.Travis BR, Ciaramitaro BP, Borhan B. Preparation of purified KHSO5.H2O and nBu4NHSO5 from Oxone by simple and efficient methods. Eur J Org Chem. 2002:3429–3434. [Google Scholar]
- 34.Altland HW, Briffa BF., Jr 1,4-Addition of triazolium thiolates to quinones. J Org Chem. 1985;50:433–437. [Google Scholar]
- 35.Johnston CI. Angiotensin receptor antagonists: Focus on losartan. Lancet. 1995;346:1403–1407. doi: 10.1016/s0140-6736(95)92411-6. [DOI] [PubMed] [Google Scholar]
- 36.Patani GA, LaVoie EJ. Bioisosterism: A rational approach in drug design. Chem Rev (Washington, D C) 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.