

Abstract
Potent ROCK inhibitors of a new class of 1-benzyl-3-(4-pyridylthiazol-2-yl)ureas have been identified. Remarkable differences in activity were observed for ureas bearing a benzylic stereogenic center. Derivatives with hydroxy, methoxy and amino groups at the meta position of the phenyl ring give rise to the most potent inhibitors (low nM). Substitutions at the para position result in substantial loss of potency. Changes at the benzylic position are tolerated resulting in significant potency in the case of methyl and methylenehydroxy groups. X-Ray crystallography was used to establish the binding mode of this class of inhibitors and provides an explanation for the observed differences of the enantiomer series. Potent inhibition of ROCK in human lung cancer cells was shown by suppression of the levels of phosphorylation of the ROCK substrate MYPT-1.
Introduction
Rho associated protein kinases (ROCKs) are Ser/Thr protein kinases, activated by small GTPases of the Rho family that act as molecular switches to mediate cell signaling. The Rho/ROCK signaling pathway is known to participate in the regulation of numerous cellular functions such as actin cytoskeleton organization, contraction, cell adhesion, motility, morphology, proliferation, cytokinesis, gene expression, and angiogenesis. Two isoforms, ROCK1 and ROCK2, have been identified sharing 64% and 79% overall sequence identity and similarity respectively and 92% identity and 97% similarity in their kinase domains. The two isoforms have been found to possess differential tissue distribution. ROCK1 is expressed in lung, liver, stomach, spleen, kidney and testis, whereas ROCK2 is highly expressed in brain, heart and muscle tissues.1 Despite the differential tissue distribution, little is known about the functional differences between the two ROCK isoforms.2–10
ROCKs have been subjected to growing attention, having been implicated in a range of therapeutic areas including cardiovascular diseases,11–15 CNS disorders,16,17 inflammation,18 and cancer.19–37 Co-overexpression of Rho and ROCK proteins in cancer cells has been reported in ovarian cancer, pancreatic, testicular, and bladder cancer.19,20 Metastasis requires changes in the migratory, invasive and adhesive properties of tumor cells. These processes which depend on the proper assembly/disassembly of actin-cytoskeleton are regulated by Rho/ROCK pathway and play an important role in the development and progression of cancer.21 The implication of Rho/ROCK signalling pathway in invasion,22,23 angiogenesis,24 and metastasis25 has been amply documented. In light of these findings, the pharmacological inhibition of ROCKs has been suggested as a promising strategy in the prevention of cell invasion, a central event in the process of metastasis.25–28
The potential of ROCK inhibitors as anticancer agents was demonstrated by the identification of ATP competitive inhibitors, Y27632 (1), and Wf536 (2) (Fig. 1).25,29,30,32 Specifically, 1 was reported to reduce metastasis in animal model systems,25 and 2 has shown efficacy in preventing tumor metastasis in vivo models by inhibiting tumor-induced angiogenesis as well as tumor motility.29,30,32 Han and coworkers have also investigated the ability of Fasudil (3) (the only ROCK inhibitor clinically approved in Japan for the treatment of cerebral vasospasm) to inhibit progression of human and rat tumors in animal models.31
Fig. 1.

Representative known Rho kinase inhibitors with anticancer properties.
Due to potential therapeutic applications, significant research efforts have been directed towards the identification of more potent and more selective ROCK inhibitors,38–43 including isoquinolinamines44,45 triazines,46 isoquinolinones,47,48 quinazolinones,49 benzothiazoles50 and diaminopyrazines51 and their use for the treatment of cardiovascular diseases and CNS disorders. To the best of our knowledge the antitumor and antimetastatic properties of these inhibitors has yet to be shown or published. Our group has been actively engaged in the design and synthesis of ROCK1 inhibitors to provide powerful in vitro and in vivo tools to probe the pharmacological inhibition of ROCK1, to further establish its function in cancer and metastasis and further validate it as an anticancer target.
The aminothiazole derivative CID5056270 (4) (Fig. 2) has been reported42 to potently inhibit ROCK2 enzymatic activity with an IC50 values <3 nM [Molecular Libraries Screening Centers Network (MLSCN),52–54 assay ID 644]. It displayed high potency in our in-house (FRET)-based Z′-Lyte biological assay55,56 (ROCK2 IC50 0.56 nM) and also inhibited ROCK1 with an IC50 of 13 nM (Fig. 2). In view of its potency against both ROCK isoforms and selectivity over Aurora-A (IC50 > 100 µM) (Fig. 2), 4 was chosen as a starting point for the design of a focused library of pyridylaminothiazole-based ROCK inhibitors. As part of our inhibitor design we were intrigued as to whether a benzylurea group would act as a surrogate of the benzyloxyacetamide chain embedded within 4, whilst retaining the pyridylthiazole ‘head’ group. Ureas have previously been incorporated into ROCK inhibitors,57,58 but not ones with the 4-pyridylthiazole hinge-binding group. The aminothiazole 5a was docked to the catalytic domain of ROCK1 (using GLIDE, and ROCK1 structure from pdb 2ETR; see Molecular Modeling Section)59 (Fig. 3) to determine a possible binding mode and to study the structural features responsible for binding of 5a to ROCK1. The model suggests that the pyridine ring binds to the hinge region via a hydrogen bond between the pyridyl nitrogen atom and the backbone amide NH of Met156 in a similar mode to the isoquinoline ring of Fasudil evident in its co-crystal complex with ROCK1.59 In the model the carbonyl oxygen of 5a forms a hydrogen bond with the side chain NH of Lys-105 and the terminal phenyl ring of the benzylurea occupies a deep hydrophobic cleft under the P-Loop (Fig. 3).
Fig. 2.

Hit generation.
Fig. 3.
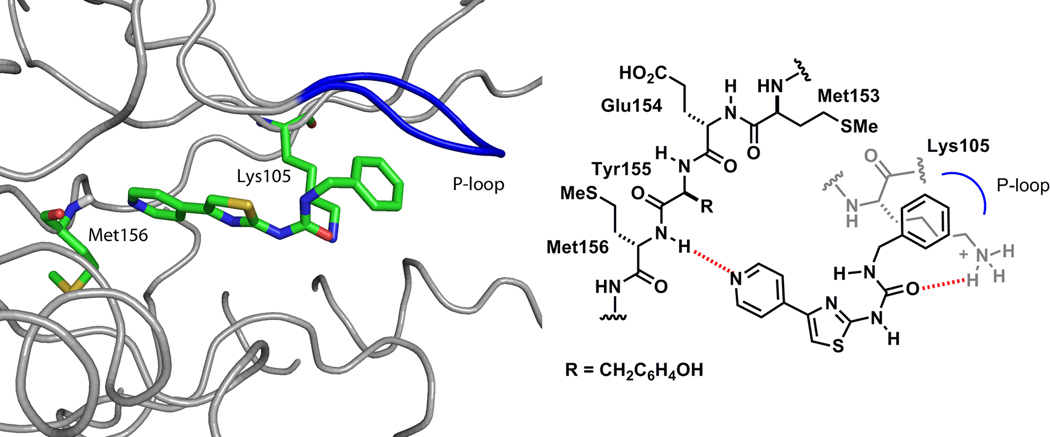
Structure of 5a docked in the catalytic domain of the ROCK1 model (green).
The pyridylthiazole urea 5a (Fig. 2) inhibited ROCK1 (IC50 170 nM) and provided a new starting point for optimization. We focused our attention on exploring, in turn, the SAR around the phenyl ring, branching and substitution at the benzylic position, urea linkage of 5a, while retaining the hinge binding 4-(4-pyridinyl)-2-thiazolyl] group (Fig. 4).
Fig. 4.

SAR around the scaffold of 5.
Chemistry
We initially prepared the new hit 5a and additional analogs 5c, 5q, 5r, 5s, 5u and 5u, via microwave heating of aminothiazoles 7 or with isocyanates 9 following the synthetic route shown in Scheme 1. Microwave-assisted condensation of the commercially available 4-(bromoacetyl)pyridine (6) with thiourea and N-methylthiourea afforded the desired aminothiazoles 7 and 8, respectively. The aminothiazoles 7 and 8 were then reacted with commercially available isocyanates 9 to afford urea library 5 in poor to moderate yields. The initial SAR revealed the potential of the new urea analogs as a promising class of ROCK1 inhibitors. We therefore sought to develop an alternative synthetic route to improve the yields. Additionally, a significant drawback of the initial synthetic route was the lack of inexpensive commercially available isocyanates, which would limit the SAR around the benzyl group of compound 5a. The carbamate 10, prepared from the aminothiazole 7 and phenyl chloroformate provided a key intermediate and offered an alternative way to introduce significant chemical and structural diversity at the benzyl terminus of 5a via coupling with inexpensive and readily available benzylamines, amino acids, anilines, and aliphatic amines (Scheme 2). This proved to be successful, allowing us to expand the library 5 under much milder conditions. As shown in Scheme 2, libraries 5 and 14 could be easily prepared by heating the reaction mixture in a sealed tube (Scheme 2, conditions d, i). By means of a heating block parallel station the generation of the library was performed in a combinatorial fashion. Moreover, microwave heating provided an efficient and convenient alternative to conventional heating for the synthesis of the desired library 5 (Scheme 2, conditions e–f). As a general note, longer reaction times or higher temperatures were required in the cases of less reactive amines. Diisopropylethylamine (DIPEA) or triethylamine were employed when the amine-containing reagents were available as the corresponding HCl salts (Scheme 2). To validate the new synthetic protocol, the original hit 5a and 5q were synthesized using the new synthetic route (Scheme 2). The different batches displayed comparable analytical data and comparable potency in the ROCK1 (FRET)-based Z′-Lyte kinase assay. Two compounds 12 and 13 were prepared to assess the effect of linker chain length. Finally, the corresponding mesylate and HCl salts of selected library members (5b, 5d and 5g) were also synthesized (see supplementary information for further details).
Scheme 1.

First approach to the synthesis of ureas 5.
Scheme 2.

Second route to urea libraries 5 and 14.
The structures of all the final compounds were confirmed by analysis of NMR and mass spectroscopic data. In addition, HPLC methods (typically two methods) were used to determine the purity (generally >96%) of the compounds. Additionally, HPLC methods were developed for the chiral compounds to determine the enantiomeric purity (generally >95%). All compounds were screened against ROCK1 and ROCK2. IC50 values were systematically determined only for compounds that inhibited ROCK1 activity by at least 40% at a compound concentration of 50 µM. The results are summarized in Tables 1, 2, 3, and 4 and discussed below.
Table 1.
Effect of the substitution of the aromatic ring Aa
 | ||||
|---|---|---|---|---|
| Compound | Method and Yield (%) | R2 | IC50 ± SD (µM) ROCK1 | IC50 ± SD (µM) ROCK2 |
| 5a | b, 35%; h, 69% | 0.17 ± 0.01 (6) | 0.05 ± 0 (6) | |
| 5b | e, 63% | 0.027 ± 0.005 (8) | 0.011 ± 0.002 (6) | |
| 5c | b, 31% | 1.40 ± 0.11 (6) | 0.47 ± 0.03 (6) | |
| 5d | j, 74% | ROCK1% inhibition @ 50 µM: 9.1 ± 1.6 | ND | |
| 5e | e, 35% | 0.05 ± 0.009 (12) | 0.02 ± 0.008 (12) | |
| 5f | e, 82% | 37.8 ± 7.8 (6) | 31.1 ± 9.1 (6) | |
| 5g | g, 59% | 0.008 ± 0.001 (10) | 0.006 ± 0.001 (9) | |
| 5h | e, 77% | 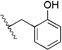 |
0.62 ± 0.019 (6) | 0.28 ± 0.05 (6) |
| 5i | e, 74% | 0.38 ± 0.046 (6) | 0.14 ± 0.029 (6) | |
| 5j | i, 90% | 1.2 ± 0.4 (3) | 0.18 ± 0.04 (3) | |
| 5k | e, 44% | 0.56 ± 0.10 (3) | 0.10 ± 0.03 (3) | |
| 5l | e, 22% | 1.3 ± 0.2 (3) | 0.13 ± 0.02 (3) | |
| 5m | e, 50% | 0.50 ± 0.05 (6) | 0.24 ± 0.03 (6) | |
| 5n | e, 87% | 2.1 ± 0.6 (3) | 0.29 ± 0.17 (3) | |
| 5o | e, 80% | 3.36 ± 1.00 (3) | 0.68 ± 0.43 (3) | |
| 5p | e, 84% | 13.0 ± 1.4 (3) | 3.44 ± 0.87 (3) | |
| 5q | b, 5%; e, 73% | 0.10 ± 0.02 (6) | 0.05 ± 0.01 (6) | |
| 5r | b, 64% | 0.40 ± 0.03 (6) | 0.12 ± 0.01 (6) | |
| 5s | b, 16% | 0.57 ± 0.05 (6) | 0.19 ± 0.00 (6) | |
| 5t | e, 54% | 0.14 ± 0.02 (6) | 0.06 ± 0.01 (6) | |
| 5u | b, 12% | 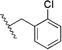 |
0.34 ± 0.02 (6) | 0.11 ± 0.01 (6) |
| 5v | e, 86% | 2.4 ± 0.5 (6) | 1.1 ± 0.2 (6) | |
| 5w | e, 59% | 0.46 ± 0.08 (6) | 0.12 ± 0.03 (6) | |
| 5x | e, 48% | 0.15 ± 0.03 (6) | 0.12 ± 0.02 (6) | |
| 5y | e, 77% | 5.2 ± 0.5 (6) | 2.0 ± 0.15 (6) | |
Key: number of repeats shown in parentheses.
Table 2.
Effect of the substitution and branching at the benzylic positiona
 |
 |
||||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | Method, yield | IC50 ± SD (µM) | Method, yield | IC50 ± SD (µM) | ||
| Me | H | (R)-14a | e, 67% | ROCK1 0.043 ± 0.007, (6) | (S)-14a | e, 55% | ROCK1 2.9 ± 0.4, (6) |
| ROCK2 0.012 ± 0.002, (6) | ROCK2 1.9 ± 0.5, (6) | ||||||
| CH2OH | H | (S)-14b | e, 55% | ROCK1 0.03 ± 0.018, (16) | (R)-14b | e, 39% | ROCK1 5.2 ± 0.6, (12) |
| ROCK2 0.01 ± 0.01, (15) | ROCK2 2.5 ± 1, (12) | ||||||
| Eta | H | (R)-14c | e, 37% | ROCK1 0.09 ± 0.012, (12) | (S)-14c | e, 65% | ROCK1 52.2 ± 3.6, (6) |
| ROCK2 0.03 ± 0.01, (12) | ROCK2 22.0 ± 7.8, (6) | ||||||
| CONH2 | H | (S)-14d | f, 30% | ROCK1 0.08 ± 0.01, (12) | (R)-14d | j, 55% | ROCK1 3.7 ± 0.5, (6) |
| ROCK2 0.042 ± 0.016, (12) | ROCK2 2.1 ± 0.36, (6) | ||||||
| CH2OMe | H | (S)-14e | e, 70% | ROCK1 0.07 ± 0.005, (12) | (R)-14e | e, 59% | ROCK1 29.5 ± 3.3, (6) |
| ROCK2 0.03 ± 0.009, (12) | ROCK2 12.0 ± 1.2, (6) | ||||||
| Me | OMe | (R)-14f | e, 50% | ROCK1 0.030 ± 0.022, (15) | (S)-14f | e, 54% | ROCK1 18.7 ± 2.1, (9) |
| ROCK2 0.009 ± 0.0075, (15) | ROCK2 9.3 ± 1.0, (9) | ||||||
| Meb | F | (R)-14g | e, 83% | ROCK1 0.12 ± 0.03, (6) | (S)-14g | e, 78% | ROCK1 30.0 ± 10.0, (4) |
| ROCK2 0.042 ± 0.016, (6) | ROCK2 6.3 ± 0.3, (2) | ||||||
| Me | Cl | (R)-14h | e, 65% | ROCK1 0.64 ± 0.25, (6) | (S)-14h | e, 40% | ROCK1 >50 |
| ROCK2 0.078 ± 0.014, (6) | ROCK2 ND | ||||||
Key: number of repeats shown in parentheses;
(±)-14b, ROCK1 IC50 0.06 ± 0.02 µM (n = 12) and ROCK2 IC50 0.03 ± 0.02 µM (prepared by e, 27%);
(±)-14g, ROCK1 IC50 0.43 ± 0.13 µM (n = 6) and ROCK2 ND (prepared by method e, 83%);
ND = not determined.
Table 3.
Effect of the N-substitutiona
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R | R1 | R2 | Method, yield | IC50 ± SD (µM) ROCK1 | IC50 ± SD (µM) ROCK2 |
| 5z | H | Me | e, 63% | 0.11 ± 0.003, (6) | 0.07 ± 0.015 (6) | |
| 5aa | Me | H | b, 53% | 0.03 ± 0, (6) | 0.007 ± 0.0002, (6) | |
| 5ab | H | Me | d, 68% | 0.04 ± 0.01, (12) | 0.015 ± 0.004, (12) | |
| (R)-14i | H | Me | 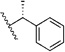 |
d, 75% | 0.12 ± 0.04, (12) | 0.047 ± 0.018, (12) |
| (S)-14i | H | Me | 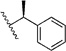 |
d, 58% | 6.4 ± 1.7, (12) | 2.7 ± 0.9, (12) |
Key: n = number of repeats.
Table 4.
Focused kinase profiling of 5g-Mes
| Kinase | % Enzyme activity (relative to DMSO controls) at 1 µMa |
IC50 (nM) Staurosporine |
|---|---|---|
| ACK1 | 96.9 | 14.27 |
| AKT1 | 44.0 | 2.16 |
| Aurora A | 61.4 | <1.0 |
| CK1a1 | 98.1 | 4698.00 |
| DMPK | 98.9 | 34.06 |
| GSK3b | 91.1 | 7.77 |
| IKKe/IKBKE | 108.0 | <1.0 |
| JAK2 | 105.5 | <1.0 |
| LIMK1 | 99.3 | 1.74 |
| MLCK/MYLK | 94.5 | 15.63 |
| MRCKa/CDC42BPA | 49.6 | 2.87 |
| p70S6K/RPS6KB1 | 38.1 | <1.0 |
| PAK1 | 100.0 | <1.0 |
| PKA | 15.5 | <1.0 |
| PKN1/PRK1 | 19.5 | <1.0 |
| ROCK1 | 3.8 | <1.0 |
| ROCK2 | 2.0 | <1.0 |
| RSK1 | 64.3 | <1.0 |
| SGK1 | 79.3 | 5.00 |
| TBK1 | 100.8 | <1.0 |
Average of two values.
Results and discussion
The initial hit 5a displayed good activity against ROCK1 (IC50 170 nM) as measured in the Z-lyte assay.55,56 The aniline-derived analog 12 was poorly active (10% inhibition at 50 µM). The phenethyl urea 13 (IC50 480 nM) was less active than 5a, we therefore centered our synthetic efforts on studying and systematically evaluating the effect of the substitution on the phenyl ring A (Fig. 4) of the ureas of type 5. The SAR around the phenyl ring A (Table 1), revealed that a hydroxyl at the meta position as in 5g (IC50 8 nM) enhanced by 21 fold the potency relative to the parent compound 5a (IC50 170 nM). Furthermore, the position of the hydroxyl group is critical as evident from the 77- and 47-fold loss of potency of derivatives 5h (IC50 620 nM) and 5i (IC50 380 nM) with the hydroxyl group in the ortho and para positions, respectively, relative to the meta-hydroxy containing 5g (IC50 8 nM). Table 1 also shows that, in general, derivatives with substitutions at the meta position are much more potent than those with substitutions in the para and to a lesser extent in the ortho position. For example, derivatives with methoxy, methyl, chloro or fluoro at the meta position are between 1850 fold and 2 fold more potent than their corresponding para and ortho derivatives (Table 1). Furthermore, the nature of the substituent at the meta position was also important with the hydroxyl derivative (5g) being the most potent followed by methoxy (5b), amino (5e), fluoro (5q) chloro (5t) and methyl (5w) derivatives. However, larger alkoxy groups such as ethoxy (5n), propoxy (5o), and iso-propoxy (5p) and ethoxymethoxy (5m) were less tolerated, resulting in progressively less activity with increased size and lipohilicity of the alkyl group. These results generally indicate that only polar groups adding a limited steric hindrance at the meta position do not lead to a significant loss of potency compared to the parent compound 5g. The binding affinity of 5b could derive from the ability of the methoxy group to act as a hydrogen bond acceptor, the greater potency of 5g might be due to both the H-bond donor and acceptor properties of phenol. The pair of meta and para methoxy isomers 5b (IC50 27 nM) and 5d (less than 10% inhibition at 50 µM) exquisitely highlights the intolerance of groups at the para position.
Next, we investigated the bioisosteric replacement of the 3-hydroxy group with the aim of retaining the high binding affinity and improving the pharmacological properties and metabolic stability of 5g and further probing the importance of the OH group. First, the OH → NH2 (5e) (IC50 50 nM) replacement resulted in a 6-fold decrease in potency compared to 5g (IC50 8 nM). The indole analog 5f displayed a near 5000-fold decrease in inhibitory activity. Next we selected groups at the meta position that have both hydrogen bond donor and acceptor properties. Examples were limited to urea 5j (IC50 1.2 µM), sulfonamide 5k (IC50 0.56 µM), and acetamide 5l (IC50 1.3 µM) however all three analogs are less active than 5g.
Di-substitution of the phenyl ring A (e.g. di-methoxy, di-chloro, di-fluoro) or its replacement with several heterocycles (e.g furan, pyridyl) resulted in a dramatic loss of potency (data not shown). In these examples, no significant improvement was observed for any of the substituents placed at the ortho and para positions. It was only when changes were made at the meta position that we observed improved activity.
We next focused on the effect of branching and substitution of the benzylic position of 5a (Table 2). A set of α-substituted benzylureas 14 was prepared and tested. Within this series, we observed a dramatic difference in ROCK potency of enantiomers when a stereogenic center was introduced at the benzylic position. The (R)-α-methylbenzylurea (R)-14a was found to be 4-fold more potent than the parent compound 5a, and 67-fold more potent than its enantiomer (S)-14a. The (R)-α-ethylbenzylurea (R)-14c was 2-fold more active than 5a and again much more active (579-fold) than its enantiomer (S)-14c. Further, the (S)-α-methylhydroxy analog (S)-14b showed an IC50 of 30 nM for ROCK1 inhibition. The S-enantiomer (S)-14b is clearly a more potent inhibitor than its R-enantiomer (R)-14b (IC50 = 5.2 µM). The IC50 of the racemate (±)-14b (IC50 = 60 nM) also indicates that the inhibitory activity results from the S-enantiomer alone. The methoxymethyl substituted (S)-14e (IC50 70 nM) was also more potent than 5a, but less active than (S)-14b. This result suggests that the hydroxy group is serving as hydrogen bonding donor. The urea (S)-14d (IC50 80 nM) possessing a CONH2 group as an alternative H-bond donor was less active than the methylhydroxy analog (S)-14b but nevertheless more potent than 5a.
For the three analogs 14f, 14g and 14h bearing an α-methyl group and a substituent on the 3-position of the phenyl ring did not result in improvement in their activity compared to their unsubstituted analogs. Within this series, the same sense of enantiomeric selectivity is preserved with chiral selectivity ranging between 630- and 270-fold. The activity of compound (R)-14f (ROCK1 IC50 30 nM) remains impressive.
The importance of the urea NH functionalities was addressed, limiting the examples to N-methylation (Table 3). Replacement of the urea hydrogen atoms was generally tolerated as shown by analogs 5z (IC50 110 nM) and 5ab (IC50 40 nM) that bear an N-2 methyl group and have similar activities to their NH counterparts 5a (IC50 170 nM) and 5b (IC50 27 nM), respectively. Urea 5aa bearing an N-1 methyl group showed similar activity to its isomer 5ab and parent 5a. The chiral ureas with an N-2 methyl group displayed the same difference in activities of the enantiomers. The (R)-enantiomer (R)-14i (IC50 120 nM) was less active than its NH parent (R)-12a (IC50 43 nM). Overall, methylation in this small series results in relatively small differences in activity.
Finally, to impart improved water solubility to 5g, 5b and 5d, the corresponding HCl and mesylate salt were prepared. As shown in Table SI2, all the salts possess in vitro similar potency to the corresponding free base (see supplementary information). Interestingly, the mesylate salt of 5d showed an increased activity when compared to the corresponding free base and HCl salt that probably can be attributed to the increase water solubility in the aqueous buffer used in the assay.
The most potent compound 5g-Mes was subjected to limited profiling using the Reaction Biology33P kinase HotspotSM kinase profiling service (Table 4). The IC50 of 5g-Mes for ROCK1 and ROCK2, using this assay format was 0.53 and 0.45 nM respectively. At 1 µM 5g-Mes was the most potent against ROCK1 (96% inhibition) and ROCK2 (98% inhibition). No other kinase was inhibited by >85%. We included the AGC kinases PKA, AKT1, SGK and PKN1 in the panel. Of all the other kinases tested, PKA was inhibited the most with 1 µM 5g inhibiting PKA by 84.5%.
To determine the mode of action of this inhibitor series, we cocrystallized (R)-14f with the kinase domain of human ROCK1 (residues 6–415). The complex crystallized in space group C2221 with two ROCK1 dimers in the asymmetric unit. The structure was refined to 2.75 Å resolution with Rcryst and Rfree values of 21% and 25%, respectively (Table S1, supplementary information). Fig. 5a shows that (R)-14f is a Type I inhibitor that binds to the hinge region of the ATP site through one hydrogen bond formed between the pyridine ring nitrogen and the main chain amide NH of Met156 (Fig. 5a). The electron density of the inhibitor is well defined, except for the methyl group of the stereogenic centre, which does not appear to interact with the enzyme. The inhibitor extends from the hinge region along Asp216 of the DFG motif towards the β-turn comprising residues Arg84 – Gly88. The urea moiety interacts with the ε-amino group of Lys105 and the carboxyl group of Asp216 with one NHCO unit in a trans conformation and the other highly twisted in a near orthogonal arrangement. The pyridine, thiazole and phenyl moieties form multiple van der Waals (hydrophobic) interactions with surrounding residues (3.4 Å < d < 4 Å). The methoxyphenyl group is sandwiched between the side chain of Lys105 and Gly85 of the P-loop (magenta) (Fig. 5c). The ring points towards residues Leu106 and Leu107. Since the para position is ~4 Å distant from Phe87 and the two leucine residues, substitutions with groups larger than fluorine are likely to cause substantial steric clashes with the enzyme. The close proximity of the meta position to Glu89 forces the ring to adopt a conformation in which the methoxy group or any other meta substituent is exposed to solvent. Modeling of the enantiomer (S)-14f into the binding site indicates steric clashes with the main chain of Arg84-Gly85 as a plausible cause for the low inhibitory activity of this compound (Fig. 5d).
Fig. 5. Molecular mode of action of (R)-14f determined by X-ray crystallography at 2.75 Å resolution.

(a) Surface representation of the ROCK1 dimer in complex with (R)-14f. The inset shows the binding interactions of the inhibitor in the ATP site. The hinge region is indicated in orange, the DFG segment in cyan, and inhibitor in yellow. Displayed in blue is the 2Fo-Fc electron density, contoured at 1σ around the inhibitor. The data and refinement statistics and the Fo-Fc electron density map with the inhibitor omitted during refinement are provided in the supplementary information. The hydrogen bonding and van der Waals (hydrophobic) interactions are shown as black and green dotted lines, respectively. (b) Schematic presentation of the binding interactions between (R)-14f and the enzyme. (c) Interactions of the methoxyphenyl ring of (R)-14f and residues Glu89-Gly85 of the P-loop residues (magenta) and the Lys105 residue. (d) Model of the enantiomer (S)-14f (blue) bound to the inhibitor site. Two possible rotamers are shown, both of which are likely to cause steric clashes with the main chain of Arg84-Gly85 (red dotted lines).
Benzylurea pyridylaminothiazole-based ROCK inhibitors are potent suppressors of the phosphorylation of the ROCK substrate MYPT-1 in human lung cancer cells
We next determined the ability of the compounds to inhibit ROCK in intact human cancer cells. To this end, we treated H1299 lung cancer cells with various doses of the compounds and processed the cells for western immunoblotting to determine their effects on the levels of phosphorylated MYPT-1 (P-MYPT-1) and the total levels of MYPT-1 as described under Methods. Fig. 6 shows that (5a) treatment of H1299 cells decreased the levels of P-MYPT-1, but not total MYPT-1, in a concentration-dependent manner. The enantiomer [(R)-14f] was more potent at decreasing P-MYPT-1 at all concentrations. This is consistent with the ability of these compounds to inhibit ROCK1 in vitro [IC50 values of 170 and 30 nM for 5a (Table 1) and (R)-14f (Table 2), respectively]. The enantiomer (S)-14f which was less potent in vitro (Table 2) was also less active at decreasing the levels of P-MYPT-1 in intact human cancer cells (Fig. 6). Furthermore, the potent meta-methoxy 5b (IC50 27 nM; Table 1) decrease the levels of P-MYPT-1 in a concentration-dependent manner. In contrast, the much less active para-methoxy isomer 5d (less than 10% in vitro ROCK inhibition at 50 µM, Table 1) did not decrease the levels of P-MYPT-1. These studies clearly demonstrate that the benzylurea pyridylaminothiazole-based ROCK inhibitors are able to penetrate cells and effectively inhibit their target in intact cells.
Fig. 6.

Pyridylthiazole-based ureas potently inhibit phosphorylation of a ROCK substrate MYPT1 in human lung cancer cells. H1299 lung cancer cells were treated with increasing concentrations of (a) 5a, (R)-14f, (S)-14f and (b) 5b and 5d and then tested by Western blot assays for phospho-MYPT1. The ROCK inhibitor Y-27632 was used as positive control (10 µM in both experiments).
Conclusions
In the search for novel ROCK selective inhibitors, chemical modifications of 5a to improve in vitro potency, and determine the structural features responsible for the activity, led us to identify benzyl pyridylthiazole ureas as novel and potent inhibitors of ROCK displaying, low nanomolar potency in vitro and sub-micromolar activity in intact human cancer cells. No significant ROCK 1 and 2 isoform selectivity was observed with this series. In summary, meta substitution of the phenyl ring appeared to be optimal for good potency. Only small and polar groups are tolerated and hydroxy, methoxy, amino group give rise to better activity. Changes at the benzylic position of 5a are tolerated resulting in significant potency in the case of methyl and methylenehydroxy groups. Significant differences in the activities of the enantiomers of this series were observed. The co-crystal structure of the first pyridylthiazole ROCK inhibitor established clear evidence for the proposed binding mode of this class of inhibitors and provides an explanation for the observed differences of the enantiomers in the substituted benzyl urea series. Furthermore, potent inhibitors in vitro were also able to potently inhibit ROCK in human lung cancer cells as demonstrated by suppression of the levels of phosphorylation of the ROCK substrate MYPT-1. The differences in the activities of the enantiomers in vitro were also respected in intact cancer cells.
Experimental
Molecular modeling
Compound docking was carried out using the GLIDE60,61 (Grid Based Ligand Docking from Energetics) program from Schrödinger, L.L.C. The Jorgensen OPLS-2005 force field was employed for GLIDE docking simulations. The optimal binding geometry (pose) for each modeled compound was obtained using GLIDE which employs Monte Carlo sampling techniques coupled with energy minimization. GLIDE also uses a scoring method based on ChemScore62 but with additional terms added to the scoring function for greater accuracy. GLIDE 4.5 SP (Standard Precision mode) was used to dock each chemical structure of these compounds followed by GLIDE 4.5 XP (Extra Precision mode) docking to find probable docking poses. An X-ray crystal structure of human ROCK1 in complex with a small molecule inhibitor Y-27632 at 2.60 Å resolution (2ETR.pdb)59 was used for ROCK1 docking.
Kinase assay
Kinase inhibition was measured using the Invitrogen Z-Lyte® FRET kinase assay with Ser/Thr 13 peptide substrate (Invitrogen, cat. PV3793) based on the myosin light chain sequence KKRPQRRYSNVF. Compounds were tested on three separate days with 8 point dilutions performed in duplicate to determine average IC50 values. The assay conditions were optimized to 15 µl of kinase reaction volume with 5 ng of enzyme in 50 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM EGTA, and 0.01% Brij-35. The reaction was incubated for 1 h at room temperature in the presence of 1.5 µM of peptide substrate with 12.5 µM of ATP (for ROCK1) or 2 µM of substrate with 50 µM of ATP (for ROCK2). The reaction was then stopped and the ratio of phosphorylated to unphosphorylated peptides was determined by selective cleavage of only the unphosphorylated peptide as described by the manufacturer (Invitrogen, cat. PV3793). This was followed by excitation of coumarin at 400 nm resulting in emission at 445 nm and energy transfer to fluorescein and final emission at 520 nm. The substrate contains both coumarin and fluorescein and only uncleaved phosphorylated substrate will undergo FRET. The ratio of the signals at 445 nm and 520 nm was measured using a Perkin-Elmer Wallac EnVision Plate Reader, model 2102 plate-reader. The kinases ROCK1 (residues 1–535, N terminal GST) and ROCK2 (residues 1–552, N terminal GST) used in the assay were purchased from Invitrogen (PV3691 and PV3759 respectively). IC50 values were determined using fitted curves with GraphPad Prism 5 software.
Biochemical and crystallographic methods
Reagents and compounds for biochemical and crystallographic experiments were purchased from Sigma–Aldrich (St. Louis, MO) and Hampton Research (Aliso Viejo, CA) unless otherwise indicated.
Enzyme purification
The gene encoding the kinase domain of human ROCK1 (residues 6–415) was synthesized, cloned into the pFB-Dual-PBL bacmid to provide an N-terminal His-Puritin-tag, and expressed in SF9 insect cells after 72 h infection (Blue Sky Biotech, Worcester, MA). All purification steps were performed by FPLC at 4 °C. Harvested insect cells were resuspended in 100 mM Na/K phosphate buffer (pH 7.4) containing 300 mM NaCl, 10 mM MgCl2, 10 mM imidazole, 0.5 mg mL−1 lysosyme, and 0.01% Triton X-100 at 4 °C for 1 h. After sonication and centrifugation (1 h at 29000 × g), the supernatant was purified by immobilized Ni2+-ion affinity chromatography (GE LifeSciences, Piscataway, NJ). Following incubation of peak fractions with TEV protease (20 : 1) at 4 °C, the cleaved His-Puritin-tag was separated by size exclusion chromatography using a Superdex 75 (26/60) column, and eluted with 50 mM HEPES buffer (pH 7.4) containing 150 mM NaCl, 10 mM MgCl2, 1 mM DTT. Purified ROCK1 (6–415) was buffer exchanged into 50 mM HEPES buffer (pH 7.4) containing 1 mM DTT and concentrated to 20 mg mL−1 for crystallization.
Protein crystallography
Crystallization was performed at 18 °C using the sitting drop vapor diffusion method. Crystals of the ROCK1-(R)-14f complex were grown from 0.1 M HEPES pH 7.0, 5% tacsimate pH 7.0, and 10% PEG 5000 MME, in the presence of 0.25 mM RKI1342. Crystals were harvested in cryo-protectant (0.1 M HEPES pH 7.0, 5% tacsimate pH 7.0, and 10% PEG 5000 MME, 25% (v/v) ethylene glycol, 0.5 mM inhibitor) prior to data collection. X-ray diffraction data were recorded at −180 °C using Cu-Kα radiation generated by a Rigaku Micro-Max 007-HF rotating anode (MSC, The Woodlands, TX) using a CCD Saturn 944+ in the Moffitt Structural Biology Core facility. Data were reduced with XDS,63 PHENIX64 was employed for phasing and refinement, and model building was performed using Coot.65 The structure was solved by molecular replacement using the MolRep program from CCP466 using the monomeric ROCK1 kinase domain of pdb entry 2ETR as a starting model. Figures were prepared using PYMOL.67
Determination of the effects of the inhibitors on the phosphorylation levels of the ROCK substrate MYPT-1 in intact cancer cells
H1299 human lung cancer cells were treated for 1 h with various ROCK inhibitors. The cells were then lysed and processed for western immunoblotting as described by us previously.68 The levels of phosphorylation of MYPT1 and total MYPT1 were determined by immunoblotting with the following antibodies: P-MYPT1 and total MYPT1(Cell Signaling, Danvers, Ma).
Supplementary Material
Acknowledgements
This research was supported by an NIH grant [U19 CA067771-11 (SMS)]: We thank the Chemical Biology Core at the Moffitt Cancer Center and Research Institute for assistance with the Z-lyte kinase assay (Dr Wesley Brooks), mass spectrometry and HPLC analysis. We also thank the Moffitt Structural Biology Core for use of the X-ray crystallography facility. The coordinates and structure factors of ROCK1 in complex with (R)-14f have been deposited in the Protein Data bank (code 3TV7).
Footnotes
Electronic supplementary information (ESI) available: Experimental details of the compound synthesis and characterization. 1H and 13C NMR, HPLC traces for the final compounds. See DOI: 10.1039/c2md00320a
References
- 1.Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. FEBS Lett. 1996;392:189–193. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- 2.Sapet C, Simoncini S, Loriod B, Puthier D, Sampol J, Nguyen C, Dignat-George F, Anfosso F. Blood. 2006;108:1868–1876. doi: 10.1182/blood-2006-04-014175. [DOI] [PubMed] [Google Scholar]
- 3.Chang J, Xie M, Shah VR, Schneider MD, Entman ML, Wei L, Schwartz RJ. Proc. Natl. Acad. Sci. U. S. A. 2006;103:14495–14500. doi: 10.1073/pnas.0601911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sebbagh M, Hamelin J, Bertoglio J, Solary E, Breard J. J. Exp. Med. 2005;201:465–471. doi: 10.1084/jem.20031877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thumkeo D, Keel J, Ishizaki T, Hirose M, Nonomura K, Oshima H, Oshima M, Taketo Makoto M, Narumiya S. Mol. Cell. Biol. 2003;23:5043–5055. doi: 10.1128/MCB.23.14.5043-5055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimizu Y, Thumkeo D, Keel J, Ishizaki T, Oshima H, Oshima M, Noda Y, Matsumura F, Taketo Makoto M, Narumiya S. J. Cell Biol. 2005;168:941–953. doi: 10.1083/jcb.200411179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y-M, Bo J, Taffet GE, Chang J, Shi J, Reddy AK, Michael LH, Schneider MD, Entman ML, Schwartz RJ, Wei L. FASEB J. 2006;20:916–925. doi: 10.1096/fj.05-5129com. 910 1096/fj 1005-5129com. [DOI] [PubMed] [Google Scholar]
- 8.Rikitake Y, Oyama N, Wang C-YC, Noma K, Satoh M, Kim H-H, Liao JK. Circulation. 2005;112:2959–2965. doi: 10.1161/CIRCULATIONAHA.105.584623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Nat. Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- 10.Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Nat. Cell Biol. 2001;3:346–352. doi: 10.1038/35070019. [DOI] [PubMed] [Google Scholar]
- 11.Shimokawa H, Rashid M. Trends Pharmacol. Sci. 2007;28:296– 302. doi: 10.1016/j.tips.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 12.Xing X-q, Gan Y, Wu S-J, Chen P, Zhou R, Xiang X-d. Drug News Perspect. 2006;19:517–522. doi: 10.1358/dnp.2006.19.9.1050426. [DOI] [PubMed] [Google Scholar]
- 13.Liao JK, Seto M, Noma K. J. Cardiovasc. Pharmacol. 2007;50:17–24. doi: 10.1097/FJC.0b013e318070d1bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimokawa H, Takeshita A. Arterioscler., Thromb., Vasc. Biol. 2005;25:1767–1775. doi: 10.1161/01.ATV.0000176193.83629.c8. [DOI] [PubMed] [Google Scholar]
- 15.Dong M, Yan BP, Yu C-M. Cardiovasc. Hematol. Agents Med. Chem. 2009;7:322–330. doi: 10.2174/187152509789541891. [DOI] [PubMed] [Google Scholar]
- 16.Kubo T, Yamaguchi A, Iwata N, Yamashita T. Ther. Clin. Risk Manage. 2008;4:605–615. doi: 10.2147/tcrm.s2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kubo T, Yamashita T. Recent Pat. CNS Drug Discovery. 2007;2:173–179. doi: 10.2174/157488907782411738. [DOI] [PubMed] [Google Scholar]
- 18.LoGrasso Philip V, Feng Y. Curr. Top. Med. Chem. 2009;9:704–723. doi: 10.2174/156802609789044452. [DOI] [PubMed] [Google Scholar]
- 19.Suwa H, Ohshio G, Imamura T, Watanabe G, Arii S, Imamura M, Narumiya S, Hiai H, Fukumoto M. Br. J. Cancer. 1998;77:147–152. doi: 10.1038/bjc.1998.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamai T, Yamanishi T, Shirataki H, Takagi K, Asami H, Ito Y, Yoshida K-I. Clin. Cancer Res. 2004;10:4799–4805. doi: 10.1158/1078-0432.CCR-0436-03. [DOI] [PubMed] [Google Scholar]
- 21.Schmitz AAP, Govek E-E, Bottner B, Van Aelst L. Exp. Cell Res. 2000;261:1–12. doi: 10.1006/excr.2000.5049. [DOI] [PubMed] [Google Scholar]
- 22.Imamura F, Mukai M, Ayaki M, Akedo H. Cancer Sci. 2000;91:811–816. doi: 10.1111/j.1349-7006.2000.tb01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Somlyo AV, Bradshaw D, Ramos S, Murphy C, Myers CE, Somlyo AP. Biochem. Biophys. Res. Commun. 2000;269:652–659. doi: 10.1006/bbrc.2000.2343. [DOI] [PubMed] [Google Scholar]
- 24.Uchida S, Watanabe G, Shimada Y, Maeda M, Kawabe A, Mori A, Arii S, Uehata M, Kishimoto T, Oikawa T, Imamura M. Biochem. Biophys. Res. Commun. 2000;269:633–640. doi: 10.1006/bbrc.2000.2315. [DOI] [PubMed] [Google Scholar]
- 25.Itoh K, Yoshioka K, Akedo H, Uehata M, Ishizaki T, Narumiya S. Nat. Med. (N.Y.) 1999;5:221–225. doi: 10.1038/5587. [DOI] [PubMed] [Google Scholar]
- 26.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 27.Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, Narumiya S. Mol. Pharmacol. 2000;57:976–983. [PubMed] [Google Scholar]
- 28.Narumiya S, Ishizaki T, Uehata M. Methods Enzymol. 2000;325:273–284. doi: 10.1016/s0076-6879(00)25449-9. [DOI] [PubMed] [Google Scholar]
- 29.Nakajima M, Hayashi K, Egi Y, Katayama K-i, Amano Y, Uehata M, Ohtsuki M, Fujii A, Oshita K-i, Kataoka H, Chiba K, Goto N, Kondo T. Cancer Chemother. Pharmacol. 2003;52:319–324. doi: 10.1007/s00280-003-0641-9. [DOI] [PubMed] [Google Scholar]
- 30.Nakajima M, Hayashi K, Katayama K-i, Amano Y, Egi Y, Uehata M, Goto N, Kondo T. Eur. J. Pharmacol. 2003;459:113–120. doi: 10.1016/s0014-2999(02)02869-8. [DOI] [PubMed] [Google Scholar]
- 31.Ying H, Biroc SL, Li W-w, Alicke B, Xuan J-A, Pagila R, Ohashi Y, Okada T, Kamata Y, Dinter H. Mol. Cancer Ther. 2006;5:2158–2164. doi: 10.1158/1535-7163.MCT-05-0440. [DOI] [PubMed] [Google Scholar]
- 32.Somlyo AV, Phelps C, Dipierro C, Eto M, Read P, Barrett M, Gibson JJ, Burnitz MC, Myers C, Somlyo AP. FASEB J. 2003;17:223–234. doi: 10.1096/fj.02-0655com. [DOI] [PubMed] [Google Scholar]
- 33.Hampson L, He XT, Oliver AW, Hadfield JA, Kemp T, Butler J, McGown A, Kitchener HC, Hampson IN. Br. J. Cancer. 2009;101:829–839. doi: 10.1038/sj.bjc.6605208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Igishi T, Mikami M, Murakami K, Matsumoto S, Shigeoka Y, Nakanishi H, Yasuda K, Gutkind JS, Hitsuda Y, Shimizu E. Int. J. Oncol. 2003;23:1079–1085. [PubMed] [Google Scholar]
- 35.Liu S, Goldstein RH, Scepansky EM, Rosenblatt M. Cancer Res. 2009;69:8742–8751. doi: 10.1158/0008-5472.CAN-09-1541. [DOI] [PubMed] [Google Scholar]
- 36.Ogata S, Morishige K-I, Sawada K, Hashimoto K, Mabuchi S, Kawase C, Ooyagi C, Sakata M, Kimura T. Int. J. Gynecol. Cancer. 2009;19:1473–1480. doi: 10.1111/IGC.0b013e3181c03909. [DOI] [PubMed] [Google Scholar]
- 37.Zohrabian VM, Forzani B, Chau Z, Murali R, Jhanwar- Uniyal M. Anticancer Res. 2009;29:119–123. [PubMed] [Google Scholar]
- 38.Chen YT, Bannister TD, Weiser A, Griffin E, Lin L, Ruiz C, Cameron MD, Schuerer S, Duckett D, Schroeter T, LoGrasso P, Feng Y. Bioorg. Med. Chem. Lett. 2008;18:6406–6409. doi: 10.1016/j.bmcl.2008.10.080. [DOI] [PubMed] [Google Scholar]
- 39.Sessions EH, Yin Y, Bannister TD, Weiser A, Griffin E, Pocas J, Cameron MD, Ruiz C, Lin L, Schurer SC, Schroter T, LoGrasso P, Feng Y. Bioorg. Med. Chem. Lett. 2008;18:6390–6393. doi: 10.1016/j.bmcl.2008.10.095. [DOI] [PubMed] [Google Scholar]
- 40.Iwakubo M, Takami A, Okada Y, Kawata T, Tagami Y, Sato M, Sugiyama T, Fukushima K, Taya S, Amano M, Kaibuchi K, Iijima H. Bioorg. Med. Chem. 2007;15:1022–1033. doi: 10.1016/j.bmc.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 41.Goodman KB, Cui H, Dowdell SE, Gaitanopoulos DE, Ivy RL, Sehon CA, Stavenger RA, Wang GZ, Viet AQ, Xu W, Ye G, Semus SF, Evans C, Fries HE, Jolivette LJ, Kirkpatrick RB, Dul E, Khandekar SS, Yi T, Jung DK, Wright LL, Smith GK, Behm DJ, Bentley R, Doe CP, Hu E, Lee D. J. Med. Chem. 2007;50:6–9. doi: 10.1021/jm0609014. [DOI] [PubMed] [Google Scholar]
- 42.Feng Y, Yin Y, Weiser A, Griffin E, Cameron MD, Lin L, Ruiz C, Schurer SC, Inoue T, Rao PV, Schroter T, LoGrasso P. J. Med. Chem. 2008;51:6642–6645. doi: 10.1021/jm800986w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sehon CA, Wang GZ, Viet AQ, Goodman KB, Dowdell SE, Elkins PA, Semus SF, Evans C, Jolivette LJ, Kirkpatrick RB, Dul E, Khandekar SS, Yi T, Wright LL, Smith GK, Behm DJ, Bentley R, Doe CP, Hu E, Lee D. J. Med. Chem. 2008;51:6631–6634. doi: 10.1021/jm8005096. [DOI] [PubMed] [Google Scholar]
- 44.Ray P, Wright J, Adam J, Bennett J, Boucharens S, Black D, Cook A, Brown AR, Epemolu O, Fletcher D, Haunso A, Huggett M, Jones P, Laats S, Lyons A, Mestres J, de Man J, Morphy R, Rankovic Z, Sherborne B, Sherry L, van Straten N, Westwood P, R. Zaman GZ. Bioorg. Med. Chem. Lett. 2011;21:97–101. doi: 10.1016/j.bmcl.2010.11.060. [DOI] [PubMed] [Google Scholar]
- 45.Ray P, Wright J, Adam J, Boucharens S, Black D, Brown AR, Epemolu O, Fletcher D, Huggett M, Jones P, Laats S, Lyons A, Man Jd, Morphy R, Sherborne B, Sherry L, Straten Nv, Westwood P, York M. Bioorg. Med. Chem. Lett. 2011;21:1084–1088. doi: 10.1016/j.bmcl.2010.12.104. [DOI] [PubMed] [Google Scholar]
- 46.Ho K-K, Beasley JR, Belanger L, Black D, Chan J-H, Dunn D, Hu B, Klon A, Kultgen SG, Ohlmeyer M, Parlato SM, Ray PC, Pham Q, Rong Y, Roughton AL, Walker TL, Wright J, Xu K, Xu Y, Zhang L, Webb M. Bioorg. Med. Chem. Lett. 2009;19:6027–6031. doi: 10.1016/j.bmcl.2009.09.046. [DOI] [PubMed] [Google Scholar]
- 47.Bosanac T, Hickey ER, Ginn J, Kashem M, Kerr S, Kugler S, Li X, Olague A, Schlyer S, R. Young ER. Bioorg. Med. Chem. Lett. 2010;20:3746–3749. doi: 10.1016/j.bmcl.2010.04.069. [DOI] [PubMed] [Google Scholar]
- 48.Ginn JD, Bosanac T, Chen R, Cywin C, Hickey E, Kashem M, Kerr S, Kugler S, Li X, Prokopowicz Iii A, Schlyer S, Smith JD, Turner MR, Wu F, Young ERR. Bioorg. Med. Chem. Lett. 2010;20:5153–5156. doi: 10.1016/j.bmcl.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 49.Fang X, Chen YT, Sessions EH, Chowdhury S, Vojkovsky T, Yin Y, Pocas JR, Grant W, Schröter T, Lin L, Ruiz C, Cameron MD, LoGrasso P, Bannister TD, Feng Y. Bioorg. Med. Chem. Lett. 2011;21:1844–1848. doi: 10.1016/j.bmcl.2011.01.039. [DOI] [PubMed] [Google Scholar]
- 50.Yin Y, Lin L, Ruiz C, Cameron MD, Pocas J, Grant W, Schröter T, Chen W, Duckett D, Schröter S, LoGrasso P, Feng Y. Bioorg. Med. Chem. Lett. 2009;19:6686–6690. doi: 10.1016/j.bmcl.2009.09.115. [DOI] [PubMed] [Google Scholar]
- 51.Henderson AJ, Hadden M, Guo C, Douglas N, Decornez H, Hellberg MR, Rusinko A, McLaughlin M, Sharif N, Drace C, Patil R. Bioorg. Med. Chem. Lett. 2010;20:1137–1140. doi: 10.1016/j.bmcl.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 52.Thomas CJ, Auld DS, Huang R, Huang W, Jadhav A, Johnson RL, Leister W, Maloney DJ, Marugan JJ, Michael S, Simeonov A, Southall N, Xia M, Zheng W, Inglese J, Austin CP. Curr. Top. Med. Chem. 2009;9:1181–1193. doi: 10.2174/156802609789753644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Austin CP, Brady LS, Insel TR, Collins FS. Science. 2004;306:1138–1139. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 54.Huryn DM, P. Cosford ND. Annu. Rep. Med. Chem. 2007;42:401–416. doi: 10.1016/S0065-7743(07)42026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang NS, Lee GN, Kim CH, Bae MA, Kim I, Cho YS. Bioorg. Med. Chem. Lett. 2009;19:533–537. doi: 10.1016/j.bmcl.2008.10.120. [DOI] [PubMed] [Google Scholar]
- 56.Koresawa M, Okabe T. Assay Drug Dev. Technol. 2004;2:153– 160. doi: 10.1089/154065804323056495. [DOI] [PubMed] [Google Scholar]
- 57.Morwick T, Büttner FH, Cywin CL, Dahmann G, Hickey E, Jakes S, Kaplita P, Kashem MA, Kerr S, Kugler S, Mao W, Marshall D, Paw Z, Shih C-K, Wu F, Young E. J. Med. Chem. 2010;53:759–777. doi: 10.1021/jm9014263. [DOI] [PubMed] [Google Scholar]
- 58.Yin Y, Cameron MD, Lin L, Khan S, Schroter T, Grant W, Pocas J, Chen YT, Schurer S, Pachori A, Lo Grasso P, Feng Y. ACS Med. Chem. Lett. 2010;1:175–179. doi: 10.1021/ml1000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jacobs M, Hayakawa K, Swenson L, Bellon S, Fleming M, Taslimi P, Doran J. J. Biol. Chem. 2006;281:260–268. doi: 10.1074/jbc.M508847200. [DOI] [PubMed] [Google Scholar]
- 60.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 61.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 62.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. J. Comput.-Aided Mol. Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 63.Kabsch W. J. Appl. Crystallogr. 1993;26:795–800. [Google Scholar]
- 64.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse- Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Emsley P, Cowtan K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 66.Collaborative Computational Project Number 4. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994;50:760–763. [Google Scholar]
- 67.DeLano WL. Palo Alto, CA, USA: DeLano Scientific LLC; 2008. http://www.pymol.org. [Google Scholar]
- 68.Kazi A, Carie A, Blaskovich MA, Bucher C, Thai V, Moulder S, Peng H, Carrico D, Pusateri E, Pledger WJ, Berndt N, Hamilton A, Sebti SM. Mol. Cell. Biol. 2009;29:2254–2263. doi: 10.1128/MCB.01029-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.