

Abstract
The ruthenium catalyst generated in situ from H2Ru(CO)(PPh3)3, (S)-SEGPHOS and a TADDOL-derived phosphoric acid promotes butadiene hydrohydroxyalkylation to form enantiomerically enriched products. Notably, the observed diastereo- and enantioselectivity is opposite of that observed using BINOL-derived phosphate counterions in combination with (S)-SEGPHOS, the same enantiomer of chiral ligand. Match-mismatch effects between chiral ligand and chiral TADDOL-phosphate counterion are described. For the first time, single crystal X-ray diffraction data is reported for a ruthenium complex modified by a chiral phosphate counterion.
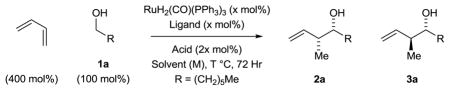
In the course of developing C-C bond forming hydrogenations beyond hydroformylation, we have found that hydrogen transfer between primary alcohols and π-unsaturated reactants generates organometal-aldehyde pairs that combine to form products of carbonyl addition, enabling a departure from stoichiometric organometallic reagents.1 During this work, iridium catalysts for enantioselective carbonyl crotylation from the alcohol oxidation level that employ α-methyl allyl acetate as a crotyl donor were developed.2,3 Butadiene hydrohydroxyalkylation is an alternate strategy for carbonyl crotylation. In initial studies from our laboratory, iridium and ruthenium catalysts that displayed the essential reactivity and regioselectivity were identified, but poor stereoselectivity was observed.4a,b While stereoselectivity can be enforced through the use of 2-silyl-butadienes,4c direct regio- and stereoselective hydrohydroxyalkylations of butadiene itself remained elusive until ruthenium catalysts modified by chiral phosphate counterions were explored.4d,5,6,7 Using the indicated H8-BINOL-derived phosphate counterion, ruthenium catalyzed butadiene hydrohydroxyalkylation with primary benzylic alcohols delivered products of crotylation with good levels of anti-diastereo- and enantioselectivity. To achieve stereoselectivity in corresponding reactions of primary aliphatic alcohols, chiral phosphate counterions were assayed in combination with the chiral phosphine ligands (R)- and (S)-SEGPHOS.8 Here, we disclose that ruthenium catalysts modified by H8-BINOL and TADDOL-derived phosphate counterions enforce opposite diastereo- and enantioselectivity even when the same enantiomer of chiral phosphine ligand is employed. Based on these findings, a protocol for syn-diastereo- and enantioselective carbonyl crotylation via butadiene hydrohydroxyalkylation was achieved (Figure 1).
Figure 1.

Ruthenium catalyzed diastereo- and enantioselective crotylation via butadiene hydrohydroxyalkylation.
To develop stereoselective ruthenium catalyzed butadiene hydrohydroxyalkylations applicable to primary aliphatic alcohols, structural classes of phosphate counterions beyond BINOL-derived systems were explored. As demonstrated previously,4d,9 the acid-base reaction of H2Ru(CO)(PPh3)3 with the chiral phosphoric acid HX conveniently attaches the chiral anion to the metal center. Remarkably, upon initial evaluation of TADDOL-derived phosphoric acids A1–A5 in the coupling of butadiene with heptanol 1a using DPPF, an achiral phosphine ligand, a modest preference for the syn-diastereomer 2a was observed (Table 1, entries 1–5). Enantioselectivities increased with increasing size of the TADDOL aryl substituent, and using the 3,5-xylyl substituted acid A5 a 78% enantiomeric excess was observed for the syn-diastereomer 1a (Table 1, entry 5). These data suggested the feasibility of developing a protocol for carbonyl syn-crotylation via butadiene hydrohydroxyalkylation. Toward this end, match-mismatch effects between the chiral phosphate counterion derived from A5 and the chiral phosphine ligands (R)- and (S)-SEGPHOS8 were explored (Table 1, entries 6 and 7). For the matched case involving the combination of acid A5 and (S)-SEGPHOS, an 89% enantiomeric excess was observed for the syn-diastereomer 1a, although syn-diastereoselectivity remained modest (Table 1, entry 7). However, upon use of phosphoric acids A6 accompanied by further variation of solvent, concentration and temperature, the syn-diastereomer 1a was generated in 82% yield, 95% enantiomeric excess and 4.4:1 syn-diastereoselectivity (Table 1, entries 8–12).
Table 1.
Selected optimization experiments in the ruthenium catalyzed syn-diastereo- and enantioselective hydrohydroxyalkylation of butadiene with heptanol 1a.a
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Acid | Ligand | Ru (mol%) | T(°C) | Solvent (M) | Yield (%) | ee(%) | 2a:3a |
| 1 | A1 | DPPF | 5 | 105 | THF (2.0) | 64 | 1 | 1:1 |
| 2 | A2 | DPPF | 5 | 105 | THF (2.0) | 82 | 18 | 1.6:1 |
| 3 | A3 | DPPF | 5 | 105 | THF (2.0) | 50 | 29 | 1.4:1 |
| 4 | A4 | DPPF | 5 | 105 | THF (2.0) | 58 | 63 | 1.8:1 |
| 5 | A5 | DPPF | 5 | 105 | THF (2.0) | 85 | 78 | 1.4:1 |
| 6 | A5 | (R)-Segphos | 5 | 105 | THF (2.0) | 48 | 31 | 1.1:1 |
| 7 | A5 | (S)-Segphos | 5 | 105 | THF (2.0) | 57 | 89 | 1.7:1 |
| 8 | A5 | (S)-Segphos | 5 | 95 | t-BuOH (2.0) | 23 | 90 | 2.7:1 |
| 9 | A5 | (S)-Segphos | 5 | 95 | Me2CO (2.0) | 46 | 90 | 3.0:1 |
| 10 | A6 | (S)-Segphos | 5 | 95 | Me2CO (2.0) | 55 | 94 | 4.1:1 |
| 11 | A6 | (S)-Segphos | 5 | 95 | Me2CO (1.0) | 57 | 94 | 4.6:1 |
| ⇨12 | A6 | (S)-Segphos | 7 | 95 | Me2CO (1.0) | 82 | 95 | 4.4:1 |
|
| ||||||||
|
| ||||||||
Yields are of material isolated by silica gel chromatography. DPPF = 1,1′-bis(diphenylphosphino)ferrocene; SEGPHOS = 5,5′-
bis(diphenylphosphino)-4,4′-bi-1,3-benzodioxole. See Supporting Information for further details.
To evaluate scope, optimal conditions identified for the syn-crotylation of 1a were applied to primary alcohols 1b–1j. The desired products of syn-crotylation 2b–2j were obtained in good yield with syn-diastereoselectivities ranging between 4–5:1 and uniformly high levels of enantioselectivity (Table 2). Interestingly, the observed diastereo- and enantioselectivity is opposite to that observed using BINOL-derived phosphate counterions in combination with DPPF or even (S)-SEGPHOS (Scheme 1). To gain insight into the origins of stereoselectivity, a crystal of the ruthenium complex modified by (S)-SEGPHOS and the phosphate counterion derived from the acid A2 was subjected to X-ray diffraction analysis. As anticipated, the phosphate counterion exists in a trans-relationship with respect to the carbonyl ligand.9 Based on the connectivity revealed in the crystal structure and the observed stereoselectivities, a preliminary stereochemical model was formulated and reconciled with the indicated catalytic mechanism (Scheme 2). The unusual syn-diastereoselectivity may arise as a consequence of kinetically preferred hydrometallation of the s-cis conformer of butadiene to furnish the anti-π-crotylruthenium isomer.10 It is postulated that the steric demand of the TADDOL-based phosphate counterion retards the rate of isomerization to the syn-π-crotylruthenium isomer, which would mandate intervention of an even more sterically congested secondary σ-crotylruthenium species. In this way, the kinetic stereoselectivity of the hydrometallation event is preserved, and carbonyl addition occurs by way of the (Z)-σ-crotylruthenium haptomer by way of closed Zimmerman-Traxler type transition structure11 to furnish the syn-diastereomer.
Table 2.
Ruthenium catalyzed crotylation via butadiene hydrohydroxyalkylation using aliphatic alcohols 1a–1j a

|
Yields are of material isolated by silica gel chromatography. See Supporting Information for further details.
5 mol% catalyst-ligand and 10 mol% acid.
Scheme 1.

Chiral phosphate counterion dependent inversion of diastereo- and enantioselectivity using the chiral catalyst modified by (S)-SEGPHOS.
aReaction performed at 105 °C.
Scheme 2.

Catalytic mechanism for ruthenium catalyzed crotylation via butadiene hydrohydroxyalkylation.
In summary, we report the chiral anion dependent inversion of diastereo- and enantioselectivity in butadiene hydrohydroxyalkylation to form products of carbonyl syn-crotylation, as well as the first X-ray crystal structure of a ruthenium complex modified by a chiral phosphate counterion. These studies provide important insight into the structural and interactional features of the catalyst, which should accelerate the design of improved second generation protocols. More broadly, the merged redox-construction events described herein minimize the degree of separation between reagent and feedstock, and represent a departure from premetallated reagents in chemistry carbonyl addition; a cornerstone of synthetic organic chemistry.
Supplementary Material
Figure 2.

Structure of Ru(CO)(OAc)(A2-phosphate)[(S)-SEGPHOS)] as determined by single crystal X-ray diffraction analysis. Displacement ellipsoids are scaled to the 50% probability level.
Acknowledgments
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) are acknowledged for partial support of this research.
Footnotes
Supporting Information Available: Experimental procedures and spectral data. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For recent reviews of C-C bond forming hydrogenation and transfer hydrogenation, see: Bower JF, Krische MJ. Top Organomet Chem. 2011;43:107. doi: 10.1007/978-3-642-15334-1_5.Hassan A, Krische MJ. Org Proc Res Devel. 2011;15:1236. doi: 10.1021/op200195m.Moran J, Krische MJ. Pure Appl Chem. 2012;84:1729. doi: 10.1351/PAC-CON-11-10-18.
- 2.For iridium catalyzed carbonyl crotylation from the alcohol oxidation level employing α-methyl allyl acetate as the crotyl donor, see: Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w.Gao X, Townsend IA, Krische MJ. J Org Chem. 2011;76:2350. doi: 10.1021/jo200068q.Gao X, Han H, Krische MJ. J Am Chem Soc. 2011;133:12795. doi: 10.1021/ja204570w.
- 3.For selected reviews on enantioselective carbonyl allylation and crotylation, see: Yamamoto Y, Asao N. Chem Rev. 1993;93:2207.Ramachandran PV. Aldrichim Acta. 2002;35:23.Kennedy JWJ, Hall DG. Angew Chem, Int Ed. 2003;42:4732. doi: 10.1002/anie.200301632.Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.Yu CM, Youn J, Jung HK. Bull Korean Chem Soc. 2006;27:463.Marek I, Sklute G. Chem Commun. 2007:1683. doi: 10.1039/b615042j.Hall DG. Synlett. 2007:1644.Li J, Menche D. Synthesis. 2009:2293.Leighton JL. Aldrichim Acta. 2010;43:3.Yus M, González-Gómez JC, Foubelo F. Chem Rev. 2011;111:7774. doi: 10.1021/cr1004474.
- 4.For iridium and ruthenium catalyzed hydrohydroxyalkylations of butadiene to form products of crotylation, see: Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:6338. doi: 10.1021/ja801213x.Zbieg JR, Fukuzumi T, Krische MJ. Adv Synth Catal. 2010;352:2416. doi: 10.1002/adsc.201000599.Zbieg JR, Moran J, Krische MJ. J Am Chem Soc. 2011;133:10582. doi: 10.1021/ja2046028.Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ. Science. 2012;336:324. doi: 10.1126/science.1219274.
- 5.For selected reviews on chiral phosphate counterions in metal catalysis, see: Shao Z, Zhang H. Chem Soc Rev. 2009;38:2745. doi: 10.1039/b901258n.Zhong C, Shi X. Eur J Org Chem. 2010:2999.Rueping M, Koenigs RM, Atodiresei I. Chem Eur J. 2010;16:9350. doi: 10.1002/chem.201001140.Phipps RJ, Hamilton GL, Toste FD. Nat Chem. 2012;4:603. doi: 10.1038/nchem.1405.Parra A, Reboredo S, Martin Castro AM, Aleman J. Org Biomol Chem. 2012;10:5001. doi: 10.1039/c2ob25479d.
- 6.For selected examples of the use of chiral phosphate counterions in metal catalysis, see: Alper H, Hamel N. J Am Chem Soc. 1990;112:2803.Komanduri V, Krische MJ. J Am Chem Soc. 2006;128:16448. doi: 10.1021/ja0673027.Hamilton GL, Kang EJ, Mba M, Toste FD. Science. 2007;317:496. doi: 10.1126/science.1145229.Rueping M, Antonchick AP, Brinkmann C. Angew Chem Int Ed. 2007;46:6903. doi: 10.1002/anie.200702439.Mukherjee S, List B. J Am Chem Soc. 2007;129:11336. doi: 10.1021/ja074678r.Liao S, List B. Angew Chem Int Ed. 2010;49:628. doi: 10.1002/anie.200905332.Jiang G, List B. Chem Commun. 2011;47:10022. doi: 10.1039/c1cc12499d.Xu B, Zhu SF, Xie XL, Shen JJ, Zhou QL. Angew Chem Int Ed. 2011;50:11483. doi: 10.1002/anie.201105485.Liao S, List B. Adv Synth Catal. 2012;354:2363.
- 7.The Akiyama-Terada type phosphoric acids derived from BINOL inspired initial efforts described in reference 6b: Akiyama T, Itoh J, Yokota K, Fuchiba K. Angew Chem Int Ed. 2004;116:1566. doi: 10.1002/anie.200353240.Uraguchi D, Terada M. J Am Chem Soc. 2004;126:5356. doi: 10.1021/ja0491533.
- 7.Saito T, Yokozawa T, Ishizaki T, Moroi T, Sayo N, Miura T, Kumobayashi H. Adv Synth Catal. 2001;343:264. [Google Scholar]
- 8.(a) Dobson A, Robinson SR, Uttley MF. J Chem Soc, Dalton Trans. 1974:370. [Google Scholar]; (b) Zbieg JR, McInturff EL, Leung JC, Krische MJ. J Am Chem Soc. 2011;133:1141. doi: 10.1021/ja1104156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xue P, Bi S, Sung HHY, Williams ID, Lin Z, Jia G. Organometallics. 2004;23:4735. [Google Scholar]
- 10.Although the s-trans conformer of butadiene is more stable than the s-cis conformer, the latter typically forms more stable metal complexes, accounting for the observed kinetic preference in the hydrometallation event. For a brief discussion of diene coordination modes, see: Murakami M, Itami K, Ito Y. Organometallics. 1999;18:1326.
- 11.Zimmerman HE, Traxler MD. J Am Chem Soc. 1957;79:1920. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.