

Abstract
2-Amino-1,7-dimethylimidazo[4,5-g]quinoxaline (MeIgQx) is a recently discovered heterocyclic aromatic amine (HAA) that is formed during the cooking of meats. MeIgQx is an isomer of 2-amino-3,8-dimethylmidazo[4,5-f]quinoxaline (MeIQx), a rodent carcinogen and possible human carcinogen that also occurs in cooked meats. MeIgQx is a bacterial mutagen but knowledge about its metabolism and carcinogenic potential is lacking. Metabolism studies on MeIgQx and MeIQx were conducted with human and mouse liver microsomes, and recombinant human P450s. DNA binding studies were also investigated in mice to ascertain the genotoxic potential of MeIgQx in comparison to MeIQx. Both HAAs underwent comparable rates of N-oxidation to form genotoxic N-hydroxylated metabolites with mouse liver microsomes (0.2 - 0.3 nmol/min/mg protein). The rate of N-oxidation of MeIQx was 4-fold greater than the rate of N-oxidation of MeIgQx with human liver microsomes (1.7 vs 0.4 nmol/min/mg protein). The rate of N-oxidation, by recombinant human P450 1A2, was comparable for both substrates (6 pmol/min/pmol P450 1A2). MeIgQx also underwent N-oxidation by human P450s 1A1 and 1B1 at appreciable rates, whereas MeIQx was poorly metabolized by these P450s. The potential of MeIgQx and MeIQx to form DNA adducts was assessed in female C57BL/6 mice given [14C]-MeIgQx (10 μCi, 9.68 mg/kg body wt) or [14C]-MeIQx (10 μCi, 2.13 mg/kg body wt). DNA adduct formation in liver, pancreas and colorectum was measured by accelerator mass spectrometry at 4, 24 or 48 h post-treatment. Variable levels of adducts were detected in all organs. The adduct levels were similar for both HAAs, when adjusted for dose, and ranged from 1 to 600 adducts per 107 nucleotides per mg/kg dose. Thus, MeIgQx undergoes metabolic activation and binds to DNA at levels that are comparable to MeIQx. Given the high amounts of MeIgQx formed in cooked meats, further investigations are warranted to assess the carcinogenic potential of this HAA.
Introduction
More than 20 heterocyclic aromatic amines (HAAs) have been identified in cooked beef, poultry, and fish.1,2 The formation of HAAs is dependent upon the type of meat and method of cooking: the concentrations of HAAs can range from less than 1 part-per-billion (ppb), up to 500 ppb in meat or poultry that is cooked well done.2-4 The HAAs thus far assayed have been shown to induce tumors at multiple sites in rodents.1 Several prevalent HAAs are classified as probable or possible human carcinogens (Group 2A and 2B), based on toxicity data reviewed by the International Agency for Research on Cancer.5,6 The Report on Carcinogens, 11th edition, of the National Toxicology Program, also concluded that prevalent HAAs are “reasonably anticipated” to be human carcinogens.7 Thus, there is much concern about the health risk associated with the exposure to these chemicals.
2-Amino-3,8-dimethylimidazo[4,5-f]quinoxaline (MeIQx) is one of the most abundant HAAs formed in cooked meats. MeIQx is a powerful bacterial mutagen and induces tumors at multiple sites in rodents during long-term feeding studies.1 It is noteworthy that MeIQx and other known HAAs account for less than 30% of the mutagenicity attributed to this class of genotoxicants in well-done grilled meats and other uncharacterized HAAs are undoubtedly present.8 We recently discovered 2-amino-1,7-dimethylimidazo[4,5-g]quinoxaline (MeIgQx), a linear tricyclic ring isomer of MeIQx in urine of meat-eaters,9 and subsequently identified MeIgQx in cooked beef.9-11 MeIgQx is the most mass-abundant HAA formed in cooked ground beef, steaks and gravies;4 it occurs at 5-fold or greater levels than the amounts of MeIQx or 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Both MeIQx and PhIP are believed to contribute to diet related cancers.1 MeIgQx is a less potent bacterial mutagen than MeIQx and PhIP; however, studies have not been conducted on the metabolism of MeIgQx, and the carcinogenic potential of MeIgQx is unknown.11
In this investigation, we have examined the capacities of human and mouse liver microsomes, and recombinant human cytochrome P450s to metabolize MeIgQx and MeIQx to electrophilic metabolites that are capable of binding to DNA. We also compared the potential of [14C]-MeIgQx and [14C]-MeIQx to bind to DNA in liver, pancreas and colorectum, potential target organs of HAA carcinogenicity, in C57BL/6 mice. These data provide a preliminary estimate of the genotoxic potential of MeIgQx in vivo.
Materials and Methods
Chemicals
MeIQx and [2-14C]-MeIQx (50 mCi/mmol, >98% radiochemical purity) were purchased from Toronto Research Chemicals (Ontario, Canada). KCN and Br2 were was purchased from Sigma (St. Louis, MO). K[14C]N (54 mCi/mmol) was purchased from Moravek Chemicals (Brea, CA). Human liver samples were from Tennessee Donor Services, Nashville, TN, and kindly provided by Dr. F.P. Guengerich, Vanderbilt University. Recombinant human P450s 1A1, 1A2 and 1B1 contained in supersomes from baculovirus-infected insect cells, and furafylline were purchased from Becton Dickinson (San Jose, CA).
General Methods
Mass spectra of synthetic derivatives and metabolites were acquired on a Finnigan TSQ Quantum Ultra triple stage quadrupole mass spectrometer (Thermo Fisher, San Jose, CA). The MS analyses were conducted in the positive ionization mode and employed an Advance CaptiveSpray™ source from Michrom Bioresource Inc. (Auburn, CA). Typical instrument tune parameters used were as follows: capillary temperature 200 °C, source spray voltage 1.4 kV, tube lens offset 95 V, capillary offset 35 V, and source fragmentation 5 V. Argon, set at 1.5 mTorr, was used as the collision gas and the collision energy was set between 15 to 30 eV; no sheath gas, sweep gas or auxiliary gas was employed.
Synthesis
The synthesis of [2-14C]-MeIgQx was done as previously described for the synthesis of the unlabelled compound with minor modifications.11 K[14C]N (0.093 mmol, 54 mCi/mmol) was diluted with KCN (0.4 mmol) in CH3OH (0.6 mL), sonicated for 10 min, and then slowly added to Br2 (0.50 mmol) in CH3OH (0.5 mL) on ice. The reaction was allowed to proceed for 20 min. Thereafter, 4-fluoro-5-nitrobenzene-1,2-diamine (41 mg, 0.24 mmol) in C2H5OH (0.5 mL) was added to the newly formed [14C]NBr, and the reagents were mixed overnight at room temperature. The 2-[14C]-5-fluoro-6-nitro-1H-benzimidazole-2-amine intermediate was reacted with NH4OH to displace the fluorine moiety, followed by reduction with NaBH4, ring-closure with methylglyoxal, and lastly by methylation with CH3I, as previously described.11 [2-14C]-MeIgQx and its 6-methylated homologue were resolved by semi-preparative HPLC employing a Waters XBridge phenyl column (10 × 250 mm, 5 μm particle size). An isocratic solvent composed of 23% CH3OH in 5 mM NH4CH3CO2 (pH 8.7) was used for chromatography. The flow rate was 3 mL/min, and the run time was 70 min. The chemical and radiochemical purities of MeIgQx exceeded 98.5% as judged by HPLC with UV diode array detection and liquid scintillation counting. The specific activity of MeIgQx was determined by ESI-MS, by measuring the ratio of the 14C/12C isotopes of the protonated molecules [M+H]+ at m/z 216.1 and 214.1: the specific activity was estimated at 11 mCi/mmol.
2-Hydroxyamino-3,8-dimethylimidazo[4,5-f]quinoxaline (HONH-MeIQx) was prepared by reduction of 2-nitro-3,8-dimethylimidazo[4,5-f]quinoxaline with hydrazine and Pd/C serving as a catalyst.12 2-Amino-8-(hydroxymethyl)-3-methylimidazo[4,5-f]quinoxaline (8-CH2OH-IQx) was prepared by the oxidation of MeIQx with SeO2 as previously described.13N-(deoxyguanosin-8-yl)-MeIQx (dG-C8-MeIQx), and [13C10]-dG-C8-MeIQx, and 5-(deoxyguanosin-N2-yl)-MeIQx (dG-N2-MeIQx) were synthesized by reaction of the N-acetoxy derivative of MeIQx with either dG or [13C10]-dG as previously reported.14
Microsomal metabolism studies
The metabolism studies were conducted with liver microsomes prepared from female C57BL/6J mice,15 the mouse strain used for in vivo DNA binding studies, or with human liver microsomes, or recombinant human P450s 1A1, 1A2, and 1B1. Liver microsomal protein (1 mg/mL) or recombinant P450 (100 pmol/mL), in 100 mM potassium phosphate (pH 7.6) containing 0.5 mM EDTA, 5 mM glucose 6-phosphate, glucose 6-phosphate dehydrogenase (1 unit/mL), 1 mM NADPH, 1 mM NAD+ were incubated with MeIgQx or MeIQx (200 μM); the N-oxidation rates are at Vmax for MeIQx at this substrate concentration.15 The enzymes plus cofactors were incubated for 3 min prior to the addition of HAA substrates. The metabolism was done for 5 or 10 min, and metabolite formation was a linear function with time. Some metabolism studies with human liver microsomes were conducted for time periods up to 30 min, to produce large amounts of metabolites for characterization by mass spectrometry. The enzyme reactions were terminated by the addition of 2 vol CH3OH. The precipitated proteins were removed by centrifugation and the supernatants were assayed by HPLC with an Agilent 1100 Chemstation equipped with an UV photodiode array detector. The metabolites were resolved with an Aquasil C18 column (4.6 × 250 mm, 5 μm particle size, Thermo Scientific), employing a linear gradient starting from 10% CH3CN in 50 mM NH4CH3CO2 (pH 6.8) and reaching 100% CH3CN at 20 min at a flow rate of 1 mL/min.
The estimates of formation of 2-hydroxamino-1,7-dimethylimidazo[4,5-g]quinoxaline (HONH-MeIgQx) and 2-hydroxyamino-3,8-dimethylimidazo[4,5-f]quinoxaline (HONH-MeIQx), and other oxidation products were determined by HPLC with UV detection. The metabolites of MeIgQx were monitored at 370 or 390 nm and metabolites of MeIQx were monitored at 274 nm. We assumed that the molar extinction coefficients of N-hydroxylated and ring-oxidized metabolites were the same values as the molar extinction coefficients of the parent compounds at their respective maximal absorbance wavelengths. The UV responses at these wavelengths were used to estimate rates of metabolite formation.
Liquid chromatography-electrospray ionization/mass spectrometry (LC-ESI/MS) characterization of metabolites
The N-oxidized and ring-oxidized metabolites were collected from the HPLC effluent and directly characterized by electrospray ionization tandem mass spectrometry (ESI/MS2) with the triple stage quadrupole mass spectrometer. The metabolites were also characterized by ESI/MS2 following reaction with nitrosobenzene (10 μg, in 0.1% HCO2H in CH3OH) at 50 °C for 1 h.
In Vivo DNA Binding Studies
DNA binding studies were conducted with female C57BL/6J mice (Taconic Farm, Albany, NY). Animals were housed in polycarbonate cages and allowed to acclimate for 1 week prior to commencement of the study.
Animal Dosing
Mice (20 g, N = 5 animals per time point) were dosed by gavage with 14C-MeIgQx (10 μCi, 9.68 mg/kg body wt) or 14C-MeIQx (10 μCi, 2.13 mg/kg body wt) in a volume of 0.25 mL water. At T 4, 24, or 48 h, animals were sacrificed by asphyxiation with CO2 gas, followed by cervical dislocation; blood was retrieved via the inferior vena cava. The liver and pancreas were harvested, rinsed with chilled PBS and snapped-frozen in liquid nitrogen. The colorectal tissue (ileo-cecal junction to the rectum) was retrieved, the fecal contents were removed, and the tissue segment was incubated with cold, freshly prepared EDTA buffer (phosphate-buffered saline containing 1.5 mM Na2-EDTA, 6000 U sodium heparin, DTT (8 mg) and phenylmethanesufonyl fluoride (4 mg, 100 μl of 40 mg/mL DMSO, per 100 ml of buffer). After 20 min, the mucosal layer containing the colonocytes was retrieved by placing the intestine segment in a Petri dish and the loosened mucosal layer was displaced by gently scraping the outer intestinal segment with the edge of a glass slide to eject the colonocytes. The colonocytes were centrifuged at 3,000 g at 4 °C. Pelleted colonocytes were resuspended in EDTA buffer (2 mL) and centrifuged again. This washing procedure was repeated a third time and the colonocytes were quick-frozen in liquid nitrogen.
Radioactivity in plasma
Radioactivity was measured with a Beckman Coulter liquid scintillation counter (Fullerton, CA) with external calibration.
Isolation of DNA
The entire liver, pancreas, or colonocytes were homogenized in 2 mL TE Buffer (50 mM Tris-HCl, 10 mM EDTA, pH 8.0) and centrifuged for 10 min at 3,000 g. DNA was isolated from the nucelar pellets of the liver and kidney (equivalent of 200 mg tissue), and from the entire nuclear pellet of the colorectum, by solvent extraction as described by Gupta.16 The purified DNA underwent a second solvent extraction procedure and ethanol precipitation step, to further remove non-covalently bound radioactivity.
The DNA was dissolved in distilled-deionized H2O and the concentration was determined by UV spectroscopy, assuming a concentration of DNA (50 μg/mL) is equal to 1.0 absorbance unit at 260 nm. The A260:280 ratios were on average greater than 1.75.
Accelerator Mass Spectrometry
AMS analyses were conducted at the MIT BEAMS Lab using procedures described elsewhere in detail.17 Sample aliquots (1.50 μL) were applied directly to a CuO matrix, and introduced into a laser-induced combustion interface for subsequent AMS analysis. Each run contained two quantitation standards ([14C-methyl] bovine serum albumin, 0.003 dpm/μL), followed by two blanks (1 μg/μL human serum albumin), 10 samples, and another two standards. Quantitation was performed by integrating peaks produced in the continuous trace of 14C detector count rate versus time generated during operation of the combustion interface, which produces and delivers the CO2 of combustion to the AMS ion source, and taking the product of the sample/standard peak area ratio multiplied by the standard concentration as the concentration of the sample. Blank values, if measurable, were subtracted from the sample peak areas prior to calculating sample concentration.
DNA Adduct Measurements by LC-ESI/MS3
DNA (100 μg) in 5 mM Bis-Tris-HCl buffer (pH 7.1, 50 μL) underwent enzymatic digestion with DNAse I for 1.5 h, followed by incubation with nuclease P1 for 3 h, and then digested with alkaline phosphatase and phosphodiesterase for 18 h.18 These enzyme digestion conditions were shown to be highly efficient in the recovery of the dG-C8-MeIQx from calf thymus DNA modified with N-acetoxy-MeIQx.14,19 The digest was added to 2 vol of chilled C2H5OH and placed on ice for 1 h, followed by centrifugation at 15,000 g to pellet salt and protein. The supernatants were transferred to silylated capillary LC vials and concentrated to dryness by vacuum centrifugation. The extracts were dissolved in 1:1 H2O:DMSO (20 μL).
The adducts were separated from non-modified nucleosides with a NanoAcquity™ UPLC system (Waters Corporation, Milford, MA) interfaced with a linear quadrupole ion trap mass spectrometer (LTQ MS, Thermo Fisher, San Jose, CA). A Waters Symmetry trap column (180 μm x 20 mm, 5 μm particle size) was employed for online solid phase enrichment of the DNA adducts. The analytical column was a Michrom C18 AQ (0.3 × 150 mm, 3 μm particle size, Michrom Bioresources Inc., Auburn, CA). The DNA digests were injected onto the trap column and washed with 0.2% HCO2H in 5% CH3CN at a flow rate of 12 μL/min for 5 min. Thereafter, the DNA adducts were back-flushed onto the C18 AQ Michrom AQ column. A linear gradient was employed to resolve the DNA adducts, starting at 0.01% HCO2H containing 5% CH3CN and arriving at 0.01% HCO2H in 95% CH3CN at 20 min. The flow rate was set at 5 μL/min.
Mass spectrometric measurements were performed at the MS3 scan stage in the positive ionization mode. The ion source was an Advance CaptiveSpray™ source from Michrom Bioresource Inc. (Auburn, CA). Representative optimized instrument tuning parameters were as follows: capillary temperature, 200 °C; source spray voltage, 2.0 kV; no sheath gas, sweep gas or auxiliary gas was employed; capillary voltage, 40 V; tube lens voltage, 110 V; and in-source fragmentation 10 V. The automatic gain control settings were full MS target 30000 and MSn target 10000, and the maximum injection time was 10 ms. The MS/MS scan mode produced the aglycone ion BH +2 adducts [M+H-116]+ or [M+H-121]+ from the protonated dG-C8-MeIQx and dG-C8-[14C]-MeIQx adducts or [13C10]dG-C8-MeIQx adduct, respectively. The ions monitored were as follows: dG-C8-MeIQx (m/z 479.1 > 363.1 >); dG-C8-[14C]-MeIQx (m/z 481.1 > 365.1 >); and [13C10]-dG-C8--C8-MeIQx (m/z 489.1 > 368.1 >). The product ion spectra of the aglycone adducts at the MS3 scan stage were acquired from m/z 125 to 400. The normalized collision energies were set at 24 and 34 eV, and the isolation widths were set at an m/z = 4.0 and 1.0, respectively, for the MS2 and MS3 scan modes. The activation Q was set at 0.35 and the activation time was 10 ms for both transitions. Helium was used as the collision damping gas in the ion trap and was set at a pressure of 1 mTorr. One μscan was used for data acquisition. The same mass spectral parameters were employed to screen for putative dG-C8 adducts of MeIgQx.
Results
Metabolism of MeIQx and MeIgQx with human and mouse liver microsomes and recombinant human P450s
HAAs undergo bioactivation by cytochrome P450-mediated N-oxidation of the exocyclic amine group, to form electrophilic N-hydroxy-HAA metabolites that covalently bind to DNA.20-22 The liver is the most active organ in the metabolism of HAAs,20-22 and P450 1A2 is the major hepatic P450 involved in the N-oxidation of most HAAs.15,23,24 We previously characterized the principal oxidation products of MeIQx formed by rat and human liver microsomes, rat P450 1A2, and recombinant human P450 1A2.15 In this study, we have examined the pathways of P450-mediated metabolism and compared the rates of N-oxidation of MeIQx and MeIgQx, by employing microsomes prepared from liver of female CN 57/BL mice, the species used for in vivo DNA adduct binding studies. The capacities of human liver microsomes, recombinant human P450 1A2, P450 1A1, and P450 1B1 to bioactivate both compounds were also investigated. Both P450 1A1 and P450 1B1 are prominently expressed in extrahepatic tissues25,26 and catalyze the N-oxidation of several HAAs.27,28 The chemical structures of MeIQx and MeIgQx and their proposed pathways of metabolism by human P450 1A2 (vide infra) are depicted in Figure 1.
Figure 1.

Chemical structures of MeIQx and MeIgQx and proposed pathways of P450-mediated metabolism. The * depicts the site of incorporation of the 14C isotope.
The HPLC-UV profiles of the metabolites of MeIQx and MeIgQx formed by human liver microsomes are shown in Figure 2, along with the UV spectra of the parent amines and their principal N-oxidized metabolites. The major pathway of metabolism of MeIQx occurs by N-oxidation of the exocyclic amine group, to form HONH-MeIQx. The UV spectrum of HONH-MeIQx is identical the spectra of the metabolite and synthetic derivative previously reported;29 the absorbance maximum (272 nm) displays a slight hypsochromic shift relative to the absorbance maximum of MeIQx (274 nm) (Figure 2).29 The dihydroxylated product, 2-(hydroxyamino)-8-hydroxymethyl-3-methylimidazo[4,5-f]quinoxaline (HONH-8-CH2OH-IQx) is the second most abundant metabolite, followed by the alcohol, 8-CH2OH-IQx, which is a minor metabolite.30 Human liver microsomes also produced three oxidation products (M-1, M-2, and M-3) of MeIgQx (Figure 2). Their UV and mass spectral data are discussed below.
Figure 2.

HPLC UV profiles of MeIQx and MeIgQx metabolites formed with human liver microsomes. The UV spectra of the parent compounds and major N-oxidized metabolites of each HAA are presented.
The metabolites of MeIQx and MeIgQx were collected from the HPLC effluent and directly infused into the triple stage quadrupole mass spectrometer. The product ion spectra of HONH-MeIQx and its azoxy derivative, obtained by reaction of HONH-MeIQx with nitrosobenzene, are shown in Figure 3. The product ion spectrum of HONH-MeIQx ([M+H]+ at m/z 230.1) showed prominent fragment ions at m/z 213.1 and 212.1, indicative of the loss of 17 and 18 Da, which are attributed to loss of HO· and H2O. These fragment ions are typically observed in product ion spectra of HONH-HAA metabolites, but they are not present in the product ion spectra of ring-oxidized HAA metabolites, which often display a neutral loss of 28 Da (CO).30,31 The product ion spectrum of the azoxy derivative confirms the structure as HONH-MeIQx.29,32 The product ion spectra of HONH-8-CH2OH-IQx and 8-CH2OH-IQx (Figure 4) are also in excellent agreement to the spectra of the reference compounds previously reported.30
Figure 3.

The product ion spectra of HONH-MeIQx and HONH-MeIgQx (M-3) (right panel) and product ion spectra of the azoxy conjugates formed with nitrosobenzene (left panel).
Figure 4.

ESI/MS/MS product ion spectra of 8-CH2OH-IQx, 8-CH2OH-HONH-IQx, and the monohydroxylated (M-2) and dihydroxylated (M-1) metabolites of MeIgQx.
The mass spectrum of principal metabolite of MeIgQx (M–3) displayed a protonated ion [M+H]+ at m/z 230.1, a mass which is 16 Da greater than MeIgQx. The ESI product ion spectra of M-3 and its adduction product with nitrosobenzene were very similar to the product ion spectra of HONH-MeIQx and its azoxy adduct (Figure 3). M-3 underwent oxidation with time, as did HONH-MeIQx, to form the presumed nitroso derivative: a prominent ion [M+H]+ was observed at m/z 228.1. The product ion spectra of these intermediates displayed prominent fragment ions at m/z 213.1 and 198.1, which are ascribed, respectively, to the loss of CH3· and NO moieties (data not shown). Thus, the structure of M-3 is assigned as 2-(hydroxamino)-1,7-dimethylimidazo[4,5-f]quinoxaline (HONH-MeIgQx).
A large bathochromic shift was observed in the UV spectrum of HONH-MeIgQx compared to the spectrum of MeIgQx. The maximum of absorbance of M-3 was situated at 385 nm, whereas the maximum absorbance of MeIgQx occurred at 365 nm (Figure 2). This large bathochromic shift is not observed in the UV spectrum of HONH-MeIQx, nor is this bathochromic shift observed in the UV spectra of the structurally related N-hydroxy-HAA derivatives of 2-amino-3-methylimidazo[4,5-f]quinoline or PhIP. However, the UV spectrum of the proposed N3-glucuronide conjugate of 2-(hydroxyamino)-1-methyl-6-phenylimidazo[4,5-b]pyridine (HONH-PhIP), which exists as an oxime, also displays a bathochromic shift of ~20 nm in its maximum absorbance in comparison to HOHN-PhIP.31,33 We surmise that HONH-MeIgQx exists preferentially as the oxime, and not as the N-hydroxylamine tautomer. The carcinogen 4-hydroxyamino quinoline 1-oxide was also reported to preferentially exist in its 4-hydroxyimino tautomeric form.34
The second most abundant metabolite of MeIgQx (M-2) displayed a UV spectrum similar that of MeIgQx. The mass spectrum of M-2 displayed a protonated ion at [M+H]+ at m/z 230.0, indicative of another mono-oxidation metabolite. Prominent ions were observed in the product ion spectrum at m/z 201.0, 200.0, and 186.1 (Figure 4). These fragment ions are proposed to arise through losses of HCO•, H2CO, and HCO• + CH3• . The product ion spectrum of M-2 is consistent with a ring-hydroxylated metabolite of MeIgQx: the site of oxidation may have occurred at the C-4 or C-9 position of the quinoxaline skeleton (Figure 1). If oxidation had occurred at the 7-CH3-group, a prominent fragment ion would have been expected at m/z 212.1 [M+H -18]+, due to the loss of H2O, as is observed in the product ion spectrum of 8-CH2OH-IQx (Figure 4).
The third metabolite of MeIgQx (M-1) displayed a UV spectrum that was very similar to spectrum of HONH-MeIgQx (M-3), with a maximum absorbance situated at 385 nm. The full scan mass spectrum of M-1 displayed a protonated ion [M+H]+ at m/z 246.1, indicating that the metabolite is a dihydroxylated product of MeIgQx. Prominent ions are observed in the product ion spectrum at m/z 228.1 and 229.1, indicative of the loss of 17 and 18 Da and suggestive of the presence of the N-hydroxylamine moiety (Figure 4). The base peak, observed at m/z 200.1, is ascribed to the loss H2O, followed by loss of CO. Thus, the product ion spectrum is consistent with the structure of a dihydroxlyated metabolite of MeIgQx that contains the N-hydroxylamine moiety with the second site of hydroxylation possibly occurring at the C-4 or C-9 position of the quinoxaline ring.
8-CH2OH-HONH-IQx and the dihydroxylated metabolite of MeIgQx (M-1) were reacted with nitrosobenzene to form azoxy adducts. Protonated molecules [M+H]+ were observed at m/z 335.2, a mass that is consistent with the molecular weight of the proposed azoxy conjugates (Figure S-1, Supporting Information). The product ion spectrum of the azoxy derivative of 8-CH2OH-HONH-IQx displays prominent ions at m/z 317.1 and 299.3, which are ascribed to loss of one and two molecules of H2O, whereas, the product ion spectrum of the azoxy derivative of M-1 displays prominent fragment ions at m/z 317.2 and 289.2, which are consistent with the loss of H2O and H2O+CO. These mass spectral data provide additional support for the secondary site of oxidation proposed to occur at the quinoxaline ring and not the 7-methyl group of MeIgQx.
Estimated Rates of P450-Mediated Oxidation of MeIQx and MeIgQx
The estimated rates of N- and C-oxidation of MeIQx and MeIgQx, by mouse and human liver microsomes and recombinant human P450s, are summarized in Table 1. The N-oxidation rates of MeIgQx and MeIQx with mouse liver microsomes were comparable, whereas the rate of N-oxidation of MeIQx was about 4-fold higher than for MeIgQx with human liver microsomes. MeIgQx underwent N-oxidation by recombinant human P450 1A2, but also by extrahepatic P450s 1A1 and 1B1. Moreover, the rate of MeIgQx N-oxidation of P450 1A1 was ~3-fold greater than the rate of N-oxidation carried out by P450 1A2. Human P450 1A2 appears to be involved only in N-oxidation (bioactivation), whereas P450 1A1 and P450 1B1 catalyze both N-oxidation and ring-oxidation (detoxication) of MeIgQx. In contrast to MeIgQx, the N-oxidation of MeIQx by P450s 1A1 and 1B1 was low to not detectable.
Table 1.
Estimated N-Oxidation and C-oxidation rates of MeIg Qx and MelQx with liver microsomes or recombinant human P450s
| nmol/min/mg microsomal protein or 100 pmol P450 | ||||||
|---|---|---|---|---|---|---|
| Enzyme | HONH-MeIg Qx | HO-MeIg Qx | HONH-(HO)-MeIg Qx | HONH-MeIQx | HONH-8-CH2OH-IQx | 8-CH2OH-IQx |
| Mouse liver microsomes | 0.3 ± 0.1 | 0.5 ± 0.2 | ND | 0.2 ± 0.01 | ND | ND |
| Human liver microsomes | 0.4 ± 0.02 | 0.1 ± 0.01 | ND | 1.7 ± 0.2 | 0.09 ± 0.01 | ND |
| Recombinant human P450 1A1 | 14.1 ± 4.9 | 6.9 ± 2.4 | ND | 0.3 ± 0.1 | ND | ND |
| Recombinant human P450 1A2 | 5.4 ± 2.1 | ND | ND | 6.1 ± 0.9 | 1.7 ± 0.5 | ND |
| Recombinant human P450 1B1 | 3.9 ± 0.6 | 2.5 ± 0.3 | ND | ND | ND | ND |
Substrate = 200 μ M; Mean ± SD, N = 3 or 4 independent measurements
ND = not detected (< 0.05 nmol/min/mg protein or <0.05 nmol/min/100 pmol P450)
In Vivo DNA Binding Studies in Mice
Plasma Clearance
The study design was not intended to completely describe the rate of absorption and clearance profile of these chemicals. However, the data obtained from the 3 time points show that the kinetics of absorption and elimination of both 14C-MeIgQx and 14C-MeIQx were rapid. The peak plasma levels in radioactivity associated with both compounds occurred at 4 h, and greater than 95% of the radioactivity was cleared from plasma within 48 h (Figure 5).
Figure 5.
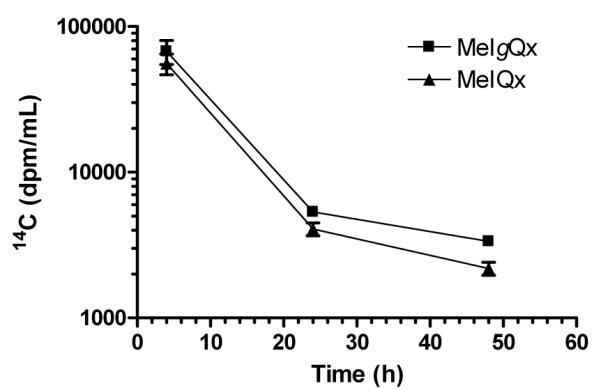
Plasma clearance of [14C]-MeIQx and [14C]-MeIgQx. Data represent the means and the standard deviations of 5 animals per time point.
DNA Adducts
The apparent levels of [14C]-MeIgQx- and [14C]-MeIQx-DNA adduct formation and removal in liver, pancreas and colorectal DNA are presented in Figure 6. The clearance of 14C-isotope associated with DNA was less rapid than the clearance of 14C-radioactivity associated with either carcinogen from plasma. The [14C]-MeIgQx isotope associated with liver DNA at T 24 and T 48 h was 33% and 27% of the 14C-isotope detected at 4 h, whereas the isotope level associated to pancreas DNA was 44% and 17% of the level of 14C-isotope bound to DNA at 4h. The [14C]-MeIQx isotope associated with liver and pancreas DNA also appeared to persist: up to 74% and 25% of the 14C-isotope observed in DNA of these organs at 4 h remained, respectively, at 48 h. The [14C]-MeIgQx isotope in colorectal DNA reached its maximum at 24 h, while the highest level of the [14C]-MeIQx isotope in colorectal DNA was observed at 4 h. The disposition and elimination of MeIgQx in the mouse is not known. The fecal content may contain high levels of 14C isotope, resulting in high levels of exposure to this carcinogen, perhaps at greater levels than most other tissues.
Figure 6.

The level of apparent [14C]-MeIQx and [14C]-MeIgQx DNA adduct formation in liver, pancreas and colon as a function of time. Data represent the means and the standard error of the means of 5 animals per time point. The values of adduct formation were normalized to adducts per nucleotide/mg HAA/kg body weight .
LC-ESI/MS3 of DNA Adducts
The formation of DNA adducts of [14C]-MeIQx and [14C]-MeIgQx was examined by LC-ESI/MS3 with a linear quadrupole ion trap mass spectrometer. The ion trap MS permits multi-stage tandem scanning for structural characterization and identification of the aglycone adducts [BH2]+.18,35 Two isomeric dG adducts of MeIQx, dG-C8-MeIQx and dG-N2-MeIQx are formed in vivo in liver of rats19 and in human hepatocytes.36 In addition, a third adduct is formed with dA; the structure has been tentatively assigned as 5-(deoxyadenosin-N6-yl)-MeIQx (dA-N6-MeIQx) (Figure 7).37
Figure 7.

Chemical structures of dG-C8-MeIQx, dG-N2-MeIQx and dA-N6-MeIQx
The stable isotope dilution method was used to measure the levels of dG-C8-MeIQx formed in mouse liver at the 24 h time point. Reconstructed ion chromatograms for dG-C8-MeIQx, dG-C8-[14C]-MeIQx, [13C10]-dG-C8-MeIQx and a putative isomeric dG-C8-MeIgQx at the MS3 scan stage are presented in Figure 8. Both the dG-C8-MeIQx and dG-C8-[14C]-MeIQx adducts are observed. The product ion spectra of the unlabeled and labeled adducts, at the MS3 scan stage, confirmed the structure of the adducts.19,35
Figure 8.

Reconstructed ion chromatograms at the MS3 scan stage for (A) dG-C8-MeIQx, dG-C8-[14C]-MeIQx, and [13C10]-dG-C8-MeIQx, (B) putative dG-C8-MeIgQx and dG-C8-[14C]-MeIgQx with [13C10]-dG-C8-MeIQx employed as an internal standard. The product ion spectra at the MS3 scan stage of dG-C8-MeIQx, dG-C8-[14C]-MeIQx and [13C10]-dG-C8-MeIQx are depicted in panel C.
The estimate of total [14C]-MeIQx-DNA adduct formation in liver 24 h post treatment, based upon AMS measurements, was 11.4 ± 7.6 adducts per 108 nucleotides versus to 3.8 ± 0.6 dG-C8-MeIQx adducts per 108 nucleotides, based on quantitative LC-ESI/MS3 measurements. The contribution of dG-C8-MeIQx to total 14C bound to DNA ranged from 13 - 72% of the radioactivity. The relative response of the signals for dG-N2-MeIQx and dA-N6-MeIQx adducts, by LC-ESI/MS3, are ~5% of the response of the signal observed for dG-C8-MeIQx.37 The dG-N2-MeIQx and dA-N6-MeIQx adducts are formed in rat liver at levels that are respectively ~30 - 50% and ~5% of the level of dG-C8-MeIQx, 24 h following oral dosing with MeIQx (10 mg/kg).19,37 If the relative level of formation of isomeric dG-MeIQx and dA-MeIQx adducts are similar in rat and mouse liver, the weak response in signals of dG-N2-MeIQx and dA-N6-MeIQx would preclude their detection, by LC-ESI/MS3, in mouse liver DNA. Thus, the contribution of dG-N2-MeIQx and dA-N6-MeIQx adduct formation to the amount of [14C]-MeIQx bound to DNA has not been taken into account, and the 14C-isotope associated to the DNA as covalently bound adducts likely exceeds the contribution made by the dG-C8-MeIQx adduct. In a previous AMS study conducted with male C57BL/6 mice treated with [14C]-MeIQx, the formation of liver adducts was shown to be linearly dependent on dose from an exposure of 5 mg down to 500 ng per kg of body weight).38 The amount [14C]-MeIQx bound to liver DNA was estimated at about 20 adducts per 108 nucleotides at 24 h post-treatment with [14C]-MeIQx (5 mg/kg). This estimate in 14C-adduct formation is remarkably similar to the level of [14C]-MeIQx bound to liver DNA, when adjusted per dose, in our study.
In contrast to MeIQx, a putative dG-C8-MeIgQx adduct was not detected, when scanning at the MS2 or MS3 scan stages. The potential formation of a dA adduct of MeIgQx ([M+H]+ at m/z 463.2) was also monitored, but no peaks were detected above background level. The data-dependent constant neutral loss-triple stage mass spectrometry scanning mode (CNL/MS3) was employed to monitor for the loss of the deoxyribose moiety, followed by MS3 of the putative aglycone adducts [BH2]+.37 However, the CNL/MS3 mode also failed to detect any adducts of MeIgQx.
Carcinogen Binding Index (CBI)
The CBI is a measure of the amount of adduct formed from a given dose; it is calculated by dividing the amount of adduct per unit of DNA by the dose per unit of body mass (μmol adduct bound per mole nucleotide)/(mmol chemical administered per kg animal).39 The CBI of MeIgQx and MeIQx in liver, pancreas and colorectum are presented in Table 2. The CBI data are reported at the T 4, 24 and 48 h time points. The CBI values for liver DNA adduct formation of MeIgQx and MeIQx were not significantly different at T 4 and 24 h (P > 0.4, student’s unpaired t test); however, the CBI values were significantly different at T 48 h (P <0.005). These data are suggestive of a faster rate of apparent MeIgQx-DNA adduct removal than for MeIQx-DNA adducts in liver; additional time points are required to accurately determine the rate of adduct elimination. For extrahepatic tissues, there was a significant difference (P < 0.01) between MeIgQx and MeIQx CBI values of pancreas at 24 h and colorectal tissue at 48 h.
Table 2.
Covalent Binding Index of MeIQx and MeIg Qx
| Liver: (μ mol/mol dNp)/(mmol carcinogen/kg bw) | ||
|---|---|---|
| Time | MeIgQx | MeIQx |
| 4 | 21.7 ± 8.6 | 27.7 ± 8.3 |
| 24 | 7.0 ± 3.2 | 11.5 ± 7.7 |
| 48 | 5.8 ± 0.6 | 20.5 ± 8.4 |
| Pancreas: (μ mol/mol dNp)/(mmol carcinogen/kg bw) | ||
| Time | MeIgQx | MeIQx |
| 4 | 2.4 ± 2.4 | 0.8 ± 0.2 |
| 24 | 1.1 ± 0.6 | 0.2 ± 0.1 |
| 48 | 0.2 ± 0.2 | 0.1 ± 0.1 |
| Colon: (μ mol/mol dNp)/(mmol carcinogen/kg bw) | ||
| Time | MeIgQx | MeIQx |
| 4 | 12.0 ± 6.5 | 113 ±119 |
| 24 | 138 ±114 | 10.4 ± 11.9 |
| 48 | 28.7 ± 18.5 | 2.1 ± 0.9 |
The mean ± SD of 4 or 5 animals per time point
Discussion
This study was designed to examine the capacity of MeIgQx to undergo metabolic activation to form DNA adducts in comparison with its isomer, MeIQx, a multi-site carcinogen in rodents.1 The metabolism and DNA binding data provide a preliminary assessment of the genotoxic potential of MeIgQx relative to MeIQx, which also induces lacI mutations in liver and colon40 and aberrant crypt foci in C57BL/6N mice,41 the species employed for our DNA binding studies. The estimated rates of P450-mediated N-oxidation of MeIgQx with mouse liver microsomes and the in vivo 14C-binding data of MeIgQx, when adjusted for dose, are comparable to those data obtained for MeIQx. We employed the highly sensitive AMS method for adduct detection,17,38 which enabled us to estimate DNA adduct formation based upon 14C isotope bound to DNA, since the structures of the MeIgQx adducts are unknown.
MeIQx and other structurally related HAAs studied induce tumors at multiple sites in rodents during long-term feeding studies. The target organs include the oral cavity, liver, stomach, colon, pancreas, and the prostate gland in males, and the mammary gland in females.1,42 The total dose required to induce tumor formation (TD50) varies for each HAA and is host species-dependent. The TD50 values of the individual HAAs have been reported to range from 0.1 to 64.6 mg/kg/day in rodents.1 MeIQx is a liver carcinogen in CDF1 mice:1 the TD50 value of MeIQx in this mouse strain is 24 mg/kg (http://potency.berkeley.edu/chempages/MeIQx.html).
The CBI approach has been used in rodent models as a means to assess the carcinogenic potential of genotoxic chemicals.43 A CBI value of 1-10 is characteristic of weak carcinogens, while moderate carcinogens display CBI values of about 100.39 The CBI for both MeIQx and MeIgQx in liver were comparable at T4 and 24 h (CBI = 7 - 28). These findings suggest that MeIgQx would be a weak to moderate liver carcinogen in this animal model. The [14C]-MeIgQx isotope is also associated with DNA in pancreas and colorectal tissue, suggesting that tumors could form in these organs as well. However, the presence of DNA adducts alone is insufficient to predict the development of cancer as carcinogenesis involves numerous biological events.44 Moreover, dietary, environmental, and genetic factors can greatly impact the biological potency of HAAs and other genotoxicants.1,45,46
Thus far, we have not successfully identified MeIgQx-DNA adducts by LC-ESI/MSn. We surmise that the linear fused tricyclic ring structure of MeIgQx inhibits the efficacy of enzymatic digestion of the MeIgQx-adducted to DNA, and the MeIgQx adducts may be present as incompletely digested oligomers, which escaped detection by LC-ESI/MSn. Alternatively, the instrument tuning parameters of the mass spectrometer may not have been optimal for detection of MeIgQx-DNA adducts, or the sensitivity in response of MeIgQx-DNA adducts is at least 10-fold weaker than the signal of response for dG-C8-MeIQx. We have not been able to synthesize HONH-MeIgQx by methods previously employed for the production of other HONH-HAAs.12 Studies that examine the reactivity of synthetic HONH-MeIgQx with deoxynucleosides and DNA are required to identify and elucidate the structures of MeIgQx-DNA adducts.
HAAs have been classified as probable human carcinogens by IARC and by the National Toxicology Program, sponsored by NIEHS.6,7 MeIgQx is the most mass abundant HAA formed in cooked beef.4,11 MeIgQx is a bacterial mutagen; however its potency in the Ames reversion assay is about 4,000-fold lower than MeIQx, in S. typhimurium tester strain TA98. The genotoxic potency of MeIgQx is 16-fold greater in S. typhimurium strain YG1024 than in S. typhimurium strain TA98. The YG1024 strain is derived from strain TA98 and contains high O-acetyltransferase activity that enhances the mutagenicity of some HAAs, presumably through the formation of reactive N-acetoxy intermediates, which covalently binds to DNA.47 The higher potency of MeIgQx in the YG1024 bacterial tester strain compared to TA98, indicates that the HONH-MeIgQx undergoes O-acetylation to form the N-acetoxy intermediate, as does MeIQx.11 MeIgQx displays about 10-fold weaker mutagenic potency than the structurally related carcinogen PhIP (3,015 rev/μg) in S. typhimurium strain YG1024. It is noteworthy that the mutagenic potency of MeIgQx is comparable to the potency of 4-aminobiphenyl, a known human carcinogen, in TA98 and YG1024 (frameshift-specific) and YG1029 (primarily point mutation-specific) tester strains.48-50 The strong mutagenic potencies of MeIQx and several other HAAs of angular tricyclic ring structure in the S. typhimurium TA98 strain have been attributed to the ability of these HAAs to frequently induce reversions about 9 base pairs upstream of the original CG deletion in the hisD+ gene in a run of GC repeats.51 This sequence context may not be a hot-spot for MeIgQx and PhIP adduct formation, or the capacities of MeIgQx and PhIP adducts to induce mutations is weaker than the potencies of adducts formed by MeIQx and other angular tricyclic HAAs. The mutagenicity of MeIgQx has not been assessed in mammalian cell systems. Such studies would provide additional data about the biological effects of MeIgQx in comparison to carcinogenic HAAs, such as MeIQx and PhIP, and aid in assessing the health risk of this newly discovered HAA.
Both MeIgQx and MeIQx undergo N-oxidation by mouse liver microsomes and by recombinant human P450 1A2 at similar rates. Moreover, recombinant human P450s 1A1 and 1B1, which are expressed in extrahepatic tissues,25,26 are far more efficient at catalyzing the N-oxidation of MeIgQx than the N-oxidation of MeIQx (Table 1). The differences in chemical structures of MeIgQx and MeIQx appear to affect the regioselectivities of P450-mediated N-oxidation of the exocyclic amine groups, as well as the quinoxaline ring structures and methyl groups of both HAAs. The higher CBI values of putative MeIgQx adducts formed in pancreas in comparison to MeIQx may be attributed to more efficient bioactivation of MeIgQx by extrahepatic P450s.
Historically, short-term bacterial mutagenesis assays have been an effective screening tool for the identification of some mutagenic HAAs in complex food matrices, but they do not reliably predict carcinogenic potency in mammals.52 MeIgQx forms apparent DNA adducts at levels that are comparable to the levels of adducts formed by its carcinogenic isomer, MeIQx, when adjusted for dose. Given the elevated concentrations of MeIgQx formed in cooked meats, further studies on the genetic toxicology and health risk of this novel HAA are clearly warranted.
Supplementary Material
Acknowledgements
Funding Source: This research was supported by grant R03ES016613 (E.E.B and R.J.T.) from the National Institute of Environmental Health Sciences, and by grant number 05B025 from the American Institute for Cancer Research (R.J.T.), and 2-PO1-ES006052 (R.L and P.L.S) from the National Institute of Environmental Health Sciences.
Abbreviations
- AMS
accelerator mass spectrometry
- CBI
carcinogen binding index
- CNL/MS3
constant neutral loss-triple stage mass spectrometry scanning mode
- HAA
heterocyclic aromatic amine
- LC-ESI/MS
Liquid chromatography-electrospray ionization/mass spectrometry
- MeIgQx
2-amino-1,7-dimethylimidazo[4,5-g]quinoxaline
- HONH-MeIgQx
2-hydroxamino-1,7-dimethylimidazo[4,5-g]quinoxaline
- MeIQx
2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline
- HONH-MeIQx
2-hydroxyamino-3,8-dimethylimidazo[4,5-f]quinoxaline
- 8-CH2OH-IQx
2-amino-8-(hydroxymethyl)-3-methylimidazo[4,5-f]quinoxaline
- HONH-8-CH2OH-IQx
2-(hydroxyamino)-8-hydroxymethyl-3-methylimidazo[4,5-f]quinoxaline
- PhIP
2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- dG-C8-MeIQx
N-(deoxyguanosin-8-yl)-MeIQx, dG-N2-MeIQx, 5-(deoxyguanosin-N2-yl)-MeIQx
- dA-N6-MeIQx
5-(deoxyadenosin-N6-yl)-MeIQx
- ppb
part per billion
Footnotes
Supporting Information Available: Additional information as noted in text (Figure S-1). This material is available free of charge via the Internet at http://pubs.acs.org.
Reference List
- 1.Sugimura T, Wakabayashi K, Nakagama H, Nagao M. Heterocyclic amines: Mutagens/carcinogens produced during cooking of meat and fish. Cancer Sci. 2004;95:290–299. doi: 10.1111/j.1349-7006.2004.tb03205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Felton JS, Jagerstad M, Knize MG, Skog K, Wakabayashi K. Contents in foods, beverages and tobacco. In: Nagao M, Sugimura T, editors. Food Borne Carcinogens Heterocyclic Amines. John Wiley & Sons Ltd.; Chichester, England: 2000. pp. 31–71. [Google Scholar]
- 3.Sinha R, Rothman N, Salmon CP, Knize MG, Brown ED, Swanson CA, Rhodes D, Rossi S, Felton JS, Levander OA. Heterocyclic amine content in beef cooked by different methods to varying degrees of doneness and gravy made from meat drippings. Food Chem. Toxicol. 1998;36:279–287. doi: 10.1016/s0278-6915(97)00162-2. [DOI] [PubMed] [Google Scholar]
- 4.Ni W, McNaughton L, LeMaster DM, Sinha R, Turesky RJ. Quantitation of 13 heterocyclic aromatic amines in cooked beef, pork, and chicken by liquid chromatography-electrospray ionization/tandem mass spectrometry. J. Agric. Food Chem. 2008;56:68–78. doi: 10.1021/jf072461a. [DOI] [PubMed] [Google Scholar]
- 5.International Agency for Research on Cancer . IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Tobacco smoking. International Agency for Research on Cancer; Lyon, France: 1986. [PMC free article] [PubMed] [Google Scholar]
- 6.IARC Monographs on the Evaluation of Carcinogenic Risks to Humans . Some naturally occurring substances: food items and constituents, heterocyclic aromatic amines and mycotoxins. International Agency for Research on Cancer; Lyon, France: 1993. [Google Scholar]
- 7.National Toxicology Program. National Toxicology Program . Report on Carcinogenesis. Eleventh Edition U.S. Department of Health and Human Services; Public Health Service, Research Triangle Park, N.C.: 2005. [Google Scholar]
- 8.Knize MG, Dolbeare FA, Carroll KL, Moore DH, Felton JS. Effect of cooking time and temperature on the heterocyclic amine content of fried beef patties. Food Chem. Toxicol. 1994;32:595–603. doi: 10.1016/0278-6915(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 9.Holland RD, Taylor J, Schoenbachler L, Jones RC, Freeman JP, Miller DW, Lake BG, Gooderham NJ, Turesky RJ. Rapid biomonitoring of heterocyclic aromatic amines in human urine by tandem solvent solid phase extraction liquid chromatography electrospray ionization mass spectrometry. Chem. Res. Toxicol. 2004;17:1121–1136. doi: 10.1021/tx049910a. [DOI] [PubMed] [Google Scholar]
- 10.Turesky RJ, Taylor J, Schnackenberg L, Freeman JP, Holland RD. Quantitation of carcinogenic heterocyclic aromatic amines and detection of novel heterocyclic aromatic amines in cooked meats and grill scrapings by HPLC/ESI-MS. J Agric. Food Chem. 2005;53:3248–3258. doi: 10.1021/jf048290g. [DOI] [PubMed] [Google Scholar]
- 11.Turesky RJ, Goodenough AK, Ni W, McNaughton L, LeMaster DM, Holland RD, Wu RW, Felton JS. Identification of 2-amino-1,7-dimethylimidazo[4,5-g]quinoxaline: an abundant mutagenic heterocyclic aromatic amine formed in cooked beef. Chem Res. Toxicol. 2007;20:520–530. doi: 10.1021/tx600317r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turesky RJ, Lang NP, Butler MA, Teitel CH, Kadlubar FF. Metabolic activation of carcinogenic heterocyclic aromatic amines by human liver and colon. Carcinogenesis. 1991;12:1839–1845. doi: 10.1093/carcin/12.10.1839. [DOI] [PubMed] [Google Scholar]
- 13.Langouët S, Welti DH, Kerriguy N, Fay LB, Huynh-Ba T, Markovic J, Guengerich FP, Guillouzo A, Turesky RJ. Metabolism of 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline in human hepatocytes: 2-amino-3-methylimidazo[4,5-f]quinoxaline-8-carboxylic acid is a major detoxification pathway catalyzed by cytochrome P450 1A2. Chem. Res. Toxicol. 2001;14:211–221. doi: 10.1021/tx000176e. [DOI] [PubMed] [Google Scholar]
- 14.Turesky RJ, Rossi SC, Welti DH, Lay JO, Jr., Kadlubar FF. Characterization of DNA adducts formed in vitro by reaction of N-hydroxy-2-amino-3-methylimidazo[4,5-f]quinoline and N-hydroxy-2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline at the C-8 and N2 atoms of guanine. Chem. Res. Toxicol. 1992;5:479–490. doi: 10.1021/tx00028a005. [DOI] [PubMed] [Google Scholar]
- 15.Turesky RJ, Constable A, Richoz J, Varga N, Markovic J, Martin MV, Guengerich FP. Activation of heterocyclic aromatic amines by rat and human liver microsomes and by purified rat and human cytochrome P450 1A2. Chem. Res. Toxicol. 1998;11:925–936. doi: 10.1021/tx980022n. [DOI] [PubMed] [Google Scholar]
- 16.Gupta RC. 32P-postlabelling analysis of bulky aromatic adducts. IARC Sci. Publ. 1993;124:11–23. [PubMed] [Google Scholar]
- 17.Liberman RG, Tannenbaum SR, Hughey BJ, Shefer RE, Klinkowstein RE, Prakash C, Harriman SP, Skipper PL. An interface for direct analysis of (14)C in nonvolatile samples by accelerator mass spectrometry. Anal. Chem. 2004;76:328–334. doi: 10.1021/ac030181y. [DOI] [PubMed] [Google Scholar]
- 18.Goodenough AK, Schut HA, Turesky RJ. Novel LC-ESI/MS/MSn method for the characterization and quantification of 2′-deoxyguanosine adducts of the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by 2-D linear quadrupole ion trap mass spectrometry. Chem. Res. Toxicol. 2007;20:263–276. doi: 10.1021/tx0601713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paehler A, Richoz J, Soglia J, Vouros P, Turesky RJ. Analysis and quantification of DNA adducts of 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline in liver of rats by liquid chromatography/electrospray tandem mass spectrometry. Chem. Res. Toxicol. 2002;15:551–561. doi: 10.1021/tx010178e. [DOI] [PubMed] [Google Scholar]
- 20.Kato R, Yamazoe Y. Metabolic activation and covalent binding to nucleic acids of carcinogenic heterocyclic amines from cooked foods and amino acid pyrolysates. Jpn. J. Cancer Res. 1987;78:297–311. [PubMed] [Google Scholar]
- 21.Kato R. Metabolic activation of mutagenic heterocyclic aromatic amines from protein pyrolysates. CRC Crit. Rev. Toxicol. 1986;16:307–348. doi: 10.3109/10408448609037466. [DOI] [PubMed] [Google Scholar]
- 22.Turesky RJ. Interspecies metabolism of heterocyclic aromatic amines and the uncertainties in extrapolation of animal toxicity data for human risk assessment. Mol. Nutr. Food Res. 2005;49:101–117. doi: 10.1002/mnfr.200400076. [DOI] [PubMed] [Google Scholar]
- 23.Butler MA, Iwasaki M, Guengerich FP, Kadlubar FF. Human cytochrome P-450PA (P450IA2), the phenacetin O-deethylase, is primarily responsible for the hepatic 3-demethylation of caffeine and N-oxidation of carcinogenic arylamines. Proc. Natl. Acad. Sci. U. S. A. 1989;86:7696–7700. doi: 10.1073/pnas.86.20.7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guengerich FP, Turvy CG. Comparison of levels of several human microsomal cytochrome P-450 enzymes and epoxide hydrolase in normal and disease states using immunochemical analysis of surgical liver samples. J. Pharmacol. Exp. Ther. 1991;256:1189–1194. [PubMed] [Google Scholar]
- 25.Sutter TR, Tang YM, Hayes CL, Wo Y-YP, Jabs EW, Li X, Yin H, Cody CW, Greenlee WF. Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2*. J. Biol. Chem. 1994;269:13092–13099. [PubMed] [Google Scholar]
- 26.Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu. Rev. Pharmacol. Toxicol. 2003;43:149–173. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- 27.Hammons GJ, Milton D, Stepps K, Guengerich FP, Kadlubar FF. Metabolism of carcinogenic heterocyclic and aromatic amines by recombinant human cytochrome P450 enzymes. Carcinogenesis. 1997;18:851–854. doi: 10.1093/carcin/18.4.851. [DOI] [PubMed] [Google Scholar]
- 28.Crofts FG, Sutter TR, Strickland PT. Metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by human cytochrome P4501A1, P4501A2 and P4501B1. Carcinogenesis. 1998;19:1969–1973. doi: 10.1093/carcin/19.11.1969. [DOI] [PubMed] [Google Scholar]
- 29.Turesky RJ, Bracco-Hammer I, Markovic J, Richli U, Kappeler A-M, Welti DH. The contribution of N-oxidation to the metabolism of the food-borne carcinogen 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline in rat hepatocytes. Chem. Res. Toxicol. 1990;3:524–535. doi: 10.1021/tx00018a006. [DOI] [PubMed] [Google Scholar]
- 30.Turesky RJ, Parisod V, Huynh-Ba T, Langouët S, Guengerich FP. Regioselective differences in C(8)- and N-oxidation of 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline by human and rat liver microsomes and cytochromes P450 1A2. Chem. Res. Toxicol. 2001;14:901–911. doi: 10.1021/tx010035s. [DOI] [PubMed] [Google Scholar]
- 31.Langouët S, Paehler A, Welti DH, Kerriguy N, Guillouzo A, Turesky RJ. Differential metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in rat and human hepatocytes. Carcinogenesis. 2002;23:115–122. doi: 10.1093/carcin/23.1.115. [DOI] [PubMed] [Google Scholar]
- 32.Yamazoe Y, Abu-Zeid M, Manabe S, Toyama S, Kato R. Metabolic activation of a protein pyrolysate promutagen 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline by rat liver microsomes and purified cytochrome P-450. Carcinogenesis. 1988;9:105–109. doi: 10.1093/carcin/9.1.105. [DOI] [PubMed] [Google Scholar]
- 33.Kadlubar FF, Kaderlik KR, Mulder GJ, Lin D-X, Butler MA, Teitel CH, Minchin RF, Ilett KF, Friesen MD, Bartsch H, Nagao M, Esumi E, Sugimura T, Lang NP. Metabolic activation and DNA adduct detection of PhIP in dogs, rats, and humans in relation to urinary bladder and colon carcinogenesis. In: Adamson RH, Gustafsson J-A, Ito N, Nagao M, Sugimura T, Wakabayashi K, Yamazoe Y, editors. Heterocyclic Amines in Cooked Foods: Possible Human Carcinogens; 23rd Proceedings of the Princess Takamatusu Cancer Society; Princeton, NJ: Princeton Scientific Publishing Co., Inc.; 1995. pp. 207–213. [PubMed] [Google Scholar]
- 34.Kawazoe Y, Ogawa O, Huang G-F. Studies on chemical carcinogens XVII: Structure of carcinogenic 4-hydroxaminoquinoline N-1 oxide derivatives. Tetrahedron. 1980;36:2933–2938. [Google Scholar]
- 35.Bessette EE, Spivack SD, Goodenough AK, Wang T, Pinto S, Kadlubar FF, Turesky RJ. Identification of carcinogen DNA adducts in human saliva by linear quadrupole ion trap/multistage tandem mass spectrometry. Chem. Res. Toxicol. 2010;23:1234–1244. doi: 10.1021/tx100098f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nauwelaers G, Bessette EE, Gu D, Tang Y, Rageul J, Fessard V, Yuan JM, Yu MC, Langouet S, Turesky RJ. DNA adduct formation of 4-aminobiphenyl and heterocyclic aromatic amines in human hepatocytes. Chem Res. Toxicol. 2011;24:913–925. doi: 10.1021/tx200091y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bessette EE, Goodenough AK, Langouet S, Yasa I, Kozekov ID, Spivack SD, Turesky RJ. Screening for DNA adducts by data-dependent constant neutral loss-triple stage mass spectrometry with a linear quadrupole ion trap mass spectrometer. Anal. Chem. 2009;81:809–819. doi: 10.1021/ac802096p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turteltaub KW, Felton JS, Gledhill BL, Vogel JS, Southon JR, Caffee MW, Finkel RC, Nelson DE, Proctor ID, Davis JC. Accelerator mass spectrometry in biomedical dosimetry: relationship between low-level exposure and covalent binding of heterocyclic amine carcinogens to DNA. Proc. Natl. Acad. Sci. U.S.A. 1990;87:5288–5292. doi: 10.1073/pnas.87.14.5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lutz WK. In vivo covalent binding of organic chemicals to DNA as a quantitative indicator in the process of chemical carcinogenesis. Mutat. Res. 1979;65:289–356. doi: 10.1016/0165-1110(79)90006-x. [DOI] [PubMed] [Google Scholar]
- 40.Itoh1 T, Suzuki T, Nishikawa A, Furukawa F, Takahashi M, Xue W, Sofuni T, Hayashi M. In vivo genotoxicity of 2-amino-3,8-dimethylimidazo[4, 5-f]quinoxaline in lacI transgenic (Big Blue) mice. Mutat. Res. 2000;468:19–25. doi: 10.1016/s1383-5718(00)00036-x. [DOI] [PubMed] [Google Scholar]
- 41.Okonogi H, Ushijima T, Shimizu H, Sugimura T, Nagao M. Induction of aberrant crypt foci in C57BL/6N mice by 2-amino-9H-pyrido[2,3-b]indole (AalphaC) and 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (MeIQx) Cancer Lett. 1997;111:105–109. doi: 10.1016/s0304-3835(96)04505-3. [DOI] [PubMed] [Google Scholar]
- 42.Shirai T, Sano M, Tamano S, Takahashi S, Hirose M, Futakuchi M, Hasegawa R, Imaida K, Matsumoto K, Wakabayashi K, Sugimura T, Ito N. The prostate: A target for carcinogenicity of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) derived from cooked foods. Cancer Res. 1997;57:195–198. [PubMed] [Google Scholar]
- 43.Otteneder M, Lutz WK. Correlation of DNA adduct levels with tumor incidence: carcinogenic potency of DNA adducts. Mutat. Res. 1999;424:237–247. doi: 10.1016/s0027-5107(99)00022-6. [DOI] [PubMed] [Google Scholar]
- 44.Sugimura T. Multistep carcinogenesis: A 1992 perspective. Science. 1992;258:603–607. doi: 10.1126/science.1411570. [DOI] [PubMed] [Google Scholar]
- 45.Ghoshal A, Preisegger KH, Takayama S, Thorgeirsson SS, Snyderwine EG. Induction of mammary tumors in female Sprague-Dawley rats by the food-derived carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and effect of dietary fat. Carcinogenesis. 1994;15:2429–2433. doi: 10.1093/carcin/15.11.2429. [DOI] [PubMed] [Google Scholar]
- 46.Ubagai T, Ochiai M, Kawamori T, Imai H, Sugimura T, Nagao M, Nakagama H. Efficient induction of rat large intestinal tumors with a new spectrum of mutations by intermittent administration of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in combination with a high fat diet. Carcinogenesis. 2002;23:197–200. doi: 10.1093/carcin/23.1.197. [DOI] [PubMed] [Google Scholar]
- 47.Schut HA, Snyderwine EG. DNA adducts of heterocyclic amine food mutagens: implications for mutagenesis and carcinogenesis. Carcinogenesis. 1999;20:353–368. doi: 10.1093/carcin/20.3.353. [DOI] [PubMed] [Google Scholar]
- 48.Scribner JD, Fisk SR, Scribner NK. Mechanisms of action of carcinogenic aromatic amines: an investigation using mutagenesis in bacteria. Chem.-Biol. Interact. 1979;26:11–25. doi: 10.1016/0009-2797(79)90090-5. [DOI] [PubMed] [Google Scholar]
- 49.Thompson PA, DeMarini DM, Kadlubar FF, McClure GY, Brooks LR, Green BL, Fares MY, Stone A, Josephy PD, Ambrosone CB. Evidence for the presence of mutagenic arylamines in human breast milk and DNA adducts in exfoliated breast ductal epithelial cells. Environ. Mol. Mutagen. 2002;39:134–142. doi: 10.1002/em.10067. [DOI] [PubMed] [Google Scholar]
- 50.Radomski JL, Hearn WL, Radomski T, Moreno H, Scott WE. Isolation of the glucuronic acid conjugate of N-hydroxy-4-aminobiphenyl from dog urine and its mutagenic activity. Cancer Res. 1977;37:1757–1762. [PubMed] [Google Scholar]
- 51.Fuscoe JC, Wu R, Shen NH, Healy SK, Felton JS. Base-change analysis of revertants of the hisD3052 allele in Salmonella typhimurium. Mutat. Res. 1988;201:241–251. doi: 10.1016/0027-5107(88)90131-5. [DOI] [PubMed] [Google Scholar]
- 52.Sugimura T. Successful use of short-term tests for academic purposes: Their use in identification of new environmental carcinogens with possible risk for humans. Mutat. Res. 1988;205:33–39. doi: 10.1016/0165-1218(88)90006-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.