

Abstract
Mass spectrometry was used to probe the preferred locations of trans-4-hydroxy-2-nonenal (HNE) addition to the cysteine, histidine, and lysine residues of human serum albumin (HSA). Considering only those modified peptides supported by high mass accuracy Orbitrap precursor ion measurements (high confidence hits), with HNE:HSA ratios of 1:1 and 10:1, 3 and 15 addition sites, respectively, were identified. Using less stringent criteria, a total of 34 modifications were identified at the higher concentration. To gain quantitative data, iTRAQ labeling studies were completed. Previous work had identified Cys34, the only free cysteine, as the most reactive residue in HSA and we have found that Lys199, His242/7, and His288 are the next most reactive residues. Although the kinetic data indicate the lysines and histidines can react at relatively similar rates, the results show that lysine addition is much less favorable thermodynamically; under our reaction conditions, lysine addition generally does not go to completion. This suggests that under physiological conditions, HNE addition to lysine is only relevant in situations where unusually high HNE concentrations or access to irreversible secondary reactions are found.
Introduction
There has been great interest in the development of biomarkers that could be used to identify and measure either generalized or localized oxidative stress and potentially provide useful clinical data related to the onset of disease states.[1-6] Protein carbonylation has been an attractive target for these studies because this form of oxidative damage introduces a functional group not normally found in proteins, an aldehyde, and therefore offers a variety of opportunities in terms of detection, isolation, or concentration strategies.[7-13] It has been assumed that protein carbonyls are generally formed by two processes: (1) direct oxidative modification of one of the protein's amino acid side chains to give an aldehyde or (2) condensation of the protein with an α,β-unsaturated aldehyde, either introduced externally or formed by other oxidative processes such as lipid peroxidation.[14, 15] The former pathway is often associated with the conversion of arginine and lysine residues to glutamic semialdehyde and 2-aminoadipic semialdehyde, respectively, by the action of a strong oxidant like the hydroxyl radical. The latter pathway is commonly associated with the reaction of nucleophilic amino acid side chains with α,β-unsaturated aldehydes such as trans-4-hydroxy-2-nonenal (HNE), a key lipid peroxidation byproduct. It has been assumed that cysteine, histidine, and lysine are the most active amino acids in reactions with HNE.[16-19]
Modifications by HNE are known to have wide-ranging biological effects including the attenuation of enzyme activity, apoptosis, and neurotoxicity. The presence of HNE modifications correlates with many disease states including Alzheimer's, diabetes, and atherosclerosis. In addition, an increase in HNE protein modifications has been observed with aging. Finally, HNE has been implicated in signaling pathways, but details of its impact in this role are just emerging.[20] Overall, HNE has been viewed as a prime, potential biomarker for diseases or processes that induce oxidative stress[21-23] and has been used in several mass spectrometric studies of protein modification.[24-31]
For these modifications to become useful biomarkers, an important step is a careful characterization of the chemical processes, which includes an understanding of the selectivity and kinetics of the protein modification reactions. In 2006, Aldini and Liebler presented separate studies aimed at characterizing the reaction of HNE with human serum albumin (HSA).[32, 33] HSA is a natural choice for model studies because it is present in high concentration in serum, is known to form complexes and adducts with a variety of species, including HNE, is a known target of oxidative stress, and is readily available.[34-37] In the studies by Aldini and Liebler, the site selectivity and kinetics of the HNE additions were evaluated using mass spectrometric methods. Different approaches were taken by the two groups, but generally the data were in accord; however, there were some noteworthy exceptions. Specifically, Aldini identified additional highly active modification sites not seen in the Liebler study. Furthermore, both groups assumed that the reactions were kinetically controlled and based their analyses on that assumption. However, it is possible that some processes reach equilibrium under the reaction conditions and this would have a significant impact on the interpretation of kinetic data.
In the present work, we revisit the reaction of HNE with HSA and track the process as a function of time using an iTRAQ-labeling strategy.[38-40] We find that the kinetics are more complicated than may have been appreciated in previous work and have evidence that some processes may be controlled by thermodynamics rather than kinetics under conditions typically used for in vitro studies.
Experimental Procedures
Chemicals
Essentially fatty acid and globulin free HSA (Product A3782), iodoacetamide, 1,4-dithio-DL-threitol (DTT) and 10× PBS concentrate were purchased from Sigma-Aldrich (St. Louis, MO). Ammonium bicarbonate was obtained from J.T. Baker (Phillipsburg, NJ) and HNE was obtained from Cayman Chemical (Ann Arbor, MI). Sequencing grade modified trypsin was from Promega (Madison, WI). BCA Protein Assay kits were purchased from Pierce (Rockford, IL) and iTRAQ reagent kits were obtained from Applied Biosystems (Foster City, CA).
Experiments
HSA modification
HSA at 15 μM in 1× PBS buffer (pH 7.4) was incubated with HNE at various final HNE:HSA molar ratios (1:4, 1:2, 1:1, 2:1, 5:1, 10:1, 50:1, 100:1). All reactions were carried out at 37°C with gentle shaking. The reaction time was 3 h for all experiments except those in which the reaction time was intentionally varied. Modified proteins were stabilized by adding NaBH4 to 5 mM and incubating for 60 min at room temperature. Repeated washes employing centrifugal filter devices (VWR, West Chester, PA) were used to remove reagents and to exchange to 50 mM ammonium bicarbonate buffer (pH 8.0) for enzymatic digestion; the molecular weight cutoff for the centrifugal filter devices used was 30 kDa.
Enzymatic digestion
Modified HSA samples in 50 mM ammonium bicarbonate buffer were first reduced by incubating with 30 mM DTT for 20 min at 50°C and then alkylated by incubating with 55 mM iodoacetamide for 30 min at room temperature in the dark. Excess DTT and iodoacetamide were removed using the centrifugal filter devices (three washes were performed using 50 mM ammonium bicarbonate buffer). Sequencing grade trypsin (substrate to enzyme weight ratio 40:1) was added and the mixture incubated for 24 h at 37°C. Digestion was terminated by adding 1% formic acid (final pH = 2∼3).
iTRAQ reagent labeling
1. Effect of varying HNE:HSA molar ratio
After digestion with trypsin, the concentrations of the HNE-modified peptide mixtures were measured using the BCA protein assay (error has been reported to be less than 5%[41]). Peptide mixtures prepared under four conditions (50 μg each) were placed in four different microcentrifuge tubes (two control samples, i.e., no HNE in prep, plus samples from 50:1 and 100:1, HNE:HSA experiments). Peptide mixtures were then dried using a centrifugal evaporator and reconstituted in 25 μl iTRAQ dissolution buffer. The iTRAQ reagents (114-117) were dissolved in 70 μl ethanol separately. Each iTRAQ reagent aliquot was added to one of the peptide mixtures in the following sequence: 114 to a control sample; 115 to 50:1 ratio sample; 116 to 100:1 ratio sample; and 117 to a control sample. After incubation for 2 h at room temperature, all four peptide mixtures were combined and then purified using a Waters (Milford, MA) Oasis MCX solid phase extraction cartridge. The final sample was dried using a centrifugal evaporator and then resuspended in HPLC equilibration mobile phase for μLC-MS/MS analysis.
2. Effect of varying reaction time
HSA was incubated with HNE at a HNE:HSA molar ratio of 100:1. The reactions were quenched by adding NaBH4 at 1, 3, and 24 h. After digestion, peptide mixtures from different time frames (containing 50 μg total peptides) were labeled separately using the four iTRAQ reagents: 114 for the 0 h sample (control); 115 for the 1 h sample; 116 for the 3 h sample; and 117 for the 24 h sample. Labeling and subsequent processing were performed as described for the varying molar ratio experiment.
μLC-MS/MS analysis
1. HNE adduct identification
Adduct identification was performed using a Thermo (San Jose, CA) LTQ XL linear ion trap mass spectrometer, equipped with electron-transfer dissociation (ETD), and a Thermo LTQ Orbitrap Velos mass spectrometer. The LTQ XL was interfaced with a Thermo Surveyor capillary HPLC system. Peptides were separated on a reversed-phase, C18 column (150 μm × 10 cm, 5 μm particles, 300 Å pores; Column Technology, Fremont, CA) at a flow rate of ∼1 μl min−1 using 0.1% formic acid in water as mobile phase A and 0.1% formic acid in methanol as mobile phase B. Approximately 2 μg peptides were injected and a Michrom (Auburn, CA) CapTrap trapping column was used for rapid sample injection. The gradient started from 2% B, then increased to 15% B over 5 min, then increased to 80% B over 70 min, and finally increased to 95% B over 15 min. The eluted peptides were introduced into the LTQ XL with a nanospray source operating at a spray voltage of 2.1 kV, a capillary voltage of 21 V, and a capillary temperature of 200°C. A full scan in the m/z range 300-2000 was performed to obtain precursor ions, followed by six data-dependent MS/MS scans (consisting of alternating collision-induced dissociation (CID) and ETD scans) for the three most abundant precursor ions in the full scan. Dynamic exclusion was used, that is, if the same precursor ion was picked for fragmentation twice within a 30 s window, it was excluded from further analysis for 180 s. For the Thermo LTQ Orbitrap Velos system, separations were performed on a Waters nanoACQUITY reversed-phase, C18 column (100 μm × 10 cm; 1.7 μm particles). Elution was achieved using a gradient of 0.1% formic acid in acetonitrile (B) versus 0.1% formic acid in water (A) at a flow rate of 0.4 μL min−1. Approximately 2 μg peptides were injected, with the loading and equilibration mobile phase being 1% B. The linear gradient ran to 35% B over the first 30 min and then to 85% B over the next 5 min. The nanospray ion source was operated at 3.5 kV.
2. iTRAQ reagent-labeled peptide quantification
For iTRAQ reagent-labeled samples, the linear ion trap-based system was operated largely as described above for HNE adduct identification except that pulsed-Q dissociation (PQD) replaced CID and ETD. The top four most abundant ions in each precursor ion scan were subjected to PQD fragmentation. Settings for PQD were normalized collision energy at 36%, activation Q at 0.7, and activation time at 0.1 ms. A targeted mass list was used and, therefore, dynamic exclusion was not enabled. Similarly, for the Orbitrap-based system, most operating parameters remained the same as those used for HNE adduct identification except that higher-energy C-trap dissociation (HCD)[42] replaced CID. For HCD, the normalized collision energy was 40 and the activation time was 0.1 ms; the top eight most abundant ions in each precursor ion scan were subjected to HCD fragmentation. Dynamic exclusion was not enabled and the same targeted mass list was used. The chromatographic gradient was also lengthened; after loading at 1% B there was an initial increase to 15% B over 25 min, followed by an increase to 25% B over 35 min, followed by an increase to 35% B over 40 min, followed by an increase to 85% B over 20 min.
Database searching and data processing
Peptide sequences and modifications were identified using the BioWorks version 3.3.1, SP1 implementation of Sequest (Thermo). No scan grouping was performed in preparing peak lists for database searching. The protein sequence database used consisted of the NCBI RefSeq version of the complete human proteome and the UniProt sequence for porcine trypsin (Accession Number P00761); reversed versions of all sequences were also included to permit false discovery rate estimation. Sequences were downloaded on November 20, 2010, and the final database contained 68040 entries. Only fully-tryptic peptides were considered and up to two missed cleavage sites were allowed. Precursor ion tolerances were ±2 Da for linear ion trap measurments and ±15 ppm for Orbitrap measurements. Fixed mass shifts were applied for alkylated cysteines (+57 Da) while differential amino acid mass shifts were incorporated for NaBH4-reduced Michael adducts at histidine and lysine (+158 Da) and at cysteine (+101 Da when the fixed mass shift at cysteine is considered), NaBH4-reduced Schiff base adducts at lysine (+140 Da), and oxidized methionines (+16 Da). In searches with the iTRAQ labels present, fixed mass shifts of +144 Da were used for the peptide N-terminus and non-carbonylated lysine residues, which resulted in the differential mass shifts associated with HNE modification at lysine being changed to +14 Da (Michael adducts) and −4 Da (Schiff base adducts). Addition of the iTRAQ tag at tyrosine (+144 Da differential modification) was also considered, but was found not to be common. Mass shifts were added to Sequest parameters files at high-precision (see Table S1 for non-iTRAQ and Table S2 for iTRAQ experiments) for compatibility with highly accurate Orbitrap precursor ion measurements and a maximum of three variable modifications were permitted for each peptide. Sequest output was refined using the Trans-Proteomic Pipeline (version 4.4; Institute for Systems Biology, Seattle, WA) software package. Specifically, PeptideProphet[43] was used, in semi-supervised mode[44], to improve identification confidence. A PeptideProphet score threshold of 0.9 was applied. In addition, MS/MS spectra for modified peptides were manually examined and any found to be inconsistent with the proposed identification were rejected (see Supplementary Material for sample spectra). Intensity measurements for iTRAQ reporter ions (114-117) were processed using Microsoft Access. Independently for each MS/MS, raw reporter ion counts were converted to relative percentages (that summed to 100% for each MS/MS). Then, for each identified peptide, results from supporting MS/MS were combined by averaging.
Results
As noted in the introduction, the major mode of action of HNE with proteins is Michael addition to the nucleophilic amino acid side chains of cysteine, histidine, and lysine to give stable adducts. HNE can also form a Schiff base with lysine.[17, 18] The relevant reaction products are given in Scheme 1. All of the Michael additions lead to products bearing an aldehyde functional group whereas the Schiff base contains an imine instead. HSA is a 66 kDa protein with 35 cysteines, 16 histidines, and 59 lysines in its secreted form. There are 17 disulfide bridges, which leaves a single free cysteine in the protein.[37] The net result is the possibility of 135 different single addition modifications by HNE; however, some would be highly unlikely if the nucleophilic residue does not have reasonable surface accessibility or is located in an environment that is too sterically crowded to accept the added HNE group.
Scheme 1. Typical HNE Modifications.

Modification Sites
Our first goal was to identify the key modification sites when HSA is treated with HNE. Starting with an HSA concentration of 15 μM, HNE:HSA ratios from 1:4 to 100:1 were surveyed and data are reported for the 1:1 and 10:1 ratios (Table 1). The exposure of HSA to HNE experiment was repeated five times at the 1:1 and 10:1 ratios. Four repeats were analyzed using the linear ion trap system while one was investigated using the Orbitrap system. In all cases, three replicate LC-MS/MS runs were recorded for each preparation. Modifications found using the linear ion trap instrument and validated by manual inspection of the MS/MS spectra were regarded as medium confidence; those confirmed with complementary data from the Orbitrap system were regarded as high confidence. These data are also presented in terms of modified peptide (rather than modification site as found in Table 1) as Table S3, for counts of LC-MS/MS runs in which the modified peptide was detected, and Table S4, for counts of modified peptide identifications across all runs. Most site identifications were supported by CID data, but numerous acceptable ETD detections were also obtained. Sample MS/MS spectra for each of the modified peptides are provided in the Supplementary Material.
Table 1.
High (bolded) and medium (italicized) confidence modification site identifications. Medium confidence hits based on low mass accuracy precursor ion measurements (linear ion trap) while high confidence hits included high mass accuracy precursor ion measurements (Orbitrap). Manual MS/MS validation was required in all cases.
| Modification Sitea | Typeb | 1:1c | 10:1c |
|---|---|---|---|
| Cys34 | MA | 9/1/0 | 9/3/3 |
| His67 | MA | 12/0/3 | 12/5/3 |
| His128 | MA | 0/2/0 | 2/3/0 |
| His146 | MA | 12/11/0 | 12/12/3 |
| His242 | MA | 8/7/0 | 11/11/0 |
| His247 | MA | not detected | 11/10/0 |
| His288 | MA | 10/0/0 | 12/1/3 |
| His338 | MA | 4/6/0 | 12/11/3 |
| His367 | MA | not detected | 9/0/0 |
| His510 | MA | 1/8/0 | 12/11/0 |
| Lys73 | MA | not detected | 3/0/0 |
| Lys106 | SB | not detected | 0/1/0 |
| Lys137 | MA | not detected | 1/0/0 |
| Lys159 | MA | not detected | 0/2/1 |
| Lys162 | MA | not detected | 12/2/2 |
| Lys162 | SB | not detected | 10/1/0 |
| Lys199 | MA | 6/0/2 | 11/0/3 |
| Lys212 | MA | not detected | 12/0/1 |
| Lys233 | MA | not detected | 9/4/0 |
| Lys240 | MA | 8/0/0 | 3/0/0 |
| Lys262 | MA | not detected | 5/3/0 |
| Lys351 | MA | 10/0/0 | 9/0/0 |
| Lys351 | SB | not detected | 3/1/0 |
| Lys359 | MA | not detected | 1/3/0 |
| Lys378 | MA | not detected | 5/1/0 |
| Lys402 | MA | not detected | 4/0/2 |
| Lys414 | MA | not detected | 11/1/3 |
| Lys414 | SB | 8/0/0 | 12/1/3 |
| Lys475 | MA | not detected | 1/0/0 |
| Lys519 | MA | not detected | 1/0/0 |
| Lys525 | MA | not detected | 0/1/3 |
| Lys525 | SB | 3/0/3 | 8/3/3 |
| Lys545 | MA | 4/7/0 | 11/11/3 |
| Lys545 | SB | not detected | 4/7/0 |
Secreted protein numbering; add 24 for nascent protein
MA indicates Michael adduct formation; SB indicates Schiff base formation
Applied HNE:HSA ratio. Values are counts of LC-MS/MS runs in which the modified site was identified using linear ion trap CID scans (maximum = 12)/linear ion trap ETD scans (maximum = 12)/linear ion trap CID scans associated with high mass accuracy Orbitrap precursor ion mass measurements (maximum = 3)
Naturally, as the amount of HNE was increased, it was possible to identify more modification sites. At a 1:1 ratio, only three sites were identified at high confidence: His67, Lys199, and Lys525 (the residue numbers are for the secreted protein; to convert to nascent protein numbering add 24). His67 and Lys199 were Michael adducts while Lys525 was a Schiff base. At 10:1, there are 15 high confidence modifications at 13 different residues (Table 1). In general, the identifications were very reproducible, but it is clear that in some cases the peptides were at a concentration that was close to the threshold for our identification criteria. In Liebler's work, ten modifications were identified with an HNE:HSA ratio of over 600:1.[33] Our full set (medium and high confidence) includes all of their set with the exception of Lys51 and His105. Michael addition at Lys51 was only detected in one out of ten experiments in Liebler's study while the presence of an alternative Michael addition site at Lys106 makes confident detection of addition at His105 challenging; however, modification at His105 can be confidently detected in experiments where iTRAQ labels are present and quantitative data were obtained for this site (see below). Aldini detected eleven modifications at their highest ratio (5:1).[32] In our studies, at a ratio of 10:1, we detected the majority of them at high confidence with the exceptions being Michael additions at His242 and His510, which we detected at medium confidence, and Schiff base formations at Lys195 and Lys199, which we did not detect. Identification of His242 is challenging because the underlying tryptic peptide contains a second addition site at His247, requiring very high quality MS/MS for certainty in modification localization. Aldini's identification of Schiff base formation at Lys195 required a careful manual search for new peaks in the HNE-treated sample's chromatogram (searches were conducted in chromatograms covering a series of mass ranges). With our global approach for identifying modifications, the observation of this modification site, which appeared to be a minor one, was much less likely.
It is not surprising that our approach apparently detected more modifications than the one used by Aldini, 15/34 (high/medium confidence) vs. 11, because we used a somewhat higher concentration (10:1 vs. 5:1) and a longer reaction time (3 h vs. 2 h). In addition, they relied to some extent on manual matching in the chromatograms so there was a possibility that some modification combinations were not considered, the full sensitivity of their instrument was not realized, or peaks were obscured in some way. On the other hand, their approach has advantages in some situations and led to identifications that we did not detect. It is more surprising that we identified significantly more sites than Liebler despite using methodologies that were fairly similar. It is unclear why so few modifications were identified in their study given the high concentrations employed, but nonetheless, there is reasonable consistency across the three studies of HNE/HSA reactivity in terms of the preferred sites of modification.
It is interesting to note that at a 1:1 ratio, the HNE addition appears to be relatively selective. Only two of the 59 lysines and one of the sixteen histidines are identified at high confidence as modified (considering both high and medium confidence identifications, there are four histidine and eight lysine modification sites at this concentration). It might be tempting to conclude that the reactions with histidine are less selective because a higher percentage of them are modified, but the larger number of modification sites is also driven by the fact that the reaction is more favorable (see below) and, therefore, at a given ratio, more of the HNE is naturally adducted to histidines.
Finally, there is evidence for HNE addition at the single free cysteine in HSA (Cys34) at both concentrations. Although it has been accepted that Cys34 is the most reactive Michael addition site,[34, 45] the MS/MS data for this site are not as consistent as those for other modifications. This does not appear to be the result of a low level of modification, but instead is probably due to cysteinylation at Cys34, a common post-translational modification that is often found in HSA preparations.[46] However, for all sites, poor detectability could be due to the ionizability of the corresponding peptide as well as the quality of its CID fragmentation pattern. Therefore, some care needs to be exercised in analyzing data from these types of experiments because effects other than concentration can have a major impact on the ability to identify modifications. As a result, more direct concentration measures, such as those from an iTRAQ-labeling scheme are needed for ranking reactivities.[40]
Relative Modification Levels Based on iTRAQ Labeling
In the iTRAQ labeling approach, peptides derived from proteins subjected to different conditions are tagged with a set of isobaric labeling reagents, one for each condition studied. The labeling reagents are identical, except for the distribution of isotopes in the tag. This difference leads to fragment ions (immonium) with unique masses that correlate with the conditions. The beauty of the approach is that peptides from each of the conditions will have the same mass and should coelute together, but once selected and fragmented will give characteristic peaks that are suitable for relative quantitation. One drawback of the approach is that the characteristic ions appear at low mass (114-117 in our case). In a quadrupole ion trap, such as that found in the LTQ XL instrument used in this work, it is very difficult to simultaneously trap low-mass iTRAQ reporter ions and the b- and y-ions needed for peptide identification because the mass window is fundamentally limited. However, in the LTQ XL, PQD can be used in place of CID to significantly widen the window and to simultaneously trap high- and low-mass ions (the approach shifts the qz value during the activation and fragmentation timeframes).[47] Although PQD allows implementation of the iTRAQ approach with ion trap instrumentation, the intensities of the label ions are rather low and extensive signal averaging is needed to obtain reproducible results (based on our results, single PQD scans for both peptide identification and reporter ion intensity measurements were more effective than collecting, for each precursor ion, a CID scan for identification and a PQD scan for reporter ion measurements). Consequently, the most efficient approach in these systems is to use a targeted mass list of anticipated peptides (modified and unmodified) rather than dynamically determining masses for fragmentation. By targeting masses and not employing a dynamic exclusion protocol, many more MS/MS spectra containing iTRAQ reporter ions can be recorded and averaged during a chromatographic run.
The mass list approach was also used with the LTQ Orbitrap Velos instrument, both to allow comparisons to be made and to improve accuracy by collecting as many measurements as possible. A list of the 21 modified peptides targeted in these studies is given in Table 2; 20 modifications at 18 residues were considered (two variants are present for one modification to take account of the possibility of methionine oxidation). In addition, unmodified versions of the listed modified peptides and peptides resulting from cleavage with trypsin of unmodified versions of the listed modified peptides were also targeted (a full list of all targeted peptides with m/z values used is given as Table S5). Since it was not practical to monitor all possible modification sites in HSA in this way (there are hundreds of potential peptide masses), sites that had been identified previously by other workers as well as sites identified in the present study whose spectra suggested significant modification levels were included.
Table 2.
List of modified peptides included in the targeted mass list
| Modified Peptidea | Modification Siteb | Typec |
|---|---|---|
| ALVLIAFAQYLQQC#PFEDHVK | Cys34 | MA |
| SLH@TLFGDK | His67 | MA |
| NECFLQH@K | His105 | MA |
| VH@TECCHGDLLECADDR | His242 | MA |
| VHTECCH@GDLLECADDR | His247 | MA |
| SH@CIAEVENDEM*PADLPSLAADFVESK | His288 | MA |
| SH@CIAEVENDEMPADLPSLAADFVESK | His288 | MA |
| CCAAADPH@ECYAK | His367 | MA |
| EFNAETFTFH@ADICTLSEK | His510 | MA |
| LVNEVTEFAKˆTCVAD | Lys51 | MA |
| YKˆAAFTECCQAADK | Lys162 | MA |
| LKˆCASLQK | Lys199 | MA |
| AFKˆAWAVAR | Lys212 | MA |
| AEFAEVSKˆLVTDLTK | Lys233 | MA |
| ADLAKˆYICENQDSISSK | Lys262 | MA |
| LAKˆTYETTLEK | Lys351 | MA |
| VFDEFKˆPLVEEPQNLIK | Lys378 | MA |
| KˆVPQVSTPTLVEVSR | Lys414 | MA |
| K∼VPQVSTPTLVEVSR | Lys414 | SB |
| KˆQTALVELVK | Lys525 | MA |
| K∼QTALVELVK | Lys525 | SB |
Unmodified versions of the listed modified peptides and peptides resulting from cleavage with trypsin of unmodified versions of the listed modified peptides were also targeted; C# indicates HNE Michael addition at Cys followed by reduction; H@ indicates HNE Michael addition at His followed by reduction; Kˆ indicates HNE Michael addition at Lys followed by reduction; K∼ indicates Schiff base formation with HNE at Lys followed by reduction; M* indicates oxidation at Met
Secreted protein numbering; add 24 for nascent protein
MA indicates Michael addition; SB indicates Schiff base formation
Effect of HNE Concentration on the Level and Distribution of Modifications
Using 3 h incubations at 37°C, we report pilot data here on two HNE:HSA ratios: 50:1 and 100:1. Linear ion trap data alone was collected for this comparison (more extensive data, including Orbitrap measurements, are provided in the next section). These ratios provide sufficient conversion at 3 h to limit the inherent uncertainty in using PQD to assess iTRAQ labels. The data are the result of three separate LC-MS/MS replicate runs on a single sample preparation. Using duplicate (no HNE present) controls allows the consistency between iTRAQ counts to be evaluated: the log10-transformed average 114:117 ratio (masses of the duplicate, control iTRAQ labels) was 0.0068, but the log10-transformed standard deviation was 0.33 (single standard deviation 114:117 ratio range is from 0.47 to 2.18). As noted above, a highly targeted peptide list (Table 2) was used in the analysis to maximize the iTRAQ detection count obtained for each peptide. Although 20 modifications were targeted, useful data were obtained for only 15 of them. Furthermore, of the 15 identified modifications, detections counts for Cys34, Lys199, and Lys262 were very low (see Table S6). Results for the histidines and lysines are presented in Figure 1. In panels (a) and (c), depletion plots are shown for the unmodified histidines and lysines, respectively, scaled to 100% for the control samples. In panels (b) and (d) are the corresponding accumulation plots in a column format for modified residues (relative intensities are used; raw reporter ion counts for the two controls were averaged before relative intensities were calculated). The measurements supporting Figure 1 are listed in tabular form in Table S6.
Figure 1.

Effect of varying HNE:HSA ratio on reaction progress measured using iTRAQ reporter ion intensity (linear ion trap/PQD) for targeted histidine and lysine sites. The targeted mass list was used. Depletion plots are scaled to 100% for the control while relative intensities are given in the accumulation bar plots. Relative abundance measurements were not obtained for His242, His247, His288, Lys51, and Lys212. MA indicates Michael addition while SB indicates Schiff base formation. When more than one unmodified peptide for a particular modification site was available, the most frequently-detected unmodified peptide was used.
In Figures 1a and 1c, one sees a reasonable dose/response relationship for the unmodified histidines and lysines—as the HNE concentration increases, there is a consistent drop in the intensity of the parent peptides. Looking at Figures 1a and 1c, the data indicate that His67 and His510 are the most reactive of the histidines and that Lys199 and Lys525 are the most reactive lysines under these conditions. In Figures 1b and 1d, the accumulation bar plots present a somewhat different picture in terms of relative reactivity. For the histidines, His67 remains a candidate for most reactive site (i.e., exhibits the most accumulation at the 50:1 HNE:HSA ratio data point), in agreement with the depletion plots, but His510 now appears to be least reactive, though the differences are modest. In the accumulation bar plots for the lysine series, Lys199 no longer stands out as the most reactive; however, the peptide supporting this modification was only detected once (see Table S6). Lys525 appears to still be relatively reactive, but the signal is split between the formation of a Michael adduct and a Schiff base, complicating interpretation. Looking for the least reactive targeted lysines, the accumulation bar plots suggest Lys262 and Lys233 (most relative intensity is found for the 100:1 HNE:HSA ratio data points), which is broadly in agreement with the depletion plots. In general, the availability of only relative intensity for the accumulation plots makes then particularly hard to interpret; we have only the relative concentrations and no knowledge of how close the reaction is to completion other than changes in the accumulation profile. Furthermore, the accumulation plots contain high degrees of uncertainty because signals are often low for the modified peptides, particularly at low conversions (intensities appear to be systematically low for the HNE-modified peptides, probably because they offer alternative fragmentation pathways). In contrast, the depletion plots are much easier to compare since the degree of modification (i.e., depletion) must be zero if no HNE has been added. Therefore, the depletion plots would seem to be the best measure of relative reactivity. A critical point with respect to the data in Figure 1 is the impact of whether or not the systems are reaching equilibrium in this time frame. This issue is addressed with a more comprehensive analysis in the following section.
Effect of Incubation Time on the Level and Distribution of HNE Modifications
Incubation times of 1, 3, and 24 h were used with an HNE:HSA ratio of 100:1. The linear ion trap- and Orbitrap-based instruments were used with the same list of targeted peptides (Table 2) to maximize the number of MS/MS collected for each peptide, thus enhancing the signal-to-noise ratio in the iTRAQ data. The data presented are the result of three complete repeat preparations, for each of which three replicate LC-MS/MS runs performed on each instrumental platform. We anticipated that the longest timeframe would lead to extensive modification and potentially an equilibrium mixture.
Data for targeted histidines are shown in Figure 2. All targeted histidines were detected except His247, which is difficult to confidently identify because it is located on the same tryptic peptide as His242. Modified and unmodified peptides for the His288 site were detected both with and without an oxidized methionine; the non-oxidized form was detected more frequently and was used in constructing these plots. Figures 2a and 2b illustrate the depletion of parent peptides and the accumulation of modified peptides, respectively, for the linear ion trap while Figures 2c and 2d provide the same information for the Orbitrap. The same unmodified peptides were used in constructing depletion plots for both instrumental platforms (see Table S7). Looking first at the depletion plots, there is good agreement between the methods on the least reactive targeted histidines (His105 and His367). This result is in good agreement with the HNE:HSA ratio versus unmodified peptide depletion comparison given in Figure 1a. His67 and His510, which were most reactive in the concentration study, are again among the most reactive sites. However, the two additional detections, His242/7 and His288, appear most reactive, although they display unusual curve shapes: His242/7, by both methods, shows a greater degree of depletion at 1 h than 3 h; His288 appears as expected in the linear ion trap data but shows a lower degree of depletion at longer reaction times in the Orbitrap data. Moving to the modified peptide accumulation bar plots, His105 and His367 are again confirmed as being among the least reactive targeted histidines (relative intensity is weighted towards longer reaction times). Again, in agreement with the depletion plots, His67, His288, and His510 are confirmed as being the most reactive targeted histidines. His242 displays a more confusing picture, but this is probably caused by the low number of identifications obtained. This is due, as mentioned above, to two modification sites (His242 and His247) being present on the same tryptic peptide (very high quality MS/MS are required to unambiguously identify one particular site when another site on the same peptide must also be considered). Taking all Figure 2 plots together, it appears that the modification reaction is moving towards completion, but a reaction time of greater than 24 h is required.
Figure 2.

Effect of varying reaction duration, at a fixed HNE:HSA ratio of 100:1, on reaction progress measured using iTRAQ reporter ion intensity for targeted histidine sites. Linear ion trap/PQD and Orbitrap/HCD data are presented. The targeted mass list was used. Depletion plots are scaled to 100% for the control while relative intensities are given in the accumulation bar plots. Relative abundance measurements were not obtained for targeted site His247, although this site shares the same unmodified peptide with His242. MA indicates Michael addition while SB indicates Schiff base formation. When more than one unmodified peptide for a particular modification site was available, the most frequently-detected unmodified peptide was used.
The data for the lysines are presented in Figure 3. Eleven of twelve targeted modifications were identified using the Orbitrap, with the exception being Lys51; with the linear ion trap, Lys51 and Lys212 were missed. Figures 3a and 3b show the depletion of parent peptides and the accumulation of modified peptides, respectively, for the linear ion trap while Figures 3c and 3d provide the same information for the Orbitrap. The same unmodified peptides were used in constructing depletion plots for both instrumental platforms (see Table S8); furthermore, the same unmodified peptides were used in constructing Figures 1, 2, and 3. The lysines present a different picture to the histidines. Not only are the extents of depletion lower, but the plots indicate that lysine modifications become saturated and level off at what appear to be equilibrium levels. The curve shapes for the lysine depletion plots are clearly different to those observed for the histidine depletion plots. A group of three sites, Lys233, Lys262, and Lys378, show very little depletion by both instrumental methods and appear to represent the set of least reactive targeted lysines. The most reactive targeted lysine is clearly Lys199 while the second most reactive is Lys525. The accumulation bar plots again provide a roughly complementary picture, but they tend to suggest that there is a slow accumulation in some modification sites in the 3-24 h time frame that is not reflected in the depletion plots. This is most evident for Lys233 and it is unclear why there is a substantial increase in modified peptide signal here despite the small drop in parent peptide intensity for this site; however, it is important to note that the column graphs represent signal percentages and often correlate to very low signal intensities, particularly for sites that exhibit low depletions, so large fractional changes may correspond to small absolute changes in signal intensity. In any case, the drift up in the 3-24 h range for the all of the other peptides suggests a rate that is well below that indicated for the first three hours and suggests the process is slowing, presumably to an equilibrium level. Interestingly, the Schiff base products show reduced relative abundance at longer reaction times, possibly indicating that the Michael adduct is the thermodynamically favored product.
Figure 3.

Effect of varying reaction duration, at a fixed HNE:HSA ratio of 100:1, on reaction progress measured using iTRAQ reporter ion intensity for targeted lysine sites. Linear ion trap/PQD and Orbitrap/HCD data are presented. The targeted mass list was used. Depletion plots are scaled to 100% for the control while relative intensities are given in the accumulation bar plots. Relative abundance measurements were not obtained for Lys51 by either instrumental approach and were obtained for Lys212 only using the Orbitrap approach. MA indicates Michael addition while SB indicates Schiff base formation. When more than one unmodified peptide for a particular modification site was available, the most frequently-detected unmodified peptide was used.
These experiments were not designed to provide quantitative kinetic data—the iTRAQ system employed provides too few data points and the uncertainties are significant. However, it is possible to extract crude rate constants from the data in Figures 2 and 3. In this analysis, a pseudo first-order approach was used, focusing on the first three hours of reaction. PQD and HCD measurements were averaged at equal weight when calculating rate constants. For the slowest targeted histidine, His105, a rate constant of 0.020 ± 0.003 M−1 s−1 was obtained. In order of increasing rate constant, the results for His367, His67, and His510 were 0.029 ± 0.001, 0.085 ± 0.008, and 0.0087 ± 0.008 M−1 s−1 (the listed uncertainties only consider the precision of the kinetic plots – including other experimental factors, uncertainties in the 10-20% range are expected). For the most reactive histidines, His242/7 and His288, the errors were much larger in the kinetic plots and the rate constants for both sites are probably best expressed as 0.2 ± 0.1 M−1 s−1. Errors were perhaps more significant because very low intensity reporter ions were being masked by noise in the mass spectrum. This approach is not possible with the lysines because they are approaching equilibrium and we do not have enough data points at early times. Nonetheless, the plots suggest that the most reactive lysine, Lys199, has a rate constant comparable to His242/7 and His288.
Discussion
Comparison to Previous Work
As noted above, the present data share many of the same features as those obtained by Aldini in terms of modification sites.[32] Comparisons focused on the relative reactivity of the sites are more difficult to make because the most reactive sites reported in the study by Aldini were difficult sites to quantify with our methods as well as with Aldini's methods. Aldini reports the following order of reactivity: Cys34 > Lys199 > His146. In the case of Cys34 and His146, they were not able to identify the modification products in tryptic digests, but had to rely on trypsin/chymotrypsin combination digests to identify them. Their conclusion was based on a combination of depletion data, comparisons of selected-ion chromatograms, and titrations identifying initial adducts at low HNE:HSA ratios. More recently, Aldini determined a rate constant of 30 M−1 s−1 for the reaction of Cys34 with HNE,[45] a value about two orders of magnitude greater than our approximate value for the most reactive histidines, suggesting that the free cysteine is exceptionally reactive. While we were able to identify adducts at Cys34, His146, and Lys199 at high confidence in our initial survey, there were problems with each of these sites. His146 was not included in the targeted mass list because this modification was observed in control runs where no HNE had been added and was not detected in initial Orbitrap test runs performed without a mass list; however, the chief peptide supporting His146, RH@PYFYAPELLFFAK, is supported by convincing MS/MS, with peak intensities conforming to the ‘proline effect’.[48] Cys34 and Lys199 were added to the mass list, but for both residues relatively small numbers of acceptable MS/MS were obtained. Lys199 has been included in the accumulation and depletion plots, but Cys34 was not due to the probable presence of extensive cysteinylation at this site in HSA as mentioned above. Although Aldini did not rank the reactivity of their other modification sites, the limited quantitative data that they provide is consistent with the more extensive results shown here in Figures 1-3.
Comparisons with Liebler's data are more difficult because the detected sites are somewhat different than those presented here.[33] First, at an HNE:HSA ratio of 10:1, we find 15/34 (high/medium confidence) modifications whereas Liebler reports only ten at an HNE:HSA ratio of 640:1. The differences in the kinetic data are also significant. Liebler obtained the following order of reactivity for the histidines: His242 > His67 ∼ His510 > His367 > His247. Their rate constants vary by a factor of 600, from 0.0005 for His247 to 0.3 M−1s−1 for His242 (Sigma HSA numbers); in contrast, using the depletion of unmodified peptides, we see a much smaller variation in rate (from 0.02 for His105 to circa 0.2 M−1s−1 for His242/7 and His288). Due to the requirement for very high quality MS/MS to confidently identify isobaric modifications on the same peptide and the consequent low numbers of detections for His242 and His247, we can only realistically use the depletion of the unmodified peptide to provide a combined rate for His242 and His247. Our results indicate the following ranking that is broadly in agreement with Liebler: His242/7 ∼ His288 > His67 ∼ His510 > His105 ∼ His367. Liebler identified but did not provide kinetic data for His105 and did not detect His288. Overall, the major difference in the kinetic data centers on the rate constant of the less reactive histidines, which were found to be much slower in the Liebler study The origin of this difference is probably rooted in the methodologies that were used. In the Liebler study, the intensity of the HNE addition product was tracked as a function of time with the assumption that at 24 h there would be 100% modification at each site. The danger in this approach is that if the modification does not go to completion for some reason, such as equilibrium formation, or if the kinetics are more complex than expected, the approach can be skewed by the use of an errant reference point (i.e., assumption of 100% conversion at longest reaction time). In addition, if the modification site undergoes any secondary reactions, it is not included as a productive reaction and would lead to underestimation of the reaction rate. In our approach, we have based our kinetics on the disappearance of the unmodified peptide. In this case, the reference point (i.e., 0% conversion in the control) is well defined. However, it is possible to overestimate the rate of a particular reaction if there are hidden pathways depleting the signal for the unmodified peptide. As a result, the present data should be viewed as upper-limits on the rates. In any case, neither our accumulation of modified peptide nor depletion of unmodified peptide data are consistent with His242 having exceptional reactivity among the histidines. Liebler gives kinetic data for only one lysine, Lys233, which like in our results, they identify as being a relatively less reactive site. Overall, the present data are broadly consistent with those from Liebler, but suggest that other histidines compete much more effectively with His242 than the previous data indicated.
Reactivity of Amino Acids
As noted above, it has been established that Cys34 is the most reactive site in HSA. This is not surprising in that it is the only free cysteine and sulfur is known to be highly nucleophilic in Michael additions.[17] Comparison of the lysines and histidines is more complicated because the lysines do not react to completion and are under a mix of kinetic and thermodynamic control with our reaction conditions. Nonetheless, some general conclusions can be drawn from the data at 1 h because the reactions, for the most part, have not reached equilibrium or completion at this point. If we look at the three most reactive lysines that were characterized, averaging the linear ion trap and Orbitrap data at equal weight, we see approximately 96% (Lys199), 68% (Lys525), and 33% (Lys351) depletion at 1 h. For the four most reactive histidines, again combining linear ion trap and Orbitrap data, we see approximately 81% (His242/7), 79% (His288), 48% (His67), and 46% (His510) depletion at 1 h. These data indicate that histidines and lysines exhibit roughly the same kinetic reactivity with HNE and that local, specific interactions are mainly responsible for the variation in their rates of addition. However with lysine, some sites are thermodynamically unfavorable and therefore will not be competitive because their reactions do not proceed to completion despite having reasonable intrinsic addition rates (i.e., they must also have fairly high off-rates). The next question is what local environmental factors control the reactivity of the histidines and lysines.
Relative Histidine Reactivity
The targeted histidines divide clearly into a fast set (His67, His242/7, His288, and His510) and a slower set (His105 and His367). His464 is a site at which no evidence of modification was found, making it suitable as a negative control for environmental factors that affect HNE reactivity. In examining the crystal structure of HSA,[49] one finds that in general the most active sites have good, but not exceptional, surface accessibility. This can be quantified by a simple calculation of the solvent accessible surface area (SASA) associated with the residue.[50, 51] Although not a perfect measure of the environment, it provides some insight. Using the GETAREA program,[52] a plot of side chain and backbone SASAs for the histidines in HSA can be generated (Figure 4a). Although there is not an absolute pattern in this small data set, the general tendency is for moderate accessibility on the side chain and low relative accessibility on the backbone. This corresponds with sites that are near the surface, but recessed in pockets or seams on the protein surface. The obvious exception is His242. In fact, His242, matches the characteristics of the negative control, His464. Liebler and co-workers noted the unusual location of His242 and suggested its reactivity was linked to it being buried in a hydrophobic pocket with its imidazole having very low basicity. We cannot clearly separate the reactivity of His242 and His247 in our data so definite conclusions cannot be made. Of all the modification sites we have identified (His and Lys), none has exhibited a side chain SASA as low as His242 (see below), so high reactivity here would be unique for the protein.
Figure 4.
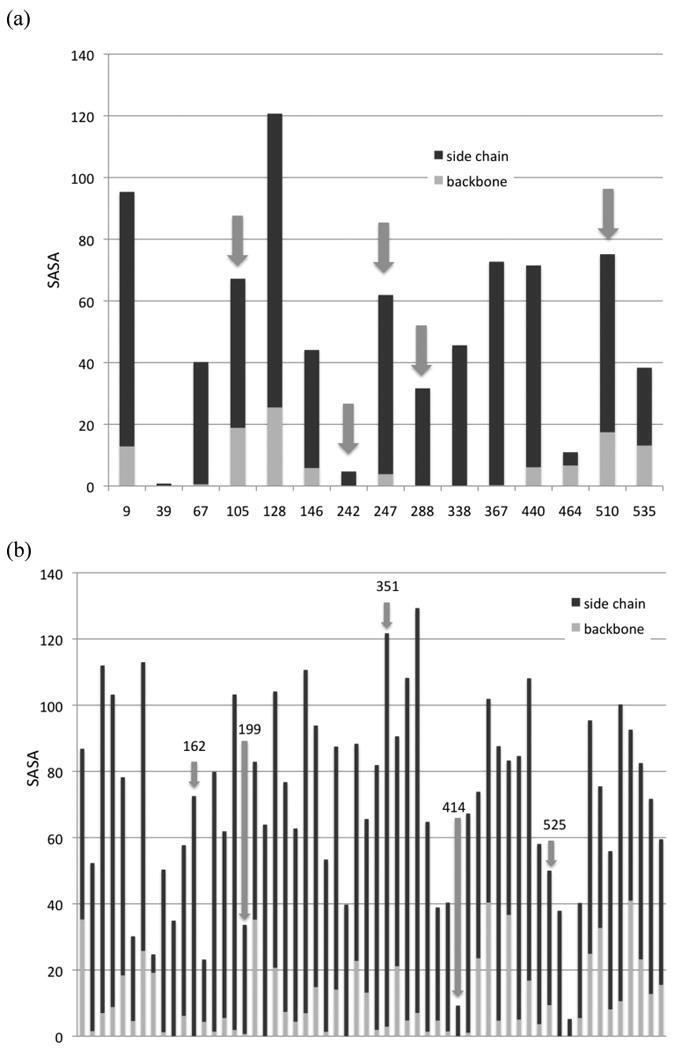
SASA in Å2 for the (a) histidines and (b) lysines of HSA. Red bars are the backbones and blue bars are the side chains. Arrows indicate sites that were identified as being particularly reactive. In panel (b), the data is too extensive to include residue numbers, but highly reactive sites are labeled.
Relative Lysine Reactivity
The lysines are more interesting in that we have identified larger variations in rate/equilibrium constants and a larger data set is available. A similar pattern emerges in the plot of SASAs (Figure 4b). The most stable lysine adducts that we identified are Lys162, Lys199, Lys351, Lys414, and Lys525. These residues follow the same general pattern as the histidines—one sees good to moderate accessibility on the side chains and very low accessibility on the backbone in the more reactive sites. Lys414 appears to be an exception, but it is in a groove and will have reasonable accessibility in some side-chain conformations. In fact, Lys414 is known to play a role in fatty acid binding (see below); in a crystal structure with myristic acid,[53] its SASA rises to 31.6 Å2, a value consistent with other reactive lysines. It is interesting to note that of the 12 lysines that the GETAREA algorithm defines as exposed on the basis of their SASA values (side chain SASA > 82 Å2 for lysine), only two were found to be modified in this study, Lys262 (a low reactivity site) and Lys351, suggesting that high accessibility poorly correlates with the probability of HNE modification.
Overall Reactivity Patterns
We have a relatively small sample set, but it is possible to draw some general conclusion about the factors that control the relative reactivity of histidine and lysine residues with HNE. As noted above, some degree of surface accessibility is necessary, but highly exposed sites are generally unreactive. The presence of nearby hydrophobic grooves or patches is a common theme in the more reactive residues identified in this study. This is not completely surprising given the fact that HNE does have a moderately hydrophobic tail and could benefit from hydrophobic interactions. Liebler noted this in his study and suggested a strong link between the hydrophobicity of the environment and reactivity.[33] It is well known that HSA has multiple binding sites for fatty acids and crystal structures are available for complexes.[53, 54] These binding sites provide a guide for locations that offer favorable hydrophobic interactions linked to a salt bridge interaction with a basic site. Interestingly, two of the strong lysine HNE binding sites, Lys414 and Lys525, correspond directly with myristate binding sites (the carboxylate of a myristate binds to the ammoniums of these lysine residues).[53] In addition, two other strong HNE binding sites, Lys162 and His288, are located adjacent to the hydrophobic pockets occupied by myristic acid. There certainly is not a one-to-one correspondence between the HNE and myristate binding sites (and one should not be expected given the differences in structure/binding), but the overlap that we have observed is at least suggestive and indicates that hydrophobicity might be playing a significant role in the HNE binding preferences. Liebler also suggested that the pKa of a histidine played an important role in its relative reactivity.[33] This conclusion was driven by their identification of His242 as the most reactive histidine and an estimate of its imidazolium's pKa that indicated that it was unusually acidic. As shown above, although we see evidence of high reactivity in the peptide containing His242 and His247, the consensus of our data is not consistent with His242 being exceptionally reactive among the histidines (His288 is similar); therefore, the low, predicted pKa does not appear to be a strong predictor of HNE reactivity.
Conclusions
The addition of a prototypical α,β-unsaturated aldehyde, HNE, to HSA exhibits relatively high selectivity with only three of the potential 135 single addition modifications being identified at high confidence at an HNE:HSA ratio of 1:1. Even at high HNE:HSA ratios (100:1), our data suggests that only ten sites reach modification levels of 50% or more (assuming that most of the depletion of the unmodified peptide containing His242 and His247 is due to the generation of His242). Our data indicate that aside from the highly reactive Cys34, the next most kinetically reactive sites are Lys199, His242/7, and His288. Although this reaction system has been studied in the past, it has not previously been recognized that the lysine modifications are under thermodynamic rather than kinetic control under typical in vitro experimental conditions and do not go to completion even with HNE:HSA ratios as high as 100:1. This result suggests that lysine modifications by HNE will only have biological relevance when local HNE concentrations are anomalously high or if secondary reactions make the additions irreversible. In contrast, the monitored histidine reactions in the study appeared to be going to completion under our conditions. Finally, the work demonstrates that care must be taken in the quantification of HNE modified residues by mass spectrometry because the modification can greatly alter the ionization/detection efficiency of the peptide. In this respect, the iTRAQ approach, with monitoring of both the modified and unmodified peptides, offers a powerful tool for quantifying the reaction progressions.
Supplementary Material
Acknowledgments
Support from the National Institutes of Health, Institute on Aging is acknowledged: R01AG034167. Orbitrap mass spectrometry was performed by Dr. K.T. Nelson at the Virginia Commonwealth University Chemical and Proteomic Mass Spectrometry Core Facility.
Footnotes
Supplementary Material Available: An expanded description of the computational data analysis procedure and a complete list of all HNE-modified peptides identified in the initial survey runs are provided as supplementary material. Annotated MS/MS are included for all sites and spectrum counts are given in addition to counts of runs in which the modified peptide was detected. The full targeted m/z ratio list and tables of detected peptides from the iTRAQ runs with detection counts and averaged relative reporter ion intensities are also provided.
References
- 1.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 2.Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A. Biomarkers of oxidative damage in human disease. Clin Chem. 2006;52:601. doi: 10.1373/clinchem.2005.061408. [DOI] [PubMed] [Google Scholar]
- 3.Dalle-Donne I, Scaloni A, Giustarini D, Cavarra E, Tell G, Lungarella G, Colombo R, Rossi R, Milzani A. Proteins as biomarkers of oxidative/nitrosative stress in diseases: The contribution of redox proteomics. Mass Spectrom Rev. 2005;24:55. doi: 10.1002/mas.20006. [DOI] [PubMed] [Google Scholar]
- 4.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 5.Nyska A, Kohen R. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol Pathol. 2002;30:620. doi: 10.1080/01926230290166724. [DOI] [PubMed] [Google Scholar]
- 6.Stadtman ER. Protein Oxidation and Aging. Science. 1992;257:1220. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 7.Mirzaei H, Regnier F. Affinity chromatographic selection of carbonylated proteins followed by identification of oxidation sites using tandem mass spectrometry. Anal Chem. 2005;77:2386. doi: 10.1021/ac0484373. [DOI] [PubMed] [Google Scholar]
- 8.Levine RL. Carbonyl modified proteins in cellular regulation, aging, and disease. Free Radical Biol Med. 2002;32:790. doi: 10.1016/s0891-5849(02)00765-7. [DOI] [PubMed] [Google Scholar]
- 9.Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends Mol Med. 2003;9:169. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 10.Dalle-Donne I, Aldini G, Carini M, Colombo R, Rossi R, Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med. 2006;10:389. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta. 2003;329:23. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 12.Mirzaei H, Regnier F. Identification and quantification of protein carbonylation using light and heavy isotope labeled Girard's P reagent. J Chromatogr A. 2006;1134:122. doi: 10.1016/j.chroma.2006.08.096. [DOI] [PubMed] [Google Scholar]
- 13.Meany DL, Xie HW, Thompson LV, Arriaga EA, Griffin TJ. Identification of carbonylated proteins from enriched rat skeletal muscle mitochondria using affinity chromatography-stable isotope labeling and tandem mass spectrometry. Proteomics. 2007;7:1150. doi: 10.1002/pmic.200600450. [DOI] [PubMed] [Google Scholar]
- 14.Nystrom T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005;24:1311. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Brien PJ, Siraki AG, Shangari N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit Rev Toxicol. 2005;35:609. doi: 10.1080/10408440591002183. [DOI] [PubMed] [Google Scholar]
- 16.Porter NA. Chemistry of Lipid-Peroxidation. Methods Enzymol. 1984;105:273. doi: 10.1016/s0076-6879(84)05035-7. [DOI] [PubMed] [Google Scholar]
- 17.Esterbauer H, Schaur RJ, Zollner H. Chemistry and Biochemistry of 4-Hydroxynonenal, Malonaldehyde and Related Aldehydes. Free Radical Biol Med. 1991;11:81. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 18.Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radical Biol Med. 2004;37:937. doi: 10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 19.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 20.Forman HJ, Fukuto JM, Miller T, Zhang HQ, Rinna A, Levy S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch Biochem Biophys. 2008;477:183. doi: 10.1016/j.abb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kruman I, BruceKeller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A. 1996;93:2696. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sayre LM, Zelasko DA, Harris PLR, Perry G, Salomon RG, Smith MA. 4-hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 24.Tang XX, Sayre LM, Tochtrop GP. A mass spectrometric analysis of 4-hydroxy-2-(E)-nonenal modification of cytochrome c. J Mass Spectrom. 2011;46:290. doi: 10.1002/jms.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rauniyar N, Prokai L. Detection and identification of 4-hydroxy-2-nonenal Schiff-base adducts along with products of Michael addition using data-dependent neutral loss-driven MS3 acquisition: Method evaluation through an in vitro study on cytochrome c oxidase modifications. Proteomics. 2009;9:5188. doi: 10.1002/pmic.200900116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rauniyar N, Stevens SM, Prokai-Tatrai K, Prokai L. Characterization of 4-Hydroxy-2-nonenal-Modified Peptides by Liquid Chromatography-Tandem Mass Spectrometry Using Data-Dependent Acquisition: Neutral Loss-Driven MS3 versus Neutral Loss-Driven Electron Capture Dissociation. Anal Chem. 2009;81:782. doi: 10.1021/ac802015m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stevens SM, Rauniyar N, Prokai L. Rapid characterization of covalent modifications to rat brain mitochondrial proteins after ex vivo exposure to 4-hydroxy-2-nonenal by liquid chromatography-tandem mass spectrometry using data-dependent and neutral loss-driven MS3 acquisition. J Mass Spectrom. 2007;42:1599. doi: 10.1002/jms.1349. [DOI] [PubMed] [Google Scholar]
- 28.Carini M, Aldini G, Facino RM. Mass spectrometry for detection of 4-hydroxy-trans-2-nonenal (HNE) adducts with peptides and proteins. Mass Spectrom Rev. 2004;23:281. doi: 10.1002/mas.10076. [DOI] [PubMed] [Google Scholar]
- 29.Bolgar MS, Gaskell SJ. Determination of the sites of 4-hydroxy-2-nonenal adduction to protein by electrospray tandem mass spectrometry. Anal Chem. 1996;68:2325. [Google Scholar]
- 30.Bruenner BA, Jones AD, German JB. Direct Characterization of Protein Adducts of the Lipid-Peroxidation Product 4-Hydroxy-2-Nonenal Using Electrospray Mass-Spectrometry. Chem Res Toxicol. 1995;8:552. doi: 10.1021/tx00046a009. [DOI] [PubMed] [Google Scholar]
- 31.Rauniyar N, Prokai-Tatrai K, Prokai L. Identification of carbonylation sites in apomyoglobin after exposure to 4-hydroxy-2-nonenal by solid-phase enrichment and liquid chromatography-electrospray ionization tandem mass spectrometry. J Mass Spectrom. 2010;45:398. doi: 10.1002/jms.1725. [DOI] [PubMed] [Google Scholar]
- 32.Aldini G, Gamberoni L, Orioli M, Beretta G, Regazzoni L, Facino RM, Carini M. Mass spectrometric characterization of covalent modification of human serum albumin by 4-hydroxy-trans-2-nonenal. J Mass Spectrom. 2006;41:1149. doi: 10.1002/jms.1067. [DOI] [PubMed] [Google Scholar]
- 33.Szapacs ME, Riggins JN, Zimmerman LJ, Liebler DC. Covalent adduction of human serum albumin by 4-hydroxy-2-nonenal: Kinetic analysis of competing alkylation reactions. Biochemistry (Mosc) 2006;45:10521. doi: 10.1021/bi060535q. [DOI] [PubMed] [Google Scholar]
- 34.Aldini G, Vistoli G, Regazzoni L, Gamberoni L, Facino RM, Yamaguchi S, Uchida K, Carini M. Albumin is the main nucleophilic target of human plasma: A protective role against pro-atherogenic electrophilic reactive carbonyl species? Chem Res Toxicol. 2008;21:824. doi: 10.1021/tx700349r. [DOI] [PubMed] [Google Scholar]
- 35.Colombo G, Aldini G, Orioli M, Giustarini D, Gornati R, Rossi R, Colombo R, Carini M, Milzani A, Dalle-Donne I. Water-Soluble alpha,beta-Unsaturated Aldehydes of Cigarette Smoke Induce Carbonylation of Human Serum Albumin. Antioxid Redox Sign. 2010;12:349. doi: 10.1089/ars.2009.2806. [DOI] [PubMed] [Google Scholar]
- 36.Giustarini D, Milzani A, Colombo R, Dalle-Donne I, Rossi R. Nitric oxide and S-nitrosothiols in human blood. Clin Chim Acta. 2003;330:85. doi: 10.1016/s0009-8981(03)00046-9. [DOI] [PubMed] [Google Scholar]
- 37.He XM, Carter DC. Atomic-Structure and Chemistry of Human Serum-Albumin. Nature. 1992;358:209. doi: 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- 38.Ross PL, Huang YLN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 39.Wiese S, Reidegeld KA, Meyer HE, Warscheid B. Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. Proteomics. 2007;7:340. doi: 10.1002/pmic.200600422. [DOI] [PubMed] [Google Scholar]
- 40.Griffin TJ, Xie HW, Bandhakavi S, Popko J, Mohan A, Carlis JV, Higgins L. iTRAQ reagent-based quantitative proteomic analysis on a linear ion trap mass spectrometer. J Proteome Res. 2007;6:4200. doi: 10.1021/pr070291b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Umrethia M, Kett VL, Andrews GP, Malcolm RK, Woolfson AD. Selection of an analytical method for evaluating bovine serum albumin concentrations in pharmaceutical polymeric formulations. J Pharm Biomed Anal. 2010;51:1175. doi: 10.1016/j.jpba.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 42.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4:709. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 43.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 44.Choi H, Nesvizhskii AI. Semisupervised model-based validation of peptide identifications in mass spectrometry-based proteomics. J Proteome Res. 2008;7:254. doi: 10.1021/pr070542g. [DOI] [PubMed] [Google Scholar]
- 45.Aldini G, Regazzoni L, Orioli M, Rimoldi I, Facino RM, Carini M. A tandem MS precursor-ion scan approach to identify variable covalent modification of albumin Cys34: a new tool for studying vascular carbonylation. J Mass Spectrom. 2008;43:1470. doi: 10.1002/jms.1419. [DOI] [PubMed] [Google Scholar]
- 46.Kleinova M, Belgacem O, Pock K, Rizzi A, Buchacher A, Allmaier G. Characterization of cysteinylation of pharmaceutical-grade human serum albumin by electrospray ionization mass spectrometry and low-energy collision-induced dissociation tandem mass spectrometry. Rapid Commun Mass Spectrom. 2005;19:2965. doi: 10.1002/rcm.2154. [DOI] [PubMed] [Google Scholar]
- 47.Schwartz JC, Syka JP, Quarmby ST. Improving the fundamentals of Msn on 2D ion traps: new ion activation and isolation techniques. 53rd ASMS Conference on Mass Spectrometry; San Antonio, Texas. 2005. [Google Scholar]
- 48.Tabb DL, Friedman DB, Ham AJL. Verification of automated peptide identifications from proteomic tandem mass spectra. Nat Protoc. 2006;1:2213. doi: 10.1038/nprot.2006.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhattacharya AA, Curry S, Franks NP. Binding of the general anesthetics propofol and halothane to human serum albumin - High resolution crystal structures. J Biol Chem. 2000;275:38731. doi: 10.1074/jbc.M005460200. [DOI] [PubMed] [Google Scholar]
- 50.Richmond TJ. Solvent Accessible Surface-Area and Excluded Volume in Proteins - Analytical Equations for Overlapping Spheres and Implications for the Hydrophobic Effect. J Mol Biol. 1984;178:63. doi: 10.1016/0022-2836(84)90231-6. [DOI] [PubMed] [Google Scholar]
- 51.Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem. 1998;19:319. [Google Scholar]
- 52.Fraczkiewicz R, Braun W. University of Texas Medical Branch; Galveston, Texas: 2008. http://curie.utmb.edu/getarea.html. [Google Scholar]
- 53.Curry S, Mandelkow H, Brick P, Franks N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat Struct Biol. 1998;5:827. doi: 10.1038/1869. [DOI] [PubMed] [Google Scholar]
- 54.Petitpas I, Grune T, Bhattacharya AA, Curry S. Crystal structures of human serum albumin complexed with monounsaturated and polyunsaturated fatty acids. J Mol Biol. 2001;314:955. doi: 10.1006/jmbi.2000.5208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.