

Abstract
Amelogenesis imperfecta (AI) is a clinically and genetically heterogeneous group of inherited defects of enamel formation. In isolated AI (no additional segregating features), mutations in at least 7 genes are known so far, causing dominant, recessive or X-linked AI and allowing the identification of the molecular etiology in 40–50% of affected families. We report on 2 siblings (an 11-year-old female and a 7-year-old male) born to consanguineous Turkish parents, with AI and mild, proportionate short stature. Both parents have normal teeth, but mother, maternal grandmother and great-grandfather are/were also of short stature. A spine X-ray performed in the girl excluded brachyolmia. Affymetrix GenomeWide SNP6.0 Array analysis identified no pathogenic copy number changes, but showed sharing of large homozygous regions, including chromosome band 15q21.3 containing the WDR72 gene. WDR72 sequence analysis in both siblings revealed homozygosity for a novel stop mutation in exon 10 (c.997A>T, p.Lys333X) explaining the AI phenotype. Mutations in WDR72 are a very rare cause of autosomal-recessive hypomaturation type of isolated AI. The mutation described in our patients specifies the diagnosis AI IIA3 and represents only the sixth WDR72 mutation reported so far. The WDR72 protein is critical for dental enamel formation, but its exact function is still unknown.
Key Words: AI IIA3, Autosomal recessive amelogenesis imperfecta, Short stature, WDR72
Amelogenesis imperfecta (AI) is a clinically variable and genetically heterogeneous group of inherited defects of dental enamel formation. It affects the quality and/or amount of enamel and usually involves all teeth in the primary and secondary dentition but with a variable phenotype within families or even within different teeth of the same person [e.g. Crawford et al., 2007; El-Sayed et al., 2009]. AI can occur as an isolated form (no additional segregating signs, i.e. nonsyndromic forms) [Bailleul-Forestier et al., 2008a] or as one sign in a phenotypic spectrum of a specific syndrome (i.e. syndromic forms) [Bailleul-Forestier et al., 2008b; Parry et al., 2009; O'sullivan et al., 2011; Cho et al., 2012; Mory et al., 2012; Schossig et al., 2012].
Healthy dental enamel is the hardest and most highly mineralized human tissue. It is acellular, nonvital and without intrinsic repair capacity, made of highly organized hydroxyapatite crystals [Bailleul-Forestier et al., 2008a]. Enamel is epithelial-derived and formed by ameloblasts during tooth development, which is a complex process regulated by epithelial-mesenchymal interactions [Hu et al., 2007; Simmer et al., 2010]. Defects in amelogenesis are correlated with a specific stage of enamel formation leading to a highly variable phenotype [Simmer et al., 2010; Gibson, 2011]. The different phenotypes have been used over the last decades as classification systems, and various schemes have been proposed [see Aldred et al., 2003; Crawford et al., 2007, for overview]. The currently most widely used AI classification scheme was proposed by Witkop [1988], dividing AI into 4 main types based on the enamel defects (AI I–IV), and then further subdividing them into 14 subgroups based on clinical appearance (phenotype) and mode of inheritance (autosomal dominant, autosomal recessive or X-linked):
Type AI I: Hypoplastic type (reduced amount of enamel, secretion defect)
Type AI II: Hypomaturation type (defect of protein processing and crystallite maturation)
Type AI III: Hypocalcified type (insufficient degree of calcification of the formed organic matrix, mineralization defect)
Type AI IV: Hypomaturation-Hypoplasia with Taurodontism
Hypoplastic enamel is hard, translucent and of reduced thickness, hypomature/hypocalcified enamel is typically discolored, shows an opaque, yellowish or brown appearance and has weak consistency with a reduced mechanical resistance [Crawford et al., 2007; Gadhia et al., 2012]. However, this classification system has its limitation because of the high clinical variability as well as the missing correlation to the underlying molecular defect. Therefore, identification of underlying genes will require modified classification schemes [Aldred et al., 2003; Crawford et al., 2007].
Reported prevalences of isolated AI vary widely depending on the studied population (possibly also influenced by some ascertainment bias) with a range from about 1:230 in Turkey [Altug-Atac and Erdem, 2007], 1:370 in India [Gupta et al., 2011], 1:700 in Sweden [Bäckman and Holm, 1986], 1:1,000 in Argentina [Sedano, 1975], 1:8,000 in Israel [Chosack et al., 1979] to about 1:14,000 in the USA [Witkop and Sauk, 1976].
In isolated AI, mutations in at least 7 genes are known so far, allowing the identification of the underlying molecular etiology in about 40–50% of affected families [Lee et al., 2010; Chan et al., 2011; Wright et al., 2011]. Although the distribution of the different types of isolated AI depends on the population, AI due to recessive gene mutations seems to be very rare. Here we describe a novel WDR72 stop mutation diagnosed in Turkish siblings with autosomal recessive hypomaturation type of AI.
Patient Reports
We report on 2 siblings, an 11-year-old female and a 7-year-old male, both referred to our genetic department because of amelogenesis imperfecta, a mild, proportionate short stature and mild speech delay (especially in the boy).
They were born to consanguineous Turkish parents (second degree cousins). Both parents have normal teeth, and no dental problems were reported in other family members. The father has a height of 170 cm (10–25th centile), and the mother is 153 cm (3rd–10th centile) tall. Family history was unremarkable apart from a mild short stature in the maternal family with a height of 150 cm in the maternal grandmother and great-grandfather. No other consanguineous relation was known in the extended family history.
The girl was born at term with normal birth measurements [weight 3,180 g (−0.69 SDS), length 49 cm (−1.23 SDS), and OFC 33.5 cm (−1.08 SDS)]. As soon as the primary teeth erupted, dental enamel problems became obvious and amelogenesis imperfecta was suspected. Early motor milestones were normal (walking without support at 13 months) as well as her bilingual speech development. She attends a regular school and has some problems with reading and orthography, but formal testing revealed normal cognitive skills for her age.
At physical examination at the age of almost 11 years, her height was 130 cm (−2.4 SDS), her weight 24 kg, resulting in a normal BMI of 14.2, and her OFC was 51.5 cm (−0.61 SDS). She had a mild, proportionate short stature. There was no facial dysmorphism apart from a mild proptosis and slightly blue sclerae. Clinically, her teeth showed defective enamel with a yellowish-brown appearance (see fig. 1a). Deciduous dentition was complete whereas orthopantographic studies at mixed dentition stages (at 8 years and 11 years, fig. 2a, b, respectively) revealed agenesis of both second inferior premolars (35 and 45) and retention of their deciduous. Pulp chambers and roots appeared normal with no hint of taurodontism.
Fig. 1.

Clinical appearance of the hypomaturation type amelogenesis imperfecta in both siblings. Mixed dentition of the female at the age of 11 years (a) and mixed dentition of the male at the age of 7 years (b).
Fig. 2.

Orthopantomograms of the girl at mixed dentition stage, age 8 years (a) and 11 years (b), revealed agenesis of both second inferior premolars (35 and 45) and retention of their deciduous. Pulp chambers and roots appeared normal.
Endocrinological evaluation performed because of short stature including growth hormones was normal. Chromosome analysis was performed and revealed a normal result (46,XX) as well as molecular analysis of the SHOX gene for deletions/duplications and for mutations. An X-ray of the left hand taken at the age of 10 years 6 months, showed normal bone morphology, but bone age was delayed by 2 years.
To exclude amelogenesis combined with brachyolmia (OMIM 601216), X-rays of the thoracic and lumbar spine were performed at the age of 11 years 6 months. This showed normal vertebral bodies; brachyolmia could be excluded (see fig. 3a, b).
Fig. 3.
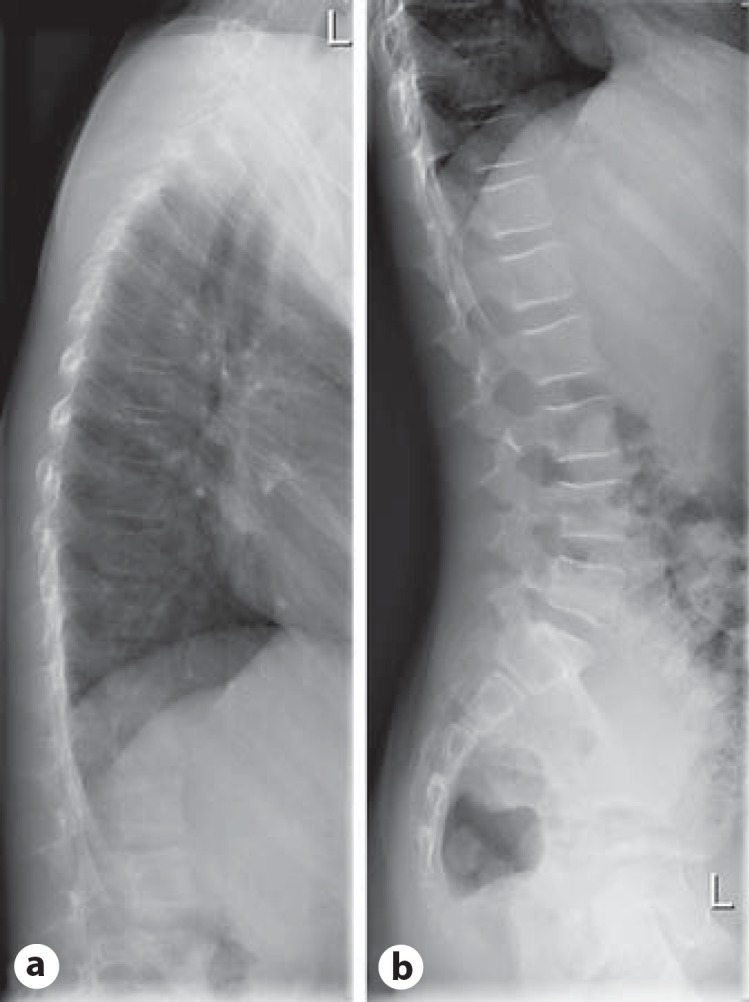
X-rays of the thoracic (a) and lumbar spine (b) performed in the girl at the age of 11 years excluded brachyolmia.
The younger brother was born at 39 weeks of gestation with low-normal birth measurements [weight 2,690 g (−1.84 SDS), length 48 cm (−1.7 SDS), OFC not documented]. Early motor milestones were normal (walking without support at 13–14 months), but speech development (bilingual) was delayed. He received speech therapy and spoke his first words at the age of 3 years. He now attends a regular primary school and his cognitive development is normal for his age. In infancy, he suffered from frequent respiratory infections. With eruption of the first deciduous teeth, amelogenesis imperfecta was obvious.
At the age of almost 7 years, he had a height of 109 cm (−2.84 SDS), a weight of 16 kg, resulting in a normal BMI of 13.5, and an OFC of 51 cm (−0.82 SDS). He had mild, proportionate short stature and a discrete proptosis. Amelogenesis imperfecta was present affecting the deciduous and the permanent teeth (see fig. 1b). Orthopantographic studies (not shown) excluded agenesis of teeth in the permanent dentition and showed normal pulp chambers and roots.
Endocrinological evaluation was initially normal, but at the age of 7 years a low level of IGF-1 was diagnosed, and growth hormone therapy started recently. A hand X-ray performed at the age of 6 years 4 months showed no morphological abnormality, but the bone age was delayed by more than 3 years.
Materials and Methods
DNA Isolation
DNA was extracted from venous blood using the FlexiGene DNA Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions.
Molecular Karyotyping
A genome-wide copy number microarray analysis was performed using the Affymetrix genome-wide human SNP array 6.0 (Affymetrix, Santa Clara, Calif., USA) according to the manufacturer's instructions. Data analysis was carried out using the Affymetrix Genotyping Console software (GTC v4.1), and the data interpretation was based on the February 2009 human genome sequence assembly (GRCh37/hg19). Conspicuous regions were compared with known CNVs, as provided by the Database of Genomic Variants (http://projects.tcag.ca/variation/).
Sequencing
The coding exons of WDR72 (NM 182758.2) and adjacent intronic sequences were amplified and sequenced by standard procedures (Sanger sequencing). The primer sequences are available upon request. Downstream analyses were performed with the SeqPilot software (JSI Medical Systems, Kippenheim, Germany).
Results
Molecular Karyotyping
Affymetrix GenomeWide SNP6.0 Array analysis excluded relevant pathogenic copy number changes but showed that both siblings share several large regions of homozygous markers. Homozygous regions larger than 2 Mb are depicted in figure 4 (bars next to the respective chromosome, boy left, girl right). Comparing the localization of all known nonsyndromic AI genes (especially the autosomal recessive ones) to the homozygous stretches that both siblings have in common, one region located on chromosome 15q21.3 (rs4776138–rs8036481, position 53,658,913–56,047,547; GRCh37/hg19) became obvious. This region (shown in fig. 5; girl M49567 and boy M49568) contains, among others, the WDR72 gene, previously identified to cause autosomal recessive hypomaturation type amelogenesis imperfecta AI IIA3 [El-Sayed et al., 2009]. Since both siblings share the same haplotype at this locus and the inheritance pattern for AI in this family is most likely autosomal recessive, WDR72 (OMIM 613214) was regarded as a promising candidate for mutation analysis.
Fig. 4.

Result of the array analysis (Affymetrix GenomeWide SNP6.0 Array) performed in both siblings. No pathogenic copy number changes were detected, but both siblings shared several stretches of homozygous markers. Those regions larger than 2 Mb are depicted next to the respective chromosome (left: boy, right: girl). Localizations of all genes known so far to cause nonsyndromic AI [ENAM (4q13.3), FAM83H (8q24.3), MMP20 (11q22.2), WDR72 (15q21.3), DLX3 (17q21.33), KLK4 (19q13.41), AMELX (Xp22.2)] are marked by an arrow, those with autosomal recessive inheritance pattern with a filled arrow head. One of the homozygous regions is located on chromosome 15q21.3 (positions 53,658,913–56,047,547 according to UCSC hg19), comprises about 2.39 Mb in size and contains the WDR72 gene (filled arrowhead with asterisk).
Fig. 5.

Detailed view of the array data of the 15q21.3 locus (M49567 – girl and M49568 – boy).
Sequencing
In both siblings, WDR72 sequence analysis revealed homozygosity for a novel base substitution in exon 10 (c.997A>T, p.Lys333X) (see fig. 6).
Fig. 6.

Result of sequencing of the WDR72 gene. WDR72 sequence analysis in both siblings revealed homozygosity for a novel stop mutation in exon 10 (c.997A>T, p.Lys333X). Both parents are heterozygous carriers of the mutation.
Analysis in both parents demonstrated the expected heterozygous carrier status for this mutation. Since this homozygous mutation segregates with the phenotype and causes a premature translation stop, it must be regarded as pathogenic. This confirms the clinical diagnosis of autosomal recessive hypomaturation type amelogenesis imperfecta and specifies subtype AI IIA3 in both siblings.
Discussion
The first gene discovered to cause isolated AI was the X-linked AMELX (amelogenin) [Lagerström et al., 1991], followed by ENAM (enamelin) [Rajpar et al., 2001] in autosomal dominant AI families. Two other autosomal dominant genes were identified, DLX3 (distal-less homeobox 3) [Dong et al., 2005], and FAM83H (family with sequence similarity 83, member H) [Kim et al., 2008]. Up to now, 4 genes are described to underlie autosomal recessive AI: ENAM [Hart et al., 2003], KLK4 (kallikrein-4) [Hart et al., 2004], MMP20 (matrix metalloproteinase-20) [Kim et al., 2005], and recently WDR72 (WD repeat containing domain 72) [El-Sayed et al., 2009].
Mutations in WDR72 (15q21.3, OMIM 613214) causing isolated AI IIA3 have been first identified by El-Sayed et al. [2009]. In contrast to the initial candidate genes for AI (e.g. AMELX, ENAM, KLK4, and MMP20) which encode for enamel matrix proteins, WDR72 was one of the genes that previously has not been expected to be involved in dental enamel formation [Lee et al., 2010]. It consists of 19 coding exons spanning around 250 kb [El-Sayed et al., 2009].
The WDR72 protein is critical for dental enamel formation, but its exact function is still unknown. It is localized in the cytoplasm [Lee et al., 2010] and is supposed to act as a scaffold for protein-protein interactions. El-Sayed et al. [2009] hypothesized that WDR72 might play a role in calcium-dependent vesicle turnover, an important step in maturation-stage ameloblasts, but up to now there is no data to support this hypothesis. WDR72 is expressed in multiple tissues with highest expression in bladder, kidney and mouth. Therefore, it is not clear yet why in a homozygous mutant state the only recognized pathologic phenotype is just isolated AI [Lee et al., 2010].
Up to now, a total of only 5 different WDR72 mutations (summarized in table 1) have been identified in 8 independent families making it a very rare cause of autosomal recessive AI. All mutations were homozygous truncating mutations and occurred in individuals born to consanguineous parents. The novel mutation in exon 10 (c.997A>T, p.Lys333X) identified in the present siblings is also a homozygous stop mutation, explaining the AI phenotype in this family. So far, there is no obvious hotspot region; the mutations are localized in different exons of the gene (exons 10, 12, 15, 16, and 17).
Table 1.
Overview of disease causing WDR72 mutations
| Reference | Exon | Notes | ||
|---|---|---|---|---|
| Present study | 10 | c.997A>T | p.Lys333X | |
| Lee et al., 2010 | 12 | c.l467_1468delAT | p.Ser489SerfsX10 | detected in 2 unrelated families |
| Wright et al., 2011 | 12 | c.l467_1468delAT | p.Ser489SerfsX10 | |
| El-Sayed et al., 2009 | 15 | c.2348C>G | p.Ser783X | detected in 2 unrelated families |
| El-Sayed et al., 2011 | 15 | c.2686C>T | p.Arg896X | |
| El-Sayed et al., 2009 | 16 | c.2857delA | p.Ser953ValfsX20 | |
| El-Sayed et al., 2009 | 17 | c.2934G>A | p.Trp978X |
Positions based on CCDC 10151.1 (http://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi).
In the few AI patients published with WDR72 mutations, no other medical problems or symptoms are mentioned. So, there is no information on other individuals with WDR72 mutations and a short stature. Therefore, it is not clear in our 2 siblings whether this is an associated finding or an independent segregating trait in this family. Given the fact, that the short stature involves several consecutive generations without any further consanguineous relation, a coincidently occurring second condition following an autosomal dominant trait (of unknown cause) seems to be likely.
A literature search for AI and short stature revealed clinical reports of patients, in whom this combination of findings was described due to a brachyolmia (i.e. a generalized platyspondyly without significant epiphyseal or metaphyseal involvement; platyspondyly with AI, OMIM 601216) [Verloes et al., 1996, 2011; Bertola et al., 2009]. Since the spine X-ray in the girl showed no abnormalities (fig. 3a, b), this differential diagnosis is no longer relevant.
In conclusion, a previously unreported family with a novel homozygous WDR72 mutation is described. In contrast to patients in the literature, the affected siblings described here present with AI in combination with short stature. The description of more patients with WDR72 mutations will allow a further characterization of the associated phenotype.
Acknowledgements
We are grateful to the family for their cooperation. Furthermore, we thank Elke Jürgens and Regina Kubica for excellent technical assistance and Bernhard Horsthemke for continuous support.
References
- Aldred MJ, Savarirayan R, Crawford PJ. Amelogenesis imperfecta: a classification and catalogue for the 21st century. Oral Dis. 2003;9:19–23. doi: 10.1034/j.1601-0825.2003.00843.x. [DOI] [PubMed] [Google Scholar]
- Altug-Atac AT, Erdem D. Prevalence and distribution of dental anomalies in orthodontic patients. Am J Orthod Dentofacial Orthop. 2007;131:510–514. doi: 10.1016/j.ajodo.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Bäckman B, Holm AK. Amelogenesis imperfecta: prevalence and incidence in a northern Swedish county. Community Dent Oral Epidemiol. 1986;14:43–47. doi: 10.1111/j.1600-0528.1986.tb01493.x. [DOI] [PubMed] [Google Scholar]
- Bailleul-Forestier I, Molla M, Verloes A, Berdal A. The genetic basis of inherited anomalies of the teeth. Part 1: clinical and molecular aspects of non-syndromic dental disorders. Eur J Med Genet. 2008a;51:273–291. doi: 10.1016/j.ejmg.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Bailleul-Forestier I, Berdal A, Vinckier F, de Ravel T, Fryns JP, Verloes A. The genetic basis of inherited anomalies of the teeth. Part 2: syndromes with significant dental involvement. Eur J Med Genet. 2008b;51:383–408. doi: 10.1016/j.ejmg.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Bertola DR, Antequera R, Rodovalho MJ, Honjo RS, Albano LM, et al. Brachyolmia with amelogenesis imperfecta: further evidence of a distinct entity. Am J Med Genet A. 2009;149A:532–534. doi: 10.1002/ajmg.a.32661. [DOI] [PubMed] [Google Scholar]
- Chan HC, Estrella NM, Milkovich RN, Kim JW, Simmer JP, Hu JC. Target gene analyses of 39 amelogenesis imperfecta kindreds. Eur J Oral Sci 119 Suppl. 2011;1:311–323. doi: 10.1111/j.1600-0722.2011.00857.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SH, Seymen F, Lee KE, Lee SK, Kweon YS, et al. Novel FAM20A mutations in hypoplastic amelogenesis imperfecta. Hum Mutat. 2012;33:91–94. doi: 10.1002/humu.21621. [DOI] [PubMed] [Google Scholar]
- Chosack A, Eidelman E, Wisotski I, Cohen T. Amelogenesis imperfecta among Israeli Jews and the description of a new type of local hypoplastic autosomal recessive amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol. 1979;47:148–156. doi: 10.1016/0030-4220(79)90170-1. [DOI] [PubMed] [Google Scholar]
- Crawford PJ, Aldred M, Bloch-Zupan A. Amelogenesis imperfecta. Orphanet J Rare Dis. 2007;2:17. doi: 10.1186/1750-1172-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Amor D, Aldred MJ, Gu T, Escamilla M, MacDougall M. DLX3 mutation associated with autosomal dominant amelogenesis imperfecta with taurodontism. Am J Med Genet A. 2005;133A:138–141. doi: 10.1002/ajmg.a.30521. [DOI] [PubMed] [Google Scholar]
- El-Sayed W, Parry DA, Shore RC, Ahmed M, Jafri H, et al. Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am J Hum Genet. 2009;85:699–705. doi: 10.1016/j.ajhg.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed W, Shore RC, Parry DA, Inglehearn CF, Mighell AJ. Hypomaturation amelogenesis imperfecta due to WDR72 mutations: a novel mutation and ultrastructural analyses of deciduous teeth. Cells Tissues Organs. 2011;194:60–66. doi: 10.1159/000322036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadhia K, McDonald S, Arkutu N, Malik K. Amelogenesis imperfecta: an introduction. Br Dent J. 2012;212:377–379. doi: 10.1038/sj.bdj.2012.314. [DOI] [PubMed] [Google Scholar]
- Gibson CW. The amelogenin proteins and enamel development in humans and mice. J Oral Biosci. 2011;53:248–256. doi: 10.2330/joralbiosci.53.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SK, Saxena P, Jain S, Jain D. Prevalence and distribution of selected developmental dental anomalies in an Indian population. J Oral Sci. 2011;53:231–238. doi: 10.2334/josnusd.53.231. [DOI] [PubMed] [Google Scholar]
- Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons D, et al. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet. 2004;41:545–549. doi: 10.1136/jmg.2003.017657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Gorry MC, Michalec MD, Ryu OH, et al. Novel ENAM mutation responsible for autosomal recessive amelogenesis imperfecta and localised enamel defects. J Med Genet. 2003;40:900–906. doi: 10.1136/jmg.40.12.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JC, Chun YH, Al Hazzazzi T, Simmer JP. Enamel formation and amelogenesis imperfecta. Cells Tissues Organs. 2007;186:78–85. doi: 10.1159/000102683. [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hart TC, Hart PS, Ramaswami MD, et al. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet. 2005;42:271–275. doi: 10.1136/jmg.2004.024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Lee SK, Lee ZH, Park JC, Lee KE, et al. FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am J Hum Genet. 2008;82:489–494. doi: 10.1016/j.ajhg.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerström M, Dahl N, Nakahori Y, Nakagome Y, Bäckman B, et al. A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1) Genomics. 1991;10:971–975. doi: 10.1016/0888-7543(91)90187-j. [DOI] [PubMed] [Google Scholar]
- Lee SK, Seymen F, Lee KE, Kang HY, Yildirim M, et al. Novel WDR72 mutation and cytoplasmic localization. J Dent Res. 2010;89:1378–1382. doi: 10.1177/0022034510382117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mory A, Dagan E, Illi B, Duquesnoy P, Mordechai S, et al. A nonsense mutation in the human homolog of Drosophila rogdi causes Kohlschutter-Tonz syndrome. Am J Hum Genet. 2012;90:708–714. doi: 10.1016/j.ajhg.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan J, Bitu CC, Daly SB, Urquhart JE, Barron MJ, et al. Whole-exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome. Am J Hum Genet. 2011;88:616–620. doi: 10.1016/j.ajhg.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry DA, Mighell AJ, El-Sayed W, Shore RC, Jalili IK, et al. Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. Am J Hum Genet. 2009;84:266–273. doi: 10.1016/j.ajhg.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ. Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum Mol Genet. 2001;10:1673–1677. doi: 10.1093/hmg/10.16.1673. [DOI] [PubMed] [Google Scholar]
- Schossig A, Wolf NI, Fischer C, Fischer M, Stocker G, et al. Mutations in ROGDI cause Kohlschütter-Tönz syndrome. Am J Hum Genet. 2012;90:701–707. doi: 10.1016/j.ajhg.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedano HO. Congenital oral anomalies in Argentinian children. Community Dent Oral Epidemiol. 1975;3:61–63. doi: 10.1111/j.1600-0528.1975.tb00281.x. [DOI] [PubMed] [Google Scholar]
- Simmer JP, Papagerakis P, Smith CE, Fisher DC, Rountrey AN, et al. Regulation of dental enamel shape and hardness. J Dent Res. 2010;89:1024–1038. doi: 10.1177/0022034510375829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verloes A, Jamblin P, Koulischer L, Bourguignon JP. A new form of skeletal dysplasia with amelogenesis imperfecta and platyspondyly. Clin Genet. 1996;49:2–5. doi: 10.1111/j.1399-0004.1996.tb04315.x. [DOI] [PubMed] [Google Scholar]
- Verloes A, Capri A, Gellé MP, van Wull W. Brachyolmia and amelogenesis imperfecta: a rare dento-osseous disorder. 2011. Abstract 22nd European Dysmorphology Meeting, Bischenberg, September.
- Witkop CJ. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited, problems in classification. J Oral Pathol. 1988;17:547–553. doi: 10.1111/j.1600-0714.1988.tb01332.x. [DOI] [PubMed] [Google Scholar]
- Witkop CJ, Sauk JJ. Heritable defects of enamel. In: Stewart R, Prescott G, editors. Oral Facial Genetics. St. Louis: Mosby Company; 1976. pp. 151–226. [Google Scholar]
- Wright JT, Torain M, Long K, Seow K, Crawford P, et al. Amelogenesis imperfecta: genotype-phenotype studies in 71 families. Cells Tissues Organs. 2011;194:279–283. doi: 10.1159/000324339. [DOI] [PMC free article] [PubMed] [Google Scholar]