

Abstract
Populations exposed to Plasmodium infection develop genetic mechanisms of protection against severe disease. The clinical manifestation of malaria results primarily from the lysis of infected erythrocytes and subsequent immune and inflammatory responses. Herein, we review the genetic alterations associated with erythrocytes or mediators of the immune system, which might influence malaria outcome. Moreover, polymorphisms in genes related to molecules involved in mechanisms of cytoadherence and their influence on malaria pathology are also discussed. The results of some studies have suggested that the combinatorial effects of a set of genetic factors in the erythrocyte-immunology pathway might be relevant to host resistance or susceptibility against Plasmodium infection. However, these results must be interpreted with caution because of the differences observed in the functionality and frequency of polymorphisms within different populations. With the recent advances in molecular biology techniques, more robust studies with reliable data have been reported, and the results of these studies have identified individual genetic factors for consideration in preventing severe disease and the individual response to treatment.
1. Introduction
Malaria is one of the most important and prevalent infectious diseases in the world. The World Health Organization (WHO) estimated 225 million malaria cases worldwide with 781,000 deaths due to Plasmodium infection per year [1]. Four species of Plasmodium (P. falciparum, P. vivax, P. malariae, and P. ovale) are responsible for almost all human infections [2].
Malaria has been associated to gene selective pressure in the human genome, and it has been associated as an evolutionary force of some genetic diseases, such as sickle cell disease (SCD), thalassemia, glucose-6-phosphate dehydrogenase (G6PD) deficiency, and other red blood cell (RBC) genetic anemia with Mendelian inheritance. Haldane (1949) suggested a “balanced polymorphism” where the hemoglobin S (HbS) homozygote disadvantage is recompensed through the resistance of the heterozygote (HbAS) in regions where malaria is endemic [3]. Reports associating several genetic disorders with malaria susceptibility or resistance are on the rise, and studies of heritability indicate that approximately 25% of the risk for severe malaria progression is determined through human genetic factors [4].
Genetic epidemiology may help in pointing out major molecular pathways of some infectious diseases, such as malaria, which involve a robust immune and inflammatory response and the participation of erythrocytes and other blood cells in its pathogenesis. The aim of this paper is to review the major genetic alterations in the human host associated with the clinical spectrum of malaria infection and disease development. We specifically address the areas of inherited disorders in red blood cells (RBC) and mutations in the genes of key molecules during the immune response that confer an increase of susceptibility or resistance against malaria. The multiplication of Plasmodium inside the RBC and its subsequent rupture have been implicated in several phenomena present in the malarial syndrome. A protective effect against malaria infection has been associated with genetic disorders involving the RBC, such as cytoskeleton disorders, surface antigen gene mutations, enzymatic machinery deficiencies, or hemoglobin alterations [5]. The immune response is critical for controlling Plasmodium infection, and the balance between proinflammatory (Th1-type) and anti-inflammatory (Th2-type) cytokines has been implicated in both the control of parasite multiplication and the development of symptoms. The genetic background of the affected individual might also influence cytokine expression and disease outcomes [6, 7]. Notably, the frequency of genetic alterations differs depending on the population origin and structure, and some mutations might differentially influence the disease outcome in different patterns.
Understanding the genetic alterations involving RBC disorders and the immune response might provide insight into the development of new strategies for host-genotype treatment and/or the prevention of malaria.
2. Inherited Disorders of Red Blood Cells and Malaria
2.1. Membrane and Enzymatic Disorders of Erythrocytes
Several membrane-inherited disorders of RBC provide additional information concerning the pathogenesis of malaria. Hereditary spherocytosis is a disorder characterized by the loss of membrane lipid surface. This common hemolytic anemia also reflects ineffective integral protein interactions and is associated with lower parasitemia [8]. Other RBC membrane inherited disorders include hereditary ovalocytosis, elliptocytosis, pyropoikilocytosis, and acanthocytosis. Elliptocytosis has been associated to resistance against invasion by P. falciparum in humans and P. knowlesi in experimental models [9]. However, ovalocytosis is a RBC-inherited cytoskeleton disorder most commonly associated with malaria. A particular type of southeast Asian ovalocytosis (SAO), also known as Melanesian elliptocytosis or stomatocytic elliptocytosis, is characterized by an inherited dominant trait related to heterozygosity for a 27-pair deletion in the gene encoding protein band 3 (SLC4A1Δ27) in the erythrocyte membrane [10]. Although SAO homozygosity has been associated with embryo mortality, its heterozygosity is associated with the absence of clinical symptoms and a lack of hemolysis. Ovalocytes are characterized as rigid and more resistant to changes in the shape as a result of low osmotic fragility and the low expression of several RBC antigens [11]. SAO ovalocytes characteristics have been associated with resistance to malaria infection, particularly against P. falciparum merozoites invasion [11]. Patients with elliptocytosis exhibit a similar degree of parasitemia independent of disease severity [11–13].
The Duffy, also called Duffy antigen/chemokine receptor (DARC), Fy glycoprotein, or CD234, is a RBC antigen encoded by the DARC human gene that is considered to be a nonspecific receptor for several chemokines. The P. vivax merozoite uses the Fy antigen to invade RBC [14]. The Fy antigen possesses two distinct alleles known as Fya and Fyb, which result from a single point mutation in codon 42 (rs2814778) that results in a glycine to asparagine substitution within the protein. Another polymorphism (−33T>C, no rs designation available) in the promoter region of the DARC gene ablates DARC expression on the surface of erythrocytes. Erythrocytes expressing Fya exhibited 41–50% lower binding to P. vivax compared with Fyb, and individuals with the Fya+b-phenotype showed a 30–80% lower risk of developing clinical symptoms of vivax malaria [15]. The RBC of individuals with the Fy-33 genotype are not susceptible to P. vivax merozoites invasion and are refractory to the erythrocytic stage of the disease. However, the hepatic malaria stage has been observed in these individuals, making them reservoirs for the disease [14]. With regard to P. falciparum infection, more than one receptor on the surface of RBC is responsible for merozoite infection, which include glycophorin A, B, and C (GPA, GPB, and GPC), protein band 3 and others (Y receptors, E, Z, and X), whose molecular identity has not yet been determined [16]. The genetic polymorphisms in the band 3 or GΥPC gene are highly prevalent in malaria endemic areas (Papua New Guinea) and confer resistance to severe diseases [17]. Polymorphisms identified in other receptors genes, such as GΥPA and GΥPB, have been shown to confer only partial protection against the Plasmodium invasion of RBC [18, 19]. Furthermore, in the Brazilian Amazon, an SNP in the GΥPB receptor gene (rs7683365) was associated with host susceptibility to P. falciparum infection [17].
The G6PD deficiency and low levels of pyruvate kinase are the most prevalent genetic alterations in RBC that can influence malaria outcomes. G6PD is a metabolic enzyme that catalyzes the first reaction in the pentose phosphate pathway, providing energy for the RBC in the form of nicotinamide adenine dinucleotide phosphate (NADPH). NADPH enables RBC to counterbalance oxidative stress through oxidant agents [20]. The G6PD gene is located on the X chromosome and is therefore more prevalent in men [21]. The G6PD deficiency is the most common cause of hereditary hemolytic anemia and is more prone to oxidative stress from the decreased production of NADPH. The clinical picture of G6PD deficiency involves different degrees of disease severity, which might include hemolytic anemia, neonatal hyperbilirubinemia, and asymptomatic cases [22]. Approximately 400 million people living in tropical and sub-tropical areas exhibit a G6PD deficiency, with a high diversity of variants, including the common G6PD B (wild type), G6PD A (nondeficient type), and G6PD A- (African deficient type) [23]. It has been suggested that G6PD deficient RBC might reduce intracellular parasite growth [24]. Moreover, studies have shown that infected erythrocytes deficient of G6PD were more phagocytosed by monocytes, which might be associated with the reduction of the parasitic load of the disease [24, 25]. Another enzyme associated with energy production is pyruvate kinase (PK), which is also an important factor in the susceptibility to malaria, and its deficiency has been associated with the reduced survival and increased phagocytosis of parasite-infected erythrocytes [26]. PK deficiency is the second most common cause of hereditary nonspherocytic hemolytic anemia in humans [27]. PK catalyzes the rate-limiting step of glycolysis, and the energy for erythrocytes is derived from glycolysis, as RBC lack mitochondria. The PK gene is highly pleomorphic and includes 59 SNPs and several loss-of-function variants that might be associated with decreased resistance to malaria [27].
2.2. Hemoglobin Alterations
Hemoglobin (Hb), the main compound of erythrocytes, is a tetrameric protein that consists of two pairs of unlike globin chains; each globin chain is associated with one prosthetic group, called a heme group. Hemoglobinopathies are inherited disorders of Hb that can be classified into two major groups. The first group involves structural alterations or variants of Hb, such as HbS, HbC, and HbE; the second group of hemoglobinopathies is classified as synthesis defects of Hb and has been associated with a decrease or absence of globin chain synthesis, with the most common alteration related to the alpha and beta-globin chains (alpha- and beta-thalassemia, resp.).
The alpha-thalassemia (alpha-thal) is the most common genetic disorder in the human population and is caused by the decreased or absent synthesis of the alpha globin chain due to the deletion or nondeletion mutation of one or both alpha-globin genes (HBA1 and HBA2), located on chromosome 16 [28, 29]. A 3.7-kilobase (Kb) deletion determines the most common form of alpha-thal, also called alpha+-thal, and there is unique potential for a rightward crossover between the HBA genes. One study reported that alpha+-thal increased the incidence of mild malaria [5]. However, several subsequent studies reported that alpha+-thal was associated with a reduced risk of uncomplicated malaria episodes [30] or a protective effect against severe forms of malaria [31–35].
Beta-thalassemia is characterized by the decreased synthesis of the beta-globin chain through a genetic alteration in the beta-globin gene (HBB) of the human globin gene cluster located on chromosome 11 [36]. The heterozygote of this inherited trait is associated with a mild anemia and an ineffective erythropoiesis, while the homozygote mutant is associated with severe anemia and the risk of early death. The beta-thalassemia trait is associated with a relative resistance against P. falciparum malaria [37] and protection against severe malaria forms [38].
The presence of a single nucleotide mutation (rs334) in the beta-globin gene (HBB) is associated with a structural modification of the beta polypeptide chain Glu6Val (HbS), resulting in the variant HbS, which can be found in the asymptomatic heterozygous state. This variant is commonly known as the sickle cell trait (HbAS). In 1978, Friedman suggested that the mechanism of resistance against Plasmodium of RBC with HbS might be solely due to intraerythrocytic conditions [39]. HbS polymerizes under deoxygenate conditions, and parasites become severely affected; HbS RBC become sickled, with the increased phagocytosis of infected erythrocytes [40, 41]. The presence of HbS in severe malaria patients is associated with less hemolysis and reduced levels of free heme [42, 43]. Many studies have described an association between the heterozygote HbAS and protection against malaria, with more than 90% protection against severe forms [44, 45]. A large genome-wide association (GWA) analysis of severe malaria cases in four different ethnic groups in Gambia has confirmed the role of HbS variant in resistance to malaria [46]. However, this study did not find new genetic associations as a result of the need for population-specific data on genome sequence variation. Therefore, it is difficult to design effective multicenter replication studies without information about sequence variation and haplotype structure in those African populations [46]. Moreover, in another GWA study carried out in Ghana, besides confirmation of previous reports on protective effects of HbS and blood group O, two novel resistance loci were described for severe malaria [47]. One of the loci was identified on chromosome 1q32 within the ATP2B4 gene, which encodes the main calcium pump of erythrocytes; and the other locus was indicated by an SNP on chromosome 16q22.2, possibly linked to a gene encoding the tight-junction MARVELD3, which may have a role in microvascular damage caused by endothelial adherence of parasitized erythrocytes [47].
Hemoglobin C (HbC) occurs from a point mutation leading to a Glu6Lys substitution at the sixth position in the beta-globin polypeptide chain (rs33930165) [48], and hemoglobin E (HbE) results from a substitution of Glu26Lys at position 26 in the beta-globin polypeptide chain (rs33950507) [49]. HbC homozygotic individuals exhibit mild hemolysis and splenomegaly, while the heterozygotic state is asymptomatic [50]. The RBC from HbE homozygous individuals is microcytic, with low hemoglobin concentrations, which reduces the possibility of merozoite invasion and impairs parasite growth within the variant RBC [51]. The presence of both protective factors, HbC and HbE, has been associated with a lower risk of developing severe forms of malaria [48, 49, 51–54].
Genetic alterations in erythrocytes were probably the first to be discovered as a result of the evolutionary pressure of malaria in the human genome. Mutations affecting several protection mechanisms related to RBC have been described for HbS [55]. Furthermore, several polymorphisms within the membrane cytoskeleton, surface antigen, and enzymatic machinery and other hemoglobin alterations within RBC influence the susceptibility and resistance against malaria.
2.3. Systemic Regulations of Heme
During the erythrocytic stage of malaria, merozoites multiply inside RBC, which result in the rupture of this structure and subsequent release of free Hb into the circulating blood. In the presence of reactive oxygen species (ROS), Hb releases its heme prosthetic group. In addition, the Plasmodium is responsible for the degradation of 60% to 80% of the total Hb [56]. Hb degradation contributes to heme release and ROS generation, which are harmful to both erythrocytic schizonts and the host [57]. Free heme is harmful to cells and tissues and can induce oxidative stress, inflammation, cytotoxicity [58], and cell death [59]. The Plasmodium has developed a series of protective mechanisms against the deleterious effects of free heme through the polymerization of heme in hemozoin, a malarial pigment that counteracts the pro-oxidative effects of iron (Fe) present in protoporphyrin IX [56]. The host also displays mechanisms of protection against free Hb cytotoxicity. Under homeostatic conditions, free Hb is released from the intravascular lysis of RBC and rapidly binds to haptoglobin (Hp). The CD163 receptor, expressed on monocytes/macrophages in the red pulp of the spleen, recognizes and internalizes the Hb/Hp complex [60]. Following internalization of the Hb/Hp complex, heme is degraded through the enzymatic action of heme-oxygenase (HO-1), producing biliverdin, iron, and carbon monoxide (CO). The beta chain (β) of Hp is approximately 40 kDa, and the alpha 1 chain (α1), which is synthesized by the allele variant Hp1, is approximately 8.86 kDa and is subdivided into 1S and 1F; the alpha 2 chain (α2), which is synthesized by the allele variant Hp2, is approximately 17.3 kDa [61]. These alleles have different affinities for free Hb (Hp1.1 > Hp1.2 > Hp2.2) and for CD163 (Hp2.2 > Hp1.2 > Hp1.1) [60]. The presence of the Hp2.2 genotype has been associated with increased iron redox activity and oxidative stress compared with the Hp1.1 genotype [62, 63]. Furthermore, monocytes can internalize the Hb/Hp2.2 complex, but not Hb/Hp1, and stimulate the release of proinflammatory cytokines [64]. Thus, the Hp2.2 has been associated with an increased susceptibility to various inflammatory conditions, including malaria. We have recently reported that subjects with the Hp2.2 genotype display a higher risk of developing symptomatic mild malaria (as opposed to asymptomatic) upon Plasmodium infection [65].
The induction of HO-1 in experimental models of malaria has been associated with increased resistance to malaria as a result of HO-1 in controlling heme-induced nonspecific tissue damage and inflammation [55, 66]. There is an (GT)n repeat polymorphism microsatellite in the promoter of the HMOX1 gene, which is associated with the increased or decreased synthesis of HO-1 in response to different stimuli [67]. Individuals with lower (GT)n dinucleotide repeats have a higher expression of HO-1, while higher (GT)n dinucleotide repetition is associated with the decreased synthesis of the protein [67]. This polymorphism has been described in several chronic degenerative diseases [68–73], but its role in malaria is controversial. The results of some studies have shown that the presence of lower (GT)n repeats in HMOX1 gene is associated with a greater chance of developing severe malaria, suggesting that the increased expression of HO-1 is deleterious for human malaria [74, 75]. We have shown that subjects with the long form (≥30 GT repeats) of the HMOX1 gene polymorphism have greater susceptibility to developing malaria and higher inflammatory scores than individuals with the short form [65]. However, studies in mice have shown that HO-1 is highly beneficial for malaria by conferring host tolerance to Plasmodium infection. Sickle Hb induces the expression of HO-1, which leads to CO production. CO binds with high affinity to free Hb and prevents the release of heme from hemoglobin, which reduces systemic levels of deleterious free heme [55, 66]. Other HMOX1 gene polymorphisms have been described, which correspond to the single nucleotide polymorphisms −1135G>A (no rs designation available) and −413A>TG (no rs designation available). However, only the last SNP seems to have functional importance [67].
Heme reduces the production of prostaglandin E2 (PGE2) and TGF-β from mononuclear cells through superoxide dismutase-1 (SOD-1), an enzyme responsible for the detoxification of harmful superoxide [76]. SOD-1 is a powerful predictor of malaria severity in individuals infected with P. vivax with higher sensitivity and sensibility than TNF-α levels [77], confirming the importance of this enzyme in malaria pathogenesis. The preliminary data from our group showed an association between several SNPs in the SOD-1 gene and different expressions of this enzyme in subjects with malaria. Furthermore, those SOD-1 SNPs were associated with malaria symptoms in individuals infected with P. vivax, indicating that the genetic predisposition of the individual might alter the response of these subjects against ROS.
3. Immune Response
3.1. Toll-Like Receptors (TLR)
Toll-like receptors are a family of transmembrane proteins present in monocytes, macrophages, and dendritic cells, which play a key role in the innate immune response. TLR recognize pathogen associated molecular patterns (PAMPs) through extracellular receptor modules and initiate the inflammatory cascade through the transcription of inflammatory cytokines, type 1 interferon, and chemokines through NF-kB or interferon regulatory factor dependent pathways [78, 79]. Furthermore, the stimulation of TLRs also leads to dendritic cell maturation and the induction of the adaptive immune response [80]. Each TLR has a unique pattern of expression, intracellular localization, and signaling pathway, resulting in different immune responses [81, 82]. The intracellular signaling of TLR is mediated through at least five different adapter proteins, including toll-interleukin 1 receptor domain containing adaptor protein (TIRAP), myeloid differentiation primary response gene 88 (MyD88), and toll-like receptor adaptor molecule 1 (TRIF) [83]. TLR1, 2, 4, 5, 6, and 10 are found on the extracellular surface of cells, whereas TLR3, 7, 8, and 9, each of which is a nucleic acid sensor, are located within the endoplasmic reticulum and cytoplasmic vesicles [84]. In the context of malaria infection, TLR2 and TLR4 have been reported to recognize P. falciparum glycosylphosphatidylinositol (GPI), while TLR9 has been reported to recognize Plasmodium DNA or the hemozoin pigment [85–87].
Some common SNPs in TLR genes are functionally important and affect the recognition of ligands and intracellular signaling. These SNPs have been associated with numerous infectious and parasitic diseases [88]. Several studies have associated TLR gene polymorphisms with clinical malaria and parasitemia levels. Two polymorphisms have been described in the 5′ untranslated region (UTR) of the TLR2 gene, a 22-base pair deletion in the first untranslated exon (Δ22), and an (GT)n dinucleotide repeat in the second intron [89]. Both polymorphisms, the deletion and shorter (GT)n repeats, are associated with reduced TLR2 reporter activity and TLR2 expression [90]. However, only the Δ22 heterozygous genotype was associated with protection from cerebral malaria [91]. Other SNPs of TLR2 (Arg677Trp, no rs designation available and Arg753Gln, rs5743708) were not identified in the Plasmodium-infected population [92]. In the case of the TLR4 gene, the more frequent genetic alterations studied were two nonsynonymous cosegregating SNPs (Asp299Gly, rs4986790 and Thr399Ile, rs4986791) that modify the ligand-binding site of the receptor [93]. The TLR4 Asp299Gly was associated with an increased risk of maternal anemia and infant low-birth weight in pregnant women with malaria [94]. In addition, the risk of severe malaria in children was increased 1.5-fold in the presence of TLR4 Asp299Gly and 2.6-fold with TLR4 Thr399Ile [92].
The TLR9 gene has been associated with the pathogenesis of severe malaria in humans and experimental models. Studies have demonstrated that TLR9-deficient mice survived more during cerebral malaria and that the antagonist-mediated TLR9 inhibition conferred protection against cerebral malaria in mice [95, 96]. A human study analyzing the (rs187084, −1486C>T) polymorphism in the promoter region of TLR9 gene showed an increased risk of low birth weight among infants from pregnant women with malaria, whereas increased parasitemia was observed in adults with mild malaria [84, 94]. However, in a large family and population-based association study, Malawi and Gambia showed that the effects of the four most common SNPs in the TLR9 gene, rs187084 (−1486C>T), rs5743836 (1237C>T), rs352139 (1174G>A), and rs352140 (2848G>A), were not associated with severe malaria [97]. Smaller population studies in Brazil, Iran, and Ghana showed associations between these polymorphisms and mild clinical malaria in their respective populations, raising the possibility that the TLR9 gene polymorphisms might be associated with a milder form of the disease [84, 98, 99]. The TIRAP/Mal interaction with tumor necrosis factor receptor-associated factor 6 (TRAF6) is responsible for mediating the downstream signaling of TLR2 and TLR4 to induce a proinflammatory response [100]. An SNP mutation in the TIRAP gene (rs8177374; S180L) has been associated with both protection against malaria [101] and susceptibility to the development of mild malaria [97], but the association of the TIRAP gene polymorphism with malaria remains controversial.
The importance of TLR in malaria infection has been recently described, particularly with regard to TLR2, 4, and 9. Genetic alterations in TLR and their signaling pathways remain controversial. Thus far, no conclusive evidence of polymorphisms in these receptors that might influence the disease outcome and effect host-genotype treatment have been identified.
3.2. Cytokines
Malaria infection is marked by changes in cytokine expression resulting from individual immune responses. Proinflammatory Th1-type cytokines (IL-1, IL-6, IL-8, IL-12, IFN-gamma, and TNF-α) are critical for controlling the erythrocytic and hepatic stages of Plasmodium infection [6, 7], but the excessive production of these cytokines might also contribute to disease manifestations and/or tissue damage, such as the brain in cases of cerebral malaria. It has also been suggested that anti-inflammatory Th2-type cytokines (IL-4, IL-10, and TGF-beta) downregulate Th1-type cytokines and the proinflammatory response, thereby preventing subjects from severe forms of malaria [102]. Furthermore, the new concept of tolerance against diseases has demonstrated protection against malaria. Unlike resistance, tolerance does not affect the pathogen burden but reduces tissue damage and other pathological effects of disease caused by the pathogen or immune response [103]. This tolerance might be due to the genetic profile of the affected individual, and HbS and HO-1 expressions are the most described mechanisms in malaria [55, 66, 104].
TNF-α is a proinflammatory cytokine that has attracted particular interest because of its ambiguous activity in host defense and pathogenesis of cerebral malaria and other serious complications [105]. High concentrations of TNF-α are related to the pathogenesis of symptoms associated with malaria, such as fever, and severe forms of infection, such as cerebral malaria [106, 107]. However, TNF-α has also been associated with the presence of potent antiparasitic activity, and persistent high levels of the cytokine lead to a rapid improvement in fever and a reduction in parasitemia [108, 109]. Genetic alterations in the TNF gene have been described in several studies with different populations in the world and sometimes with contradictory results. Population differences in susceptibility or resistance to malaria according to TNF SNPs may be a result of diverse evolutionary pressure between ethnicities, as well as different parasite strains and incidence of severe forms of disease. In Gambia, the SNPs TNF −308G>A (rs1800629) and TNF −238G>A (rs361525) were associated with an increased risk of cerebral malaria and severe malarial anemia, respectively [110, 111]. Studies in Gabon associated the TNF −308G>A polymorphism with a shorter interval to malaria reinfection and the TNF −238G>A polymorphism with protection against mild symptomatic malaria [23, 112]. In Sri Lanka, the TNF −308A allele was associated with severe malaria and other infections [113]. In another study in Mynamar, the TNFPD allele haplotype (−238G; −308G; −857T, rs1799724; −1031T, rs1799964) was associated with increased susceptibility to cerebral malaria because the transcription factor OCT-1 binds to TNF −857T in the TNFPD allele but not to TNF −857C in the TNFPA, B and C alleles and interacts with the proinflammatory NF-κB subunit transcription factor p65 at the adjacent binding site [114]. Other studies have shown no association between TNF gene polymorphisms and severe malaria in Kenya, Malawi, Mali, Tanzania, and Indonesia [111, 115–117]. It has been shown that lymphotoxin-a (LTa), which belongs to the TNF family, plays an important role in malaria [116]. Since LTa binds to the TNF receptors TNF-R1 and TNF-R2, TNF and LTa may exert their effects via the same receptors. LTA polymorphisms may influence resistance to malaria in humans, and two SNPs have been described: LTA C+80A (rs2239704) and LTA A+252G (rs909253). The first is an SNP that allows specific binding of the transcriptional repressor ABF-1 and, therefore, considered to be a low LTa-producing allele, has been associated with lower P. falciparum parasitemia in malaria-endemic Burkina Faso but was not associated with severe malaria in Gambia [118–120]. The SNP rs909253 has been reported to influence LTa production [121], but it was not associated with severe malaria in Sri Lanka [113]. Both SNPs (rs2239704 and rs909253) were reported to not be associated with severe malaria in a study from Gambia, Kenya, Malawi, and Indonesia [111, 116].
The chromosomal region 5q31–33 contains several important genes encoding molecules, such as cytokines, growth factors, and growth factor receptors, which are involved in immunity against Plasmodium infection [122]. The 5q31–33 region contains genes encoding cytokines IL-3, IL-4, IL-5, IL-9, IL-12B, IL13, and other genes, such as the immunologically active interferon regulatory factor-1 [123]. Concerning the genetic control of blood infection levels, linkage analyses studies have demonstrated the involvement of the 5q31–q33 region with parasitemia in populations from Cameroon and Burkina Faso [124, 125]. Asymptomatic parasite density was also linked to chromosome 5q31 in a study from Senegal [126]. A genomewide linkage study revealed three strongly suggestive lines of evidence for linkage between mild malaria attacking both the 6p25.1 and the 12q22 regions and between the 20p11q11 region and the prevalence of parasite density in asymptomatic Senegalese children [127]. Furthermore, in this study, one gene associated with malaria infection in the 5q31–q33 was also detected, confirming the importance of this genetic region in the susceptibility to malaria infection [127].
Type 1 helper T lymphocytes may be protective through the release of IFN-γ, which activates macrophages to destroy parasitized erythrocytes, promotes the production of opsonizing antibodies, and helps to destroy parasites during the hepatic cycle [128]. However, IFN-γ also has proinflammatory effects that may contribute to disease severity [129, 130]. Studies have reported associations between IFNG gene polymorphisms and susceptibility to disease. The first intron of the IFNG gene contains a highly polymorphic CA-repeat microsatellite, whose 12 CA-repeat allele is associated with high levels of IFN-γ production in vitro [131], and it has been associated with an SNP allele IFNG+874T (rs62559044), which coincides with a putative NF-κβ binding site [132]. In Gambia, no evidence of a strong association between severe malaria and the 12 CA-repeat allele and IFNG+874 (rs62559044) polymorphism was observed [133]. However, 14 CA repeats (IFNG CA14) were associated with CM in P. falciparum-infected children, and IFNG −183G/T (no rs designation available) and IFNG(CA)14/(CA)14 genotypes were more frequent in children with uncomplicated malaria than in children with cerebral malaria from Mali [134].
Concerning IL-13, an SNP −1055T>C (rs1800925) has showed a significant association with protection from severe malaria in Thailand [135]. A fine association mapping in the IL-13 gene using the same malaria subjects revealed that only rs1881457 located in the promoter region, which is in linkage disequilibrium with rs1800925, showed a significant association with severe malaria [123]. Furthermore, two SNPs (rs848, rs1881457) in IL-13 gene were found to be significantly different between those who have experienced one or more malaria attacks within past 10 years and those who did not in Sri Lanka [136].
IL-12 is a proinflammatory cytokine that boosts erythropoietic responses in infections with Plasmodium parasites. Low levels of IL-12 have been associated with the pathogenesis of malaria in children and nonimmune adults through the promotion of IFN-γ release from cells of the innate immune system, while high levels of this cytokine are associated with severe malaria [137]. IL-12 cytokine is a dimer composed of a 35-kD subunit encoded by the IL12A gene (chromosome 3p12-q13.2) and a 40-kD subunit encoded by the IL12B gene (chromosome 5q31–33), which exerts its effects on the immune response through receptors encoded by IL12RB1 and IL12RB2 [138]. A mutation in the promoter region of IL12B, IL12B-pro (rs17860508) has been associated with susceptibility to cerebral malaria [139, 140]. This polymorphism has been shown to affect gene expression and the production of cytokines and nitric oxide (4 bp less) [141]. Moreover, polymorphisms in IL12A (rs2243140) and IL12RB1 (rs429774) confer protection against severe malarial anemia [138].
IL-4 is a pleiotropic cytokine with multiple immune-modulating functions in several cells [142]. IL-4 plays an important role in IgE antibody antimalarial responses and regulates the differentiation of precursor T-helper cells into the Th2 subsets that regulate humoral immunity [122, 143]. Several polymorphisms in the IL-4 gene have been described, and four polymorphisms were described in the promoter region in association with total IgE production [144–146]. Despite the IL-4 −589C>T (rs2243250) influence on IgE levels, there was no association with severe malaria [122, 147]. However, a recent study that assesses the influence of 11 polymorphism in IL4 gene on predisposition to malaria in Mali found a genetic association between IL4 VNTR (rs8179190) and others IL4 mutations (−33C/T; rs2243267; rs2243268; rs2243282) with severe disease, supporting the view that IL4 genetic alterations could be a risk factor for malaria severity [129].
IL-1 is an endogenous pyrogen that plays an important role in the innate immune response of the human host to Plasmodium infection [148]. Two different genes (IL1A and IL1B) encode IL-1, which are located in chromosomal region 2q14, an area that also contains genes for IL-1 receptor types 1 and 2 (ILR1 and IL1R2), the IL-1 receptor antagonist (IL1RN), and other homologous genes that have not been well characterized [149]. The rapid induction of IL-1β might help control invading malaria parasites through the induction of an acute inflammatory response as part of the first line of defense; however, the overproduction of IL-1β might cause severe pathogenic effects [150]. Three different SNPs in the promoter region of the IL1B gene (−3737G>A, no rs designation available; −1464G>C, no rs designation available; −511A>G, rs16944) have been associated with IL-1β plasma levels [151]. The IL1B −511A allele was associated with an increased risk of severe malarial anemia and reduced levels of IL-1β [123]. In another study conducted in Gambia, significant associations between variations in IL1A +4845G>T (rs17561) and IL1B +3954C>T (rs1143634) were associated with symptomatic malaria [148].
IL-10 is an anti-inflammatory Th2-type cytokine produced primarily by monocytes and lymphocytes, and IL-10 exhibits various effects in the regulation of the immune response, including downregulating the expression of the proinflammatory (type 1) immune response [152]. The IL-10 gene is located on chromosome 1q31-32 within the promoter region and includes the well-defined SNPs IL-10 −1082A>G (rs1800870), IL-10 −819T>C (rs1800871), and IL-10 −592A>C (rs1800872) [153]. The SNP haplotype was associated with susceptibility to severe malarial anemia and functional changes in the plasma concentrations of IL-10, TNF-α, and IL-12 [154]. However, other studies have shown no evidence of association between the polymorphisms in the IL-10 gene and malaria severity [155]. A study in Gambia showed an association between the haplotype of five SNPs (+4949G, rs3024500/+919C, rs1518110/−627G, rs1800872/−1117C, rs1800896/−3585T, rs1800890) and resistance to cerebral malaria and severe anemia [156].
Several SNPs influence the levels of pro- and anti-inflammatory cytokines in malaria infection and might lead to an imbalance between these molecules that favor increased host susceptibility to Plasmodium. Thus, polymorphisms in the immune response might influence host disease tolerance against malaria.
3.3. Immunoglobulin Receptors and Nitric Oxide (NO)
Receptors for the Fc fragment of IgG (FcyRs) provide an important link between humoral and cellular immune responses. There are three families of FcyR (I, II, and III). The primary function of FcgRs is the activation of accessory cells against pathogens; thus, FcgRs are essential molecules in the host defense against infection [157]. Among the three classes of FcgR (FcgRI, FcgRII, and FcgRIII), the low-affinity FcgRII class is the most broadly distributed [158]. The FcγRIIA gene contains an important SNP with a G>A substitution in the region responsible for encoding the ligand-binding domain in which histidine (H) replaces arginine (R) at position 131 in the extracellular domain (no rs designation available). Both allotypes bind to human IgG1 and IgG3, but the FcγRIIA H131 allotype exhibits higher binding affinity to the IgG2 and IgG3 than the FcγRIIA R131 allotype, but none effectively binds to IgG4 [159]. The FcγRIIA H131 allotype is the only FcγR that binds with high affinity to IgG2, and this allele is essential for the phagocytosis of microorganisms opsonized with IgG2 and the clearance of immune complexes containing IgG2 [160, 161]. Furthermore, a protective role for IgG2 in malaria infections has been described, which involves the activation of immune effector cells through FcγRII [162]. The RR131 genotype protects against high levels of parasitemia, whereas the HH131 genotype was associated with susceptibility to severe malaria with high parasite burden [158, 163, 164]. An additional study showed an association between the FcgRIIA-RR131 genotype and severe malaria [165].
NO is a highly diffusible, lipid soluble-free radical that mediates the resistance of host severe malaria and other diseases. The production of NO and the cellular expression of enzyme-inducible nitric oxide synthase (NOS2) are associated with protection against severe forms of malaria [166]. The protective effect of NO against Plasmodium reflects parasite killing through reactive nitrogen metabolites and a decrease of endothelial adhesion molecules [167, 168]. In humans, NO is produced through the enzymatic conversion of L-arginine to L-citrulline using three different NO synthases (NOS), and NOS2 is induced through the response to pathogens and proinflammatory cytokines [166]. Several polymorphisms in the NOS2 gene have been associated with malaria severity. In Gambia, the SNP NOS2 −954G> C (no rs designation available) has been associated with resistance to severe malaria [169], whereas in another group of Gambian subjects, short forms of the polymorphic microsatellite (CCTTT) in the NOS2 transcription start site were associated with fatal malaria [170]. However, in Tanzania, neither NOS2 −954G>C polymorphisms nor CCTTT repeats were associated with severe malaria [171]. Another SNP in NOS2, −1173C>T (no rs designation available), was associated with protection against cerebral malaria in children in Tanzania and severe anemia in malaria individuals of Kenya [172]. However, no association between NOS2 polymorphisms and susceptibility to malaria was described [166].
The FcgR receptor and NO are important molecules involved in malaria outcomes, and several studies have attempted to associate mutations in these genes with increased susceptibility to develop severe forms of malaria. However, the results are conflicting, and no conclusion has yet been determined.
4. Mechanisms of Cytoadherence
One of the peculiar characteristics of P. falciparum-mediated malaria is the adhesion of infected erythrocytes to capillary endothelium [173]. This association contributes to the pathology of falciparum malaria because it causes microvascular occlusion and inhibits the immune response against parasites [174, 175]. This adhesion is one of the possible mechanisms underlying the pathogenesis of severe forms of malaria, such as cerebral and placental malaria [176–178]. The adhesion molecules include intercellular adhesion molecule 1 (ICAM-1, CD54), platelet/endothelial cell adhesion molecule1 (PECAM-1, CD31), vascular cell adhesion molecule1 (VCAM-1), thrombospondin, E-selectin, P-selectin, CD36, and chondroitin sulfate A [179]. Another characteristic of the virulent phenotype that contributes to the pathogenesis of P. falciparum is its ability to form rosettes, a property in which parasitized erythrocytes bind uninfected erythrocytes to form clumps of these cells [180]. The mechanism responsible for the virulence of the rosettes includes the microvasculature obstruction of the bloodstream in high parasitemia, favoring the invasion of merozoites and immune evasion [181–183]. The process of rosetting is mediated through ligand binding of P. falciparum to erythrocyte membrane protein1 (PfEMP1), which is expressed on the membrane of an infected RBC among a variety of uninfected RBC receptors, such as serum components, blood group antigens A and B, glucosaminoglycans, and complement receptor 1 (CR1) [184].
Autopsy studies of patients with fatal cerebral malaria or severe malarial anemia showed the sequestration of erythrocytes infected with Plasmodium on brain vascular endothelial cells with the increased expression of adhesion molecules, particularly ICAM-1 [185]. The results of a study in Killifi (Kenya) confirmed this association, showing that the adhesion of infected erythrocytes was highest in cerebral malaria compared with the asymptomatic control group [186]. The ICAM-1 (CD54) is a member of the immunoglobulin super-family, and its role in malaria susceptibility is not limited to an interaction with PfEMP1 [187]. ICAM-1 binds lymphocyte function-associated antigen (LFA)-1, facilitating the movement of leukocytes and active nature killer cells beyond the blood brain barrier during P. falciparum infection [188, 189]. Two SNPs have been described in the ICAM-1 gene, ICAM-1 Killifi (rs5491), which results from substitution of lysine for methionine at position 56 of the coding sequence, and a less well-described polymorphism (rs5498, K469E) [187]. The ICAM-1 Killifi polymorphism has been associated with both the resistance [190, 191] and susceptibility to severe forms of malaria [192]. However, subsequent studies in Gambia, Thailand, Senegal, Nigeria, and Kenya have reported no significant association between malaria phenotypes and either the ICAM-1Kilifi or the SNP identified in exon 6 (rs5498) [187, 193–197].
Most P. falciparum antigens bind to the CD36 molecule, and thus CD36 is considered the most important endothelial receptor for the sequestration of infected erythrocytes [186]. CD36 is an 88-kDa glycoprotein expressed on endothelial cells, macrophages, and dendritic cells, among others. However, in contrast to ICAM-1, this molecule is not expressed on the endothelial cells of brain capillaries [198]. CD36 serves as a receptor for several ligands, including low-density lipoprotein cholesterol (LDL-C), collagen, thrombospodin, and anionic phospholipids and participates in macrophage fusion induced through IL-4 [199]. Mutations in the CD36 receptor gene have been associated with protection against or susceptibility to severe forms of malaria. The CD36 deficiency might be induced through the two SNPs in the CD36 gene (T1264G in exon 10, rs3211938 and G1439C in exon 12, no rs designation available), which encode the truncated proteins that were expressed at high frequency in patients with severe malaria in Gambian, Tanzanian, and Kenyan patients [199–201]. This association was confirmed in a study in India, showing an association of the presence of the mutant allele in heterozygous individuals (1264T>G in exon 10) with protection against severe malaria [202]. A screening of CD36 gene in malaria patients from Thailand identified two SNPs in the promoter region (−14T>C and −53G>T, no rs designation available) that were associated with protection against cerebral malaria and one microsatellite polymorphism in intron 3 with 12 TG repeats that has been associated with the lower risk of cerebral malaria [203]. Genetic alterations in the CD36 gene influence the malaria outcome, regardless of different conclusions concerning the polymorphisms identified in this molecule, perhaps reflecting differences among the populations and clinical spectrums of the disease.
Platelet-endothelial cell adhesion molecule 1 (PECAM-1/CD31) is expressed in hematopoietic and endothelial cells. This adhesion molecule was identified as an endothelial receptor for erythrocytes infected with P. falciparum [204]. The PECAM-1 gene is polymorphic, and several polymorphisms have previously been described in the extracellular domain (exons three rs668 L/V, exon 8 rs12953 S/N, rs1131012 exon 12 R/G) and promoter region (GATA-2) [205]. Homozygous individuals with L125V and S563N SNPs in the CD31 gene were associated with an increased risk for developing cerebral malaria in Thailand [206]. However, in Kenya and Papua New Guinea, no association of the L125V SNP with malaria was observed [205]. Furthermore, an SNP in the PECAM-1 gene (exon 3 rs668 L/V) was identified as a risk factor for malaria in an endemic region, but this gene exhibited a significant association with protection from disease in a nonendemic region [202]. The mutation in exon 3 of the CD31 gene might affect the regulation of inflammation because it is present within the first IgG-like domain of the PECAM-1 molecule, which has been associated with hemophilic adhesion and regulates leukocyte transmigration [207, 208]. Thus, despite the influence of genetic alterations in the levels of adhesion molecules, polymorphisms might also alter molecular protein structure and impair the binding affinity of other molecules involved in the immunopathogenesis of malaria.
Complement receptor type 1 (CR1/CD35) is a membrane glycoprotein expressed in various cells, including erythrocytes, monocytes, B and T cells, monocytes, and dendritic cells [209]. CR1 binds with high affinity to C3b and C4b components and plays an important role in the clearance of immune complexes [210]. CR1 also plays a role in opsonization and the control of complement activation [209]. The expression of CR1 on erythrocytes has been related to the formation of rosettes, a phenomenon that results from the adhesion of PfEMP 1 on the surface of infected erythrocytes with a variety of membrane receptors on noninfected erythrocytes [180, 211, 212]. This process contributes to the pathogenesis of severe malaria because it causes the obstruction of cerebral capillaries and increases susceptibility to severe malaria anemia [186, 213]. Furthermore, erythrocyte CR1 binds immune complexes in the bloodstream through a process of “immune adherence” and removes them through phagocyte capitation in the liver and spleen [214]. Subjects with high levels of CR1 on erythrocytes are more likely to form rosettes and contribute to the sequestration of cell clumps in the microvasculature of the brain and other vital organs [180]. Moreover, high levels of CR1 also carry immune complex, which might be recognized by monocytes and endothelial cells to produce proinflammatory mediators [184]. The levels of CR1 on erythrocytes are genetically determined and correlated with the HindIII restriction fragment length polymorphism (RFLP) mutation in the CR1 gene. Homozygous subjects for the 7.4 kb HindIII genomic fragment (the H allele) have high levels of CR1 on erythrocytes, whereas homozygous individuals for the 6.9-kb genomic HindIII fragment (the L allele) exhibit low expression, and HL heterozygous individuals show intermediary levels of CR1 in the membrane of erythrocytes [211]. The association of this polymorphism with the susceptibility or resistance to malaria is contradictory. In Gambia and other African populations, a significant association between the L allele and protection from severe malaria was not observed [193, 215]. In Thailand, the LL genotype was demonstrated as a risk factor for severe malaria [216], and in Papua New Guinea, individuals heterozygous for the L allele (HL) were correlated with protection against severe malaria [217]. A new polymorphism in the promoter region of the CR1 gene (rs9429942) was associated with higher levels of CR1 on the surface of RBC and protection against cerebral malaria in Thailand [218]. Different associations between the CR1 genotype with malaria might be associated with the endemicity of malaria in different regions and an under or overestimation of the actual CR1 levels and interactions between CR1 and other genetic alterations [184].
5. Conclusion
Over the past several years, an increase in the number of scientific publications associated with the genetic predisposition to malaria and severe forms of this disease has been observed. As a result of technological advances, studies of SNPs were exchanged for studies with sophisticated gene sequencing and analyses using advanced molecular biology software. On the basis of the discovery of new functional mutations that alter the expression of several proteins fundamentally implicated in malaria pathogenesis, it is possible to individualize patient care depending on host genotype, as previously demonstrated [219]. However, molecular epidemiology studies should always be interpreted with caution because of the differences in the functionality and frequency of the polymorphisms observed in different populations as a result of diverse evolutionary pressure between different ethnicities.
The clinical manifestation of malaria is primarily described by the lysis of infected erythrocytes and subsequent immune and inflammatory response. Thus, it is critical to understand the role of genetic alterations in this pathway that might influence the disease outcome and severity of malaria. Furthermore, it should be observed that not merely one genetic alteration but rather the combination of a set of genetic factors might influence the susceptibility or resistance to malaria (Figure 1). The results from research studies have already shown that individual genetic factors must be considered for the prevention from severe diseases and individual responses to treatment.
Figure 1.
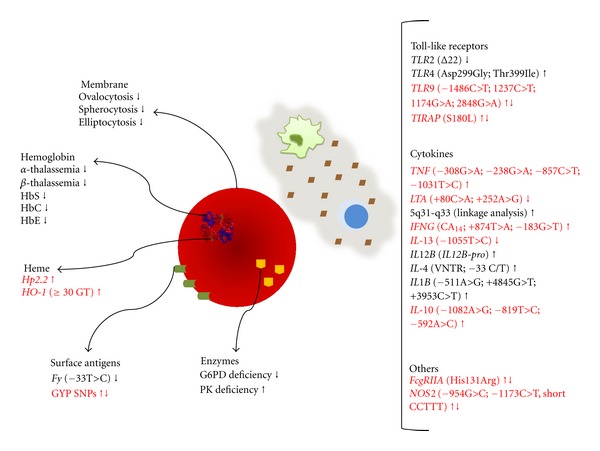
Influence of erythrocyte and immune response gene polymorphisms in malaria outcome. The diagram summarizes the major genetic alterations identified in the erythrocyte and immune response pathways that influence malaria outcome. The up arrow indicates susceptibility, and the down arrow indicates resistance to malaria. Contradictory or not confirmed results are represented by red font color. The protective effect of inherited genetic disorders involving the RBC on malaria infection has been associated with membrane cytoskeleton disorders, surface antigen mutations, enzymatic machinery deficiencies, or hemoglobin and its heme prosthetic group alterations. Considering the immune response, several polymorphisms in gene encoding the TLR receptors and in important cytokines involved in malaria immunopathology are described in the literature. Furthermore, genetic alterations in the FcgRIIA receptor and nitric oxide synthase were also associated with resistance/susceptibility to malaria.
Conflict of Interests
The authors declare that they have no conflict of interests.
Acknowledgments
The authors would like to thank the Instituto Nacional de Ciência e Tecnologia (INCT) for financial support. The authors also thank Jorge Tolentino, Marcio Santana, and Adorielze Leite for technical and logistical support. The work from our laboratory reviewed herein was supported through grants from FINEP, CNPq, and FAPESB. M. S. Goncalves and M. Barral-Netto are senior investigators from CNPq.
References
- 1.WHO—World Health Organization. http://www.who.int/malaria/en/
- 2.Cavasini MT, Ribeiro WL, Kawamoto F, Ferreira MU. How prevalent is Plasmodium malariae in Rondônia, western Brazilian Amazon? Revista da Sociedade Brasileira de Medicina Tropical. 2000;33(5):489–492. doi: 10.1590/s0037-86822000000500011. [DOI] [PubMed] [Google Scholar]
- 3.Haldane JBS. Suggestions as to quantitative measurement of rates of evolution. Evolution. 1949;3(1):51–56. doi: 10.1111/j.1558-5646.1949.tb00004.x. [DOI] [PubMed] [Google Scholar]
- 4.Mackinnon MJ, Mwangi TW, Snow RW, Marsh K, Williams TN. Heritability of malaria in Africa. PLoS Medicine. 2005;2(12, article e340) doi: 10.1371/journal.pmed.0020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams TN, Maitland K, Bennett S, et al. High incidence of malaria in α-thalassaemic children. Nature. 1996;383(6600):522–525. doi: 10.1038/383522a0. [DOI] [PubMed] [Google Scholar]
- 6.Schofield L, Villaquiran J, Ferreira A, Schellekens H, Nussenzweig R, Nussenzweig V. γ Interferon, CD8+ T cells and antibodies required for immunity to malaria sporozoites. Nature. 1987;330(6149):664–666. doi: 10.1038/330664a0. [DOI] [PubMed] [Google Scholar]
- 7.Vreden SGS, van den Broek MF, Oettinger MC, Verhave JP, Meuwissen JHET, Sauerwein RW. Cytokines inhibit the development of liver schizonts of the malaria parasite plasmodium berghei in vivo. European Journal of Immunology. 1992;22(9):2271–2275. doi: 10.1002/eji.1830220914. [DOI] [PubMed] [Google Scholar]
- 8.Schulman S, Roth EF, Cheng B, et al. Growth of Plasmodium falciparum in human erythrocytes containing abnormal membrane proteins. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(18):7339–7343. doi: 10.1073/pnas.87.18.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hadley TJ, Miller LH. Invasion of erythrocytes by malaria parasites: erythrocyte ligands and parasite receptors. Progress in Allergy. 1988;41:49–71. [PubMed] [Google Scholar]
- 10.Williams TN. Human red blood cell polymorphisms and malaria. Current Opinion in Microbiology. 2006;9(4):388–394. doi: 10.1016/j.mib.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Kidson C, Lamont G, Saul A, Nurse GT. Ovalocytic erythrocytes from Melanesians are resistant to invasion by malaria parasites in culture. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(9):5829–5832. doi: 10.1073/pnas.78.9.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallagher PG. Red cell membrane disorders. Hematology. 2005:13–18. doi: 10.1182/asheducation-2005.1.13. [DOI] [PubMed] [Google Scholar]
- 13.Genton B, Al-Yaman F, Mgone CS, et al. Ovalocytosis and cerebral malaria. Nature. 1995;378(6557):564–565. doi: 10.1038/378564a0. [DOI] [PubMed] [Google Scholar]
- 14.Maestre A, Muskus C, Duque V, et al. Acquired antibody responses against Plasmodium vivax infection vary with host genotype for duffy antigen receptor for chemokines (DARC) PLoS ONE. 2010;5(7) doi: 10.1371/journal.pone.0011437.e11437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King CL, Adams JH, Xianli J, et al. Fy(a)/Fy(b) antigen polymorphism in human erythrocyte Duffy antigen affects susceptibility toPlasmodium vivax malaria. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(50):20113–20118. doi: 10.1073/pnas.1109621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell. 2006;124(4):755–766. doi: 10.1016/j.cell.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Tarazona-Santos E, Castilho L, Amaral DRT, et al. Population genetics of GYPB and association study between GYPB∗S/s polymorphism and susceptibility to P. falciparum infection in the Brazilian Amazon. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0016123.e16123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weatherall DJ. Host genetics and infectious disease. Parasitology. 1996;112, supplement:S23–S29. [PubMed] [Google Scholar]
- 19.Miller LH. Impact of malaria on genetic polymorphism and genetic diseases in Africans and African Americans. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(7):2415–2419. doi: 10.1073/pnas.91.7.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phompradit P, Kuesap J, Chaijaroenkul W, et al. Prevalence and distribution of glucose-6-phosphate dehydrogenase (G6PD) variants in Thai and Burmese populations in malaria endemic areas of Thailand. Malaria journal. 2011;10, article 368 doi: 10.1186/1475-2875-10-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. The Lancet. 2008;371(9606):64–74. doi: 10.1016/S0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- 22.Frank JE. Diagnosis and management of G6PD deficiency. American Family Physician. 2005;72(7):1277–1282. [PubMed] [Google Scholar]
- 23.Mombo L-E, Ntoumi F, Bisseye C, et al. Human genetic polymorphisms and asymptomatic Plasmodium falciparum malaria in Gabonese schoolchildren. American Journal of Tropical Medicine and Hygiene. 2003;68(2):186–190. [PubMed] [Google Scholar]
- 24.Cappadoro M, Giribaldi G, O’Brien E, et al. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood. 1998;92(7):2527–2534. [PubMed] [Google Scholar]
- 25.Ayi K, Turrini F, Piga A, Arese P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: a common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood. 2004;104(10):3364–3371. doi: 10.1182/blood-2003-11-3820. [DOI] [PubMed] [Google Scholar]
- 26.Ayi K, Min-Oo G, Serghides L, et al. Pyruvate kinase deficiency and malaria. The New England Journal of Medicine. 2008;358(17):1805–1810. doi: 10.1056/NEJMoa072464. [DOI] [PubMed] [Google Scholar]
- 27.Berghout J, Higgins S, Loucoubar C, Sakuntabhai A, Kain KC, Gros P. Genetic diversity in human erythrocyte pyruvate kinase. Genes and Immunity. 2012;13(1):98–102. doi: 10.1038/gene.2011.54. [DOI] [PubMed] [Google Scholar]
- 28.Yuthavong Y, Wilairat P. Protection against malaria by thalassaemia and haemoglobin variants. Parasitology Today. 1993;9(7):241–245. doi: 10.1016/0169-4758(93)90065-n. [DOI] [PubMed] [Google Scholar]
- 29.Weatherall DJ. Thalassaemia and malaria, revisited. Annals of Tropical Medicine and Parasitology. 1997;91(7):885–890. doi: 10.1080/00034989760653. [DOI] [PubMed] [Google Scholar]
- 30.Enevold A, Lusingu JP, Mmbando B, et al. Reduced risk of uncomplicated malaria episodes in children with alpha +-thalassemia in Northeastern Tanzania. American Journal of Tropical Medicine and Hygiene. 2008;78(5):714–720. [PubMed] [Google Scholar]
- 31.Allen SJ, O’Donnell A, Alexander NDE, et al. α+-Thalassemia protects children against disease caused by other infections as well as malaria. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(26):14736–14741. doi: 10.1073/pnas.94.26.14736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wambua S, Mwangi TW, Kortok M, et al. The effect of α+-thalassaemia on the incidence of malaria and other diseases in children living on the coast of Kenya. PLoS Medicine. 2006;3(5, article e158) doi: 10.1371/journal.pmed.0030158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams TN, Wambua S, Uyoga S, et al. Both heterozygous and homozygous α+ thalassemias protect against severe and fatal Plasmodium falciparum malaria on the coast of Kenya. Blood. 2005;106(1):368–371. doi: 10.1182/blood-2005-01-0313. [DOI] [PubMed] [Google Scholar]
- 34.Mockenhaupt FP, Ehrhardt S, Gellert S, et al. α+-thalassemia protects African children from severe malaria. Blood. 2004;104(7):2003–2006. doi: 10.1182/blood-2003-11-4090. [DOI] [PubMed] [Google Scholar]
- 35.Gahutu J-B, Musemakweri A, Harms G, Mockenhaupt FP. Prevalence of classic erythrocyte polymorphisms among 749 children in southern highland Rwanda. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2012;106(1):63–65. doi: 10.1016/j.trstmh.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Das SK, Talukder G. A review on the origin and spread of deleterious mutants of the β-globin gene in Indian populations. Journal of Comparative Human Biology. 2001;52(2):93–109. doi: 10.1078/0018-442x-00022. [DOI] [PubMed] [Google Scholar]
- 37.Willcox M, Bjorkman A, Brohult J. A case-control study in northern Liberia of Plasmodium falciparum malaria in haemoglobin S and β-thalassaemia traits. Annals of Tropical Medicine and Parasitology. 1983;77(3):239–246. doi: 10.1080/00034983.1983.11811704. [DOI] [PubMed] [Google Scholar]
- 38.Luzzi GA, Merry AH, Newbold CI, Marsh K, Pasvol G, Weatherall DJ. Surface antigen expression on Plasmodium falciparum-infected erythrocytes is modified in α- and β-thalassemia. The Journal of Experimental Medicine. 1991;173(4):785–791. doi: 10.1084/jem.173.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedman MJ. Erythrocytic mechanism of sickle cell resistance to malaria. Proceedings of the National Academy of Sciences of the United States of America. 1978;75(4):1994–1997. doi: 10.1073/pnas.75.4.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abu-Zeid YA, Abdulhadi NH, Hviid L, et al. Lymphoproliferative responses to Plasmodium falciparum antigens in children with and without the sickle cell trait. Scandinavian Journal of Immunology. 1991;34(2):237–242. doi: 10.1111/j.1365-3083.1991.tb01542.x. [DOI] [PubMed] [Google Scholar]
- 41.Weatherall DJ. Common genetic disorders of the red cell and the ‘malaria hypothesis’. Annals of Tropical Medicine and Parasitology. 1987;81(5):539–548. doi: 10.1080/00034983.1987.11812155. [DOI] [PubMed] [Google Scholar]
- 42.Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(20):9285–9289. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balla J, Jacob HS, Balla G, Nath K, Vercellotti GM. Endothelial cell heme oxygenase and ferritin induction by heme proteins: a possible mechanism limiting shock damage. Transactions of the Association of American Physicians. 1992;105:1–6. [PubMed] [Google Scholar]
- 44.López C, Saravia C, Gomez A, Hoebeke J, Patarroyo MA. Mechanisms of genetically-based resistance to malaria. Gene. 2010;467(1-2):1–12. doi: 10.1016/j.gene.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 45.Hill AVS, Allsopp CEM, Kwiatkowski D, et al. Common West African HLA antigens are associated with protection from severe malaria. Nature. 1991;352(6336):595–600. doi: 10.1038/352595a0. [DOI] [PubMed] [Google Scholar]
- 46.Jallow M, Koram KA, Ying YT, et al. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nature Genetics. 2009;41(6):657–665. doi: 10.1038/ng.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Timmann C, Thye T, Vens M, et al. Genome-wide association study indicates two novel resistance loci for severe malaria. Nature. 2012;489(7416):443–446. doi: 10.1038/nature11334. [DOI] [PubMed] [Google Scholar]
- 48.Agarwal A, Guindo A, Cissoko Y, et al. Hemoglobin C associated with protection from severe malaria in the Dogon of Mali, a West African population with a low prevalence of hemoglobin S. Blood. 2000;96(7):2358–2363. [PubMed] [Google Scholar]
- 49.Nagel RL, Raventos-Suarez C, Rabry ME. Impairment of the growth of Plasmodium falciparum in HbEE erythrocytes. Journal of Clinical Investigation. 1981;68(1):303–305. doi: 10.1172/JCI110248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mockenhaupt FP, Ehrhardt S, Cramer JP, et al. Hemoglobin C and resistance to severe malaria in Ghanaian children. Journal of Infectious Diseases. 2004;190(5):1006–1009. doi: 10.1086/422847. [DOI] [PubMed] [Google Scholar]
- 51.Chotivanich K, Udomsangpetch R, Pattanapanyasat K, et al. Hemoglobin E: a balanced polymorphism protective against high parasitemias and thus severe P falciparum malaria. Blood. 2002;100(4):1172–1176. [PubMed] [Google Scholar]
- 52.Modiano D, Bancone G, Ciminelli BM, et al. Haemoglobin S and haemoglobin C: “Quick but costly” versus “slow but gratis” genetic adaptations to Plasmodium falciparum malaria. Human Molecular Genetics. 2008;17(6):789–799. doi: 10.1093/hmg/ddm350. [DOI] [PubMed] [Google Scholar]
- 53.Modiano D, Luoni G, Sirima BS, et al. Haemoglobin C protects against clinical Plasmodium falciparum malaria. Nature. 2001;414(6861):305–308. doi: 10.1038/35104556. [DOI] [PubMed] [Google Scholar]
- 54.Hutagalung R, Wilairatana P, Looareesuwan S, Brittenham GM, Aikawa M, Gordeuk VR. Influence of hemoglobin E trait on the severity of falciparum malaria. Journal of Infectious Diseases. 1999;179(1):283–286. doi: 10.1086/314561. [DOI] [PubMed] [Google Scholar]
- 55.Ferreira A, Marguti I, Bechmann I, et al. Sickle hemoglobin confers tolerance to plasmodium infection. Cell. 2011;145(3):398–409. doi: 10.1016/j.cell.2011.03.049. [DOI] [PubMed] [Google Scholar]
- 56.Francis SE, Sullivan DJ, Goldberg DE. Hemoglobin metabolism in the malaria parasite Plasmodium falciparium . Annual Review of Microbiology. 1997;51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- 57.Orjih AU, Banyal HS, Chevli R, Fitch CD. Hemin lyses malaria parasites. Science. 1981;214(4521):667–669. doi: 10.1126/science.7027441. [DOI] [PubMed] [Google Scholar]
- 58.Ferreira A, Balla J, Jeney V, Balla G, Soares MP. A central role for free heme in the pathogenesis of severe malaria: the missing link? Journal of Molecular Medicine. 2008;86(10):1097–1111. doi: 10.1007/s00109-008-0368-5. [DOI] [PubMed] [Google Scholar]
- 59.Gozzelino R, Soares MP. Heme sensitization to TNF-mediated programmed cell death. Advances in Experimental Medicine and Biology. 2011;691:211–219. doi: 10.1007/978-1-4419-6612-4_22. [DOI] [PubMed] [Google Scholar]
- 60.Kristiansen M, Graversen JH, Jacobsen C, et al. Identification of the haemoglobin scavenger receptor. Nature. 2001;409(6817):198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- 61.Maeda N, Yang F, Barnett DR. Duplication within the haptoglobin Hp2 gene. Nature. 1984;309(5964):131–135. doi: 10.1038/309131a0. [DOI] [PubMed] [Google Scholar]
- 62.Asleh R, Guetta J, Kalet-Litman S, Miller-Lotan R, Levy AP. Haptoglobin genotype- and diabetes-dependent differences in iron-mediated oxidative stress in vitro and in vivo. Circulation Research. 2005;96(4):435–441. doi: 10.1161/01.RES.0000156653.05853.b9. [DOI] [PubMed] [Google Scholar]
- 63.Melamed-Frank M, Lache O, Enav BI, et al. Structure-function analysis of the antioxidant properties of haptoglobin. Blood. 2001;98(13):3693–3698. doi: 10.1182/blood.v98.13.3693. [DOI] [PubMed] [Google Scholar]
- 64.Rogerson S. What is the relationship between haptoglobin, malaria, and anaemia? PLoS Medicine. 2006;3(5, article e200) doi: 10.1371/journal.pmed.0030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mendonça VRR, Luza NF, Santosa NJG, et al. Association between the haptoglobin and heme oxygenase 1 genetic profiles and soluble CD163 in susceptibility to and severity of human malaria. Infection and Immunity. 2012;80(4):1445–1454. doi: 10.1128/IAI.05933-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pamplona A, Ferreira A, Balla J, et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nature Medicine. 2007;13(6):703–710. doi: 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
- 67.Exner M, Minar E, Wagner O, Schillinger M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radical Biology and Medicine. 2004;37(8):1097–1104. doi: 10.1016/j.freeradbiomed.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 68.Kimpara T, Takeda A, Watanabe K, et al. Microsatellite polymorphism in the human heme oxygenase-1 gene promoter and its application in association studies with Alzheimer and Parkinson disease. Human Genetics. 1997;100(1):145–147. doi: 10.1007/s004390050480. [DOI] [PubMed] [Google Scholar]
- 69.Chen Y-H, Lin SJ, Lin MW, et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Human Genetics. 2002;111(1):1–8. doi: 10.1007/s00439-002-0769-4. [DOI] [PubMed] [Google Scholar]
- 70.Yamada N, Yamaya M, Okinaga S, et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. American Journal of Human Genetics. 2000;66(1):187–195. doi: 10.1086/302729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sheu C-C, Zhai R, Wang Z, et al. Heme Oxygenase-1 microsatellite polymorphism and haplotypes are associated with the development of acute respiratory distress syndrome. Intensive Care Medicine. 2009;35(8):1343–1351. doi: 10.1007/s00134-009-1504-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chin HJ, Cho HJ, Lee TW, et al. The heme oxygenase-1 genotype is a risk factor to renal impairment of IgA nephropathy at diagnosis, which is a strong predictor of mortality. Journal of Korean Medical Science. 2009;24, supplement:S30–37. doi: 10.3346/jkms.2009.24.S1.S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wagener FADTG, Toonen EJM, Wigman L, et al. HMOX1 promoter polymorphism modulates the relationship between disease activity and joint damage in rheumatoid arthritis. Arthritis and Rheumatism. 2008;58(11):3388–3393. doi: 10.1002/art.23970. [DOI] [PubMed] [Google Scholar]
- 74.Sambo MR, Trovoada MJ, Benchimol C, et al. Transforming growth factor beta 2 and heme oxygenase 1 genes are risk factors for the cerebral malaria syndrome in Angolan children. PLoS ONE. 2010;5(6) doi: 10.1371/journal.pone.0011141.e11141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Takeda M, Kikuchi M, Ubalee R, et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to cerebral malaria in Myanmar. Japanese Journal of Infectious Diseases. 2005;58(5):268–271. [PubMed] [Google Scholar]
- 76.Andrade BB, Araújo-Santos T, Luz NF, et al. Heme impairs prostaglandin E2 and TGF-β production by human mononuclear cells via Cu/Zn superoxide dismutase: insight into the pathogenesis of severe malaria. Journal of Immunology. 2010;185(2):1196–1204. doi: 10.4049/jimmunol.0904179. [DOI] [PubMed] [Google Scholar]
- 77.Andrade BB, Reis-Filho A, Souza-Neto SM, et al. Plasma superoxide dismutase-1 as a surrogate marker of vivax malaria severity. PLoS Neglected Tropical Diseases. 2010;4(4, articlee 650) doi: 10.1371/journal.pntd.0000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Basu M, Maji AK, Chakraborty A, et al. Genetic association of Toll-like-receptor 4 and tumor necrosis factor-α polymorphisms with Plasmodium falciparum blood infection levels. Infection, Genetics and Evolution. 2010;10(5):686–696. doi: 10.1016/j.meegid.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 79.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochemical and Biophysical Research Communications. 2009;388(4):621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 80.Janeway CA, Medzhitov R. Innate immune recognition. Annual Review of Immunology. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 81.Ishii KJ, Uematsu S, Akira S. ‘Toll’ gates for future immunotherapy. Current Pharmaceutical Design. 2006;12(32):4135–4142. doi: 10.2174/138161206778743484. [DOI] [PubMed] [Google Scholar]
- 82.McCall MBB, Netea MG, Hermsen CC, et al. Plasmodium falciparum infection causes proinflammatory priming of human TLR responses. Journal of Immunology. 2007;179(1):162–171. doi: 10.4049/jimmunol.179.1.162. [DOI] [PubMed] [Google Scholar]
- 83.O’Neill LAJ, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature Reviews Immunology. 2007;7(5):353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 84.Leoratti FMS, Farias L, Alves FP, et al. Variants in the toll-like receptor signaling pathway and clinical outcomes of malaria. Journal of Infectious Diseases. 2008;198(5):772–780. doi: 10.1086/590440. [DOI] [PubMed] [Google Scholar]
- 85.Krishnegowda G, Hajjar AM, Zhu J, et al. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. The Journal of Biological Chemistry. 2005;280(9):8606–8616. doi: 10.1074/jbc.M413541200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Coban C, Ishii KJ, Kawai T, et al. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. Journal of Experimental Medicine. 2005;201(1):19–25. doi: 10.1084/jem.20041836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parroche P, Lauw FN, Goutagny N, et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(6):1919–1924. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schröder NWJ, Schumann RR. Single nucleotide polymorphisms of Toll-like receptors and susceptibility to infectious disease. Lancet Infectious Diseases. 2005;5(3):156–164. doi: 10.1016/S1473-3099(05)01308-3. [DOI] [PubMed] [Google Scholar]
- 89.Noguchi E, Nishimura F, Fukai H, et al. An association study of asthma and total serum immunoglobin E levels for Toll-like receptor polymorphisms in a Japanese population. Clinical and Experimental Allergy. 2004;34(2):177–183. doi: 10.1111/j.1365-2222.2004.01839.x. [DOI] [PubMed] [Google Scholar]
- 90.Yim J-J, Lee HW, Lee HS, et al. The association between microsatellite polymorphisms in intron II of the human Toll-like receptor 2 gene and tuberculosis among Koreans. Genes and Immunity. 2006;7(2):150–155. doi: 10.1038/sj.gene.6364274. [DOI] [PubMed] [Google Scholar]
- 91.Greene JA, Sam-Agudu N, John CC, Opoka RO, Zimmerman PA, Kazura JW. Toll-like receptor polymorphisms and cerebral malaria: TLR2 Δ22 polymorphism is associated with protection from cerebral malaria in a case control study. Malaria Journal. 2012;11, article 47 doi: 10.1186/1475-2875-11-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mockenhaupt FP, Cramer JP, Hamann L, et al. Toll-like receptor (TLR) polymorphisms in African children: common TLR-4 variants predispose to severe malaria. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(1):177–182. doi: 10.1073/pnas.0506803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mockenhaupt FP, Cramer JP, Hamann L, et al. Toll-Like Receptor (TLR) polymorphisms in African children: common TLR-4 variants predispose to severe malaria. Journal of Communicable Diseases. 2006;38(3):230–245. [PubMed] [Google Scholar]
- 94.Mockenhaupt FP, Hamann L, Von Gaertner C, et al. Common polymorphisms of toll-like receptors 4 and 9 are associated with the clinical manifestation of malaria during pregnancy. The Journal of Infectious Diseases. 2006;194(2):184–188. doi: 10.1086/505152. [DOI] [PubMed] [Google Scholar]
- 95.Coban C, Uematsu S, Arisue N, et al. Pathological role of Toll-like receptor signaling in cerebral malaria. International Immunology. 2007;19(1):67–79. doi: 10.1093/intimm/dxl123. [DOI] [PubMed] [Google Scholar]
- 96.Franklin BS, Ishizaka ST, Lamphier M, et al. Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(9):3689–3694. doi: 10.1073/pnas.1015406108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Campino S, Forton J, Auburn S, et al. TLR9 polymorphisms in African populations: no association with severe malaria, but evidence of cis-variants acting on gene expression. Malaria Journal. 2009;8(1, article 44) doi: 10.1186/1475-2875-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zakeri S, Pirahmadi S, Mehrizi AA, Djadid ND. Genetic variation of TLR-4, TLR-9 and TIRAP genes in Iranian malaria patients. Malaria Journal. 2011;10, article 77 doi: 10.1186/1475-2875-10-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Omar AH, Yasunami M, Yamazaki A, et al. Toll-like receptor 9 (TLR9) polymorphism associated with symptomatic malaria: a cohort study. Malaria Journal. 2012;11(1):p. 168. doi: 10.1186/1475-2875-11-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yamamoto M, Sato S, Hemmi H, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420(6913):324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- 101.Khor CC, Vannberg FO, Chapman SJ, et al. Positive replication and linkage disequilibrium mapping of the chromosome 21q22.1 malaria susceptibility locus. Genes and Immunity. 2007;8(7):570–576. doi: 10.1038/sj.gene.6364417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wilson NO, Bythwood T, Solomon W, et al. Elevated levels of IL-10 and G-CSF associated with asymptomatic malaria in pregnant women. Infectious Diseases in Obstetrics and Gynecology. 2010;2010:7 pages. doi: 10.1155/2010/317430.317430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335(6071):936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Seixas E, Gozzelino R, Chora A, et al. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(37):15837–15842. doi: 10.1073/pnas.0903419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kwiatkowski D. Genetic susceptibility to malaria getting complex. Current Opinion in Genetics and Development. 2000;10(3):320–324. doi: 10.1016/s0959-437x(00)00087-3. [DOI] [PubMed] [Google Scholar]
- 106.Kwiatkowski D, Hill AVS, Sambou I, et al. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. The Lancet. 1990;336(8725):1201–1204. doi: 10.1016/0140-6736(90)92827-5. [DOI] [PubMed] [Google Scholar]
- 107.Karunaweera ND, Grau GE, Gamage P, Carter R, Mendis KN. Dynamics of fever and serum levels of tumor necrosis factor are closely associated during clinical paroxysms in Plasmodium vivax malaria. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(8):3200–3203. doi: 10.1073/pnas.89.8.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mordmüller BG, Metzger WG, Juillard P, et al. Tumor necrosis factor in Plasmodium falciparum malaria: high plasma level is associated with fever, but high production capacity is associated with rapid fever clearance. European Cytokine Network. 1997;8(1):29–35. [PubMed] [Google Scholar]
- 109.Depinay N, Franetich JF, Grüner AC, et al. Inhibitory effect of TNF-α on malaria pre-erythrocytic stage development: influence of host hepatocyte/parasite combinations. PLoS ONE. 2011;6(3) doi: 10.1371/journal.pone.0017464.e17464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McGuire W, Hill AVS, Allsopp CEM, Greenwood BM, Kwjatkowski D. Variation in the TNF-α promoter region associated with susceptibility to cerebral malaria. Nature. 1994;371(6497):508–511. doi: 10.1038/371508a0. [DOI] [PubMed] [Google Scholar]
- 111.Clark TG, Diakite M, Auburn S, et al. Tumor necrosis factor and lymphotoxin-α polymorphisms and severe malaria in African populations. Journal of Infectious Diseases. 2009;199(4):569–575. doi: 10.1086/596320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Meyer CG, May J, Luty AJ, Lell B, Kremsner PG. TNFα-308A associated with shorter intervals of Plasmodium falciparum reinfections. Tissue Antigens. 2002;59(4):287–292. doi: 10.1034/j.1399-0039.2002.590406.x. [DOI] [PubMed] [Google Scholar]
- 113.Wattavidanage J, Carter R, Perera KLRL, et al. TNFα∗2 marks high risk of severe disease during Plasmodium falciparum malaria and other infections in Sri Lankans. Clinical and Experimental Immunology. 1999;115(2):350–355. doi: 10.1046/j.1365-2249.1999.00804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ubalee R, Suzuki F, Kikuchi M, et al. Strong association of a tumor necrosis factor-α promoter allele with cerebral malaria in Myanmar. Tissue Antigens. 2001;58(6):407–410. doi: 10.1034/j.1399-0039.2001.580610.x. [DOI] [PubMed] [Google Scholar]
- 115.Stirnadel HA, Stöckle M, Felger I, Smith T, Tanner M, Beck HP. Malaria infection and morbidity in infants in relation to genetic polymorphisms in Tanzania. Tropical Medicine and International Health. 1999;4(3):187–193. doi: 10.1046/j.1365-3156.1999.43381.x. [DOI] [PubMed] [Google Scholar]
- 116.Randall LM, Kenangalem E, Lampah DA, et al. A study of the TNF/LTA/LTB locus and susceptibility to severe malaria in highland papuan children and adults. Malaria Journal. 2010;9(1, article 302) doi: 10.1186/1475-2875-9-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cabantous S, Doumbo O, Ranque S, et al. Alleles 308A and 238A in the tumor necrosis factor alpha gene promoter do not increase the risk of severe malaria in children with Plasmodium falciparum infection in Mali. Infection and Immunity. 2006;74(12):7040–7042. doi: 10.1128/IAI.01581-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Knight JC, Keating BJ, Kwiatkowski DP. Allele-specific repression of lymphotoxin-α by activated B cell factor-1. Nature Genetics. 2004;36(4):394–399. doi: 10.1038/ng1331. [DOI] [PubMed] [Google Scholar]
- 119.Barbier M, Delahaye NF, Fumoux F, Rihet P. Family-based association of a low producing lymphotoxin-α allele with reduced Plasmodium falciparum parasitemia. Microbes and Infection. 2008;10(6):673–679. doi: 10.1016/j.micinf.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 120.Diakite M, Clark TG, Auburn S, et al. A genetic association study in the Gambia using tagging polymorphisms in the major histocompatibility complex class III region implicates a HLA-B associated transcript 2 polymorphism in severe malaria susceptibility. Human Genetics. 2009;125(1):105–109. doi: 10.1007/s00439-008-0597-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Messer G, Spengler U, Jung MC, et al. Polymorphic structure of the tumor necrosis factor (TNF) locus: an NcoI polymorphism in the first intron of the human TNF-β gene correlates with a variant amino acid in position 26 and a reduced level of TNF-β production. Journal of Experimental Medicine. 1991;173(1):209–219. doi: 10.1084/jem.173.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gyan BA, Goka B, Cvetkovic JT, et al. Allelic polymorphisms in the repeat and promoter regions of the interleukin-4 gene and malaria severity in Ghanaian children. Clinical and Experimental Immunology. 2004;138(1):145–150. doi: 10.1111/j.1365-2249.2004.02590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Naka I, Nishida N, Patarapotikul J, et al. Identification of a haplotype block in the 5q31 cytokine gene cluster associated with the susceptibility to severe malaria. Malaria Journal. 2009;8(1, article 232) doi: 10.1186/1475-2875-8-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rihet P, Traoré Y, Abel L, Aucan C, Traoré-Leroux T, Fumoux F. Malaria in humans: Plasmodium falciparum blood infection levels are linked to chromosome 5q31-q33. American Journal of Human Genetics. 1998;63(2):498–505. doi: 10.1086/301967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Garcia A, Marquet S, Bucheton B, et al. Linkage analysis of blood Plasmodium falciparum levels: interest of the 5q31-q33 chromosome region. American Journal of Tropical Medicine and Hygiene. 1998;58(6):705–709. doi: 10.4269/ajtmh.1998.58.705. [DOI] [PubMed] [Google Scholar]
- 126.Sakuntabhai A, Ndiaye R, Casadémont I, et al. Genetic determination and linkage mapping of Plasmodium falciparum malaria related traits in Senegal. PLoS ONE. 2008;3(4) doi: 10.1371/journal.pone.0002000.e2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Milet J, Nuel G, Watier L, et al. Genome wide linkage study, using a 250K SNP map, of Plasmodium falciparum infection and mild malaria attack in a senegalese population. PLoS ONE. 2010;5(7) doi: 10.1371/journal.pone.0011616.e11616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zeyrek FY, Kurcer MA, Zeyrek D, Simsek Z. Parasite density and serum cytokine levels in Plasmodium vivax malaria in Turkey. Parasite Immunology. 2006;28(5):201–207. doi: 10.1111/j.1365-3024.2006.00822.x. [DOI] [PubMed] [Google Scholar]
- 129.Cabantous S, Poudiougou B, Oumar AA, et al. Genetic evidence for the aggravation of Plasmodium falciparum malaria by interleukin 4. Journal of Infectious Diseases. 2009;200(10):1530–1539. doi: 10.1086/644600. [DOI] [PubMed] [Google Scholar]
- 130.Torre D, Speranza F, Giola M, Matteelli A, Tambini R, Biondi G. Role of Th1 and Th2 cytokines in immune response to uncomplicated Plasmodium falciparum malaria. Clinical and Diagnostic Laboratory Immunology. 2002;9(2):348–351. doi: 10.1128/CDLI.9.2.348-351.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pravica V, Asderakis A, Perrey C, Hajeer A, Sinnott PJ, Hutchinson IV. In vitro production of IFN-γ correlates with CA repeat polymorphism in the human IFN-γ gene. European Journal of Immunogenetics. 1999;26(1):1–3. doi: 10.1046/j.1365-2370.1999.00122.x. [DOI] [PubMed] [Google Scholar]
- 132.Pravica V, Perrey C, Stevens A, Lee JH, Hutchinson IV. A single nucleotide polymorphism in the first intron of the human IFN-γ gene: absolute correlation with a polymorphic CA microsatellite marker of high IFN-γ production. Human Immunology. 2000;61(9):863–866. doi: 10.1016/s0198-8859(00)00167-1. [DOI] [PubMed] [Google Scholar]
- 133.Kock O, Rockett K, Jallow M, Pinder M, Sisay-Joof F, Kwiatkowski D. Investigation of malaria susceptibility determinants in the IFNG/IL26/IL22 genomic region. Genes and Immunity. 2005;6(4):312–318. doi: 10.1038/sj.gene.6364214. [DOI] [PubMed] [Google Scholar]
- 134.Cabantous S, Poudiougou B, Traore A, et al. Evidence that interferon-γ plays a protective role during cerebral malaria. Journal of Infectious Diseases. 2005;192(5):854–860. doi: 10.1086/432484. [DOI] [PubMed] [Google Scholar]
- 135.Ohashi J, Naka I, Patarapotikul J, Hananantachai H, Looareesuwan S, Tokunaga K. A single-nucleotide substitution from C to T at position-1055 in the IL-13 promoter is associated with protection from severe malaria in Thailand. Genes and Immunity. 2003;4(7):528–531. doi: 10.1038/sj.gene.6364010. [DOI] [PubMed] [Google Scholar]
- 136.Dewasurendra RL, Suriyaphol P, Fernando SD. Genetic polymorphisms associated with anti-malarial antibody levels in a low and unstable malaria transmission area in southern Sri Lanka. Malaria Journal. 2012;11, article 281 doi: 10.1186/1475-2875-11-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Phawong C, Ouma C, Tangteerawatana P, et al. Haplotypes of IL12B promoter polymorphisms condition susceptibility to severe malaria and functional changes in cytokine levels in Thai adults. Immunogenetics. 2010;62(6):345–356. doi: 10.1007/s00251-010-0439-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang L, Prather D, Eng JV, et al. Polymorphisms in genes of interleukin 12 and its receptors and their association with protection against severe malarial anaemia in children in western Kenya. Malaria Journal. 2010;9(1, article 87) doi: 10.1186/1475-2875-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Naka I, Patarapotikul J, Tokunaga K, Hananantachai H, Tsuchiya N, Ohashi J. A replication study of the association between the IL12B promoter allele CTCTAA and susceptibility to cerebral malaria in Thai population. Malaria Journal. 2009;8(1, article 290) doi: 10.1186/1475-2875-8-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Marquet S, Doumbo O, Cabantous S, et al. A functional promoter variant in IL12B predisposes to cerebral malaria. Human Molecular Genetics. 2008;17(14):2190–2195. doi: 10.1093/hmg/ddn118. [DOI] [PubMed] [Google Scholar]
- 141.Morahan G, Boutlis CS, Huang D, et al. A promoter polymorphism in the gene encoding interleukin-12 p40 (IL12B) is associated with mortality from cerebral malaria and with reduced nitric oxide production. Genes and Immunity. 2002;3(7):414–418. doi: 10.1038/sj.gene.6363909. [DOI] [PubMed] [Google Scholar]
- 142.Marsh DG, Neely JD, Breazeale DR, et al. Linkage analysis of IL4 and other chromosome 5q31.1 markers and total serum immunoglobulin E concentrations. Science. 1994;264(5162):1152–1156. doi: 10.1126/science.8178175. [DOI] [PubMed] [Google Scholar]
- 143.Romagnani S. Biology of human T(H)1 and T(H)2 cells. Journal of Clinical Immunology. 1995;15(3):121–129. doi: 10.1007/BF01543103. [DOI] [PubMed] [Google Scholar]
- 144.Nakayama EE, Hoshino Y, Xin X, et al. Polymorphism in the interleukin-4 promoter affects acquisition of human immunodeficiency virus type 1 syncytium-inducing phenotype. Journal of Virology. 2000;74(12):5452–5459. doi: 10.1128/jvi.74.12.5452-5459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Marsh DG, Neely JD, Breazeale DR, et al. Total serum IgE levels and chromosome 5q. Clinical and Experimental Allergy. 1995;25(supplement 2):79–83. doi: 10.1111/j.1365-2222.1995.tb00429.x. [DOI] [PubMed] [Google Scholar]
- 146.Suzuki I, Hizawa N, Yamaguchi E, Kawakami Y. Association between a C+33T polyrnorphism in the IL-4 promoter region and total serum IgE levels. Clinical and Experimental Allergy. 2000;30(12):1746–1749. doi: 10.1046/j.1365-2222.2000.00983.x. [DOI] [PubMed] [Google Scholar]
- 147.Verra F, Avellino P, Bancone G, Mangano V, Modiano D. Genetic epidemiology of susceptibility to malaria: not only academic exercises. Parassitologia. 2008;50(1-2):147–150. [PubMed] [Google Scholar]
- 148.Walley AJ, Aucan C, Kwiatkowski D, Hill AVS. Interleukin-1 gene cluster polymorphins and susceptibility to clinical malaria in a Gambian case-control study. European Journal of Human Genetics. 2004;12(2):132–138. doi: 10.1038/sj.ejhg.5201084. [DOI] [PubMed] [Google Scholar]
- 149.Aucan C, Walley AJ, Hennig BJW, et al. Interferon-alpha receptor-1 (IFNAR1) variants are associated with protection against cerebral malaria in The Gambia. Genes and Immunity. 2003;4(4):275–282. doi: 10.1038/sj.gene.6363962. [DOI] [PubMed] [Google Scholar]
- 150.Ouma C, Davenport GC, Awandare GA, et al. Polymorphic variability in the interleukin (IL)-1β promoter conditions susceptibility to severe malarial anemia and functional changes in IL-1β production. Journal of Infectious Diseases. 2008;198(8):1219–1226. doi: 10.1086/592055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rogus J, Beck JD, Offenbacher S, et al. IL1B gene promoter haplotype pairs predict clinical levels of interleukin-1β and C-reactive protein. Human Genetics. 2008;123(4):387–398. doi: 10.1007/s00439-008-0488-6. [DOI] [PubMed] [Google Scholar]
- 152.Lalani I, Bhol K, Ahmed AR. Interleukin-10: biology, role in inflammation and autoimmunity. Annals of Allergy. 1997;79(6):469–483. doi: 10.1016/S1081-1206(10)63052-9. [DOI] [PubMed] [Google Scholar]
- 153.Eskdale J, Gallagher G. A polymorphic dinucleotide repeat in the human IL-10 promoter. Immunogenetics. 1995;42(5):444–445. doi: 10.1007/BF00179416. [DOI] [PubMed] [Google Scholar]
- 154.Ouma C, Davenport GC, Were T, et al. Haplotypes of IL-10 promoter variants are associated with susceptibility to severe malarial anemia and functional changes in IL-10 production. Human Genetics. 2008;124(5):515–524. doi: 10.1007/s00439-008-0578-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Medina TS, et al. Increased interleukin-10 and interferon-ã levels in Plasmodium vivax malaria suggest a reciprocal regulation which is not altered by IL-10 gene promoter polymorphism. Malaria Journal. 2011;10, article 264 doi: 10.1186/1475-2875-10-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wilson JN, Rockett K, Jallow M, et al. Analysis of IL10 haplotypic associations with severe malaria. Genes and Immunity. 2005;6(6):462–466. doi: 10.1038/sj.gene.6364227. [DOI] [PubMed] [Google Scholar]
- 157.Omi K, Ohashi J, Patarapotikul J, et al. Fcγ receptor IIA and IIIB polymorphisms are associated with susceptibility to cerebral malaria. Parasitology International. 2002;51(4):361–366. doi: 10.1016/s1383-5769(02)00040-5. [DOI] [PubMed] [Google Scholar]
- 158.Shi YP, Nahlen BL, Kariuki S, et al. Fc-γ receptor IIa (CD32) polymorphism is associated with protection of infants against high-density Plasmodium falciparum infection. VII. Asembo bay cohort project. Journal of Infectious Diseases. 2001;184(1):107–111. doi: 10.1086/320999. [DOI] [PubMed] [Google Scholar]
- 159.Braga EM, Scopel KKG, Komatsu NT, da Silva-Nunes M, Ferreira MU. Polymorphism of the Fcgamma receptor IIA and malaria morbidity. Journal of Molecular and Genetic Medicine. 2005;1(1):5–10. doi: 10.4172/1747-0862.1000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Salmon JE, Edberg JC, Kimberly RP. Fcγ receptor III on human neutrophils. Allelic variants have functionally distinct capacities. Journal of Clinical Investigation. 1990;85(4):1287–1295. doi: 10.1172/JCI114566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sanders LAM, Feldman RG, Voorhorst-Ogink MM, et al. Human immunoglobulin G (IgG) Fc receptor IIA (CD32) polymorphism and IgG2- mediated bacterial phagocytosis by neutrophils. Infection and Immunity. 1995;63(1):73–81. doi: 10.1128/iai.63.1.73-81.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Aucan C, Traoré Y, Tall F, et al. High immunoglobulin G2 (IgG2) and low IgG4 levels are associated with human resistance to Plasmodium falciparum malaria. Infection and Immunity. 2000;68(3):1252–1258. doi: 10.1128/iai.68.3.1252-1258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ouma C, Keller CC, Opondo DA, et al. Association of FCγ receptor IIA (CD32) polymorphism with malarial anemia and high-density parasitemia in infants and young children. American Journal of Tropical Medicine and Hygiene. 2006;74(4):573–577. [PubMed] [Google Scholar]
- 164.Cooke GS, Aucan C, Walley AJ, et al. Association of Fcγ receptor IIa (CD32) polymorphism with severe malaria in West Africa. American Journal of Tropical Medicine and Hygiene. 2003;69(6):565–568. [PubMed] [Google Scholar]
- 165.Nasr A, Iriemenam NC, Troye-Blomberg M, et al. Fc gamma receptor IIa (CD32) polymorphism and antibody responses to asexual blood-stage antigens of Plasmodium falciparum malaria in Sudanese patients. Scandinavian Journal of Immunology. 2007;66(1):87–96. doi: 10.1111/j.1365-3083.2007.01947.x. [DOI] [PubMed] [Google Scholar]
- 166.Levesque MC, Hobbs MR, O’Loughlin CW, et al. Malaria severity and human nitric oxide synthase type 2 (NOS2) promoter haplotypes. Human Genetics. 2010;127(2):163–182. doi: 10.1007/s00439-009-0753-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Balmer P, Phillips HM, Maestre AE, McMonagle FA, Phillips RS. The effect of nitric oxide on the growth of Plasmodium falciparum, P. chabaudi and P. berghei in vitro. Parasite Immunology. 2000;22(2):97–106. doi: 10.1046/j.1365-3024.2000.00281.x. [DOI] [PubMed] [Google Scholar]
- 168.Serirom S, Raharjo WH, Chotivanich K, Loareesuwan S, Kubes P, Ho M. Anti-adhesive effect of nitric oxide on Plasmodium falciparum cytoadherence under flow. American Journal of Pathology. 2003;162(5):1651–1660. doi: 10.1016/S0002-9440(10)64299-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kun JFJ, Mordmüller B, Lell B, Lehman LG, Luckner D, Kremsner PG. Polymorphism in promoter region of inducible nitric oxide synthase gene and protection against malaria. The Lancet. 1998;351(9098):265–266. doi: 10.1016/S0140-6736(05)78273-8. [DOI] [PubMed] [Google Scholar]
- 170.Burgner D, Xu W, Rockett K, et al. Inducible nitric oxide synthase polymorphism and fatal cerebral malaria. The Lancet. 1998;352(9135):1193–1194. doi: 10.1016/S0140-6736(05)60531-4. [DOI] [PubMed] [Google Scholar]
- 171.Levesque MC, Hobbs M, Anstey N. Nitric oxide synthase type 2 promoter polymorphisms, nitric oxide production, and disease severity in Tanzanian children with malaria. Journal of Infectious Diseases. 2000;181(6):1994–2002. doi: 10.1086/315140. [DOI] [PubMed] [Google Scholar]
- 172.Hobbs MR, Udhayakumar V, Levesque MC, et al. A new NOS2 promoter polymorphism associated with increased nitric oxide production and protection from severe malaria in Tanzanian and Kenyan children. The Lancet. 2002;360(9344):1468–1475. doi: 10.1016/S0140-6736(02)11474-7. [DOI] [PubMed] [Google Scholar]
- 173.Miller LH, Good MF, Milon G. Malaria pathogenesis. Science. 1994;264(5167):1878–1883. doi: 10.1126/science.8009217. [DOI] [PubMed] [Google Scholar]
- 174.Patnaik JK, Das BS, Mishra SK, Mohanty S, Satpathy SK, Mohanty D. Vascular clogging, mononuclear cell margination, and enhanced vascular permeability in the pathogenesis of human cerebral malaria. The American Journal of Tropical Medicine and Hygiene. 1994;51(5):642–647. [PubMed] [Google Scholar]
- 175.Urban BC, Ferguson DJP, Pain A, et al. Plasmodium falciparuminfected erythrocytes modulate the maturation of dendritic cells. Nature. 1999;400(6739):73–77. doi: 10.1038/21900. [DOI] [PubMed] [Google Scholar]
- 176.Ockenhouse CF. The molecular basis for the cytoadherence of Plasmodium falciparum-infected erythrocytes to endothelium. Seminars in Cell and Developmental Biology. 1993;4(5):297–303. doi: 10.1006/scel.1993.1036. [DOI] [PubMed] [Google Scholar]
- 177.Ockenhouse CF, Betageri R, Springer TA, Staunton DE. Plasmodium falciparum-infected erythrocytes bind ICAM-1 at a site distinct from LFA-1, Mac-1, and human rhinovirus. Cell. 1992;68(1):63–69. doi: 10.1016/0092-8674(92)90206-r. [DOI] [PubMed] [Google Scholar]
- 178.Ockenhouse CF, Tegoshi T, Maeno Y, et al. Human vascular endothelial cell adhesion receptors for Plasmodium falciparum-infected erythrocytes: roles for endothelial leukocyte adhesion molecule 1 and vascular cell adhesion molecule 1. Journal of Experimental Medicine. 1992;176(4):1183–1189. doi: 10.1084/jem.176.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sherman IW, Eda S, Winograd E. Cytoadherence and sequestration in Plasmodium falciparum: defining the ties that bind. Microbes and Infection. 2003;5(10):897–909. doi: 10.1016/s1286-4579(03)00162-x. [DOI] [PubMed] [Google Scholar]
- 180.Rowe JA, Moulds JM, Newbold CI, Miller LH. P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature. 1997;388(6639):292–295. doi: 10.1038/40888. [DOI] [PubMed] [Google Scholar]
- 181.Nash GB, Cooke BM, Carlson J, Wahlgren M. Rheological properties of rosettes formed by red blood cells parasitized by Plasmodium falciparum . British Journal of Haematology. 1992;82(4):757–763. doi: 10.1111/j.1365-2141.1992.tb06955.x. [DOI] [PubMed] [Google Scholar]
- 182.Rowe JA, Obiero J, Marsh K, Raza A. Short report: positive correlation between rosetting and parasitemia in Plasmodium falciparum clinical isolates. American Journal of Tropical Medicine and Hygiene. 2002;66(5):458–460. doi: 10.4269/ajtmh.2002.66.458. [DOI] [PubMed] [Google Scholar]
- 183.Deans A-M, Rowe JA. Plasmodium falciparum: rosettes do not protect merozoites from invasion-inhibitory antibodies. Experimental Parasitology. 2006;112(4):269–273. doi: 10.1016/j.exppara.2005.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Rout R, Dhangadamajhi G, Mohapatra BN, Kar SK, Ranjit M. High CR1 level and related polymorphic variants are associated with cerebral malaria in eastern-India. Infection, Genetics and Evolution. 2011;11(1):139–144. doi: 10.1016/j.meegid.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 185.Turner GDH, Morrison H, Jones M, et al. An immunohistochemical study of the pathology of fatal malaria: evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. American Journal of Pathology. 1994;145(5):1057–1069. [PMC free article] [PubMed] [Google Scholar]
- 186.Newbold C, Warn P, Black G, et al. Receptor-specific adhesion and clinical disease in Plasmodium falciparum . American Journal of Tropical Medicine and Hygiene. 1997;57(4):389–398. doi: 10.4269/ajtmh.1997.57.389. [DOI] [PubMed] [Google Scholar]
- 187.Fry AE, Auburn S, Diakite M, et al. Variation in the ICAM1 gene is not associated with severe malaria phenotypes. Genes and Immunity. 2008;9(5):462–469. doi: 10.1038/gene.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1) Cell. 1987;51(5):813–819. doi: 10.1016/0092-8674(87)90104-8. [DOI] [PubMed] [Google Scholar]
- 189.Baratin M, Roetynck S, Pouvelle B, et al. Dissection of the role of PfEMP1 and ICAM-1 in the sensing of Plasmodium falciparum-infected erythrocytes by natural killer cells. PLoS ONE. 2007;2(2, article e228) doi: 10.1371/journal.pone.0000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Fernandez-Reyes D, Craig AG, Kyes SA, et al. A high frequency African coding polymorphism in the N-terminal domain of ICAM-1 predisposing to cerebral malaria in Kenya. Human Molecular Genetics. 1997;6(8):1357–1360. doi: 10.1093/hmg/6.8.1357. [DOI] [PubMed] [Google Scholar]
- 191.Kun JF, Klabunde J, Lell B, et al. Association of the ICAM:1(KILIFI) mutation with protection against severe malaria in lambarene, gabon. The American Journal of Tropical Medicine and Hygiene. 1999;61(5):776–779. doi: 10.4269/ajtmh.1999.61.776. [DOI] [PubMed] [Google Scholar]
- 192.Amodu OK, Gbadegesin RA, Ralph SA, et al. Plasmodium falciparum malaria in south-west Nigerian children: is the polymorphism of ICAM-1 and E-selectin genes contributing to the clinical severity of malaria? Acta Tropica. 2005;95(3):248–255. doi: 10.1016/j.actatropica.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 193.Bellamy R, Kwiatkowski D, Hill AVS. Absence of an association between intercellular adhesion molecule 1, complement receptor 1 and interleukin 1 receptor antagonist gene polymorphisms and severe malaria in a West African population. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1998;92(3):312–316. doi: 10.1016/s0035-9203(98)91026-4. [DOI] [PubMed] [Google Scholar]
- 194.Ohashi J, Naka I, Patarapotikul J, Hananantachai H, Looareesuwan S, Tokunaga K. Absence of association between the allele coding methionine at position 29 in the N-terminal domain of ICAM-1 (ICAM-1Kilifi) and severe malaria in the northwest of Thailand. Japanese Journal of Infectious Diseases. 2001;54(3):114–116. [PubMed] [Google Scholar]
- 195.Ndiaye R, Sakuntabhai A, Casadémont I, et al. Genetic study of ICAM1 in clinical malaria in Senegal. Tissue Antigens. 2005;65(5):474–480. doi: 10.1111/j.1399-0039.2005.00388.x. [DOI] [PubMed] [Google Scholar]
- 196.Ayodo G, Price AL, Keinan A, et al. Combining evidence of natural selection with association analysis increases power to detect malaria-resistance variants. American Journal of Human Genetics. 2007;81(2):234–242. doi: 10.1086/519221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Jenkins NE, Mwangi TW, Kortok M, Marsh K, Craig AG, Williams TH. A polymorphism of intercellular adhesion molecule-1 is associated with a reduced incidence of nonmalarial febrile illness in Kenyan children. Clinical Infectious Diseases. 2005;41(12):1817–1819. doi: 10.1086/498156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chilongola J, Balthazary S, Mpina M, Mhando M, Mbugi E. CD36 deficiency protects against malarial anaemia in children by reducing Plasmodium falciparum-infected red blood cell adherence to vascular endothelium. Tropical Medicine and International Health. 2009;14(7):810–816. doi: 10.1111/j.1365-3156.2009.02298.x. [DOI] [PubMed] [Google Scholar]
- 199.Kajeguka D, Mwanziva C, Daou M, et al. CD36 c.1264 T > G null mutation impairs acquisition of IgG antibodies to Plasmodium falciparum MSP119 antigen and is associated with higher malaria incidences in Tanzanian children. Scandinavian Journal of Immunology. 2012;75(3):355–360. doi: 10.1111/j.1365-3083.2011.02661.x. [DOI] [PubMed] [Google Scholar]
- 200.Aitman TJ, Cooper LD, Norsworthy PJ, et al. Malaria susceptibility and CD36 mutation. Nature. 2000;405(6790):1015–1016. doi: 10.1038/35016636. [DOI] [PubMed] [Google Scholar]
- 201.Pain A, Urban BC, Kai O, et al. A non-sense mutation in Cd36 gene is associated with protection from severe malaria. The Lancet. 2001;357(9267):1502–1503. doi: 10.1016/S0140-6736(00)04662-6. [DOI] [PubMed] [Google Scholar]
- 202.Sinha S, Qidwai T, Kanchan K, et al. Variations in host genes encoding adhesion molecules and susceptibility to falciparum malaria in India. Malaria Journal. 2008;7, article 250 doi: 10.1186/1475-2875-7-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Omi K, Ohashi J, Naka I, et al. Polymorphisms of CD36 in Thai malaria patients. The Southeast Asian journal of tropical medicine and public health. 2002;33(supplement 3):1–4. [PubMed] [Google Scholar]
- 204.Treutiger CJ, Heddini A, Fernandez V, Muller WA, Wahlgren M. PECAM-1/CD31, an endothelial receptor for binding Plasmodium falciparum- infected erythrocytes. Nature Medicine. 1997;3(12):1405–1408. doi: 10.1038/nm1297-1405. [DOI] [PubMed] [Google Scholar]
- 205.Casals-Pascual C, Allen S, Allen A, et al. Short report: codon 125 polymorphism of CD31 and susceptibility to malaria. American Journal of Tropical Medicine and Hygiene. 2001;65(6):736–737. doi: 10.4269/ajtmh.2001.65.736. [DOI] [PubMed] [Google Scholar]
- 206.Kikuchi M, Looareesuwan S, Ubalee R, et al. Association of adhesion molecule PECAM-1/CD31 polymorphism with susceptibility to cerebral malaria in Thais. Parasitology International. 2001;50(4):235–239. doi: 10.1016/s1383-5769(01)00082-4. [DOI] [PubMed] [Google Scholar]
- 207.Newton JP, Buckley CD, Jones EY, Simmons DL. Residues on both faces of the first immunoglobulin fold contribute to homophilic binding sites of PECAM-1/CD31. Journal of Biological Chemistry. 1997;272(33):20555–20563. doi: 10.1074/jbc.272.33.20555. [DOI] [PubMed] [Google Scholar]
- 208.Liao F, Ali J, Greene T, Muller WA. Soluble domain 1 of platelet-endothelial cell adhesion molecule (PECAM) is sufficient to block transendothelial migration in vitro and in vivo. The Journal of Experimental Medicine. 1997;185(7):1349–1357. doi: 10.1084/jem.185.7.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Krych-Goldberg M, Moulds JM, Atkinson JP. Human complement receptor type 1 (CR1) binds to a major malarial adhesin. Trends in Molecular Medicine. 2002;8(11):531–537. doi: 10.1016/s1471-4914(02)02419-x. [DOI] [PubMed] [Google Scholar]
- 210.Krych-Goldberg M, Atkinson JP. Structure-function relationships of complement receptor type 1. Immunological Reviews. 2001;180:112–122. doi: 10.1034/j.1600-065x.2001.1800110.x. [DOI] [PubMed] [Google Scholar]
- 211.Rowe JA, Rogerson SJ, Raza A, et al. Mapping of the region of complement receptor (CR) 1 required for Plasmodium falciparum rosetting and demonstration of the importance of Cr1 in rosetting in field isolates. Journal of Immunology. 2000;165(11):6341–6346. doi: 10.4049/jimmunol.165.11.6341. [DOI] [PubMed] [Google Scholar]
- 212.Stoute JA. Complement-regulatory proteins in severe malaria: too little or too much of a good thing? Trends in Parasitology. 2005;21(5):218–223. doi: 10.1016/j.pt.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 213.Heddini A, Pettersson F, Kai O, et al. Fresh isolates from children with severe Plasmodium falciparum malaria bind to multiple receptors. Infection and Immunity. 2001;69(9):5849–5856. doi: 10.1128/IAI.69.9.5849-5856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Birmingham DJ, Hebert LA. CR1 and CR1-like: the primate immune adherence receptors. Immunological Reviews. 2001;180:100–111. doi: 10.1034/j.1600-065x.2001.1800109.x. [DOI] [PubMed] [Google Scholar]
- 215.Rowe JA, Raza A, Diallo DA, et al. Erythrocyte CR1 expression level does not correlate with a HindIII restriction fragment length polymorphism in Africans; implications for studies on malaria susceptibility. Genes and Immunity. 2002;3(8):497–500. doi: 10.1038/sj.gene.6363899. [DOI] [PubMed] [Google Scholar]
- 216.Nagayasu E, Ito M, Akaki M, et al. CR1 density polymorphism on erythrocytes of falciparum malaria patients in Thailand. American Journal of Tropical Medicine and Hygiene. 2001;64(1-2):1–5. doi: 10.4269/ajtmh.2001.64.1.11425154. [DOI] [PubMed] [Google Scholar]
- 217.Cockburn IA, Mackinnon MJ, O’Donnell A, et al. A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(1):272–277. doi: 10.1073/pnas.0305306101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Teeranaipong P, Ohashi J, Patarapotikul J, et al. A functional single-nucleotide polymorphism in the CR1 promoter region contributes to protection against cerebral malaria. Journal of Infectious Diseases. 2008;198(12):1880–1891. doi: 10.1086/593338. [DOI] [PubMed] [Google Scholar]
- 219.Tobin DM, Roca FJ, Oh SF, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148(3):434–446. doi: 10.1016/j.cell.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]