

Abstract
Peripheral T-cell lymphomas are very aggressive hematologic malignancies for which there is no targeted therapy. New, rational approaches are necessary to improve the very poor outcome in these patients. Phosphatidylinositol-3-kinase is one of the most important pathways in cell survival and proliferation. We hypothesized that phosphatidylinositol-3-kinase inhibitors could be rationally selected drugs for treating peripheral T-cell lymphomas. Several phosphatidylinositol-3-kinase isoforms were inhibited genetically (using small interfering RNA) and pharmacologically (with CAL-101 and GDC-0941 compounds) in a panel of six peripheral and cutaneous T-cell lymphoma cell lines. Cell viability was measured by intracellular ATP content; apoptosis and cell cycle changes were checked by flow cytometry. Pharmacodynamic biomarkers were assessed by western blot. The PIK3CD gene, which encodes the δ isoform of phosphatidylinositol-3-kinase, was overexpressed in cell lines and primary samples, and correlated with survival pathways. However, neither genetic nor specific pharmacological inhibition of phosphatidylinositol-3-kinase δ affected cell survival. In contrast, the pan-phosphatidylinositol-3-kinase inhibitor GDC-0941 arrested all T-cell lymphoma cell lines in the G1 phase and induced apoptosis in a subset of them. We identified phospho-GSK3β and phospho-p70S6K as potential biomarkers of phosphatidylinositol-3-kinase inhibitors. Interestingly, an increase in ERK phosphorylation was observed in some GDC -0941-treated T-cell lymphoma cell lines, suggesting the presence of a combination of phosphatidylinositol-3-kinase and MEK inhibitors. A highly synergistic effect was found between the two inhibitors, with the combination enhancing cell cycle arrest at G0/G1 in all T-cell lymphoma cell lines, and reducing cell viability in primary tumor T cells ex vivo. These results suggest that the combined treatment of pan-phosphatidylinositol-3-kinase + MEK inhibitors could be more effective than single phosphatidylinositol-3-kinase inhibitor treatment, and therefore, that this combination could be of therapeutic value for treating peripheral and cutaneous T-cell lymphomas.
Introduction
Peripheral T-cell lymphomas (PTCL) are a heterogeneous group of disorders with a very low incidence and very poor prognosis.1,2 The molecular biology of PTCL is poorly understood, partially due to the rarity of the disease, the heterogeneity of subtypes and the low number of appropriate models (no representative cell lines are available and no current mouse model recapitulates the human disease). These restrictions have limited understanding of the pathogenesis of PTCL and, therefore, the development of the new targeted therapies that are necessary to improve the outcome of patients with this disease.3
The phosphatidylinositol-3-kinase (PI3K) family of proteins participates in one of the most essential pathways for cell survival, proliferation and growth. It is an important mediator of drug resistance and it has been described to be frequently altered in a wide variety of types of human cancer.4-6 Two subclasses of PI3K have been related to cancer: class IA (p110a, β and δ isoforms of the catalytic subunit) is predominantly activated by tyrosine kinases, and class IB (p110γ isoform) is mainly activated by G-protein-coupled receptors. Upon extracellular stimulation, PI3K phosphorylate phosphatidylinositol 4,5 biphosphate (PIP2) to form phosphatidylinositol 3,4,5 triphosphate (PIP3). Subsequently, PIP3 can phosphorylate AKT, which initiates a signaling cascade of protein phosphorylation leading to cell survival, growth and proliferation. The activity of PI3K can be inhibited by the phosphatase and tensin homologue (PTEN), which acts as a tumor suppressor protein.4-6
Since aberrant regulation of the PI3K pathway has been frequently observed in leukemic cells (presumably due to activating signals from the microenvironment rather than mutations in genes belonging to the pathway), PI3K is considered to be a promising target for therapy.4,6,7 Preclinical experiments indicated that the PI3K inhibitors (PI3Ki) LY294002 and wortmannin induced apoptosis in leukemic cells, but they were also toxic to normal cells, probably because of the low specificity and inhibition of other kinases. Recently, selective isoform-specific inhibitors, such as the PI3Kδ inhibitor CAL-101, have appeared to affect survival of chronic lymphocytic leukemia cells and CAL-101 has entered phase I/II clinical trials. Although the single specificity inhibitors are effective in vitro, their effect in patients appears to be modest. Therefore, combination therapy targeting different intermediates of the PI3K signaling pathway is also under investigation. Furthermore, since lymphomagenesis involves aberrant regulation of various signal transduction pathways, simultaneous inhibition of multiple deregulated targets is considered to be a promising therapeutic strategy.4
The aims of this study were to determine the efficiency of PI3K inhibition in PTCL, to look for pharmacodynamic biomarkers, and to identify markers that could distinguish responders from non-responders.
Design and Methods
The molecular signature and bioinformatic analysis of peripheral T-cell lymphoma
The PTCL molecular signature was identified by comparing the gene expression profile in a series of 38 frozen PTCL cases and six reactive lymph nodes, and further analyzed using Connectivity Map (Cmap)8 and Gene Set Enrichment Analysis (GSEA)9 programs, as described in the Online Supplementary Design and Methods. All microarray data are available at the Gene Expression Omnibus under accession number GSE36172. The research was approved by the ethical committees of the Instituto de Salud Carlos III (Madrid, Spain) and the Hospital Universitario Marqués de Valdecilla (Santander, Spain).
Cell lines, primary samples and reagents
The cell lines and primary samples are described in the Online Supplementary Design and Methods. GD -0941 was obtained from Chemdea (Ridgewood, NJ, USA). LY294002 and UO126 were purchased from Calbiochem (Darmstadt, Germany). CAL-101 was obtained from Selleck Chemicals (Houston, TX, USA). ETP-45658 was developed10 and kindly provided by the Experimental Therapeutics Program of the Spanish National Cancer Research Center (Madrid, Spain).
PIK3CD and PIK3CA genetic silencing
MyLa, SR786 and HuT78 cell lines were electroporated with specific small interfering RNA (siRNA) against the PIK3CD and PIK3CA genes, using the Neon® Transfection System (Invitrogen, Carlsbad, CA, USA), as described in the Online Supplementary Design and Methods.
Pharmacological inhibition assays
For drug cytotoxicity experiments, cell lines and primary tumor T cells were treated for 72 or 48 h, respectively, with PI3Ki, MEK inhibitors (MEKi) or their combinations. Cell viability was measured as the intracellular ATP content using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA) following the manufacturer's instructions. For drug combination experiments, the combination index (CI) was calculated according to the method of Chou and Talalay.11 The distribution of cells among different phases of the cell cycle and induction of apoptosis were evaluated by flow cytometry, as described in the Online Supplementary Design and Methods. All experiments were done in triplicate and all numerical data are expressed as the average of the values ± the standard error of the mean (SEM).
Results
Phosphatidylinositol-3-kinase as a potential therapeutic target in peripheral T-cell lymphoma
In order to test drugs that could be of therapeutic value in PTCL, we used the Cmap program and a PTCL molecular signature generated in our group of 38 PTCL cases and in six reactive lymph nodes. Cmap identified two PI3K/mTOR pathway inhibitors among the drugs that could most significantly reverse this PTCL signature (Figure 1A). We note that trichostatin A and MS-275, two histone deacetylase inhibitors [like vorinostat and romidepsin, which have already been approved by the Food and Drug Administration for the treatment of cutaneous T-cell lymphoma (CTCL)12,13], also appeared on the list.
Figure 1.

PI3K is a potential therapeutic target in PTCL. (A) Connectivity Map identified PI3K/mTOR pathway inhibitors (indicated by arrows) as the drugs that could potentially reverse (negative enrichment score) the PTCL molecular signature in a very significant manner. (B) The GSEA program revealed that three survival pathways [T-cell receptor (TCR), NF-κB and CD40 signaling] were positively and significantly (false discovery rate <0.25) correlated with PIK3CD expression in the PTCL molecular signature. (C and D) Both PIK3CA and PIK3CD genes were overexpressed in (C) PTCL and CTCL cell lines and (D) Sézary's syndrome (SS) primary T cells compared with normal T cells isolated from healthy donors (control #1-3) measured by quantitative RT-PCR.
Furthermore, using the GSEA program we observed in the PTCL molecular signature that the expression of PIK3CD (the gene encoding p110δ) was the only PI3K iso-form to be significantly correlated with several survival pathways, such as the T-cell receptor, nuclear factor-κB and CD40 pathways (Figure 1B). Moreover, we found that both PIK3CD and PIK3CA (encoding p110α) were overexpressed in the six PTCL and CTCL cell lines (Figure 1C) and in primary T cells isolated from five patients with Sézary's syndrome (Figure 1D), compared to the expression by normal T cells from healthy donors.
Since the PI3K pathway is frequently altered at the genetic level in many types of human cancer, the mutational status of several genes in the pathway was explored in six PTCL and CTCL cell lines. We did not find any known mutation in PIK3CA, AKT1, PTEN, KRAS and BRAF genes in any cell line. The described14NRAS mutation Q61K was confirmed in the HuT78 cell line, but was absent from the other PTCL cell lines (data not shown). Although no mutations have yet been reported in the PIK3CD gene,15 two described variants16 were also studied in PTCL cell lines. We found that the SR786 cell line carried a single nucleotide polymorphism (c935g → S312C, rs61755420) (data not shown). Moreover, these PIK3CD polymorphisms were analyzed in a series of 27 PTCL cases. Interestingly, we observed two cases (one angioimmunoblastic T cell lymphoma and one anaplastic large cell lymphoma ALK+) harboring the same polymorphism. Additionally, one case of PTCL-not otherwise specified was found to have another single nucleotide polymorphism in this region (c2319t → S773S, rs139848768) (data not shown).These data indicate that PI3Kδ is an attractive, potential therapeutic target in PTCL.
Effects of genetic silencing of the delta and alpha isoforms of phosphatidylinositol-3-kinase on peripheral T-cell lymphoma cell lines
To test the hypothesis that targeting PI3Kδ is of therapeutic value in PTCL, we performed genetic silencing experiments to specifically abolish P K3CD and P K3CA expression (because gain-of-function mutations in P K3CA occur very frequently in human tumors6). The efficiency of the silencing was 60-90% at the mRNA level, depending on the cell line and the time point (Online Supplementary Figure S1). We observed a slight tendency in MyLa and SR786 cell lines, but not in HuT78, to undergo apoptosis when PIK3CD (Figure 2A) or PIK3CA (Figure 2B) were knocked down, although the differences compared with the non-template control were not statistically significant in any case. No significant changes in the cell cycle profile were observed in any condition or cell line (data not shown). The results suggest that the active protein remaining after knockdown could maintain survival of PTCL cell lines.
Figure 2.

Effects of a single PI3K isoform inhibition on PTCL and CTCL cell line survival. (A) PIK3CD knockdown by siRNA slightly, but not significantly, increased induction of apoptosis in MyLa and SR786 cell lines, while no effects were observed in HuT78. * indicates a statistically significant difference (P<0.05). (B) A slight, but not significant, increase in apoptosis was found in MyLa and SR786, but not in HuT78, when PIK3CA was knocked down. (C) The specific pharmacological PI3Kδ inhibitor CAL-101 was only able to reduce PTCL cell line viability at high doses. (D) Most of the PTCL cell lines did not undergo apoptosis upon treatment with CAL-101. The Y axis indicates the percentage of annexin V+/propidium iodide- plus annexin V+/propidium iodide+ cells in the treatment with 10 μM CAL-101 minus the cell death in DMSO. (E) After exposure to 5 μM CAL-101 for 24 h most of the PTCL cell lines were not arrested in any cell cycle phase; only HH and DERL7 were slightly arrested at G1.
Effects of specific pharmacological inhibition of phosphatidylinositol-3-kinase-delta on peripheral T-cell lymphoma survival
In order to inhibit the PI3Kδ catalytic activity more efficiently, PTCL cell lines were treated with the specific pharmacological PI3Kδ inhibitor CAL-101. We observed that this drug generally reduced cell viability only at high doses (Figure 2C). Accordingly, CAL-101 was not able to induce apoptosis in any PTCL cell line, only HH suffered an 8% increase in apoptosis compared to the DMSO-treated cells, even when incubated with a dose of 10 μM for 72 h (Figure 2D). Similarly, slight cell cycle arrest at G0/G1 was found after exposure to 5 μM CAL-101 for 24 h in HH and DERL7 cell lines (Figure 2E). These results confirmed that the inhibition of PI3Kδ alone was not sufficient to reduce PTCL cell survival, suggesting that different PI3K isoforms may share redundant functions.
Effects of the pharmacological inhibition of all phosphatidylinositol-3-kinase isoforms on peripheral T-cell lymphoma survival
In order to inhibit all class I PI3K isoforms, six PTCL and CTCL cell lines were treated with three pharmacological PI3Ki (LY294002, ETP-45658 and GDC-0941). Although these drugs have different specificities (LY294002 is a dual PI3K/mTOR inhibitor and the other two are specific pan-PI3Ki10,17), all of them had similar effects on PTCL cell line survival (Online Supplementary Figure S2). We found that GD -0941 was able to reduce cell viability in all cell lines in a dose-dependent manner, but the effect was stronger in some cell lines than in others, such as HuT78 and MJ, which were considered to be resistant (Figure 3A). Furthermore, GDC-0941 promoted cell cycle arrest at the G0/G1 phase in all PTCL cell lines, being stronger in sensitive cells lines than in resistant ones (Figure 3B). These differential responses were confirmed at the level of apoptosis induction: only the sensitive cell lines underwent slight apoptosis upon treatment with pan-PI3Ki (Figure 3C), while the resistant cell lines HuT78 and MJ did not. These results indicate that the pharmacological inhibition of all PI3K isoforms was more effective than isoform-specific inhibition.
Figure 3.

Effects of the pharmacological pan-PI3Ki GDC-0941 on PTCL and CTCL cell line survival. (A) GDC-0941 reduced PTCL cell viability after 72 h treatment in all cell lines, but HuT78 and MJ cell lines were more resistant to the PI3Ki. (B) GDC-0941 promoted cell cycle arrest at the G1 phase in all studied PTCL cell lines. The arrest was stronger in sensitive cell lines than in resistant ones. (C) GDC-0941 significantly induced apoptosis in sensitive cell lines, but not in resistant ones. The Y axis indicates the percentage of annexin V+/propidium iodide- plus annexin V+/propidium iodide+ cells following treatment with 1 μM GDC-0941 minus the cell death in DMSO. *Indicates a statistically significant difference compared with DMSO (P<0.05).
Biomarkers for pan-phosphatidylinositol-3-kinase inhibitors in peripheral T-cell lymphoma
In order to test the activity of the pan-PI3Ki in PTCL cell lines, the phosphorylation status of several proteins belonging to the PI3K pathway was examined following GDC-0941 treatment. First, we found by flow cytometry that AKT phosphorylation, the most direct target of PI3K,6 was reduced overall in all cell lines after 4 h of treatment with GDC-0941 (Figure 4A), confirming that the drug was indeed inhibiting PI3K even in the resistant cell line HuT78, although the reduction was not very substantial. Next, we studied other downstream proteins and observed that GSK3b and p70S6K phosphorylation was reduced in the sensitive cell lines, but not in the resistant ones, even after high-dose treatment with GDC-0941 (Figure 4B). These data suggest that pGSK3β and p-p70S6K could act as biomarkers for the response to PI3Ki in PTCL cell lines.
Figure 4.
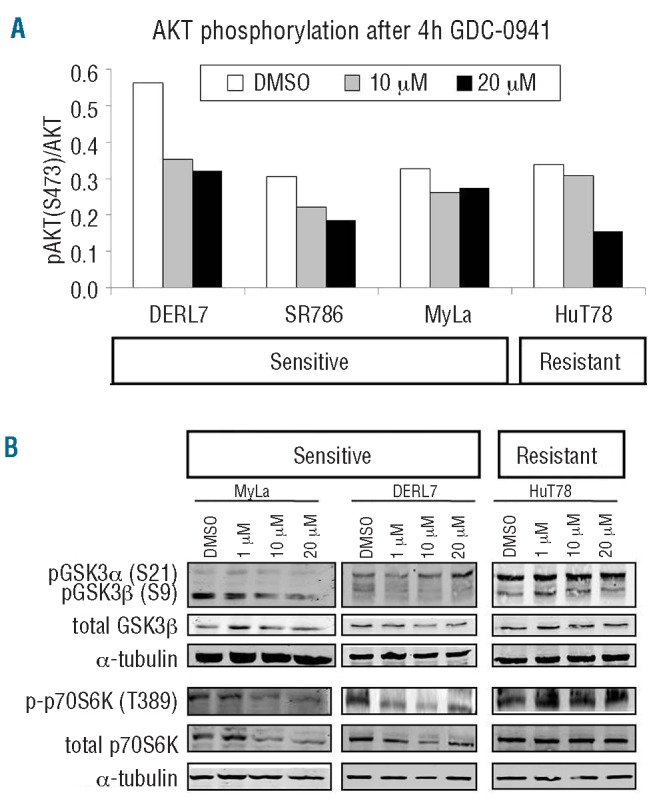
PI3Ki activity and response biomarkers in PTCL and CTCL cell lines. (A) GDC-0941 diminished AKT phosphorylation in a dose-dependent manner in all cell lines after 4 h of treatment. (B) GSK3β and p70S6K phosphorylation was reduced only in sensitive cell lines after 4 h exposure to GDC-0941, but not in resistant cell lines.
Cooperation between phosphatidylinositol-3-kinase and ERK pathways in peripheral T-cell lymphoma
Although the pharmacological pan-PI3Ki strategy seemed to be more effective than the isoform-specific inhibitor approach, the effects were not very dramatic. In order to determine whether another cellular pathway could compensate for the reduced PI3K activity and sustain PTCL survival, we studied the phosphorylation status of ERK, because the Ras/Raf/MEK/ERK pathway is one of the most essential pathways in cell survival.18 Following treatment with GDC-0941 for 24 h, strong ERK activation was observed in the MyLa and SR786 cell lines, but not in HuT78 (Figure 5A). This result could explain the marginal effects on cell survival we had observed when PI3K was pharmacologically or genetically inhibited. The differential behavior of HuT78 could be explained by its resistance to PI3Ki: perhaps the ERK pathway did not need to be activated in this cell line in the presence of GDC-0941, because even under these conditions the downstream members of PI3K were still activated, sustaining cell survival (Figure 4B). More importantly, these results could support the use of a combined pan-PI3Ki + MEKi treatment in PTCL.
Figure 5.

Cooperation between PI3K and ERK pathways in PTCL and CTCL in vitro and ex vivo. (A) ERK phosphorylation was increased in MyLa and SR786 cell lines after 24 h of treatment with GDC-0941 10 μM. (B) The combination index (CI) was calculated for different doses of the combined pan-PI3Ki (GDC-0941) + M Ki (UO126): CI <1 indicates synergism between the two drugs, CI ≈1 indicates an additive effect, CI >1 indicates antagonism. (C) The combination enhanced cell cycle arrest at G1 in all PTCL cell lines when compared with single treatments. (D) The PI3Ki + M Ki combination significantly reduced cell viability in primary tumor T cells from eight CTCL patients (5 with Sézary's syndrome and 3 with mycosis fungoides). *indicates statistically significant differences compared with DMSO (P<0.05).
PTCL and CTCL cell lines were, therefore, treated with the pan-PI3Ki GDC-0941 plus the MEKi UO126 for 72 h. We found that the combination was highly synergistic (CI <1) in four of the six PTCL cell lines at doses equivalent to IC50 values (Figure 5B). On the other hand, we observed a slight antagonism (CI >1) in the HuT78 cell line. Moreover, the combination enhanced the cell cycle arrest at G0/G1 in all PTCL cell lines (Figure 5C). In contrast, overall, the drug combination did not increase the apoptosis induced by each drug alone (Online Supplementary Figure S3). This is probably due to the levels of pAKT and pERK, which remained even in the presence of the drug combination (Online Supplementary Figure S4). Importantly, simultaneous PI3K and MEK inhibition significantly reduced cell viability in primary tumor T cells ex vivo; at higher doses, the drug combination had a stronger effect than the pan-PI3Ki alone (Figure 5D).
These results suggest that combined treatment with pan-PI3Ki + MEKi could be more effective than treatment with a PI3Ki alone and that the combination could be of therapeutic value in the treatment of PTCL and CTCL.
Discussion
Here we hypothesized that PI3K inhibition, especially inhibition of the delta isoform, could be of therapeutic value in PTCL because: (i) our in silico approach identified PI3K pathway inhibitors as drugs that potentially reverse the PTCL molecular signature; (ii) PI3Kδ has been described to play a role in the differentiation, survival and activation of normal T cells, and so is probably also important for T-cell transformation;19-21 (iii) PIK3CD was the only isoform to be significantly correlated with survival pathways in this PTCL signature; and (iv) we found that PIK3CD was much more overexpressed than PIK3CA in PTCL and CTCL cell lines and in Sézary's syndrome primary T cells.
However, our results indicated that the genetic or specific pharmacological inhibition of PI3Kδ had no effects on the survival of PTCL cell lines. CAL-101 is a PI3Kδ inhibitor that has been reported to have very promising preclinical activity in chronic lymphocytic leukemia (either alone22,23 or in combination with other agents24) and very recently, also in Hodgkin's lymphoma.25 The initial clinical data in phase I/II studies were most impressive in chronic lymphocytic leukemia, and have created some excitement about PI3K drug development. Nevertheless, some controversy exists: leukemia and lymphomas generally express all class I PI3K isoforms and rarely carry mutations in PI3K genes or have PTEN loss. In addition, the first set of preclinical studies with CAL-101 did not clearly predict the dramatic efficacy of the drug in clinical trials: cytotoxic effects on human chronic lymphocytic leukemia samples in vitro were generally achieved only at high CAL-101 doses (10 μM). These results suggest that selective PI3Kδ inhibition has a minimal impact on cell-intrinsic survival signals in B-lineage cancer cells, where it seems to act mainly by perturbing the signals received from the tumor microenvironment.26,27
Moreover, even in cancers that seem to be specifically reliant on either PI3Kα or β, there is the concern that other non-targeted PI3K isoforms might eventually compensate for the decreased activity of the targeted isoform.6 It has been described that removal of more than 90% of p85-associated PI3K activity (α, β and δ isoforms) did not compromise the proliferation in hematopoietic progenitor cells.28 These data could explain the absence of effect that we observed when a single PI3K isoform was inhibited in PTCL cell lines, because our knockdown efficiency was 80% at the mRNA level, and thus, more than 10% of the protein was still active, along with the other intact isoforms, allowing the survival of PTCL cell lines.
Accordingly, a stronger effect was found with the pan-PI3Ki GDC-0941. All cell lines were arrested at the G0/G1 phase and a subset of them underwent apoptosis. Overall, the response was mainly cytostatic, as widely reported.6,28,29 Almost complete class IA inhibition is required to produce cell death, but such full inhibition of PI3K activity might be difficult to achieve with ATP-competitive inhibitors. This could partially explain why PI3Ki do not induce apoptosis but rather cytostasis.28 However, this outcome could also be explained by alternative mechanisms of activation of downstream effectors through feedback loops,4 especially because of the inconsistent reduction of pAKT we observed after GDC-0941 treatment, which might also suggest an AKT-independent mechanism of action. PI3K pathway members are able to interact very actively with a number of proteins, creating a very complex network.30 Cooperation between the PI3K/AKT and the Ras/Raf/MEK/ERK pathways has been extensively described in many types of cell types.6,28,31 Interestingly, we found that ERK phosphorylation was increased upon PI3Ki treatment in PTCL cell lines, probably as a part of a compensatory up-regulation of interconnected pathways. More importantly, the combination of PI3Ki + MEKi was highly synergistic overall (as reported already6,28,31) in vivo and ex vivo, enhancing the cytostatic effect although without induction of apoptosis.
Unfortunately, the biological basis of the poor outcome in patients with PTCL, in contrast to that of patients with aggressive B-cell lymphomas, is not well understood.3 Biomarkers that predict the response to a given therapy are, therefore, needed. Very recently, a study in primary PTCL cases highlighted the clinical relevance of pAKT, which was negatively correlated with the response rate and survival,32 thus supporting the use of PI3Ki in PTCL therapy. Here, we present preclinical evidence suggesting that PI3Ki could be potential therapeutic agents for the treatment of PTCL. Furthermore, we found different sensitivities among the PTCL cell lines and observed that phosphorylation of GSK3β and p70S6K was only reduced in the most sensitive cell lines, while remaining unchanged in the resistant cell lines. This result suggests that these phospho-proteins could act as potential pharmacodynamic biomarkers for the response to PI3Ki in PTCL.
In addition, we looked for biomarkers that could distinguish potential responders before treatment, such as mutations in some parts of the PI3K pathway. Although mutations in PIK3CA, AKT1 and PTEN have been detected in patients with leukemia, these mutations appear to be rare in hematologic neoplasms.4,33 None of the known activating mutations in PIK3CA, AKT1, KRAS or BRAF or deletions in PTEN was found in any PTCL and CTCL cell line. Consistent with this, PTEN alterations were not observed at the genetic or protein levels in a series of cases of PTCL-not otherwise specified.34 The only gene variant found in one of the six PTCL cell lines and two of the 27 PTCL cases was PIK3CD, which harbored two single nucleotide polymorphisms (c935g = rs61755420 and c2319t = rs139848768). The first polymorphism (c935g) might be somehow related to the incidence of anaplastic large cell lymphoma ALK+, because it was present in the only anaplastic large cell lymphoma ALK+ cell line available and in one of the three cases of anaplastic large cell lymphoma ALK+. Nevertheless, a case-control study reporting a significant association in a large series would be required to explore this possibility.
In conclusion, collectively these results suggest that the simultaneous inhibition of all PI3K isoforms, instead of specific PI3Kδ inhibition, could be a therapeutic strategy in a subset of PTCL and CTCL. More importantly, the cytostatic effects can be enhanced in combination with MEKi, and might improve the poor outcome in PTCL patients, who lack efficient therapies.
Acknowledgments
The authors would like to thank the Spanish National Tumor Bank Network for their help in collecting and managing the samples from the hospitals. We would also like to thank Francisco X. Real for his contribution to the scientific discussion.
Funding: This work was supported by grants from the Asociación Española Contra el Cáncer, Fondo de Investigaciones Sanitarias (PI051623, PI052800 and PI080856), RTICC (RD06/0020/0107) and Ministerio de Ciencia e Innovación (SAF2008-0387-1). EMS is supported by a grant from the Department of Education, Universities and Research of the Basque Government (BFI08.207). MSB is supported by a Contract Miguel Servet from Fondo de Investigaciones Sanitarias (CP11/00018).
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures: Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org
References
- 1.de Leval L, Bisig B, Thielen C, Boniver J, Gaulard P. Molecular classification of T-cell lymphomas. Crit Rev Oncol Hematol. 2009;72(2):125-43 [DOI] [PubMed] [Google Scholar]
- 2.Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood. 2011;117(25):6756-67 [DOI] [PubMed] [Google Scholar]
- 3.Dunleavy K, Piekarz RL, Zain J, Janik JE, Wilson WH, O'Connor OA, et al. New strategies in peripheral T-cell lymphoma: understanding tumor biology and developing novel therapies. Clin Cancer Res. 2010;16(23):5608-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polak R, Buitenhuis M. The PI3K/PKB signaling module as key regulator of hematopoiesis: implications for therapeutic strategies in leukemia. Blood. 2012;119(4):911-23 [DOI] [PubMed] [Google Scholar]
- 5.Guo D, Teng Q, Ji C. NOTCH and phosphatidylinositide 3-kinase/phosphatase and tensin homolog deleted on chromosome ten/AKT/mammalian target of rapamycin (mTOR) signaling in T-cell development and T-cell acute lymphoblastic leukemia. Leuk Lymphoma. 2011;52(7):1200-10 [DOI] [PubMed] [Google Scholar]
- 6.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550-62 [DOI] [PubMed] [Google Scholar]
- 7.Burger JA, Hoellenriegel J. Phosphoinositide 3'-kinase delta: turning off B R signaling in chronic lymphocytic leukemia. Oncotarget. 2011;2(10):737-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929-35 [DOI] [PubMed] [Google Scholar]
- 9.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Link W, Oyarzabal J, Serelde BG, Albarran MI, Rabal O, Cebria A, et al. Chemical interrogation of FOXO3a nuclear translocation identifies potent and selective inhibitors of phosphoinositide 3-kinases. J Biol Chem. 2009;284(41):28392-400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27-55 [DOI] [PubMed] [Google Scholar]
- 12.Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs. 2007;16(7):1111-20 [DOI] [PubMed] [Google Scholar]
- 13.Mann BS, Johnson JR, He K, Sridhara R, Abraham S, Booth BP, et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin Cancer Res. 2007;13(8):2318-22 [DOI] [PubMed] [Google Scholar]
- 14.Kiessling MK, Oberholzer PA, Mondal C, Karpova MB, Zipser M, Lin WM, et al. High-throughput mutation profiling of CTCL samples reveals KRAS and NRAS mutations sensitizing tumors toward inhibition of the RAS/RAF/MEK signaling cascade. Blood. 2011;117(8):2433-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ciraolo E, Morello F, Hirsch E. Present and future of PI3K pathway inhibition in cancer: perspectives and limitations. Curr Med Chem. 2011;18(18):2674-85 [DOI] [PubMed] [Google Scholar]
- 16.Dan S, Okamura M, Seki M, Yamazaki K, Sugita H, Okui M, et al. Correlating phosphatidylinositol 3-kinase inhibitor efficacy with signaling pathway status: in silico and biological evaluations. Cancer Res. 2010;70(12):4982-94 [DOI] [PubMed] [Google Scholar]
- 17.Workman P, Clarke PA, Raynaud FI, van Montfort RL. Drugging the PI3 kinome: from chemical tools to drugs in the clinic. Cancer Res. 2010;70(6):2146-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steelman LS, Franklin RA, Abrams SL, Chappell W, Kempf CR, Basecke J, et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia. 2011;25(7):1080-94 [DOI] [PubMed] [Google Scholar]
- 19.Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297(5583):1031-4 [DOI] [PubMed] [Google Scholar]
- 20.Soond DR, Bjorgo E, Moltu K, Dale VQ, Patton DT, Torgersen KM, et al. PI3K p110delta regulates T-cell cytokine production during primary and secondary immune responses in mice and humans. Blood. 2010;115(11):2203-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rolf J, Bell SE, Kovesdi D, Janas ML, Soond DR, Webb LM, et al. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol. 2010;185(7):4042-52 [DOI] [PubMed] [Google Scholar]
- 22.Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116(12):2078-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phos-phatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herman SE, Lapalombella R, Gordon AL, Ramanunni A, Blum KA, Jones J, et al. The role of phosphatidylinositol 3-kinase-delta in the immunomodulatory effects of lenalidomide in chronic lymphocytic leukemia. Blood. 2011;117(16):4323-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meadows SA, Vega F, Kashishian A, Johnson D, Diehl V, Miller LL, et al. PI3Kdelta inhibitor, GS-1101 (CAL-101), attenuates pathway signaling, induces apoptosis, and overcomes signals from the microenvironment in cellular models of Hodgkin lymphoma. Blood. 2012;119(8):1897-900 [DOI] [PubMed] [Google Scholar]
- 26.So L, Fruman DA. PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem J. 2012;442(3):465-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fruman DA, Rommel C. PI3K inhibitors in cancer: rationale and serendipity merge in the clinic. Cancer Discovery. 2011;1(7):562-72 [DOI] [PubMed] [Google Scholar]
- 28.Foukas LC, Berenjeno IM, Gray A, Khwaja A, Vanhaesebroeck B. Activity of any class IA PI3K isoform can sustain cell proliferation and survival. Proc Natl Acad Sci USA. 2010;107(25):11381-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28(6):1075-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27(41):5527-41 [DOI] [PubMed] [Google Scholar]
- 31.Hoeflich KP, O'Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15(14):4649-64 [DOI] [PubMed] [Google Scholar]
- 32.Cai Q, Deng H, Xie D, Lin T. Phosphorylated AKT protein is overexpressed in human peripheral T-cell lymphomas and predicts decreased patient survival. Clin Lymphoma Myeloma Leuk. 2012;12(2):106-12 [DOI] [PubMed] [Google Scholar]
- 33.Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114(3):647-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gazzola A, Bertuzzi C, Agostinelli C, Righi S, Pileri SA, Piccaluga PP. Physiological PTEN expression in peripheral T-cell lymphoma not otherwise specified. Haematologica. 2009;94(7):1036-7 [DOI] [PMC free article] [PubMed] [Google Scholar]