

Abstract
Cardiac fibroblasts are key cellular effectors of cardiac repair; their phenotype and function are modulated by interactions with extracellular matrix proteins. This review manuscript discusses the effects of the extracellular matrix on the inflammatory and reparative properties of fibroblasts in the infarcted myocardium. Early generation of matrix fragments in the infarct induces a pro-inflammatory and matrix-degrading fibroblast phenotype. Formation of a fibrin/fibronectin-rich provisional matrix serves as a conduit for migration of fibroblasts into the infarcted area. Induction of ED-A fibronectin and non-fibrillar collagens may contribute to myofibroblast transdifferentiation. Upregulation of matricellular proteins promotes transduction of growth factor and cytokine-mediated signals. As the scar matures, matrix cross-linking, clearance of matricellular proteins and reduced growth factor signaling cause deactivation and apoptosis of reparative infarct fibroblasts. Understanding the effects of matrix components on infarct fibroblasts may guide the design of peptides that reproduce, or inhibit, specific matricellular functions, attenuating adverse remodeling.
Keywords: fibroblast, extracellular matrix, cardiac remodeling, myocardial infarction, myofibroblast, matricellular proteins
1. INTRODUCTION
Cardiomyocytes, fibroblasts and vascular cells (endothelial cells, vascular smooth muscle cells and pericytes) constitute the predominant cellular components of the adult mammalian heart [1], [2], [3]. In normal adult hearts, fibroblasts are the most populous non-myocyte cell type [4] and account for about 20% of the myocardial volume [5]. Although the role of cardiac fibroblasts in cardiac homeostasis remains understudied, a growing body of evidence suggests that beyond their contribution in preservation of the cardiac extracellular matrix network, fibroblasts may also directly interact with cardiomyocytes, serving as active participants in normal cardiac function [6]. The importance of fibroblasts in cardiac pathologic conditions is much better documented. Practically every form of heart failure is associated with activation of cardiac fibroblasts resulting in increased deposition of extracellular matrix and expansion of the cardiac interstitium. In the infarcted myocardium, activated myofibroblasts are the main matrix-producing cells responsible for formation of a collagen-based scar. Fibroblasts are versatile cells: in addition to secreting matrix proteins, they are capable of acting as inflammatory cells, modulate angiogenesis, transduce signals to neighboring cardiomyocytes and regulate matrix metabolism through expression of proteolytic enzymes and their inhibitors. Thus, changes in the phenotypic characteristics of cardiac fibroblasts profoundly affect geometry and function in the remodeling heart.
In response to changes in their environment, fibroblasts undergo dynamic phenotypic alterations. Due to their strategic location in the cardiac interstitial and perivascular space, fibroblasts may act as true “sentinel cells” [7] that can sense changes in the microenvironment and initiate the response to injury. Extensive myocardial necrosis ultimately activates a reparative fibroblast program leading to deposition of matrix in the injured heart. Fibroblasts are embedded in the interstitial matrix and are typically viewed as the main regulators of matrix composition. What is less appreciated is that the relation between fibroblasts and the matrix is amphidromous, as components of the extracellular matrix exert important modulatory effects on fibroblast phenotype. Our review manuscript deals with the effects of the extracellular matrix on cardiac fibroblast phenotype and function in the infarcted myocardium. After an introductory section on the role of the matrix network in homeostatic function of cardiac fibroblasts, we discuss the dramatic changes in the composition of the extracellular matrix in the infarcted heart and their effects on fibroblast activity. We will also attempt to identify promising therapeutic strategies that may attenuate post-infarction remodeling by interfering with matrix:fibroblast interactions.
2. FIBROBLASTS AND THE MATRIX NETWORK IN THE NORMAL MYOCARDIUM
In the adult mammalian heart, ventricular myocytes are arranged in layers of tightly coupled cardiomyocytes [8]; adjacent layers are separated by clefts. The laminar architecture of the myocardium is defined by an intricate network of extracellular matrix proteins, comprised primarily of fibrillar collagen. The collagen-based cardiac matrix network does not only serve as a scaffold for the cellular components, but is also important for transmission of the contractile force. Approximately 85% of total collagen is type I, primarily associated with thick fibers that confer tensile strength, whereas type III collagen represents 11% of the total collagen protein in the heart, typically forms thin fibers, and maintains the elasticity of the matrix network [9], [10]. In addition to collagens, the cardiac extracellular matrix also contains glycosaminoglycans (such as hyaluronan), glycoproteins and proteoglycans. Significant stores of growth factors and proteases are bound to the cardiac extracellular matrix and can be activated following injury.
Cardiac fibroblasts are enmeshed in the endomysial interstitial matrix that surrounds cardiomyocytes. In the developing heart cardiac fibroblasts regulate cardiomyocyte proliferation through a fibronectin/β1-integrin-mediated pathway [11]. As the predominant matrix-producing cells in the myocardium [12], fibroblasts play an important role in preserving the integrity of the matrix network.. The cardiac fibroblast population undergoes a dramatic change during the neonatal period [13]. As the fetal circulation transitions to the neonatal circulation, elevated left ventricular pressures trigger a marked expansion of the cardiac fibroblast population within the first two neonatal weeks [13]. In the young adult heart, cardiac fibroblasts remain quiescent and do not exhibit significant inflammatory or proliferative activity.
Very little is known regarding the role of the extracellular matrix in cardiac fibroblast homeostasis. During physiologic adaptive remodeling of the neonatal heart, the matricellular protein periostin may play an important role in fibroblast maturation and differentiation. This concept is supported by findings suggesting that 3 month-old periostin −/− hearts contain a large population of undifferentiated mesenchymal-like cells [14]. Moreover, microarray analysis in adult periostin −/− hearts demonstrated significant alterations in expression of numerous genes associated with fibrosis and matrix remodeling [15] suggesting an altered cardiac fibroblast gene program. However, these cell-biological consequences of periostin loss appear to have limited impact on function and geometry of the adult heart: young adult periostin −/− mice have normal systolic function accompanied by a slight reduction in chamber dimensions [15]. In the adult heart, an intact matrix network may promote a quiescent fibroblast phenotype by shielding fibroblasts from mechanical stress [16], [17]. Although this hypothesis is attractive, the in vivo significance of matrix:fibroblast interactions in stabilization of the cardiac fibroblast population and in preservation of ventricular geometry and function has not been investigated.
3. MATRIX:FIBROBLAST INTERACTIONS IN THE REMODELING INFARCTED MYOCARDIUM
3.1. Cardiac remodeling
The term cardiac remodeling was initially coined to describe the geometric and structural alterations of the myocardium following infarction [18], [19]. Extensive clinical evidence demonstrates that, following myocardial infarction, dilative cardiac remodeling is associated with increased mortality, arrhythmias and a high incidence of heart failure [20]. Over the last twenty years use of the term cardiac remodeling has been expanded to describe changes occurring in a wide variety of cardiac conditions [21]. In the infarcted heart the extent of adverse remodeling is dependent on the size of the infarct and on the qualitative characteristics of the healing wound [19].
Myocardial infarction is associated with dynamic changes in fibroblast phenotype and with dramatic alterations in the composition of the cardiac extracellular matrix. Fibroblast:matrix interactions are critical determinants of cardiac repair; impaired fibroblast responses and defects in scar formation may be associated with catastrophic complications. Excessive early degradation of the cardiac extracellular matrix and defective, or delayed, formation of the new matrix network may play an important role in the pathogenesis of cardiac rupture, a dramatic and fatal complication of acute myocardial infarction. In the later stages of healing, defects in fibroblast function and impaired deposition of extracellular matrix may alter the mechanical properties of the heart, resulting in worse dilative remodeling and dysfunction. As fibroblasts are the predominant cell type involved in matrix synthesis and metabolism, the modulatory effects of the matrix network on fibroblast phenotype may act as a feedback mechanism that prevents excessive pro-fibrotic responses in the infarcted myocardium.
For descriptive purposes, infarct healing can be divided in three distinct, but overlapping phases: the inflammatory phase, the proliferative phase, and the maturation phase [22]. Subcellular constituents releases by necrotic cells activate the complement cascade, while matrix fragments activate Toll-like receptor (TLR) signaling and tissue ischemia generates reactive oxygen species in the infarcted myocardium. These overlapping pathways induce Nuclear Factor (NF)-κB-dependent cytokine and chemokine upregulation [23], [24], [25], [26] in resident myocardial cells activating the inflammatory cascade [27], [28]. Abundant leukocytes infiltrate the infarct. Macrophages phagocytose dead cells and matrix debris, and produce growth factors that activate fibroblasts and vascular cells. During the proliferative phase of healing, repression and resolution of inflammation is followed by phenotypic modulation of fibroblasts that become myofibroblasts and secrete large amounts of extracellular matrix proteins [29], [30], [31]. At the same time there is activation of angiogenic pathways and formation of an extensive vascular network. Maturation of the scar follows: a collagen-based scar is formed while the cellular elements undergo apoptosis. After completion of the reparative response and disappearance of reparative infarct fibroblasts, a large population of resident fibroblasts remains in the non-infarcted remodeling myocardium. These cells may undergo chronic activation due to the increased wall stress producing extracellular matrix proteins and participating in matrix metabolism. Whether these fibroblasts are phenotypically and functionally distinct from the population of infarct myofibroblasts remains unknown. Our discussion will focus primarily on the reparative fibroblasts infiltrating the necrotic zone; these cells undergo dynamic changes during the healing process. During all phases of infarct healing, the composition of the extracellular matrix plays a critical role in regulating fibroblast behavior (Table) [32].
Table 1.
Table Effects of the dynamic alterations in infarct matrix composition on fibroblast phenotype and function
| Matrix component | Phase of healing | Effect on cardiac fibroblasts |
|---|---|---|
| Collagen fragments | Inflammatory | Migration |
| Fibronectin fragments | Inflammatory | Inflammatory activation, MMP expression |
| Low molecular weight hyaluronan fragments | Inflammatory | Inflammatory activation, matrix synthesis (?), activation of TGF-β signaling (?) |
| Fibrin | Inflammatory/Proliferative | Inflammatory activation, migration |
| Cellular fibronectin | Proliferative | Integrin-mediated migration |
| ED-A fibronectin | Proliferative | Myofibroblast transdifferentiation |
| Collagen VI | Proliferative | Myofibroblast transdifferentiation |
| Thrombospondin-1 | Proliferative | TGF-β activation, matrix preservation, MMP inhibition, myofibroblast transdifferentiation |
| Tenascin-C | Proliferative | De-adhesion and migration |
| SPARC | Proliferative | Matrix organization, TGF-β activation |
| Osteopontin | Proliferative | Angiotensin II-induced proliferation, anti-apoptotic effects |
| Periostin | Proliferative | Migration, differentiation, collagen fibrillogenesis |
| Cross-linked matrix proteins | Maturation | Quiescence, stress-shielding, apoptosis (?) |
3.2. Matrix:fibroblast interactions during the inflammatory phase of infarct healing
Cardiomyocyte necrosis triggers an innate immune response [33] leading to rapid activation of an inflammatory cascade in the infarcted myocardium. Activation of the Nlrp3 inflammasome induces caspase-1-mediated processing and secretion of Interleukin (IL)-1β releasing a potent pro-inflammatory stimulus in the area of necrosis. Infarct fibroblasts exhibit early activation of the inflammasome [34] and may be important cellular effectors of the inflammatory reaction. Early dramatic alterations in the extracellular matrix may potently induce an inflammatory phenotype in cardiac fibroblasts of the infarcted area. Both effects of matrix degradation products and actions mediated by components of the plasma-derived provisional matrix may play a role in inflammatory activation of cardiac fibroblasts (Figure 1).
Figure 1.

Matrix-fibroblast interactions during the inflammatory phase of infarct healing. Rapid protease activation in the infarcted heart generates collagen (Col) and fibronectin (Fn) fragments; pro-inflammatory low molecular weight hyaluronan (LMWH) fragments are also generated. Matrix fragments may induce pro-inflammatory fibroblast activation; moreover, fibronectin fragments are known to induce matrix metalloproteinase (MMP) expression. Fibrinogen extravasation into the infarcted area may also induce inflammatory signaling in fibroblasts.
Myocardial infarction is associated with rapid disruption and fragmentation of the cardiac extracellular matrix network [35], [36]. Ten minutes after coronary occlusion, Matrix Metalloproteinase (MMP) activation is noted in the cardiac interstitium [37]; protease activation results in matrix protein degradation and reduces collagen content in the infarcted heart [38]. Rapid generation of type I collagen fragments in the serum occurs within minutes after coronary occlusion [39] and reflects the extensive matrix degradation in the infarcted heart. Generation of matrix fragments plays an important role in leukocyte recruitment in sites of injury linking tissue damage with the inflammatory response [40], [41]. Non-specific collagen fragments and elastin-derived peptides are capable of inducing neutrophil, monocyte and fibroblast chemotaxis [42], [43]. Fibronectin is also rapidly degraded following myocardial infarction [44]. The 120kDa cell-binding fibronectin fragment (Fn120) is released in the cardiac extracellular space following coronary occlusion and reperfusion [44] and may exert potent modulatory actions on cardiac fibroblasts, both directly, and through activation of inflammatory pathways. In contrast to the intact fibronectin molecule, the Fn120 fragment activates inflammation potently stimulating monocyte chemotaxis [45]. Fn120 fragments activate a matrix-degrading molecular program in human fibroblasts, increasing MMP-1 expression and enhancing collagenase and stromelysin activity [46], [47]. Moreover, a 45kDa fibronectin fragment (Fn45) induced MMP-13 expression and increased MMP-3 activity in dermal fibroblasts [48]. Our understanding of the effects of fibronectin fragments on fibroblast phenotype and function is primarily based on experiments performed in human dermal fibroblasts. Considering the functional heterogeneity of fibroblasts from various sites, the significance of these interactions in myocardial infarction remains unknown.
Fragmentation of the extracellular matrix in the infarcted myocardium is not limited to proteins; glycosaminoglycans (such as hyaluronan) also undergo degradation leading to generation of low molecular weight fragments with pro-inflammatory properties [49]. Low molecular weight hyaluronan (LMWH) fragments induce inflammatory gene expression in endothelial cells, macrophages [50] and fibroblasts [51]. A growing body of evidence suggests that removal of hyaluronan fragments from the injured tissue is essential for resolution of chronic inflammation [52]. Hyaluronan participates in induction and resolution of inflammation through interactions with the transmembrane adhesion molecule CD44 [53]. Our studies demonstrated disruption of the hyaluronan matrix in the infarcted myocardium [54]. CD44 expression is markedly induced in the infarct and is predominantly localized on infiltrating leukocytes, myofibroblasts and vascular cells [54]. CD44 −/− animals have accentuated inflammatory activity in the healing infarct. CD44 absence also results in profound alterations in fibroblast phenotype; CD44 null cells exhibit decreased collagen synthesis and impaired activation of TGF-β/Smad3 signaling [54]. Whether enhanced inflammatory activity in CD44 null animals is due to loss of direct CD44-mediated effects on cardiac fibroblasts, or related to the persistent pro-inflammatory actions of LMWH fragments remains unknown.
In addition to the effects of matrix fragmentation, fibrinogen extravasated through the hyperpermeable vasculature may also induce a pro-inflammatory phenotype in cardiac fibroblasts. Fibrinogen is known to induce expression of adhesion molecules and chemokines in synovial fibroblasts [55]
3.3. Matrix:fibroblast interactions during the proliferative phase of infarct healing
During the proliferative phase of healing, fibroblasts proliferate, undergo myofibroblast transdifferentiation and acquire a matrix synthetic phenotype. These changes are directed by dynamic alterations in the composition and structure of the extracellular matrix. First, formation of a fibrin/fibronectin based provisional matrix serves as a scaffold for fibroblast migration into the infarct. Second, expression of specialized matrix proteins (such as the ED-A variant of fibronectin) in the infarcted myocardium induces myofibroblast transdifferentiation. Third, matricellular proteins are incorporated into the matrix of the infarct and serve as molecular bridges that transduce growth factor-mediate signals in myofibroblasts. When incorporated into a provisional matrix that contains large amounts of matricellular proteins, infarct fibroblasts respond to the growth factor-rich environment by synthesizing a collagen-based matrix that replaces the provisional matrix components.
3.4. The fibrin/fibronectin-based provisional matrix serves as a conduit for fibroblast infiltration
As the original matrix network is degraded during the inflammatory phase of infarct healing, extravasation of plasma proteins through the hyperpermeable vasculature results in formation of a dynamic provisional matrix network based on fibrin and plasma fibronectin (Figure 2) [49]. The plasma-derived provisional matrix is later lysed by proteolytic enzymes produced by granulation tissue cells, and is quickly replaced by an organized cell-derived “second order” provisional matrix containing cellular fibronectin and hyaluronan [56]. Healing of the infarcted heart is dependent on an orchestrated movement of cellular populations into the infarcted area. Because the normal collagen-based matrix constrains cellular movement, infiltration of the infarct with fibroblasts would have been impossible in the absence of the provisional matrix. Thus, in the healing infarct, the fibrin/fibronectin-rich matrix network serves as a conduit for migrating fibroblasts [57] and may also promote fibroblast proliferation [58]. Interactions between the migrating cells and matrix fibronectin involve α5β1 and αvβ3 integrins [59] and the transmembrane heparin sulfate proteoglycan syndecan-4 [60]; these interactions also provide signals that modulate cellular phenotype and gene expression [61]. Moreover, the composition of the matrix may modulate the fibroblast integrin expression profile. Platelet-Derived Growth Factor (PDGF)-BB induces fibroblast α3 and α5 integrins in human dermal fibroblasts cultured in a fibronectin-rich environment, but not in fibroblasts populating collagen gels [62].
Figure 2.
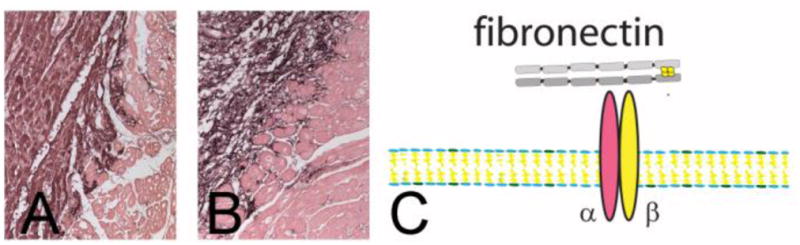
A fibrin/fibronectin-based provisional matrix serves as a scaffold for fibroblast migration into the infarct. Immunohistochemical staining shows fibrin(ogen) localization in a canine infarct after 1h of ischemia and 24h of reperfusion (A). Fibrin deposition demarcates the infarct (black staining). After 7 days of reperfusion fibrin is incorporated into an organized provisional matrix network (B). C: Fibronectin incorporation into the matrix is crucial for fibroblast migration through interactions that involve integrins.
3.5. The role of the extracellular matrix in myofibroblast transdifferentiation
Phenotypic modulation of fibroblasts into myofibroblasts is the hallmark of the proliferative phase of infarct healing. Myofibroblasts have ultrastructural and phenotypic characteristics of smooth muscle cells, exhibiting formation of contractile stress fibers [63], [64]. Expression of -smooth muscle actin (α-SMA) is the main characteristic of differentiated myofibroblasts; however, α-SMA expression is not a necessary criterion for myofibroblast identification. At the early stages of the healing process, myofibroblasts contain stress fibers composed of cytoplasmic actins, but lack α-SMA expression; these cells are termed proto-myofibroblasts. The origin of myofibroblasts in the infarcted myocardium remains controversial. Resident cardiac fibroblasts likely represent the most important source of myofibroblasts in cardiac repair [65]; however, recruitment of blood-derived fibroblast progenitors [66] and endothelial to mesenchymal transition [67] may also contribute to the infarct myofibroblast population. Regardless of their origin, myofibroblasts are responsible for infarct contraction and are the main matrix-synthetic cells in the infarcted heart.
The matrix environment co-operates with TGF-β/Smad3 signaling to induce myofibroblast transdifferentiation [32]. The splice variant ED-A of cellular fibronectin is upregulated in the infarcted heart [68] and co-operates with TGF-β in mediating acquisition of the myofibroblast phenotype [69], [70], [71]. Deposition of non-fibrillar collagens (such as collagen VI) in the infarcted myocardium may also modulate myofibroblast transdifferentiation [72]. In vitro, type VI collagen potently induces myofibroblast differentiation, but has little effect on fibroblast proliferation [72]. In vivo, collagen VI disruption attenuated fibrosis and improved cardiac function in a model of myocardial infarction. However, the beneficial effects of collagen VI loss on the infarcted heart may, in addition to inhibition of fibrosis, also involve a reduction in cardiomyocyte apoptosis [73]. Assembly of a pericellular hyaluronan coat may also promote and maintain myofibroblast transdifferentiation in response to TGF-β [74], [75]. The matrix environment also modulates myofibroblast transdifferentiation by altering mechanical stress conditions. In normal hearts, fibroblasts are generally protected from mechanical stimuli by a stable cross-linked matrix network. Once the structural integrity of the interstitial matrix is disrupted, exposure of the cells to mechanical stress contributes to proto-myofibroblast transdifferentiation [76].
3.6. Matricellular proteins: extracellular adaptor molecules between the matrix and the fibroblasts
Matricellular proteins are a family of structurally unrelated extracellular macromolecules that bind to the extracellular matrix without serving a structural role, but function as molecular links between matrix proteins and cells [77]. The family includes the thrombospondins (TSPs), tenascin-C, osteopontin (OPN), SPARC (secreted protein acidic and rich in cysteine), periostin and members of the CCN family. The highly cellular environment of the healing infarct contains an abundance of growth factors and cytokines released by platelets, leukocytes and mesenchymal cells. Matricellular proteins bind to the provisional matrix, interact with cytokines, growth factors and proteases and activate or modulate integrin responses in a variety of cell types [78]. Most matricellular proteins are not expressed in normal hearts, but are markedly induced following myocardial infarction and play an important role in cardiac remodeling. Fibroblasts are major targets for matricellular functions.
TSP-1, a prototypical matricellular protein is a potent angiostatic mediator with an essential role in TGF-β activation [79]. TSP-1 expression is selectively upregulated in the infarct border zone in experimental models of myocardial infarction [80], and is localized in an area with abundant myofibroblasts (Figure 3). TSP-1 loss resulted in increased dilative post-infarction remodeling associated with prolonged and expanded inflammation and with impaired TGF-β/Smad activation [80]. Thus, localized induction of TSP-1 in the infarct border zone may form an anti-inflammatory “barrier” preventing expansion of the inflammatory infiltrate in the non-infarcted area. Although the protective effects of TSP-1 in the remodeling infarcted heart may be due, at least in part, to effects on fibroblasts, this possibility has not been systematically studied. TSP-1-mediated TGF-β activation may promote a matrix-preserving phenotype in cardiac fibroblasts enhancing matrix deposition, while preventing dilative remodeling. TSPs also exert direct matrix-stabilizing effects through protease inhibition [81], [82]. Experiments in a model of pressure overload fibrosis suggested that TSP-1 loss is associated with increased MMP activity, impaired myofibroblast transdifferentiation and reduced fibroblast-derived collagen synthesis [83]. However, the potential involvement of these alterations in post-infarction cardiac remodeling has not been examined.
Figure 3.

The prototypical matricellular protein thrombospondin (TSP)-1 is upregulated in the infarct border zone and may regulate fibroblast responses. Immunohistochemical staining shows a strikingly selective deposition of TSP-1 in the border zone following canine myocardial infarction (A- arrows). Serial section staining combines CD31 labeling (black) to identify endothelial cells and α-SMA staining (red) to label myofibroblasts as spindle-shaped cells located outside the vascular media (B- arrows). Note the abundance of myofibroblasts in the TSP-1-rich border zone. C: TSP-1 is a crucial TGF-β activator. TSP-1 acts by directly binding the Latency-Associated Peptide (LAP) inducing a conformational change that makes the TGF-β dimer accessible to its receptor.
Tenascin-C is markedly upregulated during the proliferative phase of healing [84], and is also localized in the infarct border zone, an area exhibiting abundant myofibroblast infiltration. Tenascin-C promotes a deadhesive state and may facilitate migration of fibroblasts and other granulation tissue cells in the infarct. In a model of electrical cardiac injury [85] tenascin-C null mice exhibited delayed recruitment of myofibroblasts in the injured site. In experimental myocardial infarction tenascin-C loss significantly attenuated cardiac remodelling and diastolic dysfunction; these protective actions were associated with less pronounced fibrosis [86]. The mechanisms for the pro-fibrotic actions of tenascin-C remain unknown.
SPARC induction is also induced in healing infarcts and critically regulates fibroblast responses. SPARC −/− mice had a four-fold increase in mortality following myocardial infarction due to cardiac rupture and developed heart failure [87]. Defective repair in SPARC null animals was associated with disorganized granulation tissue formation and deposition of immature collagen. The protective effects of endogenous SPARC may be mediated through activation of TGF-β responses in infarct myofibroblasts.
OPN upregulation is also consistently found in experimental models of myocardial infarction [88]. OPN acts as a cytokine, when secreted in a soluble form, and as a matricellular protein when bound to the matrix. Loss-of-function studies demonstrated a protective role for OPN in post-infarction cardiac remodeling [89]. Absence of OPN resulted in increased left ventricular dilatation associated with reduced collagen deposition in the infarcted myocardium [90]. OPN is a highly multifunctional protein with potent effects on cell survival, adhesion and migration, and in regulation of the immune response. In vitro, OPN mediates the proliferative effects of angiotensin II in cardiac fibroblasts [91]. Moreover, OPN null cardiac fibroblasts are more susceptible to oxidant-induced apoptosis than WT cells; thus, OPN expression may protect cardiac fibroblasts from death in the hostile environment of the infarct [92]. Due to the multifunctional actions of OPN on all cell types involved in cardiac repair, the significance of its effects on fibroblasts remains unknown.
Periostin is highly expressed in the neonatal heart and plays a role in maturation and differentiation of fibroblasts. In both mouse and human myocardial infarction periostin is highly expressed in border zone myofibroblasts and critically modulates fibroblast phenotype and function [93]. Periostin null mice had impaired healing following myocardial infarction exhibiting increased incidence of cardiac rupture [15], [93] associated with decreased recruitment of myofibroblasts and impaired collagen fibrillogenesis in the infarct; adenovirus-mediated gene transfer of a spliced form of periostin protected knockout mice from rupture [93]. However, periostin −/− animals surviving the acute phase had better preserved systolic function 8 weeks after infarction [15]. Protection of periostin null mice from dysfunction during the remodeling phase was associated with attenuated inflammatory activity and significantly reduced fibrosis of the infarcted heart. The potent actions of periostin on migration, differentiation and activation of infarct myofibroblasts may, in part, explain its effects on the remodeling infarcted heart.
Induction of matricellular proteins in the infarcted myocardium is critical for fibroblast migration and differentiation, and for activation of essential growth factor-mediated pathways. However, their expression in the infarcted heart is transient; clearance of matricellular proteins from the infarcted myocardium marks the end of the proliferative phase and leads to stabilization of the scar.
3.7. Matrix:fibroblast interactions during infarct maturation
The maturation phase is characterized by cross-linking of the extracellular matrix and formation of a dense collagen-based scar with relatively low cellular content. Apoptosis appears to be the main mechanism responsible for the elimination of most myofibroblasts from the mature infarct [94]. Alterations in matrix composition may determine the fate of fibroblasts in the mature infarct. Formation of a cross-linked collagenous matrix may “shield” fibroblasts from external mechanical stress resulting in deactivation stress fiber dissolution and cellular quiescence [17], [63]. Moreover, reduced expression of growth factors and clearance of matricellular proteins may deprive the fibroblasts from essential survival signals leading to their apoptosis. As the heart continues to remodel after completion of the reparative response, fibroblasts residing in the remodeling non-infarcted myocardium may become chronically activated in response to increased wall stress. These non-reparative cells may contribute to chamber remodeling and ventricular dysfunction by producing matrix proteins and proteases.
4. TARGETING MATRIX-FIBROBLAST INTERACTIONS IN CARDIAC REMODELING
Matrix-fibroblast interactions play a crucial role in cardiac repair and in the pathogenesis of post-infarction remodeling. Thus, understanding how matrix proteins modulate fibroblast phenotype and function in the infarcted myocardium has outstanding pathophysiologic significance. Moreover, therapeutic approaches interfering with effects of matrix proteins on infarct fibroblasts may hold promise in the treatment of myocardial infarction. Strategies targeting cellular interactions with components of the provisional matrix of the infarct have been proposed as potential therapies in an attempt to attenuate the inflammatory and fibrotic response. A fibrin-derived peptide that competes with the fibrin fragment N-terminal disulfide knot-II exerted anti-inflammatory actions in mouse models of myocardial ischemia and reperfusion attenuating leukocyte infiltration and decreasing replacement fibrosis [95]. Although the protective effects of the peptide may be mediated through attenuation of endothelial-leukocyte interactions, effects on fibroblast phenotype may contribute to the beneficial actions. The effectiveness of the strategy in large animal models or in human patients has not been tested. Moreover, the protective effects of ED-A fibronectin loss in experimental models of infarction [69] suggest that ED-A inhibition may hold promise as a therapeutic strategy. Attenuation of the pro-fibrotic actions of ED-A fibronectin may prevent overactive fibrotic responses, while attenuating matrix-degrading pathways.
Matricellular proteins may also be promising therapeutic targets. Because matricellular proteins exhibit spatially and temporally-restricted expression and act in a context-dependent manner, activation or inhibition of carefully selected matricellular functions would be expected to selectively target the area of injury without affecting homeostatic functions. Loss-of-function studies have demonstrated that TSP-1, SPARC, OPN and periostin exert protective actions on the infarcted myocardium by promoting repair. Identification of the functional domains and pathways responsible for the beneficial effects of matricellular proteins allows design of peptides that selectively reproduce specific protective actions, or inhibit detrimental profibrotic effects [78], [96]. As an example, the peptide LSKL specifically inhibits TSP-1-dependent TGF-β activation and reduces fibrosis in a variety of experimental models [97]. The effects of such an approach in myocardial infarction have not been tested; in the absence of experimental evidence, the consequences are difficult to predict. Following myocardial infarction attenuation of TSP-1-driven TGF-β activity may reduce fibrotic remodeling; however, loss of matrix-preserving TGF-β actions may accentuate matrix degradation promoting chamber dilation.
Clinical translation of approaches targeting cell-matrix interactions for the treatment of patients with myocardial infarction poses several major challenges. First, little is known regarding the significance of matrix-cell interactions in human patients. Second, matrix proteins have many distinct functional domains; in most cases understanding of the functional role of specific cell-matrix interactions is incomplete. Third, post-infarction remodeling in human patients is complex and pathophysiologically heterogeneous. Patients surviving myocardial infarction exhibit varying degrees of dilative and fibrotic remodeling. Despite comparable initial injury some patients develop chamber dilation and systolic dysfunction (possibly due to overactive inflammatory and matrix-degrading responses), while others exhibit excessive fibrosis and diastolic dysfunction (presumably due to augmented pro-fibrotic signaling). Clearly, in human patient populations, the complexity of the pathophysiologic responses highlights the need for biomarker-guided personalized treatment approaches [98].
Acknowledgments
Dr Frangogiannis’ laboratory is supported by NIH grants R01 HL-76246 and HL-85440, the Wilf Family Cardiovascular Research Institute and the Edmond J Safra/Republic National Bank of New York Chair in Cardiovascular Medicine.
References
- 1.Nag AC. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28:41–61. [PubMed] [Google Scholar]
- 2.Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ Physiol. 2006;291:H1015–1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 3.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SJ. Origin of cardiac fibroblasts and the role of periostin. Circ Res. 2009;105:934–947. doi: 10.1161/CIRCRESAHA.109.201400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 6.Kakkar R, Lee RT. Intramyocardial fibroblast myocyte communication. Circ Res. 2010;106:47–57. doi: 10.1161/CIRCRESAHA.109.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith RS, Smith TJ, Blieden TM, Phipps RP. Fibroblasts as sentinel cells. Synthesis of chemokines and regulation of inflammation. Am J Pathol. 1997;151:317–322. [PMC free article] [PubMed] [Google Scholar]
- 8.Leonard BL, Smaill BH, LeGrice IJ. Structural remodeling and mechanical function in heart failure. Microsc Microanal. 2012;18:50–67. doi: 10.1017/S1431927611012438. [DOI] [PubMed] [Google Scholar]
- 9.Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. 2003;108:1395–1403. doi: 10.1161/01.CIR.0000085658.98621.49. [DOI] [PubMed] [Google Scholar]
- 10.Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637–1652. doi: 10.1016/0735-1097(89)90360-4. [DOI] [PubMed] [Google Scholar]
- 11.Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong TT, Shaw RM, Srivastava D. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev Cell. 2009;16:233–244. doi: 10.1016/j.devcel.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eghbali M, Blumenfeld OO, Seifter S, Buttrick PM, Leinwand LA, Robinson TF, Zern MA, Giambrone MA. Localization of types I, III and IV collagen mRNAs in rat heart cells by in situ hybridization. J Mol Cell Cardiol. 1989;21:103–113. doi: 10.1016/0022-2828(89)91498-3. [DOI] [PubMed] [Google Scholar]
- 13.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 2007;293:H1883–1891. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 14.Norris RA, Borg TK, Butcher JT, Baudino TA, Banerjee I, Markwald RR. Neonatal and adult cardiovascular pathophysiological remodeling and repair: developmental role of periostin. Ann N Y Acad Sci. 2008;1123:30–40. doi: 10.1196/annals.1420.005. [DOI] [PubMed] [Google Scholar]
- 15.Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW, 2nd, Conway SJ, Aronow BJ, Robbins J, Molkentin JD. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res. 2007;101:313–321. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 18.Roberts CS, Maclean D, Maroko P, Kloner RA. Early and late remodeling of the left ventricle after acute myocardial infarction. Am J Cardiol. 1984;54:407–410. doi: 10.1016/0002-9149(84)90206-6. [DOI] [PubMed] [Google Scholar]
- 19.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 20.St John Sutton M, Lee D, Rouleau JL, Goldman S, Plappert T, Braunwald E, Pfeffer MA. Left ventricular remodeling and ventricular arrhythmias after myocardial infarction. Circulation. 2003;107:2577–2582. doi: 10.1161/01.CIR.0000070420.51787.A8. [DOI] [PubMed] [Google Scholar]
- 21.Swynghedauw B. Remodeling of the heart in chronic pressure overload. Basic Res Cardiol. 1991;86(Suppl 1):99–105. [PubMed] [Google Scholar]
- 22.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost. 2007;97:738–747. [PubMed] [Google Scholar]
- 24.Frangogiannis NG, Mendoza LH, Ren G, Akrivakis S, Jackson PL, Michael LH, Smith CW, Entman ML. MCSF expression is induced in healing myocardial infarcts and may regulate monocyte and endothelial cell phenotype. Am J Physiol Heart Circ Physiol. 2003;285:H483–492. doi: 10.1152/ajpheart.01016.2002. [DOI] [PubMed] [Google Scholar]
- 25.Bujak M, Dobaczewski M, Gonzalez-Quesada C, Xia Y, Leucker T, Zymek P, Veeranna V, Tager AM, Luster AD, Frangogiannis NG. Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction. Circ Res. 2009;105:973–983. doi: 10.1161/CIRCRESAHA.109.199471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176:2177–2187. doi: 10.2353/ajpath.2010.090759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58:88–111. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, Frangogiannis NG. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173:57–67. doi: 10.2353/ajpath.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frangogiannis NG, Michael LH, Entman ML. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb) Cardiovasc Res. 2000;48:89–100. doi: 10.1016/s0008-6363(00)00158-9. [DOI] [PubMed] [Google Scholar]
- 30.Willems IE, Havenith MG, De Mey JG, Daemen MJ. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol. 1994;145:868–875. [PMC free article] [PubMed] [Google Scholar]
- 31.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 32.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010;48:504–511. doi: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mann DL. The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res. 2011;108:1133–1145. doi: 10.1161/CIRCRESAHA.110.226936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 35.Cannon RO, 3rd, Butany JW, McManus BM, Speir E, Kravitz AB, Bolli R, Ferrans VJ. Early degradation of collagen after acute myocardial infarction in the rat. Am J Cardiol. 1983;52:390–395. doi: 10.1016/0002-9149(83)90145-5. [DOI] [PubMed] [Google Scholar]
- 36.Whittaker P, Boughner DR, Kloner RA. Role of collagen in acute myocardial infarct expansion. Circulation. 1991;84:2123–2134. doi: 10.1161/01.cir.84.5.2123. [DOI] [PubMed] [Google Scholar]
- 37.Etoh T, Joffs C, Deschamps AM, Davis J, Dowdy K, Hendrick J, Baicu S, Mukherjee R, Manhaini M, Spinale FG. Myocardial and interstitial matrix metalloproteinase activity after acute myocardial infarction in pigs. Am J Physiol Heart Circ Physiol. 2001;281:H987–994. doi: 10.1152/ajpheart.2001.281.3.H987. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi S, Barry AC, Factor SM. Collagen degradation in ischaemic rat hearts. Biochem J. 1990;265:233–241. doi: 10.1042/bj2650233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villarreal F, Omens J, Dillmann W, Risteli J, Nguyen J, Covell J. Early degradation and serum appearance of type I collagen fragments after myocardial infarction. J Mol Cell Cardiol. 2004;36:597–601. doi: 10.1016/j.yjmcc.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 40.Adair-Kirk TL, Senior RM. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol. 2008;40:1101–1110. doi: 10.1016/j.biocel.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaggar A, Jackson PL, Noerager BD, O’Reilly PJ, McQuaid DB, Rowe SM, Clancy JP, Blalock JE. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180:5662–5669. doi: 10.4049/jimmunol.180.8.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riley DJ, Berg RA, Soltys RA, Kerr JS, Guss HN, Curran SF, Laskin DL. Neutrophil response following intratracheal instillation of collagen peptides into rat lungs. Exp Lung Res. 1988;14:549–563. doi: 10.3109/01902148809087827. [DOI] [PubMed] [Google Scholar]
- 43.Senior RM, Griffin GL, Mecham RP. Chemotactic activity of elastin-derived peptides. J Clin Invest. 1980;66:859–862. doi: 10.1172/JCI109926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trial J, Baughn RE, Wygant JN, McIntyre BW, Birdsall HH, Youker KA, Evans A, Entman ML, Rossen RD. Fibronectin fragments modulate monocyte VLA-5 expression and monocyte migration. J Clin Invest. 1999;104:419–430. doi: 10.1172/JCI4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark RA, Wikner NE, Doherty DE, Norris DA. Cryptic chemotactic activity of fibronectin for human monocytes resides in the 120-kDa fibroblastic cell-binding fragment. J Biol Chem. 1988;263:12115–12123. [PubMed] [Google Scholar]
- 46.Huttenlocher A, Werb Z, Tremble P, Huhtala P, Rosenberg L, Damsky CH. Decorin regulates collagenase gene expression in fibroblasts adhering to vitronectin. Matrix Biol. 1996;15:239–250. doi: 10.1016/s0945-053x(96)90115-8. [DOI] [PubMed] [Google Scholar]
- 47.Kapila YL, Kapila S, Johnson PW. Fibronectin and fibronectin fragments modulate the expression of proteinases and proteinase inhibitors in human periodontal ligament cells. Matrix Biol. 1996;15:251–261. doi: 10.1016/s0945-053x(96)90116-x. [DOI] [PubMed] [Google Scholar]
- 48.Kong W, Longaker MT, Lorenz HP. Cyclophilin C-associated protein is a mediator for fibronectin fragment-induced matrix metalloproteinase-13 expression. J Biol Chem. 2004;279:55334–55340. doi: 10.1074/jbc.M410804200. [DOI] [PubMed] [Google Scholar]
- 49.Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006;324:475–488. doi: 10.1007/s00441-005-0144-6. [DOI] [PubMed] [Google Scholar]
- 50.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 51.Campo GM, Avenoso A, D’Ascola A, Scuruchi M, Prestipino V, Nastasi G, Calatroni A, Campo S. The inhibition of hyaluronan degradation reduced pro-inflammatory cytokines in mouse synovial fibroblasts subjected to collagen-induced arthritis. J Cell Biochem. 2012;113:1852–1867. doi: 10.1002/jcb.24054. [DOI] [PubMed] [Google Scholar]
- 52.Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW. Resolution of lung inflammation by CD44. Science. 2002;296:155–158. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- 53.Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 54.Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, Haudek S, Thakker G, Frangogiannis NG. CD44 Is Critically Involved in Infarct Healing by Regulating the Inflammatory and Fibrotic Response. J Immunol. 2008;180:2625–2633. doi: 10.4049/jimmunol.180.4.2625. [DOI] [PubMed] [Google Scholar]
- 55.Liu X, Piela-Smith TH. Fibrin(ogen)-induced expression of ICAM-1 and chemokines in human synovial fibroblasts. J Immunol. 2000;165:5255–5261. doi: 10.4049/jimmunol.165.9.5255. [DOI] [PubMed] [Google Scholar]
- 56.Welch MP, Odland GF, Clark RA. Temporal relationships of F-actin bundle formation, collagen and fibronectin matrix assembly, and fibronectin receptor expression to wound contraction. J Cell Biol. 1990;110:133–145. doi: 10.1083/jcb.110.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clark RA. Wound repair. Overview and general considerations. In: RAC, editor. The Molecular and Cellular Biology of Wound Repair. New York: Plenum Press; 1995. pp. 3–50. [Google Scholar]
- 58.Rybarczyk BJ, Lawrence SO, Simpson-Haidaris PJ. Matrix-fibrinogen enhances wound closure by increasing both cell proliferation and migration. Blood. 2003;102:4035–4043. doi: 10.1182/blood-2003-03-0822. [DOI] [PubMed] [Google Scholar]
- 59.Greiling D, Clark RA. Fibronectin provides a conduit for fibroblast transmigration from collagenous stroma into fibrin clot provisional matrix. J Cell Sci. 1997;110 (Pt 7):861–870. doi: 10.1242/jcs.110.7.861. [DOI] [PubMed] [Google Scholar]
- 60.Lin F, Ren XD, Doris G, Clark RA. Three-dimensional migration of human adult dermal fibroblasts from collagen lattices into fibrin/fibronectin gels requires syndecan-4 proteoglycan. J Invest Dermatol. 2005;124:906–913. doi: 10.1111/j.0022-202X.2005.23740.x. [DOI] [PubMed] [Google Scholar]
- 61.Corbett SA, Schwarzbauer JE. Fibronectin-fibrin cross-linking: a regulator of cell behavior. Trends Cardiovasc Med. 1998;8:357–362. doi: 10.1016/s1050-1738(98)00028-0. [DOI] [PubMed] [Google Scholar]
- 62.Xu J, Clark RA. Extracellular matrix alters PDGF regulation of fibroblast integrins. J Cell Biol. 1996;132:239–249. doi: 10.1083/jcb.132.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 64.Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech. 2010;43:146–155. doi: 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 65.Yano T, Miura T, Ikeda Y, Matsuda E, Saito K, Miki T, Kobayashi H, Nishino Y, Ohtani S, Shimamoto K. Intracardiac fibroblasts, but not bone marrow derived cells, are the origin of myofibroblasts in myocardial infarct repair. Cardiovasc Pathol. 2005;14:241–246. doi: 10.1016/j.carpath.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 66.Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, Pilling D, Gomer RH, Trial J, Frangogiannis NG, Entman ML. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci U S A. 2006;103:18284–18289. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 68.Knowlton AA, Connelly CM, Romo GM, Mamuya W, Apstein CS, Brecher P. Rapid expression of fibronectin in the rabbit heart after myocardial infarction with and without reperfusion. J Clin Invest. 1992;89:1060–1068. doi: 10.1172/JCI115685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG, Peters JH, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP. Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circ Res. 2011;108:582–592. doi: 10.1161/CIRCRESAHA.110.224428. [DOI] [PubMed] [Google Scholar]
- 70.Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142:873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Naugle JE, Olson ER, Zhang X, Mase SE, Pilati CF, Maron MB, Folkesson HG, Horne WI, Doane KJ, Meszaros JG. Type VI collagen induces cardiac myofibroblast differentiation: implications for postinfarction remodeling. Am J Physiol Heart Circ Physiol. 2006;290:H323–330. doi: 10.1152/ajpheart.00321.2005. [DOI] [PubMed] [Google Scholar]
- 73.Luther DJ, Thodeti CK, Shamhart PE, Adapala RK, Hodnichak C, Weihrauch D, Bonaldo P, Chilian WM, Meszaros JG. Absence of type VI collagen paradoxically improves cardiac function, structure, and remodeling after myocardial infarction. Circ Res. 2012;110:851–856. doi: 10.1161/CIRCRESAHA.111.252734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Webber J, Meran S, Steadman R, Phillips A. Hyaluronan orchestrates transforming growth factor-beta1-dependent maintenance of myofibroblast phenotype. J Biol Chem. 2009;284:9083–9092. doi: 10.1074/jbc.M806989200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meran S, Thomas D, Stephens P, Martin J, Bowen T, Phillips A, Steadman R. Involvement of hyaluronan in regulation of fibroblast phenotype. J Biol Chem. 2007;282:25687–25697. doi: 10.1074/jbc.M700773200. [DOI] [PubMed] [Google Scholar]
- 76.Zhao XH, Laschinger C, Arora P, Szaszi K, Kapus A, McCulloch CA. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J Cell Sci. 2007;120:1801–1809. doi: 10.1242/jcs.001586. [DOI] [PubMed] [Google Scholar]
- 77.Bornstein P. Matricellular proteins: an overview. J Cell Commun Signal. 2009;3:163–165. doi: 10.1007/s12079-009-0069-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635–688. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;36:961–968. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML. The critical role of endogenous Thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 81.Rodriguez-Manzaneque JC, Lane TF, Ortega MA, Hynes RO, Lawler J, Iruela-Arispe ML. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc Natl Acad Sci U S A. 2001;98:12485–12490. doi: 10.1073/pnas.171460498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hogg PJ. Thrombospondin 1 as an enzyme inhibitor. Thromb Haemost. 1994;72:787–792. [PubMed] [Google Scholar]
- 83.Xia Y, Dobaczewski M, Gonzalez-Quesada C, Chen W, Biernacka A, Li N, Lee DW, Frangogiannis NG. Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension. 2011;58:902–911. doi: 10.1161/HYPERTENSIONAHA.111.175323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Willems IE, Arends JW, Daemen MJ. Tenascin and fibronectin expression in healing human myocardial scars. J Pathol. 1996;179:321–325. doi: 10.1002/(SICI)1096-9896(199607)179:3<321::AID-PATH555>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 85.Tamaoki M, Imanaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, Sakakura T, Yoshida T. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol. 2005;167:71–80. doi: 10.1016/S0002-9440(10)62954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nishioka T, Onishi K, Shimojo N, Nagano Y, Matsusaka H, Ikeuchi M, Ide T, Tsutsui H, Hiroe M, Yoshida T, Imanaka-Yoshida K. Tenascin-C may aggravate left ventricular remodeling and function after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2010;298:H1072–1078. doi: 10.1152/ajpheart.00255.2009. [DOI] [PubMed] [Google Scholar]
- 87.Schellings MW, Vanhoutte D, Swinnen M, Cleutjens JP, Debets J, van Leeuwen RE, d’Hooge J, Van de Werf F, Carmeliet P, Pinto YM, Sage EH, Heymans S. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J Exp Med. 2009;206:113–123. doi: 10.1084/jem.20081244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murry CE, Giachelli CM, Schwartz SM, Vracko R. Macrophages express osteopontin during repair of myocardial necrosis. Am J Pathol. 1994;145:1450–1462. [PMC free article] [PubMed] [Google Scholar]
- 89.Krishnamurthy P, Peterson JT, Subramanian V, Singh M, Singh K. Inhibition of matrix metalloproteinases improves left ventricular function in mice lacking osteopontin after myocardial infarction. Mol Cell Biochem. 2009;322:53–62. doi: 10.1007/s11010-008-9939-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Trueblood NA, Xie Z, Communal C, Sam F, Ngoy S, Liaw L, Jenkins AW, Wang J, Sawyer DB, Bing OH, Apstein CS, Colucci WS, Singh K. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ Res. 2001;88:1080–1087. doi: 10.1161/hh1001.090842. [DOI] [PubMed] [Google Scholar]
- 91.Ashizawa N, Graf K, Do YS, Nunohiro T, Giachelli CM, Meehan WP, Tuan TL, Hsueh WA. Osteopontin is produced by rat cardiac fibroblasts and mediates A(II)-induced DNA synthesis and collagen gel contraction. J Clin Invest. 1996;98:2218–2227. doi: 10.1172/JCI119031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zohar R, Zhu B, Liu P, Sodek J, McCulloch CA. Increased cell death in osteopontin-deficient cardiac fibroblasts occurs by a caspase-3-independent pathway. Am J Physiol Heart Circ Physiol. 2004;287:H1730–1739. doi: 10.1152/ajpheart.00098.2004. [DOI] [PubMed] [Google Scholar]
- 93.Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, Saito M, Fukuda K, Nishiyama T, Kitajima S, Saga Y, Fukayama M, Sata M, Kudo A. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med. 2008;205:295–303. doi: 10.1084/jem.20071297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. 2002;50:71–79. doi: 10.1177/002215540205000108. [DOI] [PubMed] [Google Scholar]
- 95.Petzelbauer P, Zacharowski PA, Miyazaki Y, Friedl P, Wickenhauser G, Castellino FJ, Groger M, Wolff K, Zacharowski K. The fibrin-derived peptide Bbeta15–42 protects the myocardium against ischemia-reperfusion injury. Nat Med. 2005;11:298–304. doi: 10.1038/nm1198. [DOI] [PubMed] [Google Scholar]
- 96.Henkin J, Volpert OV. Therapies using anti-angiogenic peptide mimetics of thrombospondin-1. Expert Opin Ther Targets. 2011;15:1369–1386. doi: 10.1517/14728222.2011.640319. [DOI] [PubMed] [Google Scholar]
- 97.Belmadani S, Bernal J, Wei CC, Pallero MA, Dell’italia L, Murphy-Ullrich JE, Berecek KH. A Thrombospondin-1 Antagonist of Transforming Growth Factor-{beta} Activation Blocks Cardiomyopathy in Rats with Diabetes and Elevated Angiotensin II. Am J Pathol. 2007 doi: 10.2353/ajpath.2007.070056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Frangogiannis NG. Biomarkers: hopes and challenges in the path from discovery to clinical practice. Transl Res. 2012;159:197–204. doi: 10.1016/j.trsl.2012.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]