

Abstract
Zn2+ and Cd2+ ion exchange between transcription factor IIIA (TFIIIA) and apo-metallothionein (MT) were studied using a combination of methods including chromatography, ultrafiltration and UV spectroscopy. Under near stoichiometric conditions, apoMT was able to remove most if not all of the zinc ions from TFIIIA, whether or not the TFIIIA was bound to the 5S DNA internal control region (ICR), and concomitantly inhibit its DNA-binding activity as indicated by an electrophoretic mobility shift assay. The kinetics of the two processes were similar. The rate of the metal exchange reaction increased with the concentrations of both reactants. A second-order rate constant of 30 ± 10 M−1 s−1 was calculated. Similar observations were made for the reaction between apoMT and Cd-substituted TFIIIA, which proceeded without observable intermediates according to a spectrophotometric analysis. A very slow metal ion exchange occurred between Cd-TFIIIA and Zn-MT, but not between Cd-MT and Zn-TFIIIA. Comparative studies on the reaction of TFIIIA with a small competing ligand, ethylenedinitrilo-tetraacetic acid (EDTA), were also conducted. Although EDTA reacts with free Zn-TFIIIA, under similar conditions it failed to compete for Zn2+ bound as Zn-TFIIIA-ICR.
Keywords: Transcription factor IIIA, Apo-metallothionein, Cadmium, Zinc, Metal exchange
1. Introduction
DNA-binding zinc-finger proteins such as transcription factor IIIA from Xenopus laevis represent one of the most common type of eucaryotic transcription factor [1]. All are characterized by a common domain conformation that includes two lengths of antiparallel β structure and an α helix, Zn2+ bound to a set of two cysteinyl sulfhydryl and histidinyl imidazole nitrogen ligands (C2H2), and a mini-hydrophobic core of three non-polar side chains [2–12]. Zinc-binding is required to achieve stable, folded fingers and specific protein–DNA-binding [1,3,12–16]. Three-dimensional structures of the first three and six Zn fingers of TFIIIA associated with their DNA-binding sites have been published, showing helical folding of finger domains 1–3 around the major groove of its DNA-binding site, and stretching of fingers 4–6 along the length of the DNA oligomer [9,12].
It has been hypothesized that under different cellular conditions, the availability of Zn2+ for Zn-finger proteins might change, leading to alterations in their functionality [17,18]. This proposal receives support from the observation that TFIIIA does not bind Zn2+ tightly, thus, potentially making its DNA-binding conformation sensitive to alterations in intracellular Zn2+ concentration through the following generalized equilibrium, in which Zn2+ is bound to cellular ligands (L) [19]:
| (1) |
A physiologically relevant example of this equilibrium may constitute the basis for the induction of apo-metallothionein synthesis. ApoMT can be induced by Zn2+ through a mechanism that involves the zinc-finger transcription factor, MTF-1 [20,21]. The in vitro DNA-binding affinity of MTF-1 for the metal response elements (MREs) of the metallothionein promoter is directly dependent on zinc concentration [22]. From this experiment, it is inferred that apo-MTF-1 does not bind to these base sequences but the folded conformation, Zn-MTF-1 does, depending on the position of the equilibrium in reaction (1).
Reaction (1) might also work in the reverse direction such that Zn-finger structures would be inactivated through the removal of Zn2+ from Zn-finger by competing metal-binding ligands. With its 20 sulfhydryl groups, apo-metallothionein is a potent Zn2+ chelating agent [23,24]. It is considered to be a central component in cellular zinc homeostasis and has been suggested to play a role in gene regulation by changing Zn2+ occupancy of various Zn-finger proteins [25,26]. For example, apoMT might feedback inhibit its own synthesis through a reaction that extracts the Zn2+ out of Zn-MTF-1.
In order to demonstrate that the Zn-finger motif might be reactive with apoMT, Kägi and coworkers [25,26] showed that preincubation of the zinc-finger transcription factors Zn-TFIIIA and Zn-Sp1, a three finger transcription factor, with apoMT interferes with specific DNA-binding by the proteins according to gel shift and in vitro transcription assays. More recently, two reports have described chemical properties of the reaction of selected Zn-finger peptides with apoMT or the α-domain of metallothionein [27,28]. The present experiments were undertaken to gain insight into the chemical properties of the reaction of Zn-TFIIIA with apoMT. In order to examine possible, physiologically significant reactions, it was important to compare the properties of this reaction in the absence or presence of its natural DNA-binding site, the internal control region of the 5S ribosomal RNA gene.
Metallothionein also plays a key detoxification role by sequestering Cd2+ which has been distributed throughout the cell [29,30]. However, little is known about the sites, such as metalloproteins, to which Cd2+ binds and the properties of the reactions of apo- or Zn-MT with such sites. Studies of the reaction of apoMT and one of its component peptide sequences with Cd-carbonic anhydrase showed that both were remarkably effective in competing for protein-bound Cd2+ [31,32]. There is evidence that, in vitro, Cd2+ can substitute for Zn2+ in zinc-finger structures and that such substitution can change the activities of the finger domains [22,28,33]. Therefore, this study has also examined how Cd-TFIIIA reacts with apo- or Zn7-MT. For comparison, the reactions of Zn- and Cd-TFIIIA with a small competing ligand, EDTA, have also been included in the investigation.
2. Materials and methods
2.1. Preparation of TFIIIA and MT
TFIIIA has nine tandem zinc fingers of the C2H2 type (–Y/F–X–C–X2,4.5–C–X3–F–X5–L–X2–H–X3,4–H–X2–6–) within its 38.5 kDa structure [34]. Methods for preparation of recombinant X. laevis TFIIIA from Escherichia coli and for the isolation of MT-2 from rabbit liver have been described in detail elsewhere [35,36]. Zn7- or Cd7-MT-2 (Ac-MDPNCSCAADGSCTCATSCKCKECKCTSCKKSCCSCCPSGCAKCAQGCICKGASDKCSCCA) have molecular masses between 6 and 7 kDa and migrate on Sephadex G-75 like a globular 10-kDa protein [37].
The purity of the TFIIIA protein was shown to be routinely 80–90% according to densitometric analysis of silver-stained 12% SDS–PAGE of various protein preparations [35]. The metal-binding stoichiometry of TFIIIA was found to vary between less than 1–9 for both Zn- and Cd-forms, depending on the details of the preparation procedure and buffering conditions [35]. Znn-TFIIIA (n = 7 ± 2) is active in DNA-binding in buffers containing 5–20 μM of excess Zn2+ [35]. Since TFIIIA with less than 9 bound metals may be relevant in vivo, protein samples of various metal stoichiometries were used in the present study and the results compared.
Metallothionein was characterized by metal (atomic absorption spectrophotometry) and sulfhydryl analysis using Ellman’s reagent, DTNB [38]. Typically, the metal ion to sulfhydryl ratio was 2.7–3.0, very close to the theoretical value of 2.86.
2.2. Apo-metallothionein preparation and use
Apo-MT was prepared by acidifying Zn7-MT to pH 2 and chromatographing the product over Sephadex G-50 at pH 2 in order to separate the protein from dissociated metal ions. Under the conditions of the experiments at pH 7, no significant oxidation of apo-MT occurred over a 60-min period. In order to minimize adventitious oxidation of thiols prior to its ligand-substitution reactions with Zn- or Cd-TFIIIA, the metal-free protein was prepared fresh before each experiment and maintained at pH 2 until its use. In addition, DTT was included in most of the reactions to maintain reducing conditions. Finally, the spectrophotometric analysis of the reaction of apo-MT with Cd-TFIIIA was conducted under anaerobic conditions to minimize the possibility of apo-MT oxidation. The stoichiometric transfer of Zn from TFIIIA to apoMT under conditions of equal concentration of Zn and apoMT Zn-binding sites confirmed the prolonged stability of apoMT’s sulfhydryl groups during the reactions.
2.3. Protein determination
The TFIIIA protein concentration was determined by using the Bradford assay with bovine serum albumin as the standard [39]. A coefficient of 0.62± 0.06 was applied in order to correct for the differential staining behavior of albumin and TFIIIA, based on comparative staining of known amounts of these proteins. The relative error of this measurement was typically 20%. The MT protein concentration was determined by employing the DTNB assay for thiol concentration and then dividing this value by 20, the number of cysteine residues in each MT molecule. The relative error of the determination was 10% based on the reproducibility of the DTNB measurement.
2.4. Metal determination
Zn and Cd concentrations were determined using flame atomic absorption spectrophotometry. The relative error of these determinations was generally less than 5%.
2.5. Ultrafiltration study of metal exchange reactions
A Centricon-30 centrifugal concentrator (Amicon) was used to separate TFIIIA, 39 kDa, from MT, 6 kDa, or other smaller ligands. In kinetic studies, reactants with a total volume of 0.5–1 mL were mixed, sealed in the reaction vessel with parafilm to prevent evaporation and contamination, incubated for the appropriate length of time and then centrifuged in the Centricon-30 concentrator at 6000 rpm for 30–60 min at 4 °C in an SS-34 rotor in a Sorvall RC-5B+ centrifuge. Control experiments showed that about 90% of the filtration was complete in 15 min and that at the end of the centrifugation 80–90% of the solution was filtered through the membrane. Retentate, containing the larger TFIIIA protein and some MT, and filtrate, containing the smaller molecular weight molecules and MT, were analyzed directly or after appropriate dilution. Controls with TFIIIA and MT, respectively, showed that, typically, less than 10% of the metals bound in TFIIIA and 90–100% of the metals bound in MT were found in the filtrate. In experiments where free Zn2+ was present in the buffer, sufficient apoMT was added to compete both for the unbound Zn2+ and Zn2+ in TFIIIA. The former binds to apoMT within the time of mixing, thereby, distinguishing it from TFIIIA-associated Zn2+.
2.6. Sephadex G-75 gel filtration separation of proteins
To follow the Zn2+ transfer between TFIIIA and MT, a 1×70 cm Sephadex G-75 (Pharmacia) gel filtration chromatographic column was used. The relative migration of the two metalloproteins was determined by chromatographing the individual proteins prior to the reaction. Their elution peak maxima are indicated on Fig. 1. Care was taken to assure the system was metal-free by pretreating the column with the metal chelator, EDTA, and all buffers were pretreated with Chelex-100 (BioRad), a resin with iminodiacetic acid moieties attached. The fractions were analyzed for metal content by atomic absorption spectrophotometry and the results presented in μg/mL (ppm).
Fig. 1.

Sephadex G-75 gel filtration chromatographic profile of the ligand-substitution reaction: Zn4-TFIIIA+apoMT (■). The reaction mixture was applied to the column immediately after mixing. The markers refer to the peak positions for the elution of samples of Zn-TFIIIA and Zn-MT. Representative Zn4-TFIIIA control sample profile scaled to the Zn-TFIIIA peak of the reaction mixture (○). Conditions: 4.5 μM TFIIIA (18 μM Zn2+) and 2.5 μM apoMT, respectively, in 20 mM Hepes buffer, pH 7.4, at 25 °C. The column (1×60 cm) was equilibrated in the running buffer, 20 mM Hepes buffer, pH 7.4.
2.7. Spectroscopic study of metal exchange reactions
UV–Vis spectroscopic studies of ligand-substitution reactions of Cd-TFIIIA were performed using a Beckman DU-70 spectrophotometer. The anaerobic reactants were mixed rapidly and observation began within about 10 s. Stock solutions of the competing ligands were made in the same buffer as that used for the protein, usually 20 mM Hepes, pH 7.4.
2.8. Kinetic analysis
For second-order reactions between reactants with the same concentration in the absence of an initial product concentration, the following equation applies:
| (2) |
where y represents the absorbance or concentration of metal ion transferred, t represents reaction time, a is the initial product concentration, b is the initial absorbance or concentration of the reactants and c = 1/kb. Then, k, representing the rate constant, is equal to 1/bc. For the kinetic analyses Znn-TFIIIA is represented by Zn2+ concentration (n× [Znn-TFIIIA]) and apo-MT by metal ion-binding site concentration (7×[apo-MT]). The data were curve-fitted using least-squares regression with TableCurve 2D (v2.0, Jandel Scientific). For second-order reactions of two reactants with different initial concentrations,
| (3) |
in which a and b are the initial concentrations of the two reactants, y is the product concentration at time t, and k is the rate constant. The data were subjected to a linear regression analysis.
2.9. Electrophoretic mobility shift assay
The electrophoretic mobility shift assay (EMSA) as well as the method for the preparation and radio-labeling of the 146 bp ICR-containing DNA probe have been described elsewhere [35]. After electrophoresis for 2 h, the gel was dried and placed in contact with X-ray film (Fuji) at −80 °C to reveal the location and concentration of free and bound ICR. Exposures of various time lengths were made in order find the linear response range for the radioactive probe. Then, defining the intensity of the free ICR as 100%, the percentage reaction of Zn-TFIIIA-DNA with apoMT was scaled to this value after measuring the intensity of the free ICR that separated during electrophoresis.
3. Results
3.1. Zn2+ transfer from TFIIIA to apoMT
Zeng et al. [25] showed that preincubation of apoMT with Zn-TFIIIA prevented subsequent binding of TFIIIA in the reaction mixture to the internal control region of the 5S rRNA gene and activation of transcription of this gene. It was inferred that apoMT successfully competed with the Zn-finger sites in TFIIIA to remove Zn2+ and unfold the protein. In order to explore the properties of the reaction, we used both gel filtration and ultrafiltration to physically separate the two proteins from each other during or after the reaction and then measured the zinc distribution between them (Figs. 1 and 2). The former provides clear proof of the metal ion transfer from Zn-TFIIIA to apoMT. The latter method has the advantage of more accurate control over reaction time and, thus, was used to follow the time course of metal transfer.
Fig. 2.

Kinetics of the ligand-substitution reaction: Znn-TFIIIA+apoMT, followed by ultrafiltration and gel shift assay. (a) The time-dependent percentage of total Zn transferred was measured by Centricon ultrafiltration (●) or by percentage of inhibition of DNA-binding activity in the electrophoretic mobility shift assay (▲) and plotted against reaction time. The curve is the best secondorder fit for all of the data (Eq. (2)). Conditions: (●) 1 μM Zn9-TFIIIA and 2.0 μM apoMT in 20 mM Hepes buffer, pH 7.4, plus 25 mM NaCl, 1% glycerol, 5 μM ZnSO4, and 0.5 mM DTT, at 25 °C; (▲) 0.75 nM ICR probe, 0.8 μM Zn7 -TFIIIA, 1.6 μM apoMT in 20 mM Tris/Cl buffer, pH 7.4, plus 70 mM KCl, 10% glycerol, 5 μM ZnSO4, 50 μg/ml poly(dI–dC), and 1 mM DTT at 25 °C. (b) Second-order rate plot for a reaction with different concentrations of starting materials. Conditions: (■) 1.4 μM for both Zn4-TFIIIA and apoMT, in 20 mM Hepes buffer, pH 7.4 (reaction is 90% complete at 250 min; data analyzed with Eq. (3)). (c) Total Zn transferred between t = 0 and 15 min as a function of the apoMT or Zn4-TFIIIA concentrations. Conditions: constant Zn4-TFIIIA (■, 1.4 μM) or apoMT (●, 1.1 μM), respectively, and 20 mM Hepes, pH 7.4 and 25 °C.
Fig. 1 shows a Sephadex G-75 gel filtration chromatographic profile of the ligand-substitution reaction mixture described in reaction (3). The presence of Zn2+ in the MT band indicates that about 50% of the Zn2+ initially bound to TFIIIA shifted to apoMT within minutes of mixing
| (4) |
In Fig. 2(a), the time course of the reaction was followed by ultrafiltration and the EMSA. It demonstrates that under stoichiometric conditions for Zn2+-binding sites in apoMT and TFIIIA-bound Zn2+ plus free Zn2+, Zn2+ transfer from TFIIIA to apoMT goes approximately to completion (70–90%) within the error of these measurements. From these data, the ultrafiltration data alone yielded a constant of 43 M−1 s−1. In these rate constants, molarity refers to metal ion-binding site concentration in apoMT. A plot of the rate data for the non-stoichiometric reaction of Zn4-TFIIIA with apoMT in Fig. 2(b) revealed a rate constant of 27 M−1 s−1. Both sets of data are consistent with a simple second-order reaction as portrayed in reaction (4). The second-order nature of the reaction was confirmed by examining the approximate initial rate of reaction as a function of Zn4-TFIIIA or apoMT concentration. The amount of Zn2+ transferred in the first 15 min of the reaction, which was assumed to provide a measure of initial reaction rate, was proportional to the concentrations of both reactants (Fig. 2(c)). Indeed, using the 15-min data to approximate initial rates and the the initial concentrations of apoMT and Zn-TFIIIA, the calculated second-order rate constants were 21 and 32 M−1 s−1 for reactions with apoMT or Zn-TFIIIA constant, respectively. Based on all these results, the reaction was considered as a simple, irreversible second-order reaction in which TFIIIA and MT denote binding sites in the two proteins. Within the sensitivity of the assays of metal ion transfer, the kinetics support the assumption that the n-Zn fingers of Znn-TFIIIA represent a set of homogeneous Zn-binding sites and that MT binds Zn in a concerted fashion such that it, too, contains a homogeneous set of binding sites. Averaging all of the results, the rate constant for ligand substitution was 30±0 M−1 s−1.
Fig. 3(a) is an electrophoretic mobility shift assay that shows the effect of apoMT on the DNA-binding activity of Zn-TFIIIA. Lanes 1 and 2 are negative and positive controls. After incubation of Zn-TFIIIA with a stoichiometric concentration of Zn2+-binding sites from apoMT, it can be seen in lanes 3–8 that with increasing reaction time, declining amounts of Zn-TFIIIA-ICR complex were detected, until, at the longest reaction time, only a small amount of the complex was observed. For reactions shown in lanes 6–8, since apoMT and TFIIIA were mixed before the 30-min DNA-binding reaction, the actual contact time for TFIIIA and apoMT should be considered to be 30, 40 and 60 min, respectively. It is apparent that apoMT largely abolished the DNA-binding capacity of TFIIIA whether or not the protein–DNA complex had been preformed. When the time course of this reaction was plotted in Fig. 2(a) in order to compare it with the kinetics of Zn2+ transfer from unbound Zn-TFIIIA to apoMT, the two profiles were similar. Data from lanes 6–8 were not plotted, since the reactants’ concentrations were different from those in the other data. This coincidence between inhibition of DNA-binding and exchange of Zn2+ strongly supports the view that the loss of DNA binding activity is, indeed, due to the removal of Zn2+ from TFIIIA by apoMT.
Fig. 3.
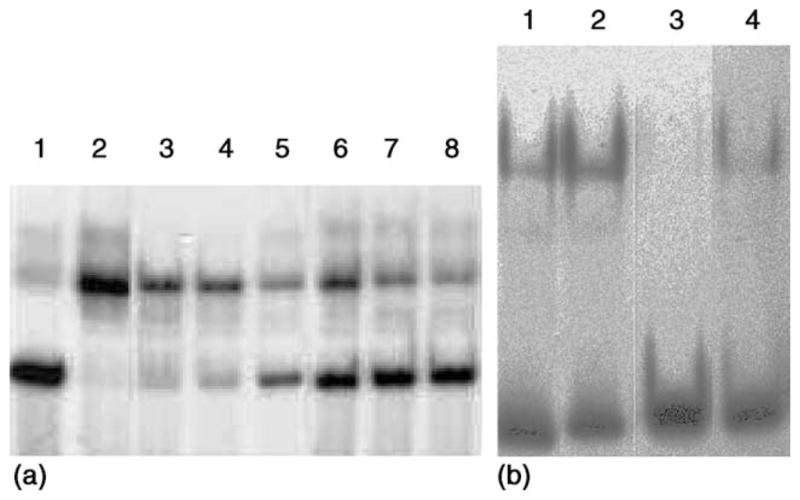
Effect of apoMT on the DNA-binding activity of Zn-TFIIIA. (a) Lane 1: 0.75 nM free probe; lane 2: 0.75 nM ICR preincubated with 0.8 μMZn7-TFIIIA; lanes 3–5: 1.6 μMZn7-TFIIIA preincubated with 0.75 nM ICR for 30 min and then reacted with 1.6 μM apoMT for 0, 10, and 30 min, respectively, before EMSA. Lanes 6–8: 0.8 μM Zn7 -TFIIIA and 1.6 μM apoMT were mixed and reacted for 0, 10, and 30 min and then for 30 min with the 0.75 nM ICR, respectively, before EMSA. (b) ApoMT, 315 μM Zn2+-binding capacity (lanes 1 and 2) or 630 μMZn2+-binding capacity (lanes 3 and 4), was added immediately before (lanes 1 and 3) or 30 min after (lanes 2 and 4) Zn9-TFIIIA (6 μM) was mixed with the ICR (2.9 nM). After 30-min incubation of Zn-TFIIIA (ICR) with apoMT, the EMSA was carried out. Conditions: 20 mM Tris/Cl buffer, pH 7.4, plus 70 mM KCl, 10% glycerol, 30 μM ZnSO4, 50 μg/ml poly(dI–dC), and 1 mM DTT at 25 °C.
In order to investigate whether preformation of the protein–DNA complex has an effect on the reaction of Zn-TFIIIA with apoMT, parallel reactions were carried out in which all the reactant concentrations and the overall contact time (30 min) between Zn-TFIIIA and apoMT were identical. One involved preformed Zn-TFIIIA-ICR complex, produced by a 30-min co-incubation, while the other proceeded during the 30-min DNA-binding reaction. As shown in Fig. 3(b), slightly more Zn-TFIIIA-ICR adduct was detected when apoMT was added after the formation of the Zn-TFIIIA-ICR complex rather than before it. This suggests that Zn2+ in TFIIIA was somewhat less reactive with apoMT after the protein was bound to ICR. However, as shown in Fig. 2(a) and in calculations based on these data, within error, the same second-order rate constant was determined under both conditions.
3.2. Cd2+ transfer from TFIIIA to apoMT
Cd2+ reacts with Zn-TFIIIA-ICR and causes the dissociation of the protein–DNA adduct [35]. Thus, it was of interest to determine whether apo- or Zn-MT might compete successfully for Cd2+ bound to TFIIIA and, thereby, provide the possibility to restore its DNA-binding capacity (reaction (4)):
| (5) |
Fig. 4 shows a Sephadex G-75 gel filtration chromatographic profile of this ligand-substitution reaction. As in the case with Zn-TFIIIA, apoMT abstracts almost all of the Cd2+ in TFIIIA under near stoichiometric conditions.
Fig. 4.

Sephadex G-75 gel filtration chromatographic profiles of the ligand-substitution reaction, Cd3-TFIIIA+apoMT. The reaction mixture was applied to the column 40 min after mixing. Conditions: concentrations of TFIIIA and apoMT, 14 and 7 μM, respectively, in 20 mM Hepes buffer, pH 7.4, at 25 °C. The column (1×60 cm) was equilibrated in the running buffer, 20 mM Hepes buffer, pH 7.4.
Since the charge transfer bands for Cd2+-thiolate bonds are more red-shifted than those for Zn2+ and, thus, easier to detect, a spectroscopic method was also employed to follow this reaction. Fig. 5 shows the spectroscopic profile of the reaction at different reaction times. A maximum at 242 nm was found for the spectral changes during the reaction between Cd-TFIIIA and apoMT. This spectral feature was caused by the changes in the apparent intensity of the charge transfer bands when Cd2+ was removed from the binding sites in TFIIIA with thiolate and imidazole ligands and bound to the binding sites in MT where all the ligands are thiolates. The small decrease around 280 nm may be caused by the variations of the apparent extinction co-efficients of the aromatic residues in TFIIIA in a changing molecular environment as a consequence of ligand-substitution reaction. The isosbestic point at 265 nm implies a simple conversion of UV absorbing reactants to products and the absence of appreciable amounts of reaction intermediates.
Fig. 5.

Reaction of Cd3-TFIIIA with apoMT. Absorption spectra of the reaction mixture: Cd3-TFIIIA+apoMT measured from 0 to 30 min. Conditions: final concentrations of TFIIIA and apoMT, 14 and 7 μM, respectively, in 20 mM Hepes buffer, pH 7.4, at 25 °C. The direction of arrows indicates reaction time. Inset. The time course of the metal transfer reaction between Cd3-TFIIIA and apoMT followed by UV spectroscopy. Conditions: final concentrations of TFIIIA and apoMT, 14 and 6 μM, respectively, in 20 mM Hepes buffer, pH 7.4, at 25 °C.
The progress of the reaction was followed by monitoring the absorption changes at 242 nm. Fig. 5 (inset) depicts a typical reaction profile. Absorbance at the beginning of the reaction (t = 0 min) coincided well with that calculated from the absorbances of the two reactants, indicating that there was no significant reaction during mixing. The kinetics of the reaction followed at 242 nm were consistent with a second-order process, first order in Cd2+ ion and Cd2+-binding sites in apoMT, with a rate constant of 40±10 M−1 s−1, which was identical with that for Zn2+ transfer in reaction (2).
3.3. Metal exchange reaction between Cd-TFIIIA and Zn-MT
Having established that apoMT was able to abstract Cd2+ out of Cd-substituted TFIIIA, the possibility of metal exchange between Zn7-MT and Cd3-TFIIIA was considered:
| (6) |
According to the binding constants of the two metal ions to MT and to TFIIIA or its zinc-finger peptide, this reaction should be thermodynamically favorable [19,23,37,40]. Indeed, some metal exchange between Zn-MT and Cd-TFIIIA was detected by chromatographic, ultrafiltration and spectrophotometric methods, but the Cd2+–Zn2+ exchange reaction was quite slow.
In the reverse direction, reaction (5) should be thermodynamically unfavorable. Consistent with this, no metal exchange between Cd7-MT and Zn7-TFIIIA was detected. Nor was Cd7-MT able to inactivate binding by Zn7-TFIIIA to ICR in the gel shift assay.
3.4. Reaction of Zn- or Cd-TFIIIA with EDTA
In Fig. 6, the time course of the reaction between near stoichiometric concentrations of Zn-TFIIIA and EDTA was followed by ultrafiltration and simulated as a second-order reaction, first order in Zn2+ and EDTA concentrations, with a rate constant of 160 M−1 s−1. Interestingly, a 200-fold excess of EDTA with respect to Cd2+ in Cd3-TFIIIA did not completely remove Cd from the protein during a 29-h incubation.
Fig. 6.

Time course of the ligand-substitution reaction, Zn4-TFIIIA+EDTA, followed by ultrafiltration. Conditions: 1.4 μM TFIIIA and 4.5 (■) or 45 μM (◇) EDTA, in 20 mM Hepes buffer, pH 7.4, at 25 °C. Percentage of Zn2+ transferred was plotted against reaction time.
Fig. 7 illustrates the effects of EDTA on the binding activity of TFIIIA in a gel shift assay. According to Fig. 7, preincubation of Zn-TFIIIA with 1.7–3.4 times the concentration of EDTA needed to stoichiometrically bind Zn2+ completely abolished the DNA-binding (lanes 3–4). This was expected since EDTA had been shown to extract all the Zn2+ in TFIIIA under similar conditions (Fig. 6). In contrast, the same concentration of EDTA had no effect on the preformed Zn-TFIIIA-ICR complex (lanes 5–6). Indeed, following 30 min incubation to form the Zn-TFIIIA-ICR adduct, addition of 25 mM EDTA (4×104 molar excess Zn2+-binding capacity) did not completely dissociate the complex after a 30-min period of reaction (data not shown). The depression of the reactivity of DNA-bound Zn-TFIIIA must be due to additional thermodynamic and/or kinetic barriers created by DNA binding. It did not result from the presence in the binding buffer of 5 mM Mg2+, which has a much lower binding affinity for EDTA [41].
Fig. 7.
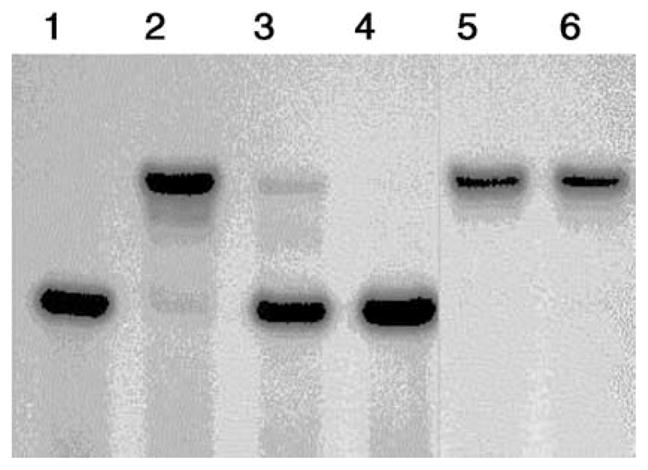
Effects of EDTA on the DNA-binding activity of Zn7-TFIIIA. Lane 1: 0.8 nM free probe; lane 2: 0.8 nM ICR probe preincubated with 0.8 μM TFIIIA; lanes 3 and 4: 0.8 μM Zn7-TFIIIA incubated for 20 min with EDTA at a final concentration of 10 and 20 μM, respectively, before an additional 20-min incubation with the ICR (0.8 nM)-binding mixture prior to EMSA. Lanes 5 and 6: 0.8 μM Zn7 -TFIIIA and the ICR (0.8 nM)-binding mixture were incubated for 20 min before EDTA was added at a final concentration of 10 and 20 μM, respectively, and this mixture incubated for another 20 min prior to EMSA.
EDTA has been reported to inhibit DNA-binding by zf123, the first three fingers of TFIIIA, which associate with the ICR with high affinity (Kd = 5.6 nM) [42]. However, 50% inhibition occurred at 25 mM EDTA and complete inhibition at 50 mM, which was more than 104 times the concentration of Zn2+ in zf123 and orders of magnitude larger than the concentrations used in the present experiment.
4. Discussion
Metallothionein, discovered 40 years ago as a Cd-binding protein in horse kidney, remains a singular metalloprotein structure that appears to have multiple functions in protecting cells from toxic insults including heavy metals and oxidants [40,43,44]. Although its concentration is up-regulated by these agents, metallothionein, including apoMT, also exists at basal levels in cells in the absence of exposure to such deleterious substances [45–47]. This and other findings have encouraged the hypothesis that metallothionein plays a role in zinc metabolism. Nevertheless, the initial reports that MT gene knock-out mice apparently suffer no problems during fetal and neonatal development and into adulthood has brought into question the essentiality of MT for the normal metabolism of Zn [48].
The lack of participation of MT in normal Zn metabolism is not clear-cut. A recent study by Andrews et al. [49] demonstrated that mild zinc deficiency during the pregnancy of MT null mice led to almost a three fold increase in birth defects in comparison with controls fed the same diet. They also showed that fetal development in MT transgenic mice resulted in lower than control rates of defects under Zn-deficient conditions. These results strongly support the function of MT in the acquisition of Zn by the fetus under mild conditions of Zn insufficiency. Furthermore, neonatal MT-null mice suckling on Zn-deficient dams rapidly developed kidney damage [50]. Thus, MT seems to play a key role in cellular Zn acquisition in rapidly developing organisms subjected to environmentally relevant conditions of nutrient Zn supply.
Considering its chemical properties, Zn7-MT is unusually reactive in ligand substitution and in metal exchange reactions in comparison with typical Zn-metalloenzymes [23]. Based on this information and a few in vitro and cellular studies, it has been proposed that MT may serve as an intracellular Zn-transfer protein that participates in the organized movement of Zn2+ from its point of entry in cells to final binding sites in Zn-proteins, either by donating or accepting Zn2+ as in reaction (7) [51–56],
| (7) |
The recognition that apoMT must form initially during protein synthesis and that steady-state concentrations of apoMT are found in a variety of cells suggests that the reverse of reaction (7) needs to be considered and that MT induction might be able to perturb intracellular Zn2+ distribution [46,47]. The attractiveness of this idea increased with the discovery of Zn-finger transcription factors. At least some of these appear to bind Zn2+ relatively weakly at kinetically reactive sites. For example, the stability constants for Zn2+-binding to TFIIIA fingers are in the range of 107 [35]. Zn-TFIIIA also readily reacts with EDTA, establishing the reactivity of its metal-binding sites [57].
Several years ago, Kägi and colleagues [25,26] demonstrated that incubation of apoMT with either Zn-TFIIIA or Zn-Sp1 prevented these transcription factors from associating with their specific DNA-binding sites. Then, Wilcox and coworkers [27] showed directly that a single Zn-finger from Sp1 reacted with the α-domain of metallothionein through a ligand-substitution process. Similarly, Roesijadi and coworkers [28] recently observed that apoMT reacted with another C2H2 Zn-finger peptide, Tramtrack, resulting in the inactivation of its capacity to bind to DNA. In that study, the authors favored the following mechanism in which there is no direct interaction between apoMT and Zn2-Tramtrack:
| (8) |
| (9) |
In the present work, the reactions of Zn-TFIIIA with apoMT or EDTA were characterized in the absence or presence of the native DNA-binding site for Zn-TFIIIA. From such experiments, one can assess the plausibility that apoMT might react with Zn-finger proteins at their native-binding sites in cells, where the DNA partner might provide a major steric or electrostatic barrier to reaction. The reactions between near stoichiometric amounts of Zn- or Cd-substituted TFIIIA and apoMT demonstrated that direct metal exchanges do occur relatively rapidly. The reaction between apoMT and Znn-TFIIIA (n = 4 and 9) is second order (Fig. 2) and proceeds with a rate constant of 30±10 M−1 s−1, independent of the Zn2+ to TFIIIA stoichiometry. Using this result, at a cellular concentration of 10 μM apoMT, a pseudo-first-order rate constant of 3×10−4 s−1 and a half-time of 40 min are calculated for this reaction. Thus, with this plausible cellular concentration of apoMT, the reaction appears to be physiologically accessible. The reaction between Cd3-TFIIIA and apoMT occurred with similar kinetics (Fig. 5).
The observation of simple kinetics indicates that all of the Zn-associated fingers in each Znn-TFIIIA (n = 4–9) sample react similarly with apoMT and that in the process apoMT reacts in a concerted manner to form holo-Zn- or Cd-thiolate clusters. The former is consistent with Znn-TFIIIA structures in which Zn2+ is bound either to specific fingers or is distributed among all of the fingers for all values of n and all of the fingers are comparably reactive with apo-MT. The concerted formation of the holo-clusters and domains is both thermodynamically and kinetically favored [58,59].
The results of the present study suggest that direct bimolecular reactions do occur between apoMT and the Zn-finger sites and that such reactions are not rate limited by first-order dissociation of Zn2+ from the fingers. In addition, it appears that all of the zinc or cadmium finger sites are independently and similarly reactive with apoMT. The apparent lack of heterogeneity of rate constants characterizing these ligand-substitution reactions differs from a previous report as well as our observations on the reaction of several chelating agents with Zn-TFIIIA, in which two classes of reacting Zn-finger sites were observed [35,60].
Induction of MT synthesis by Cd2+ leads to the formation of Cdm,Znn-MT. It is known that the final mixed metal species results from interprotein metal ion exchange [61]:
| (10) |
This remarkable reaction signals the possibility that Zn7-MT, itself, might be able to exchange metals (M) with other sites as well:
| (11) |
Supporting this contention, metal ion exchange was observed in the reaction of Zn7-MT with Cd2-Tramtrack [28]. Although some reaction was also detected between Zn7-MT and Cd3-TFIIIA, it was very slow and probably does not have physiological significance. In each case, it appears that interprotein metal ion exchange can occur between Zn-MT and other metalloprotein structures.
A comparison of the reactions of Zn- or Cd-TFIIIA with apoMT or EDTA shows that the second-order rate constant for the apoMT reaction was only a factor of 4 slower than that for EDTA (Fig. 6). A faster rate of reaction for EDTA in comparison with apoMT might be anticipated for two reasons. First, it is much smaller and, therefore, less sterically hindered in its encounters with Zn-TFIIIA. Second, since Zn-TFIIIA is a positively charged molecule, the negative charge on EDTA may facilitate their reaction, whereas, the positive charge on apoMT might electrostatically retard its interaction with the Zn-finger sites. Considering these factors, the difference in rate constant for the two reactions was surprisingly small. In another study it was shown that apoMT was actually a much more effective competing ligand than EDTA for Cd2+ bound to carbonic anhydrase [31]. The two results underscore that apoMT is a kinetically competent metal-binding ligand even when Zn2+ or Cd2+ are bound to other structures that present large steric obstructions.
This conclusion is bolstered by comparable experiments with Zn-TFIIIA prebound to its DNA-binding site. DNA protected the Zn-fingers in the Zn-TFIIIA-ICR complex from the reaction with EDTA (Fig. 7), but did not markedly hinder the reaction of the Zn-fingers with apoMT (Fig. 3). The difference in reactivity of these two agents supports the view that the rate-limiting step in the reaction with apoMT involved direct attack of apoMT on the DNA-bound Zn-fingers, not the dissociation of TFIIIA or Zn2+ from the Zn-TFIIIA-ICR adduct. Otherwise, EDTA should have been equally reactive. In addition, a significant part of the reaction took place well before 30 min of incubation time, which is the half-life for the dissociation of TFIIIA from ICR. This implied that dissociation of the adduct/complex is not rate-limiting in these reactions.
Examination of the local molecular environment of the Zn2+-binding sites in the three-dimensional structures of parts of Zn-TFIIIA bound to cognate portions of the ICR shows that substantial steric hindrance to access to bound Zn2+ is imposed by DNA [9,12]. Electrostatic repulsion between negatively charged EDTA and the protein–DNA complex which also bears an overall negative charge might contribute to the lack of reactivity of EDTA. In contrast, apoMT, which has the opposite charge from Zn-TFIIIA-ICR, remained effective as a competing ligand, despite the enhanced steric barrier to reaction of apoMT in comparison with EDTA.
It has been hypothesized that the C-terminal end of the apoMT structure is the reactive part of the molecule [32]. Between residues 50 and 61, there are four cysteinyl residues that comprise the only four consecutive ones which coordinate to a single Cd2+ or Zn2+ in either metal-thiolate cluster of the folded MT structure [62]. We have speculated that binding of metal ions to these particular sulfhydryl groups may serve as a nucleation site in the assembly of intact clusters [32]. That is, once the Cd1-MT species has formed, Cd2+ would be expected to redistribute among thiolate ligands with non-rate-limiting kinetics, resulting in the construction of intact Cd-thiolate clusters. The location of the key thiolates at the C-terminus of the MT peptide would minimize the steric barrier of reaction between two macro-molecules, apo-MT and another metal-containing protein, and, thereby, facilitate the bimolecular interaction between them. In support of this proposal, a peptide with the amino acid sequence of 49–61 is much more reactive with Cd-carbonic anhydrase than is apoMT [31].
The present work shows that apoMT can react with free or DNA-bound Zn-TFIIIA to extract Zn2+ from the transcription factor under physiologically reasonable conditions and at plausible rates. Coupled with the observation that apoMT exists under some conditions in cells, these and previous findings raise the question of the role of apoMT in the speciation or intracellular distribution of Zn2+ bound to proteins such as Zn-fingers [46,47]. In particular, the relationship between the active form of the Zn-MTF-1 transcription factor and the product of its transcriptional stimulus, apoMT, provides a tangible example of the complexities of cellular Zn speciation.
Finally, the present findings expand the understanding of the chemistry that may underlie the protective function of MT against Cd2+. In cells exposed to Cd2+, significant induction of MT synthesis requires hours so that Cd2+ distributes itself among ill-defined cellular ligands (L) prior to its binding to metallothionein [30]. In this circumstance, the efficient formation of Cd-MT requires not only that MT have a large binding constant for Cd2+, but that it be able to react relatively rapidly with these other Cd-ligand complexes. Little is known about the rates of reaction of Cd-protein complexes with apoMT. The present results as well as those showing that Cd2-Tramtrack and Cd-carbonic anhydrase are reactive with apoMT provide a chemical underpinning for the hypothesis that MT is protective against Cd2+ [28,31].
Acknowledgments
The authors gratefully acknowledge the gift of clones from Dr. David Setzer and Dr. Paul Romaniuk and their helpful discussions. We thank Dr. Michael Reddy for discussions and Barbra Wimpee for technical support. This research was supported by NIH Grants ES-04026 and ES-04184.
5. Abbreviations
- TFIIIA
transcription factor IIIA
- MT
metallothionein
- AAS
atomic absorption spectroscopy
- Abs
absorbance
- BSA
bovine serum albumin
- C2H2
two cysteine and 2 histidine ligands
- DTT
dithiothreitol
- DTNB
5,5′-dithiobis-2-nitrobenzoic acid
- EDTA
ethylenedinitrilo-tetraacetic acid
- EMSA
electrophoretic mobility shift assay
- ICR
internal control region
- Hepes
4-(2-hydroxylethyl) piperazine-1-sulfonic acid
- PAR
4-(2-pyridylazo) resorcinol
References
- 1.Miller J, McLachlan AD, Klug A. EMBO J. 1985;4:1609–1614. doi: 10.1002/j.1460-2075.1985.tb03825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green LM, Berg JM. Proc Natl Acad Sci USA. 1989;86:4047–4051. doi: 10.1073/pnas.86.11.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parraga G, Horvath S, Hood L, Young ET, Klevit RE. Science. 1988;241:1489–1492. doi: 10.1126/science.3047872. [DOI] [PubMed] [Google Scholar]
- 4.Lee MS, Gippert GP, Soman KV, Case DA, Wright PE. Science. 1989;245:635–637. doi: 10.1126/science.2503871. [DOI] [PubMed] [Google Scholar]
- 5.Klevit RE, Harriott JR, Horvath SJ. Proteins. 1990;7:215–226. doi: 10.1002/prot.340070303. [DOI] [PubMed] [Google Scholar]
- 6.Omichinski JG, Clore GM, Appella E, Sakaguchi K, Gronenborn AM. Biochemistry. 1990;29:9324–9434. doi: 10.1021/bi00492a004. [DOI] [PubMed] [Google Scholar]
- 7.Pavletich NP, Pabo CO. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 8.Fairall L, Schwabe JWR, Chapman L, Finch JT, Rhodes D. Nature. 1993;366:483–487. doi: 10.1038/366483a0. [DOI] [PubMed] [Google Scholar]
- 9.Wuttke DS, Foster MP, Case DA, Gottesfeld JM, Wright PE. J Mol Biol. 1997;273:183–206. doi: 10.1006/jmbi.1997.1291. [DOI] [PubMed] [Google Scholar]
- 10.Narayan VA, Kriwacki RW, Caradonna JP. J Biol Chem. 1997;272:7801–7809. doi: 10.1074/jbc.272.12.7801. [DOI] [PubMed] [Google Scholar]
- 11.Elrod-Erickson M, Rould MA, Neklukova L, Pabo CO. Structure. 1996;4:1171–1180. doi: 10.1016/s0969-2126(96)00125-6. [DOI] [PubMed] [Google Scholar]
- 12.Nolte RT, Conlin RM, Harrison SC, Brown RS. Proc Natl Acad Sci USA. 1998;95:2938–2943. doi: 10.1073/pnas.95.6.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanas JS, Hazuda DJ, Bogenhagen DF, Wu FYH, Wu CW. J Biol Chem. 1983;258:14120–14125. [PubMed] [Google Scholar]
- 14.Hanas JS, Bogenhagen DF, Wu CW. Proc Natl Acad Sci USA. 1983;80:2142–2145. doi: 10.1073/pnas.80.8.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith DR, Jackson IJ, Brown DD. Cell. 1984;37:645–652. doi: 10.1016/0092-8674(84)90396-9. [DOI] [PubMed] [Google Scholar]
- 16.Frankel AD, Berg JM, Pabo CO. Proc Natl Acad Sci USA. 1987;84:4841–4845. doi: 10.1073/pnas.84.14.4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chesters JK. Nutr Rev. 1992;50:217–223. doi: 10.1111/j.1753-4887.1992.tb01331.x. [DOI] [PubMed] [Google Scholar]
- 18.Berg JM, Shi Y. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 19.Makowski GS, Sunderman FW., Jr J Inorg Biochem. 1992;48:107–119. doi: 10.1016/0162-0134(92)80020-v. [DOI] [PubMed] [Google Scholar]
- 20.Westin G, Schaffner W. EMBO J. 1988;7:3763–3770. doi: 10.1002/j.1460-2075.1988.tb03260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radtke F, Heuchel R, Georgiev M, Hergersberg M, Gariglio O, Dembic Z, Schaffner W. EMBO J. 1993;12:1355–1361. doi: 10.1002/j.1460-2075.1993.tb05780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bittel D, Dalton T, Samson SL-A, Gedamu L, Andrews GK. J Biol Chem. 1998;273:7127–7133. doi: 10.1074/jbc.273.12.7127. [DOI] [PubMed] [Google Scholar]
- 23.Otvos JD, Petering DH, Shaw CF., III Comm Inorg Chem. 1989;1:1–35. [Google Scholar]
- 24.Ejnik J, Zhu J, Shaw CF, III, Petering DH. J Inorg Biochem. 2002;15:144–152. doi: 10.1016/s0162-0134(01)00393-2. [DOI] [PubMed] [Google Scholar]
- 25.Zeng J, Vallee BL, Kägi JHR. Proc Natl Acad Sci USA. 1991;88:9984–9988. doi: 10.1073/pnas.88.22.9984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeng J, Heuchel R, Schaffner W, Kägi JHR. FEBS Lett. 1991;297:310–312. doi: 10.1016/0014-5793(91)80175-3. [DOI] [PubMed] [Google Scholar]
- 27.Posewitz MC, Wilcox DE. Chem Res Toxicol. 1995;8:1020–1028. doi: 10.1021/tx00050a005. [DOI] [PubMed] [Google Scholar]
- 28.Roesijadi G, Bogumil R, Vašák M, Kägi JHR. J Biol Chem. 1998;273:17425–17432. doi: 10.1074/jbc.273.28.17425. [DOI] [PubMed] [Google Scholar]
- 29.Klaassen CD, Liu J, Choudhuri S. Ann Rev Pharmacol Toxicol. 1999;39:267–294. doi: 10.1146/annurev.pharmtox.39.1.267. [DOI] [PubMed] [Google Scholar]
- 30.Winge DR, Premakumar R, Rajagopalan KV. Arch Biochem Biophys. 1978;188:466–475. doi: 10.1016/s0003-9861(78)80031-9. [DOI] [PubMed] [Google Scholar]
- 31.Ejnik J, Muñoz A, Gan T, Shaw CF, III, Petering DH. J Biol Inorg Chem. 1999;4:784–790. doi: 10.1007/s007750050351. [DOI] [PubMed] [Google Scholar]
- 32.Muñoz A, Laib F, Petering DH, Shaw CF., III J Biol Inorg Chem. 1999;4:495–507. doi: 10.1007/s007750050335. [DOI] [PubMed] [Google Scholar]
- 33.Hanas JS, Gunn CG. Nucleic Acids Res. 1996;24:924–930. doi: 10.1093/nar/24.5.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller J, McLachlan AD, Klug A. EMBO J. 1985;4:1609–1614. doi: 10.1002/j.1460-2075.1985.tb03825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang M, Krepkiy D, Hu W, Petering DH. J Inorg Biochem. 2004;98 doi: 10.1016/j.jinorgbio.2004.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minkel DT, Poulsen K, Wielgus S, Shaw CF, III, Petering DH. Biochem J. 1980;191:475–485. doi: 10.1042/bj1910475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kägi JHR, Kojima Y. Metallothionein II, Experientia Supplementum. Vol. 52. Birkhäuser Verlag; Basel: 1987. pp. 25–61. [DOI] [PubMed] [Google Scholar]
- 38.Ellman GL. Arch Biochem Biophys. 1958;74:443–450. doi: 10.1016/0003-9861(58)90014-6. [DOI] [PubMed] [Google Scholar]
- 39.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 40.Kägi JHR, Vallee BL. J Biol Chem. 1961;236:2435–2442. [PubMed] [Google Scholar]
- 41.Sillen LG, Martell AE. Stability Constants of Metal–Ion Complexes. The Chemical Society, Burlington House; London: 1964. [Google Scholar]
- 42.Liao X, Clemens KR, Tennant L, Wright PE, Gottesfeld JM. JMB. 1992;223:857–871. doi: 10.1016/0022-2836(92)90248-i. [DOI] [PubMed] [Google Scholar]
- 43.Rossman TG, Goncharova EI. Mutat Res. 1998;402:103–110. doi: 10.1016/s0027-5107(97)00287-x. [DOI] [PubMed] [Google Scholar]
- 44.Coyle P, Philcox JC, Carey LC, Rofe AM. Cell Mol Life Sci. 2002;59:627–647. doi: 10.1007/s00018-002-8454-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Onosaka S, Cherian MG. Toxicology. 1982;23:11–20. doi: 10.1016/0300-483x(82)90037-3. [DOI] [PubMed] [Google Scholar]
- 46.Pattanaik A, Shaw CF, III, Petering DH, Garvey J, Kraker AJ. J Inorg Biochem. 1994;54:91–105. doi: 10.1016/0162-0134(94)80023-5. [DOI] [PubMed] [Google Scholar]
- 47.Yang Y, Maret W, Vallee BL. Proc Natl Acad Sci USA. 2001;98:5556–5559. doi: 10.1073/pnas.101123298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masters BA, Kelly EJ, Quaife CJ, Brinster RL, Palmiter RD. Proc Natl Acad Sci USA. 1994;91:584–588. doi: 10.1073/pnas.91.2.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andrews GK, Geiser J. J Nutr. 1999;129:1643–1648. doi: 10.1093/jn/129.9.1643. [DOI] [PubMed] [Google Scholar]
- 50.Kelly EJ, Quaife CJ, Froelick GJ, Palmiter RD. J Nutr. 1996;126:1782–1790. doi: 10.1093/jn/126.7.1782. [DOI] [PubMed] [Google Scholar]
- 51.Li TY, Kraker AJ, Shaw CF, III, Petering DH. Proc Natl Acad Sci USA. 1980;77:6334–6338. doi: 10.1073/pnas.77.11.6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Udom AO, Brady FO. Biochem J. 1980;187:329–335. doi: 10.1042/bj1870329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krezoski SK, Villalobos J, Shaw CF, III, Petering DH. Biochem J. 1988;255:483–491. [PMC free article] [PubMed] [Google Scholar]
- 54.Jacob C, Maret W, Vallee BL. Proc Natl Acad Sci USA. 1998;95:3489–3494. doi: 10.1073/pnas.95.7.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zaia J, Fabris D, Wei D, Karpel RL, Fenselau C. Protein Sci. 1998;7:2398–2404. doi: 10.1002/pro.5560071117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cano-Gauci DF, Sarkar B. FEBS Lett. 1996;386:1–4. doi: 10.1016/0014-5793(96)00356-0. [DOI] [PubMed] [Google Scholar]
- 57.Shang Z, Liao YD, Wu FY, Wu CW. Biochemistry. 1989;28:9790–9795. doi: 10.1021/bi00451a037. [DOI] [PubMed] [Google Scholar]
- 58.Nielson KB, Winge DR. J Biol Chem. 1983;258:13063–13069. [PubMed] [Google Scholar]
- 59.Ejnik J, Robinson J, Zhu J, Försterling H, Shaw CF, Petering DH. J Inorg Biochem. 2002;88:144–152. doi: 10.1016/s0162-0134(01)00393-2. [DOI] [PubMed] [Google Scholar]
- 60.Han MK, Cyran FP, Fisher MT, Kim SH, Ginsburg A. J Biol Chem. 1990;265:13792–13799. [PubMed] [Google Scholar]
- 61.Nettesheim DG, Engeseth HR, Otvos JD. Biochemistry. 1985;24:6744–6751. doi: 10.1021/bi00345a003. [DOI] [PubMed] [Google Scholar]
- 62.Braun W, Vašák M, Robbins AH, Stout CD, Wagner G, Kägi JH, Wüthrich K. Proc Natl Acad Sci USA. 1992;89:10124–10128. doi: 10.1073/pnas.89.21.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]