

Abstract
We introduce a computational pipeline and suite of software tools for the approximation of diffusion-limited binding based on a recently developed theoretical framework. Our approach handles molecular geometries generated from high-resolution structural data and can account for active sites buried within the protein or behind gating mechanisms. Using tools from the FEniCS library and the APBS solver, we implement a numerical code for our method and study two Ca2+-binding proteins: Troponin C and the Sarcoplasmic Reticulum Ca2+ ATPase (SERCA). We find that a combination of diffusional encounter and internal ‘buried channel’ descriptions provide superior descriptions of association rates, improving estimates by orders of magnitude.
1. Introduction and Motivation
Examples abound of proteins that have evolved to maximize substrate association rates. A common factor in fast association rates is the enhanced diffusion of ligands adjacent to a protein’s active site, through favorable electrostatic interactions [46] that counterbalance diffusional barriers imposed by geometry. Dating back to Smoluchowki [37], descriptions for the diffusional encounter rate, kD, between enzyme and substrate have been proposed; Bashford provides an excellent review of these in [2]. In essence, the formulations describe the interplay of the diffusion constant of the substrate with the geometry of the reacting species.
A common approach for including the contribution of electrostatics proceeds through the Smoluchowski equation given in equation (1), which relates the time-dependent change in substrate concentration due to fluxes arising from the concentration gradient and an electrostatic potential. Recently, a numerical solution to this equation was implemented, which provides an estimate for kD, given the solvent-accessible surface of a proteins and the electrostatic potential [11]. While this software represents exciting progress toward modeling substrate association at the molecular level, its focus on globular proteins limits its application to non-trivial geometries such as trans-membrane proteins and macro-structures such as DNA and microtubules.
Further complicating this picture are the roles of conformational changes [51] and binding pathways, such as channels, that present diffusional barriers to the active site [4]. The factors can depress observed association rates relative to the diffusional encounter rate between enzyme and substrate. Recent works [4, 3] proposed analytical solutions to the observed association rate, kon, which treat the association as an external, diffusion-encounter problem followed by an internal, post-encounter problem. The later stage reflects the effects of stochastic gating and potentials of mean force along a pathway bridging the encounter-surface and the active site; both of which can be estimated from molecular dynamics and Brownian dynamics.
Our computational pipeline is based on this mathematical framework developed for binding in gated channels, i.e. narrow regions of the geometry near the active site [3, 4]. We will treat the notion of ‘channel’ more generically as any binding pathway bridging the outer molecular surface and the active site. The results we present here emphasize the importance of these theoretical developments as well as demonstrate the feasibility of integrating the effects of diffusional and post-encounter effects into a suite of algorithms. We use a finite element method to compute the steady state of ionic diffusion in a spatially-realistic three dimensional region exterior the molecule with an absorbing boundary condition taken at the entrance to the channel. Inside the channel, we use a molecular dynamics simulation to approximate the local potential of mean force V (x) which can then be used to estimate the rate of transport of ions from the channel entrance to the active site. If the access to the channel is blocked stochastically by a gating mechanism, we make use of formulas describing how the approximation of the association rate should be modified according to the comparison of gating speed to the characteristic diffusion time.
We apply this numerical protocol to two proteins, Troponin C (TnC) and the Sarcoplasmic Reticulum Ca2+ ATPase (SERCA), which exemplify the concepts of the electrostatically-driven diffusional encounter and post-encounter effects in binding of Ca2+. Troponin C and SERCA serve vital roles in muscle cells by controlling the cell’s contractile function and reabsorbing cytosolic calcium, respectively [7]; molecular-level details of these proteins and Ca2+ binding mechanism could improve our understanding of calcium dynamics in contractile cells. We consider TnC both in isolation and bound to a cryo-EM reconstruction of the thin filament (Fig. 1a), as well as SERCA in its native lipid membrane environment (Fig. 1b). To the best of our knowledge, this is the first time substrate association has been modeled using proteins situated in their native environment. In both cases, we consider the contribution of molecular interactions with the proteins, which are captured in a potential of mean force calculations (Fig. 2). For the case of SERCA, we also consider the influence of an apparent gated binding event exemplified in Fig. 1b. We show that accounting for conformational gating and buried binding channels profoundly impacts the association rates.
Figure 1. Troponin C and SERCA molecular structures.

(a) Isolated troponin C molecule structure with Ca2+ binding site II (blue).(left) TnC (red, blue) on thin filament (gray) (right) (b) SERCA (gray cartoon) embedded in lipid membrane. Red to blue shading indicates relative height of membrane. Inset magnifies putative L3 pathway leading to Ca2+ binding site II; lipid coloring is rescaled to accentuate divot near L3 pathway (left) Putative gating event involving E309 at site II. (right)
Figure 2.
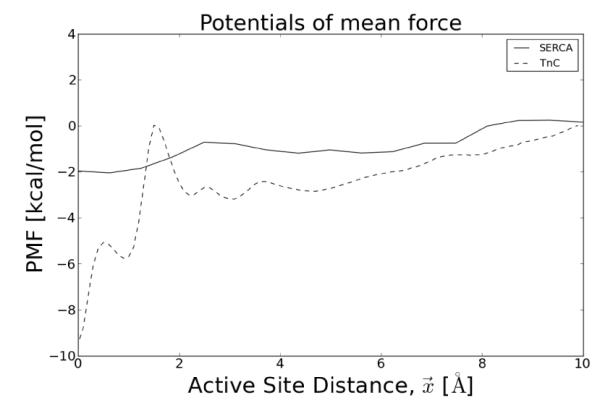
Potentials of mean force. Computed for Ca2+ binding to Asp67 in the Troponin C active site (site II) (in preparation) and Asp800 of SERCA site II [25]
The paper is outlined as follows. In Section 2, we explain our algorithms and computational tools. In Section 3, we state and discuss our results. We give concluding remarks and suggest some future research directions in Section 4.
2. Methods and Algorithms
We present a hierarchy of methods for approximating a generic substrate binding rate kon. The approximation kD ignores the presence of a buried channel and any effects of a local potential of mean force (PMF). The approximation kss accounts for a local PMF in the channel but ignores any gating effects. The approximation kg includes a local PMF and gating effects. We will describe each algorithm and the supporting mathematical theory at a high level in turn in Sections 2.1-2.3, followed by a more in depth description of the computational tools used to implement each piece. Notation for domains with or without a channel are shown in Fig. 3 and described in the caption. The mathematical framework was introduced by Zhou in [50] and expanded upon recently in [3, 4].
Figure 3.

Notation for our computational domains. (a) First, the domain is treated without a buried channel. The molecular interior Ωi has an active site Γa and a non-reactive boundary Γs. (b) When the domain is treated as a buried channel, the molecular interior Ωi is bounded by Γs ⋃ Γc ⋃ Γa. The active site Γa can be accessed from the exterior region Ωe by passing through the buried channel region Ωc. Access to Ωc is through the mouth of the channel Γm which may represent a gating mechanism. Both domain types sit inside a large sphere with boundary Γe. (c) Inside the channel, the reaction coordinate axis (shown here as a dashed line) is described by the coordinate x ranging from x = 0 (at the active site Γa) to the x = L (at the channel entrance Γm). The cross-sectional region at a particular x is called σ(x) with area denoted by ∣σ(x)∣.
2.1. Steady state rate constant computation without channels
In the case where no channel is assumed, the time-dependent Smoluchowski problem is formulated as follows: Find an ionic concentration function such that
| (1) |
in Ωe, where D is the diffusion constant, β := 1/kBT is the inverse Boltzmann energy with kB the Boltzmann constant and T the temperature, and ϕ the electrostatic potential, which is related to the electric field, , via the standard equation . The boundary conditions are taken to be
| (2) |
where is the flux of ρ, α is the relative absorption of substrate at the active site, and ρb is the bulk ionic concentration, assumed to be constant outside of Ωe, and is the surface normal along Γ. To compute the reaction rate in this context, one solves the steady state problem
| (3) |
and computes the accompanying steady state flux . The reaction rate for the steady state reaction rate with no channel (e.g. the diffusional encounter), kD is then determined by an area integral of the incident flux [38]
| (4) |
We approximate this by the following algorithm.
Algorithm 1 (Binding site not in a channel)
Input: Hard sphere model of a molecule, with marked boundaries Γs and Γa
Compute the electrostatic potential ϕ on the volumetric mesh using APBS.
Compute the steady-state solution ρ from (3) using SMOL.PY.
Compute kD from (4) using SMOL.PY.
Output: Association rate kD
2.2. Steady state rate constant computation for ungated buried channels
Now we consider the case where the active site lies at the bottom of a buried channel as shown in Fig. 3b, but no gating mechanism is assumed. Suppose that the channel is symmetrical about the x-axis such that the binding site of the molecule is located at the origin and the mouth of the channel, Γa, is contained in the plane x = L. Let denote the cross-sectional region of the channel for x ∈ [0, L] and ∣σ(x)∣ the corresponding area. Assume that the ligand dynamics are sufficiently fast to ensure local steady state on each cross section σ(x). Let V (x) denote the one-dimensional potential of mean force inside the channel; this can be computed analytically for simple channel geometry (e.g. cylindrical) or approximated using molecular dynamics simulations.
Berezhkovskii et al suggest in [4] that the steady state rate constant for the buried channel domain, kss, can be estimating using the formula
| (5) |
Here, kext denotes the steady state rate constant for ligands outside the channel to be absorbed at the channel entrance Γm. This rate can be determined by assigning a fully absorbing boundary condition ρ = 0 on Γm and using formula (4) on the modified exterior geometry Ωe shown in Fig. 3b. The term kint is the association rate to the active site inside the channel and is estimated by
| (6) |
where integration of the potential of mean force, V (x), is carried out over the length of the channel (from x = 0 to x = L), D is the bulk diffusion constant, and ∣σ(x)∣ signifies the area of the cross-sectional region at x (see Fig. 3c).
The term kbind is the chemical reaction rate and is given by the formula κ0σ(0)e–βV(0) where κ0 indicates the intrinsic binding rate to the surface of the binding site. We will assume, as is typical in this context [36], that κ0 is sufficiently large to make the last summand negligible. We thus have the following algorithm.
Algorithm 2 (Binding site in a channel)
Input: Hard sphere model of a molecule, with marked boundaries Γs, Γa, Γc, Γm, and value of diffusion constant inside channel Ωc
Compute kext using Algorithm 1 with active site replaced by channel entrance.
Compute V (x) in Ωc using an appropriate molecular dynamics simulation.
Compute σ(x), i.e. the cross-sectional area as a function of the reaction coordinate.
Compute kint from (6), then kss from (5) using CHANNEL SMOL.PY.
Output: Association rate kss
2.3. Steady state rate constant computation for gated buried channels
In the event that access to the binding site is intermittently open, we summarize the Smoluchowski equation under gating conditions. To this end we consider open (absorbing) and closed (reflective) states of the active site with a ligand-independent forward rate of ωa and backward rate of ωr, as indicated by
| (7) |
The presence of a ligand near a gate can affect the rate at which it opens and closes, hence ωa and ωr have a dependence on the spatial variable x. These rates satisfy the detailed balance condition [50, 3, 9]:
| (8) |
where Ua(x) and Ur(x) are the interaction potentials associated with the open (absorbing boundary) and closed (reflective boundary) conformational states, respectively, and are computed using
| (9) |
where ω(x) = ωa(x) + ωr(x).
Under this scenario, Barreda et al [3] suggest that the rate constant subject to gating at the active site may be estimated from the coupled PDE system [3]:
| (10) |
| (11) |
where ρa and ρr are the pair-wise distributions associated with the absorbing and reflecting boundary conditions at Γa, respectively. Note that both ρa and ρr are distributions for ρ but refer to different boundary conditions on the active site.
The association rate kg, resulting from the governing equations (10)-(11) can be expressed in a simplified form under two gating limits, which we elaborate upon in Section 6.2. These limits refer to the speed of gating relative to the characteristic diffusion time, τd, e.g. the time required for the substrate to diffuse sufficiently far away from the protein. Under a relatively slow gating speed, i.e. when , we generalize the result of Barreda et al. [3] equation (3.24a) to yield
| (12) |
where kext is as in (5) and Va(x) represents a 1D potential of mean force that quantifies the attraction of the ligand for the open state of the binding gate. Intuitively, this limit suggests that the probability of reaction is dependent on the whether the slowly gated active site is open for each rapid visit of the substrate to the vicinity of the gate. When , the gating speed is fast relative to τd, yielding
| (13) |
where Veff is determined from
| (14) |
In this gating extreme, the gate is expected to present an open state before the substrate diffuses away from the reaction vicinity. Thus, from the perspective of the substrate, the gate is always open. While we have considered here only two reacting states, the approach may be extended to an arbitrary number of states as follows from [43, Appendix A].
We summarize this in the following algorithm.
Algorithm 3 (Binding site in a gated channel)
Input: hard sphere model of a molecule, with marked boundaries Γs, Γa, Γc, Γm, and gating rates
Compute transition rate constants ωa and ωr from (7) using molecular dynamics simulations.
Compute potentials of mean force Va and Vr for open and closed states.
Compute kext using Algorithm 1.
Compute Veff using (14).
Compute kPMF using Algorithm 2, with Veff in place of V (x).
Compute from (12) if ωa + ωr >> τd or from (13) if ωa + ωr << τd via GATED.PY.
Output: Association rate kg
We now explain the details of the methods used in our implementation of Algorithms 1, 2, and 3.
2.4. Molecular mesh
The molecular surface is represented as a mesh approximating the Connolly surface and may be generated by applying the EDTSurf [48] package to Protein Data Bank [5] data of the atomic substructure of the molecule. For this manuscript, we consider three geometry types: globular, linear and planar, which correspond to our isolated TnC (Fig. 5a,b), thin filament (Fig. 5c) and SERCA cases (Fig. 6a), respectively. Using the resulting molecular mesh, we use the BLAMER suite to define an encapsulating region (sphere for a globular protein, cylinder for linear, and rectangular for planar systems). BLAMER is a version of Blender (www.blender.org) which has been bootstrapped with the python implementation of Gamer [49]. While a standalone manuscript is not available, the package has been used in [18]. For globular proteins, the radius of the outer sphere was 10 times the radius of the molecule. For linear proteins, we used a cylinder whose height was equal to the molecule length along the dominant axis, and whose radius was twice the width along the minor axis. For planar geometries, we chose a rectangular box whose x and y dimensions were equal to the dimensions of the modeled lipid bilayer, and whose height was three times the height of the molecule.
Figure 5.

Steady-state Ca2+ distributions about Troponin C. The TnC N-terminal domain in (a) absence and (b) presence of electrostatic potential, as well as (c) TnC on the actin myofilament (left) and with red contours corresponding to 40 M [Ca2+] (right)
Figure 6.

Steady state Ca2+ distributions about SERCA. (a) Ca2+ steady-state distribution perpendicular to the SERCA active site (gray). Contours of elevated Ca2+ density for the (b) charged and (c) uncharged lipids.
MolecularMesh mesh in the GAMer package [49] (www.fetk.org) can be used to automatically generate finite element-ready meshes for globular proteins. For linear and planar cases, we use the Boolean difference modifier in Blender to create a new mesh based on the encapsulating region and the molecular mesh. The active sites were chosen to coincide with the binding sites of Ca2+. For convenience, we labeled the molecular surface comprising the active site with the absorbing condition. Since we are partitioning the diffusion domain into a diffusional encounter region and a post-encounter region, it may be more suitable to position the absorbing boundary for the diffusional encounter at the outer limits of the potential of mean force domain.
Since the diffusional encounter rate scales with the radius of the absorbing center, our approximation likely underestimates the diffusional encounter rate by a factor of (rm/rpmf, where rM and rpmf reflect the location of the molecular surface and exterior problem interface. All other boundaries, including the molecular surface were assigned reflective boundary conditions. The GAMer package was used within Blender (www.blender.org) to repair and refine the mesh. The refined mesh was tetrahedralized via TETGEN (tetgen.berlios.de).
2.5. Electrostatic potential
APBS provides a standard protocol for computing the electrostatic potential due to a solvated protein via numerical evaluation of the Poisson-Boltzmann equation [1],
| (15) |
where ϕ = eΦ/kBT is the reduced form of the electrostatic potential Φ, ρ is the fixed distribution of charges corresponding to the protein, κ2 is the Debye screening parameter due to a monovalent electrolyte solution, and ∊ is the dielectric constant defined over the protein and the solvent domains. For small ψ, the sinh(ψ) contribution from the electrolyte (15) may be linearized to yield the linearized Poisson-Boltzmann equation, which we used for estimating the electrostatic potential for dilute (I = 0 mM) or near physiological (I = 200 mM) ionic strength assuming 2 Å radius monovalent ions.
We computed the electrostatic potential at several grid resolutions ranging from 0.3 Å to 25 Å resolution around the active site, in order to balance higher accuracy required around the active site with a potential defined over the entire simulation domain. This was done by using the MG-AUTO function to first compute the potential on a 1650×1650×1650 Å grid using 65 grid points along each axis, adaptively focus the solution to increasing resolutions until the desired resolution is obtained. The resulting finite-difference APBS grids were interpolated onto the finite element molecule meshes using a tri-linear interpolation algorithm distributed with SMOL [11].
The local concentration of ions, ρ(r), due to a fixed electric potential, ϕ(r), relative to the bulk concentration, ρbulk is given by the Boltzmann equation ρ(r) = ρbulke–βqϕ. With this description, the electrostatic potential from APBS predicts unphysically high concentrations of Ca2+. In reality, the counter-ion density saturates directly adjacent to the charged molecular surface, giving rise to the Stern layer [24]. Therefore, we assume that the maximum concentration of Ca2+ along the molecular surface is 55 M, and truncate the electrostatic potential to limit local [Ca2+] to the maximum concentration.
2.6. Potential of mean force
The potential of mean force, V (x), is computed via molecular dynamics simulations along a suitable reaction coordinate, x. Since the purpose of this study is to illustrate the incorporation of estimated PMF data into a numerical model, we are utilizing data from molecular dynamics studies of Troponin C (in preparation) and SERCA [26] for this experiment. In these studies, the PMF along the reaction coordinate was computed using Adaptive Force Biasing [14] and Weighted Histogram Analysis Method [17], respectively, and details of the windows and convergence are described in our manuscripts. In short, for SERCA we performed 6 ns of umbrella sampling at each 0.5 Å interval between the Ca2+ site II binding region and the mouth of the binding channel; the potential of mean force was computed with WHAM and confidence intervals were reported based on a bootstrapping procedure included with WHAM. For TnC, we performed ABF calcuations until the potential of mean force changed little between simulations (after roughly 100 ns).
We assume in both cases the the diffusion constant for Ca2+ within the binding channel is 60% of its bulk value of 780 μm2/s, findings from [6], where D was explicitly computed within the K+ binding channel. For the computation of kPMF, we integrate the V (x) and the additional spatially-dependent terms numerically using the numpy trapz trapezoidal integration function. For the case of ligand-induced gating, for simplicity we assume that Vr = 0 and Va is equal to the potential of mean force at the gate location.
2.7. Gating kinetics
Gating kinetics are determined from molecular dynamics experiments. For our system, we consider two states that are distinguished by a change in side chain position. In practice, more complex gating events could be characterized by partitioning a molecular dynamics trajectory structurally diverse sets based on k-means clustering of the root mean squared deviations. ωa and ωr are determined by counting the transitions between the states, yielding a transition probability matrix.
2.8. Numerical solution
We introduce a python-based program dubbed Smolfin, to perform the numerical approximation and computation of the problems described above. The molecular mesh, the electrostatic potential, and, as appropriate, the 1D potential of mean force and gating forward and backward rates, are provided as input. Smolfin has operations for loading the marked finite element mesh corresponding to the simulation domain and defining the Dirichlet boundary conditions at the appropriate boundaries. Smolfin then solves a weak form of the Smoluchowski equation using a piecewise linear Galerkin finite element method with the default direct linear solver. The approach is based on [38], with the exception of using DOLFIN [30], the C++/python interface to the numerical PDE solver FEniCS (http://fenics.project.org), a finite element package. Smolfin obtains kon by integrating the steady-state flux along Γa according to (4). Additional details and references on the Smoluchowski solver are given in the Supplement Section 6.3. Parameters used in this study are summarized in Tbl. 2.
3. Results
3.1. A fully-absorbing, attractively charged sphere
We validate the proposed numerical protocol using a prototypical system for which analytical estimates of the association rate are available. We begin with a neutral and uniformly reacting sphere. We created the mesh for an 12.5 Å radius ion using GAMer. The resulting mesh contained 21,706 vertices and 97,315 simplices. All numerical solutions in this paper were computed in less than 1 min on a single CPU.
For the spherical case, we chose R=12.25 [Å] for a typical globular protein, D=780 [μm2/s] for Ca2+, and an infinitely dilute solution. Using Alg. 1, we predict the steady state distribution, ρss, shown in Fig. 4a. The steady state probability density, ρss approaches the bulk value at the periphery and zero at the active site, as specified by our boundary conditions. Kimball and Collins quantified the association rate for this scenario using
| (16) |
where R is the radius of sphere and D is the diffusion constant for the reacting species [12]. Smolfin predicts kon = 7.09× 109, which is in almost exact agreement with the analytical estimate of kon = 7.23× 109 [1/Ms]. We then consider a negatively charged sphere, for which the interaction potential can by described by Coulombic potential of mean force, W(r) = q/r, where q is the effective ligand charge. In Fig. 4b we show that in the presence of an oppositely-charged electrostatic potential, the probability density scarcely deviates from the bulk value except within a few angstroms of the reacting sphere. This leads to a significantly larger concentration gradient adjacent to the active site that contributes to the higher flux at Γa and results in an increased value of kon. Assuming spherical symmetry, Song et al. [38] demonstrate that the analytical solution to the Smoluchowski equation in the presence of a spherically-symmetric electrostatic potential takes the form
| (17) |
where r1 is the radius of the reacting sphere, r2 is the radius of the outer sphere (we assume r2 → inf) and q is the effective ligand charge. Our predicted kon (3.49× 1010) is slightly larger than the analytical prediction using (17) (9.6× 109) [1/Ms]; the slight error may be due to assuming a linear interpolation scheme for mapping the APBS electrostatic potential onto the finite element mesh, or our truncation of the potential at the molecular boundary to yield a maximum 55M Ca2+ concentration. This case exemplifies the role of favorable electrostatic interactions in driving charged substrates at rates higher than permitted by diffusion alone, as is commonly observed in biological systems.
Figure 4.

Ca2+ binding predictions for a uniformly reacting sphere. Steady state ρ distribution around (a) neutral and (b) charged sphere. (c) kPMF as a function of barrier height, V0, in a linear potential (d) kg under ligand-induced slow gating (−), ligand-independent slow gating (−) and fast gating (−.) in a constant binding channel potential with a range of ligand-dependent gating potentials, Va.
In the following example, we compute the association rate resulting from the diffusional encounter with the molecular surface and subsequent diffusion along a linear potential toward the active site, which is a reasonable approximation to the binding potential of mean force in Fig. 2. We assume energetic barriers ranging from Va = −10 to Va = 10 [kcal/mol], which spans the range of PMF barriers encountered in our recent simulation studies (in preparation). We furthermore assume a cylindrical binding channel of length L=10 [Å] and radius R= 5 [Å]. Based on (5), kon is dominated by the diffusional encounter rate for attractive potentials, while kpmf dominates when unfavorable barriers are present. Indeed we observe in Fig. 4c that kss ≈ kD for attractive barriers and rapidly decreases as the barrier assumes larger unfavorable values (approximately for V > 2.5 kcal/mol).
Lastly, we consider ligand-independent versus ligand-dependent gating in the fast and slow gating limits. For the slow gating scenario, we choose ω(r) = 0.01 for r far from the protein, while ω(r) at the active site is determined based on the conformation-dependent potentials Ua and Ur in (8). In Fig. 4d, we demonstrate kon for a range of ligand-dependent potentials, spanning Ua = −5 to Ua = 0 [kcal/mol]. We note that in the fast gating limit, kon approaches the ungated limit (although still reflecting the time spent diffusing within the channel), whereas the slow gating values report drastically reduce association rates based on the open probability. The ligand-dependent gating rate offers nearly an order of magnitude higher association rate compared to the ligand- independent case for strong potentials. As the attractive Ua diminishes, the ligand-induced gating limit approaches that of the ligand-independent case.
3.2. Troponin C
Myofilaments contract owing to intercalation of the thick filaments, comprised of myosin, and the actin chains forming the thin filament (TF). Contraction is tightly regulated by Troponin proteins tethered to the TF, and in particular, Ca2+ binding to TnC sparks the initial stages of the concerted process [15]. Troponin C is a 161 residue protein consisting of two Ca2+ binding domains bridged by a linker region [45]. The C-terminal domain is bound to the TF, while the N-terminal domain (Fig. 1a) responds to physiological changes in [Ca2+] [45] and induces changes in the Troponin complex. Characterizing the dynamics of Ca2+ binding in light of molecular geometry and charge distribution, provides unique insight into muscle cell contraction. To this end, we compute Ca2+ association rates to an isolated Troponin C fragment with the active site at the solvent-exposed surface, second to a ’buried’ binding site, and lastly to Troponin C embedded to a complete thin-filament subunit.
We begin with the Ca2+-free form of cardiac troponin C (PDB code: 1SPY) determined from NMR [41], This structure consists of the N-terminal domain, which contains the alpha helix-loop EF hand responsible for binding Ca2+ at physiological concentrations [35]. We chose Glu76, a residue from site II that directly coordinates Ca2+, as the centroid of the active site and labeled the molecular surface within 5 Å of the active site as Γa. Using GAMer the resulting mesh had 59,860 vertices and 276,072 simplices.
We apply Alg. 1 to this geometry in absence of the electrostatic potential and obtain the steady-state distribution of ρ shown in Fig. 5a). Here we observe that the [Ca2+]≈0 at the active site and is nearly equal to ρbulk within 10 Å of the active site; this solution yields a kD of 4.4× 108 [1/Ms]. The kD for the uncharged Troponin C is about one order of magnitude smaller than the rate predicted for the uncharged sphere of similar radius and indicates that the non-trivial molecular geometry and smaller active region limit the association rate of Ca2+.
Introducing the electrostatic potential into the numerical solution yields the steady state distribution in Fig. 5b. In particular, we observe an accumulation of Ca2+ density several-fold higher than the bulk concentration at several regions of the protein, including the active site. Directly adjacent to the active site, we again see large concentration gradients similar to the charged sphere case. Thus the favorable electrostatic potential, which is considerably larger in magnitude than the sphere case, yields a nearly 50-fold increase in the association rate to kon = 2.14x1010 [1/Ms]. However, this estimate is nearly 1,000 times faster than experimental estimates (107 to 108 [1/Ms] [35, 19]).
We attribute the disparity between the diffusional encounter rate and experimental estimates to post-encounter interactions that greatly reduce the association rate to the active site. Moreover, prior studies evidence a change in flexibility at site II [40, 28] which we postulate may contribute to an entropic penalty to binding. These effects manifest in a complicated potential of mean force along a reaction coordinate defined as the distance between Ca2+ and site II residue D76 in a related study (in preparation).
We note that far from the protein, the potential gradually decreases with smaller values of R, which is indicative of the electrostatic attraction between substrate and TnC. At r ≈ 1.8 [Å] we observe an abrupt barrier of 2.7 [kcal/mol] that that we postulated arises from a competition between the free energy cost upon shifting from coordination state involving water to one involving the binding residues in the active site (Asp65, Asp67, Ser69, Thr71, and Glu76 [45], in preparation) . We include the contribution of this PMF to our estimate of kon using Alg. 2. For simplicity, we assume a cylindrical reaction path with a radius of 5 Å. We find that the PMF reduces the overall kon by an order of magnitude and brings our estimates in closer agreement with experiment. Our finding that the observed binding rate is strongly dependent on the potential of mean force barrier associated with dehydration of Ca2+ suggests the strong role of water coordination in shaping the kinetics of Ca2+ binding.
In an intact myofilament, Troponin C is bound to actin of the thin filament. To determine the extent to which the myofilament geometry impacts the steady state distribution and kon, we applied our computational protocol to a cryo-EM model of the myofilament from [47] (PDB Code 2W4U). The resulting mesh from Section. 2.4 contained 44,362 vertices and 178940 simplices. We assumed non-periodic boundary conditions, as we expect the contribution of the electrostatic potential from adjacent actin units to be very small, given the Debye length of (λd ≈ 6.8 Å) based on physiological ionic strength.
In Fig. 5, we compare ρ in the intact filament versus the N-terminal fragment. We note for the uncharged case that the depletion of ρ extends along the thin filament, suggesting that to some extent its geometry influences local Ca2+ dynamics (data not shown). The picture is drastically different when the electrostatic potential is introduced as seen in Fig. 5c. For this case, and in Table 1, we report a kon value of 1.7× 106 [1/Ms], which is significantly smaller than the kD value obtained for the charged, isolated Troponin C.The large difference in kD is surprising, given that we had expected that the electrostatic contribution from the thin filament would be largely shielded by the small Debye length. However, we note in Fig. 5c substantial accumulation of Ca2+ (red contours representing 40 M Ca2+) that were not observed in the uncharged case. Moreover, the large gradients at the active site observed for the isolated Troponin C case are substantially attenuated in the intact geometry, although the coarser representation of Troponin C on the filament make direct comparisons diffcult. It is possible that the accumulation regions on the thin filament behave as buffers and effectively reduce the available Ca2+ for reaction at the active site, as evidenced by the smaller gradients, which thereby reduce the effective association rate. However, it is likely that our simulations over-estimate this bu ering effect, as the counter-ions K+ and Mg2+ could also compete for these negatively-charged regions.
Table 1.
Predicted kon values for di usional encounter and post-encounter scenarios. Included are applications for TnC (intact geometry) and SERCA (neutral phospholipids).
| case | kD, q = 0 | kD, q ≠= 0 | kss | kg | applications |
|---|---|---|---|---|---|
| Sphere(1 x 109) | 7.09 | 3.49 | |||
| SERCA(1 x 109) | 0.10 | 0.56 | 0.15 | 0.03* | 2.96 |
| TnC(1 x 109) | 0.39 | 21.5 | 1.25 | NA | 0.0015 |
Computed directly from (12).
Our observation that the thin filament geometry impacts Ca2+ association rates raises the possibility that changes in the myofilament lattice spacing during rigor [16] could modulate kon. Decreased lattice spacing, e.g. the distance between adjacent thick and thin filaments, is believed to decrease during contraction and this controversy surrounds whether the decrease is correlated with an increase in Troponin C’s affinity for Ca2+[16]. At physiological ionic strengths, we do not expect any significant differences in the electrostatic potential as the thick and thin filaments are still separated by ranges much greater than the Debye length. Alternatively, conformational changes of troponin are known to occur during rigor, including Troponin C (as observed in circular dichroism experiments [31]) and shifting of tropomyosin [8]. Changes to either of these structure’s solvent-exposed surface could alter the electrostatic profile of the system and thereby impact Ca2+ association rates. We plan to examine these factors in a follow-up study.
3.3. SERCA
The Sarcoplasmic reticulum Ca2+ ATPase (SERCA) is a trans-membrane (TM) Ca2+ pump that transports two Ca2+ ions into the sarcoplasmic reticulum from the cytosol through ATP-dependent conformational transitions. Previous simulations [32, 21, 13] have explored possible pathways of Ca2+ entry. Borrowing the convention proposed in [21] the L3 region (near E309, Fig. 1b) has been suggested as a predominant entryway for Ca2+ binding based on molecular dynamics (MD) and solutions of the Poisson Boltzmann equation. For SERCA, we consider association rates to the L3 recognition domain discussed in [26]. This recognition domain is lined with several negatively-charged amino acids, including Glu51, Glu55, Glu58, Asp59 and Asp 109, which contribute to a strongly negative electrostatic potential favoring cation association. Moreover, the prevalence of charged residues along the SERCA transmembrane bundle gives rise to a deformation of the lipid bilayer near the L3 entryway consistent in other studies [39, 27].
We assume here that the effect of the lipid environment is imposed in the ’exterior’ problem (for which the conventional Smoluchowski PDE is used), as the lipids are relatively far from the active site (and outside the L3 binding channel). We created the molecular geometry using a snapshot from our molecular dynamics simulations of the Ca2+-bound structure (PDB code: 1SU4) from Obara et al [34] determined at 2.4 Å resolution; details of structure preparation, including embedding in POPC lipid, are available in [26]. For the PDE simulation domain, using Blamer we embedded the lipid head groups and upper transmembrane/cytosolic domains in a rectangular box. The active site was defined on the molecular surface within 5 Å of the mid-point between D55 and D113, which sits in the center of the L3 binding vestibule. The resulting grid contained 88,868 vertices and 360,296 simplices.
Based on this representation, we predict kon of 5.8× 108 [1/Ms] and the steady state distribution of ρ is shown in Fig. 6a. Particularly interesting is the significant accumulation of Ca2+ density along the lipid indentation, which suggests that the deformation may enhance the rate of flux into the active site. Moreover, we note substantial Ca2+ density along the lipid bilayer far from the binding site that suggests the negatively charged phosphate heads buffer Ca2+. We had anticipated that polar head groups of the lipid bilayer near the L3 site might provide an additional enhancement of the kon. To probe this, we recomputed the electrostatic potential using uncharged lipids and surprisingly found that kD improved to 3.0× 109 [1/MS] and accumulation of Ca2+ along the lipid was drastically reduced. Thus, it appears that by reducing the buffering capacity of the lipids, more free Ca2+ is available to react at the active site, as we had hypothesized for the Troponin C/thin filament system. We note that this phenomenon may serve as physiological purpose, as [7] provides evidence that the sarcolemmal membrane buffers Ca2+; while the sarcolemmal lipid composition differs from the POPC assumed in our model, the prevalence of negative charge may help guide cations toward the SERCA binding region.
While it appears that Ca2+ can freely diffuse to the binding mouth, the abundance of highly mobile, charged residues in the L3 binding vestibule raises the possibility that they may play a direct role in transferring Ca2+ to the active site. To incorporate the influence of these interactions on the association rate, we computed the potential of mean force using molecular dynamics and umbrella sampling in [26]. We found that the potential of mean force did not vary significantly far from the active region where the lipids could be expected to interact with Ca2+, which we attribute to the relatively small number of lipids lining the largely solvent-exposed opening of the binding channel. Indeed, we observed a smooth, nearly linear decrease in the potential of mean force at large distances, while a slight barrier was found just outside the Ca2+ binding site (≈0.5 [kcal/mol]) at r ≈ 2.5 Å. In [26], we attributed this barrier to a conformational change of E309 that draws Ca2+ from the L3 vestibule into the binding site. Based on this PMF, we predict a kss of 1.5× 108 [1/Ms] using Alg. 2. Hence, the association rate appears to be only modestly impacted by the molecular interactions within the binding site, which is not unexpected given the vary small barrier height.
Instead, we postulate that gating of Glu309 serves a more prominent role in modulating Ca2+ association rates. Prior studies have implicated Glu309 in controlling access to site II , as mutations of the residue to alanine [42] and glutamine [23] resulted in faster Ca2+ dissociation rates. We explored this potential gating mechanism in [26] and discovered that the Glu309 side chain oscillates between an open and closed state. In the absence of Ca2+, the Glu309 carboxyl terminus tends to favor placement within the L3 vestibule, and as Ca2+ approaches the binding site, Glu309 heavily favors the Ca2+ binding site, through a conformational change driven by nanosecond-timescale flips in the χ2 dihedral angle between Cα and Cβ. The equilibrium constant for this conformational change was ≈ 20% when Ca2+ was far from the active site (R >> 15Å) and approached ≈ 60% (data not shown) just prior to binding at site II. The difference in gating rates implies a gating potential of U(r) ≈ 0.75 [kcal/mol] based on (8). We utilize Alg. 3 to include the effect of this putative gating mechanism. Based on a free Ca2+ ion (D=780 μm2), we expect a characteristic diffusion time for Ca2+ leaving the active site (r=10Å) of τd= 0.2 ns using τd = r2/6D, which is within the slow gating regime. In this limit, our analysis would suggest that the binding site is accessible during roughly 20% of the simulation, which would further reduce the observed kon by nearly an order of magnitude. While we have not estimated gating frequencies for other common ions, such as Mg2+ or K+, we might expect a less favorable interaction potential between Glu309 and the solvated ion, which in turn would reduce the association rate as we observed in Fig. 4d. If true, this differential in association rates could constitute a mechanism for ion selectivity.
3.4. Limitations
While this numerical approach represents significant progress toward multi-scale modeling of substrate association in realistic molecular scale environments, several limitations should be addressed to improve the approach. For one, we assume in this framework that a 1-D potential of mean force is sufficient to describe diffusion within the binding channel. For SERCA, the binding channel appears to be approximately linear, but for TnC and likely for other systems, the preferred pathway to substrate binding may be very non-linear. Should this be the case, (6) could be modified to include additional degrees of freedom. However, this would likely entail modifying the boundary conditions inherent to the decomposition into interior and exterior problems, which we have not addressed in this study.
An additional complication is that the potential of mean force assumes that fluctuations orthogonal the reaction coordinate are fast relative to the reaction coordinate axis. We have observed in our molecular dynamics simulations of SERCA that gradual changes in the bundle that could invalidate this assumption. However, it may be possible to group these changes into conformational states that inter-convert with some quantifiable equilibrium constant, for which we may estimate kon in a similar fashion to the gated active site formalism.
We also assume a diffusion constant within the binding channel that is equal to the diffusion in the bulk. Clearly the interactions between Ca2+ and the binding site will perturb the diffusion constant in a spatially-dependent fashion. Strategies including estimates based on the mean free passage time [20] or the friction kernel from the velocity autocorrelation function [10] could improve our estimates of D within the binding channel. However, based on a study of the K+ channel in [6], the diffusion constant within the binding channel was at most reduced by 40% relative to the bulk value, hence our estimate of Dchannel in this study may be reasonable.
Here in this study we also assumed that the Ca2+ probability distribution is determined by its concentration gradient and a simple approximation of the potential based on the linearized Poisson-Boltzmann equation. In reality, additional factors, such as finite-size effects [22], could offer an improved estimate of the probability distribution near the active site. It is likely that including these effect would reduce our estimates of the diffusional encounter and bring our estimates of the overall association rate in line with experiment.
4. Conclusions and Future Work
This paper presents a numerical framework for integrating molecular structural and dynamic data, including binding along gated channels, into continuum models of diffusion. Incorporation of these effects helps bridge the gap between rates predicted by diffusional-encounter models and the actual binding kinetics measured by experiment. We stress that these results are generalizable to variety of ligand-protein biding events are not limited to calcium.
While we have focused here on steady-state solutions to the Smoluchowski equation, we note that the time-dependent case can be addressed by adapting the weak form to include the time-dependent term and using a finite difference time-stepping approach. This modification may offer superior estimates of the association rate when local equilibrium about the receptor is not guaranteed. The advantage of PDE-based estimation of association rates is the flexibility to include more detailed numerical models, such as the Poisson-Nernst-Planck equation to handle ion-ion interactions [33].
5. Acknowledgements
We would like to thank Gary Huber, Denis Bucher, Pan Li and Cesar de Oliveira for interesting discussions and careful reading of the manuscript. This work was supported by the National Institutes of Health, the National Science Foundation, the Howard Hughes Medical Institute, and the National Biomedical Computation Resource.
Footnotes
PACS numbers: 82.39.-k, 82.39.Rt, 87.10.Kn, 87.14.E-
References
- [1].Baker N, Sept D, Joseph S, Holst M, McCammon J. Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences. 2001;98(18):10037. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bamford C, Tipper C, Compton R. Diffusion-Limited Reactions. volume 25 of Comprehensive Chemical Kinetics. Elsevier; [Google Scholar]
- [3].Barreda J, Zhou H. Theory and simulation of diffusion-influenced, stochastically gated ligand binding to buried sites. The Journal of Chemical Physics. 2011;135:145101. doi: 10.1063/1.3645000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Berezhkovskii A, Szabo A, Zhou H. Diffusion-influenced ligand binding to buried sites in macromolecules and transmembrane channels. The Journal of chemical physics. 2011;135:075103. doi: 10.1063/1.3609973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov I, Bourne P. The Protein Data Bank. Nucleic Acids Research. 2000:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bernèche S, Roux B. A microscopic view of ion conduction through the K+ channel. Proceedings of the National Academy of Sciences. 2003 Jul;100(15):8644–8648. doi: 10.1073/pnas.1431750100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bers D. Excitation-Contraction Coupling and Cardiac Contractile Force. 1 edition volume 1. Kluwer Academic Publishers, Kluwer Academic Publishers; Nov, 2001. [Google Scholar]
- [8].Bers DM. Calcium cycling and signaling in cardiac myocytes. Annual review of physiology. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- [9].Cai L, Zhou H. Theory and simulation on the kinetics of protein–ligand binding coupled to conformational change. The Journal of Chemical Physics. 2011;134:105101. doi: 10.1063/1.3561694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chahine JJ, Oliveira RJR, Leite VBPV, Wang JJ. Configuration-dependent diffusion can shift the kinetic transition state and barrier height of protein folding. Proceedings of the National Academy of Sciences. 2007 Sept.104(37):14646–14651. doi: 10.1073/pnas.0606506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cheng Y, Suen JK, Zhang D, Bond SD, Zhang Y, Song Y, Baker NA, Bajaj CL, Holst MJ, McCammon JA. Finite element analysis of the time-dependent Smoluchowski equation for acetylcholinesterase reaction rate calculations. Biophysical Journal. 2007 May;92(10):3397–3406. doi: 10.1529/biophysj.106.102533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].COLLINS F, KIMBALL G. Diffusion-Controlled Reaction Rates. Journal of Colloid Science. 1949;4(4):425–437. [Google Scholar]
- [13].Costa V, Carloni P. Calcium, binding to the transmembrane domain of the sarcoplasmic reticulum Ca2+-ATPase: Insights from molecular modeling. Proteins-Structure Function And Bioinformatics. 2003;50(1):104–113. doi: 10.1002/prot.10219. [DOI] [PubMed] [Google Scholar]
- [14].Darve E, Pohorille A. Calculating free energies using average force. The Journal of chemical physics. 2001;115(20):9169. [Google Scholar]
- [15].Filatov V, Katrukha A, Bulargina T, Gusev N. Troponin: structure, properties, and mechanism of functioning. Biochemistry (Moscow) 1999;64(9):969–985. [PubMed] [Google Scholar]
- [16].Fuchs F, Martyn D. Length-dependent Ca 2+ activation in cardiac muscle: some remaining questions. Journal of Muscle Research and Cell Motility. 2005;26(4):199–212. doi: 10.1007/s10974-005-9011-z. [DOI] [PubMed] [Google Scholar]
- [17].Grossfield A. An implementation of WHAM: the Weighted Histogram Analysis Method.
- [18].Hake J, Edwards A, Yu Z, Kekenes Huskey P, McCammon A, McCulloch A, Michailova A. Modeling Calcium Sparks in a Three-Dimensional Reconstruction of a Cardiac Calcium Release Unit. Journal of Physiology. 2012 doi: 10.1113/jphysiol.2012.227926. (accepted) edition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hazard AL, Kohout SC, Stricker NL, Putkey JA, Falke JJ. The kinetic cycle of cardiac troponin C: Calcium binding and dissociation at site II trigger slow conformational rearrangements. Protein Science. 1998 Nov.7(11):2451–2459. doi: 10.1002/pro.5560071123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hinczewski M, von Hansen Y. Deconvolution of dynamic mechanical networks. Proceedings of the …. 2010. [DOI] [PMC free article] [PubMed]
- [21].Huang Y, Li H, Bu Y. Molecular dynamics simulation exploration of cooperative migration mechanism of calcium ions in sarcoplasmic reticulum Ca 2+-ATPase. Journal Of Computational Chemistry. 2009 Oct.30(13):2136–2145. doi: 10.1002/jcc.21219. [DOI] [PubMed] [Google Scholar]
- [22].HYON Y, Eisenberg B. A mathematical model for the hard sphere repulsion in ionic solutions. Comm. Math. Sci. 2010 [Google Scholar]
- [23].Inesi G, Ma H, Lewis D, Xu C. Ca2+ occlusion and gating function of Glu309 in the ADP-fluoroaluminate analog of the Ca2+-ATPase phosphoenzyme intermediate. The Journal of biological chemistry. 2004;279(30):31629. doi: 10.1074/jbc.M403211200. [DOI] [PubMed] [Google Scholar]
- [24].Israelachvili JN. Intermolecular and Surface Forces. Academic Press; Jun, 2011. [Google Scholar]
- [25].Kekenes-Huskey P, Cheng Y, Hake J, McCammon J, Michailova A. Modeling effects of L-type Ca2+ current and Na+-Ca2+ exchanger on Ca2+ trigger flux in rabbit myocytes with realistic t-tubule geometries (accepted) Frontiers in Computational Physiology and Medicine. doi: 10.3389/fphys.2012.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kekenes-Huskey P, Metzger V, Grant B, McCammon J. Calcium binding and allosteric signaling mechanisms for Sarcoplasmic reticulum Ca2+ ATPase (accepted) Protein Science. doi: 10.1002/pro.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lervik A, Bresme F, Kjelstrup S. Molecular dynamics simulations of the Ca2+-pump: a structural analysis. Physical Chemistry Chemical Physics. 2012;14(10):3543. doi: 10.1039/c2cp23002j. [DOI] [PubMed] [Google Scholar]
- [28].Lindert S, Kekenes-Huskey PM, Huber G, Pierce L, McCammon JA. Dynamics and Calcium Association to the N-Terminal Regulatory Domain of Human Cardiac Troponin C: A Multiscale Computational Study. The Journal of Physical Chemistry B. 2012 Feb. doi: 10.1021/jp212173f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Logg A, Mardal K-A, Wells G. Automated solution of differential equations by the finite element method automated solution of differential equations by the finite element method. Lecture Notes in Computational Science and Engineering. 2012 Springer;volume 84:1–736. [Google Scholar]
- [30].Logg A, Wells G. DOLFIN: Automated finite element computing. ACM Transactions on Mathematical Software (TOMS) 2010;37(2):20. [Google Scholar]
- [31].Martyn DA, Regnier M, Xu D, Gordon AM. Ca2+- and Cross-Bridge-Dependent Changes in N- and C-Terminal Structure of Troponin C in Rat Cardiac Muscle. Biophysical Journal. 2001 Jan.80(1):360–370. doi: 10.1016/S0006-3495(01)76020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Musgaard M, Thogersen L, Schiott B, Tajkhorshid E. Tracing Cytoplasmic Ca2+ Ion and Water Access Points in the Ca2+-ATPase. Biophysical Journal. 2012 Jan.102(2):268–277. doi: 10.1016/j.bpj.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Noskov S, Im W, Roux B. Ion permeation through the [alpha]-hemolysin channel: theoretical studies based on Brownian dynamics and Poisson-Nernst-Plank electrodiffusion theory. Biophysical Journal. 2004;87(4):2299–2309. doi: 10.1529/biophysj.104.044008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Obara K, Miyashita N, Xu C, Toyoshima L, Sugita Y, Inesi G, Toyoshima C. Structural role of countertransport revealed in Ca2+ pump crystal structure in the absence of Ca2+ Proceedings of the National Academy of Sciences. 2005;102(41):14489–14496. doi: 10.1073/pnas.0506222102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ogawa Y. Calcium binding to troponin C and troponin: effects of Mg2+, ionic strength and pH. Journal of biochemistry. 1985 Apr.97(4):1011–1023. doi: 10.1093/oxfordjournals.jbchem.a135143. [DOI] [PubMed] [Google Scholar]
- [36].Rice S. Diffusion-limited reactions. In: Bamford C, Tipper C, Compton R, editors. Comprehensive Chemical Kinetics. volume 25. Elsevier Science; 1985. [Google Scholar]
- [37].Smoluchowski M. Versuch einer mathematischen Theorie der Koagulationskinetik kolloi- derLösnngen. Apr. 1918.
- [38].Song Y, Zhang Y, Shen T, Bajaj CL, McCammon JA, Baker NA. Finite element solution of the steady-state Smoluchowski equation for rate constant calculations. Biophysical Journal. 2004 Apr.86(4):2017–2029. doi: 10.1016/S0006-3495(04)74263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sonntag Y, Musgaard M, Olesen C, Schiott B, Moller JV, Nissen P, Thogersen L. Mutual adaptation of a membrane protein and its lipid bilayer during conformational changes. Nature Communications. 2:2011. doi: 10.1038/ncomms1307. [DOI] [PubMed] [Google Scholar]
- [40].Spyracopoulos L, Gagné SM, Li MX, Sykes BD. Dynamics and Thermodynamics of the Regulatory Domain of Human Cardiac Troponin C in the Apo- and Calcium-Saturated States. Biochemistry. 1998 Dec.37(51):18032–18044. doi: 10.1021/bi9816960. [DOI] [PubMed] [Google Scholar]
- [41].Spyracopoulos L, Li MX, Sia SK, Gagné SM, Chandra M, Solaro RJ, Sykes BD. Calcium-Induced Structural Transition in the Regulatory Domain of Human Cardiac Troponin C. Biochemistry. 1997 Oct.36(40):12138–12146. doi: 10.1021/bi971223d. [DOI] [PubMed] [Google Scholar]
- [42].Sugita Y, Miyashita N, Ikeguchi M, Kidera A, Toyoshima C. Protonation of the acidic residues in the transmembrane cation-binding sites of the Ca2+ pump. J. Am. Chem. Soc. 2005;127(17):6150–6151. doi: 10.1021/ja0427505. [DOI] [PubMed] [Google Scholar]
- [43].Szabo A, Shoup D, Northrup S, McCammon J. Stochastically gated diffusion-influenced reactions. The Journal of Chemical Physics. 1982;77:4484. [Google Scholar]
- [44].Szabo A, Shoup D, Northrup S, McCammon J. Stochastically gated diffusion-influenced reactions. Journal Of Chemical Physics. 1982;77(9):4484–4493. [Google Scholar]
- [45].Takeda S, Yamashita A, Maeda K, Maéda Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature. 2003 Jul;424(6944):35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- [46].Wade RC, Gabdoulline RR, LÃdemann SK, Lounnas V. Electrostatic steering and ionic tethering in enzyme-ligand binding: insights from simulations. Proceedings of the National Academy of Sciences. 1998 May;95(11):5942–5949. doi: 10.1073/pnas.95.11.5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wu S, Liu J, Reedy M, Tregear R, Winkler H, Franzini-Armstrong C, Sasaki H, Lucaveche C, Goldman Y, Reedy M. Electron tomography of cryofixed, isometrically contracting insect flight muscle reveals novel actin-myosin interactions. PLoS ONE. 2010;5(9):e12643. doi: 10.1371/journal.pone.0012643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Xu D, Zhang Y. Generating triangulated macromolecular surfaces by euclidean distance transform. PloS one. 2009;4(12):e8140. doi: 10.1371/journal.pone.0008140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yu Z, Holst M, Cheng Y, McCammon J. Feature-preserving adaptive mesh generation for molecular shape modeling and simulation. Journal of Molecular Graphics and Modelling. 2008;26(8):1370–1380. doi: 10.1016/j.jmgm.2008.01.007. [DOI] [PubMed] [Google Scholar]
- [50].Zhou H. Theory of the diffusion-influenced substrate binding rate to a buried and gated active site. Journal of Chemical Physics. 1998;108:8146–8154. [Google Scholar]
- [51].Zhou HXH, Wlodek STS, McCammon JAJ. Conformation gating as a mechanism for enzyme specificity. Proceedings of the National Academy of Sciences. 1998 Aug.95(16):9280–9283. doi: 10.1073/pnas.95.16.9280. [DOI] [PMC free article] [PubMed] [Google Scholar]