

Abstract
Anopheles mosquitoes are vectors of malaria, a potentially fatal blood disease affecting half a billion humans worldwide. These blood-feeding insects include in their antihemostatic arsenal a potent thrombin inhibitor, the flexible and cysteine-less anophelin. Here, we present a thorough structure-and-function analysis of thrombin inhibition by anophelin, including the 2.3-Å crystal structure of the human thrombin·anophelin complex. Anophelin residues 32–61 are well-defined by electron density, completely occupying the long cleft between the active site and exosite I. However, in striking contrast to substrates, the D50-R53 anophelin tetrapeptide occupies the active site cleft of the enzyme, whereas the upstream residues A35-P45 shield the regulatory exosite I, defining a unique reverse-binding mode of an inhibitor to the target proteinase. The extensive interactions established, the disruption of thrombin’s active site charge–relay system, and the insertion of residue R53 into the proteinase S1 pocket in an orientation opposed to productive substrates explain anophelin’s remarkable specificity and resistance to proteolysis by thrombin. Complementary biophysical and functional characterization of point mutants and truncated versions of anophelin unambiguously establish the molecular mechanism of action of this family of serine proteinase inhibitors (I77). These findings have implications for the design of novel antithrombotics.
Keywords: coagulation inhibitor, macromolecular recognition, protease, X-ray structure, catalytic triad
Malaria is a life-threatening human disease caused by Plasmodium spp. parasites that infect liver and red blood cells, and it is transmitted exclusively through the bites of Anopheles mosquitoes. A. gambiae is the primary vector of malaria in sub-Saharan Africa, whereas A. stephensi is the main urban vector of the disease in India (1). Furthermore, A. darlingi and A. funestus are important vectors in Central and South America and Africa during the dry season, respectively (2, 3). Finally, A. albimanus is the principal malaria vector in most Caribbean countries. Despite important vector control and prophylactic measures, about 225 million cases of malaria and ∼800,000 deaths were reported in 2009, accounting for ∼2% of worldwide deaths and 20% of all childhood deaths in Africa (4).
Like other hematophagous animals, Anopheles mosquitoes rely on potent inhibitors of the host hemostatic and inflammatory responses for blood ingestion, storage, and digestion. Specific inhibitors of blood coagulation factors and particularly, the final peptidase in the cascade thrombin seem to be critical in this regard. The only mosquito-derived thrombin inhibitor described to date is anophelin (family I77 in the MEROPS classification; http://merops.sanger.ac.uk) (5), first isolated from A. albimanus salivary gland extracts (6, 7). Anophelin homologs (less than 50% sequence identity) were identified in A. gambiae (8), A. stephensi (1), A. darlingi (2), and A. funestus (3), but with the recent exception of one of the A. gambiae variants (9), they remain uncharacterized (Fig. 1 shows a sequence alignment of family I77 inhibitors). Interestingly, the anophelin gene is expressed in mosquito salivary glands in response to Plasmodium sporozoite invasion, suggesting a role in parasite infection (10). Moreover, recent RNAi silencing studies have shown that anophelin depletion in adult A. gambiae mosquitoes results in increased probing time and decreased blood capacity (11).
Fig. 1.

Sequence alignment of family I77 inhibitors from anopheline mosquitoes. The mature sequences of anophelins from the Old World mosquito species A. gambiae [pink eye standard (PEST) variant] (62, 63), A. stephensi (1), and A. funestus (3) and the New World species A. albimanus (6, 7) and A. darlingi (2) (vectors of malaria in sub-Saharan Africa, India, Africa, and Central and South America, respectively) were aligned with ClustalW (64). Also included is the sequence of TTI (G. morsitans morsitans; MEROPS family I76) (47). Strictly conserved residues are highlighted in green, whereas other conservative replacements are shaded red. The numbering for mature anophelinAa is given above the alignment, and the numbering for mature TTI is given above the respective sequence. The figure was prepared with Aline (65).
A. albimanus anophelin is a potent, tight-binding inhibitor of α-thrombin, the final proteinase of the blood-clotting cascade (7). Thrombin is an atypical (chymo)trypsin-like enzyme, with stringent specificity arising from its narrow active site cleft and secondary positively charged recognition surfaces (exosites) (12). Exosites are not only essential for substrate and cofactor binding (13–18) but also, are targeted by many natural anticoagulants (19–25). Thorough kinetic analyses revealed that anophelin is a dual inhibitor that binds both the exosite I and the active site of the target proteinase (6, 7). Accordingly, anophelin not only blocks physiological thrombin activities, such as fibrinogen clotting and platelet activation, but also abolishes esterolytic activity on small chromogenic substrates.
Here, we report the unique molecular mechanism of thrombin inhibition by anophelin. In contrast to previously characterized natural bivalent inhibitors that contact one of the exosites through their C-terminal regions, anophelin displays an unexpected reverse-binding mode to thrombin. This hitherto unobserved binding mechanism explains not only the remarkable affinity and specificity of anophelin but also, its resistance to proteolysis by thrombin, despite lacking disulfide bridges. The key residues for anticoagulant activity have been identified and are highly conserved across similar proteins of Anopheles mosquitoes. The structural and functional data now obtained unravel the essential role of this protein in mosquito blood feeding and open new perspectives to the rational design of improved anticoagulants.
Results
Anophelins from All Anopheles Mosquitoes Are Effective Thrombin Inhibitors.
The only thoroughly characterized member of the anophelin inhibitor family (I77) is the protein from the minor malaria vector A. albimanus (anophelinAa) (6, 7). Other family members have been identified in the genomes of A. darlingi (anophelinAd), A. stephensi (anophelinAs), A. funestus (anophelinAf), and A. gambiae (anophelinAg), all sharing less than 50% amino acid identity. It has been recently shown that one anophelin variant (cE5) from A. gambiae behaves as a specific thrombin inhibitor in vitro (9). Strictly conserved regions are limited to the N-terminal 1A-P-Q-4Y tetrapeptide, the hirudin-like motif with the consensus sequence 7G-(D/E)-X-P-X-Y-D-E-15(D/E), and the downstream 50D-P-G-53R stretch (Fig. 1 shows a sequence alignment; numbers refer to mature anophelinAa). In line with their lack of cysteines and the preponderance of polar residues, anophelins are predicted to be intrinsically disordered in solution (Fig. S1A). Indeed, the circular dichroism (CD) spectrum of anophelinAa is typical of random coils, and its deconvolution with different methods suggests a mostly disordered polypeptide (Fig. S1B).
Despite the considerable divergence of the anophelin genes, functionally important regions seem to have been preserved, because the five recombinant anophelins retained anticoagulant activity in vitro, prolonging 5- to 13-fold the time necessary for thrombin-catalyzed clotting of blood plasma [thrombin time (TT)] (Table 1). Furthermore, all anophelins behaved as tight-binding inhibitors of α-thrombin, with Ki values between 3.5 and 66 pM (Table 2), and significantly increased the thermal stability of α-thrombin on complex formation (Fig. 2A and Table S1). In addition, all five homologs were able to inhibit the exosite I-disrupted γ-thrombin (26) at high molar excess (Fig. S2).
Table 1.
Anophelin inhibits plasma clotting by prolongation of thrombin time
| Inhibitor | Concentration (μM) |
|||
| 0.1 | 0.5 | 1 | 5 | |
| AnophelinAd | 85.0 | >240 | ND | ND |
| AnophelinAg | 221.0 | >240 | ND | ND |
| AnophelinAf | 131.0 | >240 | ND | ND |
| AnophelinAs | 86.0 | >240 | ND | ND |
| AnophelinAa | 84.0 | >240 | >240 | ND |
| Anophelin32–61 | 25.5 | 88.0 | 155.5 | ND |
| Anophelin1–31 | ND | ND | 17.0 | 17.0 |
| AnophelinAa R54A | 110.0 | >240 | ND | ND |
| AnophelinAa R54N | 87.0 | >240 | ND | ND |
| AnophelinAa R54E | 67.0 | 176.5 | ND | ND |
| AnophelinAa R53A | 15.5 | 18.0 | 22.0 | 29.0 |
| AnophelinAa R53K | 59.0 | >240 | >240 | >240 |
| AnophelinAa R53Q | 30.5 | 135.0 | 237.0 | >240 |
| AnophelinAa R53H | 41.0 | 175.0 | >240 | >240 |
| AnophelinAa D50A | 19.5 | 19.0 | 25.0 | 39.0 |
| AnophelinAa D50E | 35.0 | 96.0 | 110.0 | 182.0 |
| AnophelinAa D50N | 19.0 | 27.0 | 42.0 | 71.0 |
The thrombin time (TT) determined in the absence of inhibitor was 16.5 s. TT values (in s) are the mean of three replicates. ND, not determined.
Table 2.
Kinetic parameters of α-thrombin inhibition by anophelin
| Inhibitor | Ki (nM) | IC50 (nM) | Model |
| AnophelinAd | 0.066 ± 0.005 | 0.99 | Morrison |
| AnophelinAg | 0.0035 ± 0.001 | 0.26 | Morrison |
| AnophelinAf | 0.0067 ± 0.003 | 0.28 | Morrison |
| AnophelinAs | 0.0304 ± 0.006 | 0.68 | Morrison |
| AnophelinAa | 0.034 ± 0.003 | 0.50 | Morrison |
| Anophelin32–61 | 0.304 ± 0.042 | 2.82 | Morrison |
| Anophelin1–31 | No inhibition | ND | |
| AnophelinAa R54A | 0.494 ± 0.050 | 6.12 | Morrison |
| AnophelinAa R54N | 1.090 ± 0.070 | 12.72 | Morrison |
| AnophelinAa R54E | 10.410 ± 1.020 | ND | Competitive |
| AnophelinAa R53A | No inhibition | ND | |
| AnophelinAa R53K | 2.693 ± 0.010 | 34.91 | Morrison |
| AnophelinAa R53Q | 18.050 ± 3.990 | ND | Competitive |
| AnophelinAa R53H | 6.970 ± 3.160 | ND | Competitive |
| AnophelinAa D50A | 203.600 ± 29.610 | ND | Competitive |
| AnophelinAa D50E | 44.750 ± 5.340 | ND | Competitive |
| AnophelinAa D50N | 54.790 ± 7.310 | ND | Competitive |
Ki values ± SEM given are representative of at least two independent experiments. ND, not determined.
Fig. 2.

Anophelin binds with high affinity to and stabilizes human α-thrombin. (A) Thrombin stability to thermal denaturation was measured by DSF in the presence of a 50-fold molar excess of WT anophelins, anophelin1–31, anophelin32–61, or anophelin mutants. Melting temperatures (Tm) were determined as the inflection points of the experimental curves, and they are given as mean values ± SEM. (B) Anophelin binding to immobilized human α-thrombin was measured by SPR. Sensorgrams depict kinetics experiments for WT anophelin and anophelin32–61. Each set of experimental curves (green) represents decreasing concentrations of analyte in twofold dilution steps (the highest concentration used is indicated). The black traces represent the fitted data according to the 1:1 Langmuir binding model. RU, resonance unit.
Arg53 Is Essential for Anophelin Inhibitory Activity.
Kinetic studies have suggested anophelinAa to be a bivalent inhibitor interacting with both the active center and the exosite I of thrombin (7). Comparison with the amino acid sequence of ideal thrombin substrates hinted that the AD50-AR54 tetrapeptide could fit in an antiparallel, substrate-like manner into the proteinase active site cleft; AR53 and AR54 are likely candidate P1 residues for occupying the acidic S1 pocket of thrombin (27, 28). [Substrate/inhibitor residues are denoted Pn, ..., P1, P1′, ..., Pm′, from N- to C-terminal end, where P1–P1′ is the scissile peptide bond; the corresponding proteinase subsites that accommodate these residues are termed Sn, ..., S1, S1′, ..., Sm′ according to the nomenclature in the work by Schechter and Berger (29).] The sequence is conserved in all known anophelins, except for the A. gambiae homolog, which has an asparagine at position 54 (Fig. 1). To verify the putative functional role of these basic residues in thrombin binding and inhibition, several mutants of anophelinAa were generated and characterized. Mutants R54A, R54N, and R54E prolonged TT to a roughly similar extent as WT anophelinAa (Table 1). A similar behavior was observed for R53K, whereas the TTs obtained for R53Q and R53H were about one-half the TTs of the WT protein. Remarkably, mutant R53A was unable to prolong TT at low concentrations and only marginally increased TT (1.8-fold) at concentrations as high as 5 μM.
Anophelin mutants R54A, R54N, and R53K retained the tight-binding inhibition mode of the WT inhibitor, although with 14-, 32-, and 80-fold higher Ki values than anophelinAa, respectively (Table 2). In striking contrast, the reversal-of-charge mutant R54E and variants in which R53 is replaced by polar residues (R53Q and R53H) were rapid, reversible competitive inhibitors of α-thrombin, with several orders of magnitude higher Ki values. More dramatically, R53A failed to inhibit the procoagulant proteinase (Table 2). These anophelin mutants also inhibited γ-thrombin with similar relative efficiencies (Fig. S2).
Surface plasmon resonance (SPR) analysis on immobilized thrombin revealed the formation of a very stable complex with anophelinAa (KD = 3.65 nM), whereas mutants were one (R54A, R54N, and R53K) or two (R54E, R53Q, and R53H) orders of magnitude less potent binders (Fig. 2B, Table 3, and Fig. S3). Finally, in good agreement with the inhibition studies, no appreciable binding of mutant R53A to the thrombin-coated chip surface was observed (Table 3 and Fig. S3). Taken together, these findings suggested a key role for anophelin residue R53 in thrombin binding and inhibition, likely by occupying the S1 specificity pocket. Although with a milder impact, differences in the kinetic behavior indicated that the positive charge of AR54 contributes to anophelin’s strong interaction with thrombin.
Table 3.
Kinetic parameters of anophelin binding to immobilized α-thrombin measured by surface plasmon resonance
| Analyte | ka (M−1s−1) | CV (%) | kd (s−1) | CV (%) | KD (M) | CV (%) |
| AnophelinAa | 1.04 × 105 | 1.36 | 3.80 × 10−4 | 8.56 | 3.65 × 10−9 | 7.36 |
| Anophelin32–61 | 1.18 × 105 | 18.7 | 4.48 × 10−4 | 9.47 | 3.86 × 10−9 | 9.53 |
| Anophelin1–31 | NRB | NRB | NRB | |||
| AnophelinAa R54A | 8.65 × 104 | 0.90 | 3.59 × 10−3 | 1.38 | 4.15 × 10−8 | 0.51 |
| AnophelinAa R54N | 7.02 × 104 | 0.70 | 5.46 × 10−3 | 1.69 | 7.78 × 10−8 | 2.36 |
| AnophelinAa R54E | 3.05 × 104 | 29.0 | 1.34 × 10−2 | 1.06 | 4.61 × 10−7 | 28.2 |
| AnophelinAa R53A | NRB | NRB | NRB | |||
| AnophelinAa R53K | 1.77 × 105 | 17.6 | 7.83 × 10−3 | 3.16 | 4.48 × 10−8 | 14.5 |
| AnophelinAa R53Q | 1.23 × 105 | 32.2 | 5.16 × 10−2 | 18.9 | 4.40 × 10−7 | 20.2 |
| AnophelinAa R53H | 6.22 × 104 | 5.57 | 1.90 × 10−2 | 9.33 | 3.05 × 10−7 | 3.71 |
| AnophelinAa D50A | NRB | NRB | NRB | |||
| AnophelinAa D50E | 4.40 × 104 | 27.0 | 6.50 × 10−2 | 18.0 | 1.50 × 10−6 | 9.40 |
| AnophelinAa D50N | NRB | NRB | NRB |
Binding data was fitted to the Langmuir 1:1 interaction model. Values given are the mean for the interaction of the analytes with thrombin surfaces at two different densities. CV, coefficient of variation between independent experiments; NRB, no residual binding.
Anophelin Displays a Unique Mode of Thrombin Inhibition.
To understand the molecular details of thrombin recognition and inhibition by anophelins, the equimolar human α-thrombin·anophelinAa complex was crystallized, and its structure was determined at 2.3-Å resolution (Table 4 and Fig. 3A). The final model comprises thrombin light-chain residues TE1C to TD14L and TI16 to TE247 from the catalytic domain, with the only exception of residues TW148 to TG149D of the 149 insertion loop that are not defined in the electron density maps. On complexation, thrombin suffers only minor backbone rearrangements [rmsd of 0.27 Å for 248 aligned Cα atoms between the free (30) (Protein Data Bank ID code 3U69) and anophelin-bound proteinase].
Table 4.
Data collection and refinement statistics
| Dataset | High-resolution dataset* | Low-resolution dataset* |
| Data collection | ||
| Space group | P3112 | H32 |
| Unit cell dimensions (Å) | a = b = 120.2; c = 77.8 | a = b = 209.7; c = 127.4 |
| Resolution range (Å) | 62.3–2.30 (2.43–2.30) | 46.8–3.20 (3.37–3.20) |
| Reflections (measured/unique) | 166,049/28,423 | 90,441/17,855 |
| Completeness (%) | 99.8 (99.0) | 99.9 (98.8) |
| Multiplicity | 5.8 (4.1) | 2.6 (2.5) |
| Rsym | 0.061 (0.683) | 0.143 (0.807) |
| Rpim | 0.027 (0.375) | 0.070 (0.403) |
| Mean [(I)/σ (I)] | 16.6 (2.1) | 9.9 (1.8) |
| Complexes per asymmetric unit | 1 | 1 |
| Matthews coefficient (Å3 Da−1) | 3.77 | 6.12 |
| Solvent content (%) | 67.4 | 79.9 |
| Refinement | ||
| Resolution range (Å) | 32.7–2.30 | 46.8–3.20 |
| Rfactor/free Rfactor (%) | 16.9/20.1 | 17.7/20.8 |
| No. of unique reflections (working/test set) | 26,958/1,438 | 17,853/911 |
| Water molecules | 87 | 23 |
| Ions | 1 Na+ | 1 Na+ |
| Total number of atoms | 2,611 | 2,512 |
| Average overall B factor (Å2) | 64.6 | 83.6 |
| Average protein B factor (Å2) | 64.8 | 83.5 |
| Average main-chain B factor (Å2) | 61.7 | 81.0 |
| Average side-chain B factor (Å2) | 67.8 | 86.0 |
| Average water B factor (Å2) | 54.8 | 65.4 |
| rmsd bonded Bs (Å2) | 5.64 | 8.64 |
| rmsd bond lengths (Å) | 0.007 | 0.009 |
| rmsd bond angles (°) | 1.041 | 1.256 |
*Values in parentheses correspond to the outermost resolution shell. Each dataset was collected from a single crystal.
Fig. 3.

Anophelin inhibits α-thrombin in a unique reverse-binding mode. (A) The acidic AE32-AF45 segment of anophelin (stick model with nitrogen atoms in blue, oxygen in red, and carbon in green) binds to the exosite I of thrombin (positive surface electrostatic potential in blue and negative surface electrostatic potential in red), whereas the downstream AD50-AL55 segment occupies the active site cleft of the proteinase. The unbiased Fo-Fc electron density (1.5-σ cutoff) for anophelin is displayed as a blue mesh. The thrombin molecule is shown in the standard orientation for serine proteinases (i.e., substrates would run from left to right or exactly opposite to the path followed by the Anopheles inhibitor). (B) Close-up view of the Van der Waals interactions between AE38 (anophelin colored as in A) and the pivotal TY76 at the exosite I of thrombin [gray cartoon with selected residues as sticks color-coded as anophelin except for carbon atoms (colored salmon); active site residues in ball and stick]. (C) Close-up view of the interaction between the strictly conserved AD50 and the active site residues TH57 and TS195 of thrombin (colors as in B). The invariant AR53 occupies the S1 specificity pocket. Water molecules are represented as red spheres. B, Right and C, Right provide schematic representations of the main anophelin⋅thrombin interactions. Hydrogen bonds are represented as dotted black lines. The figure was prepared with PyMOL (http://www.pymol.org) and PoseView (66).
Electron density maps were readily interpretable for mature anophelin residues AE32 to AP61 (Fig. 3A), but no significant density was detectable for the remaining N-terminal residues AA1 to AD31. However, the presence of the full-length inhibitor was detected by MS analysis of thrombin·anophelin crystals. Because the N-terminal region of anophelin is facing a solvent-filled channel, it is probably flexibly disordered. This N-terminal segment was similarly disordered in a second crystal form of the anophelin·thrombin complex solved at 3.2-Å resolution (space group H32; rmsd of 0.25 Å for 263 aligned Cα atoms) (Table 4) and also in this case, not constrained by crystal packing.
The most striking result of the structural analysis is that the cysteine-less inhibitor anophelin displays an unexpected mode of binding to thrombin, which is unique for a natural serine proteinase inhibitor. In the two independent crystal structures of the anophelin·thrombin complex, the inhibitor adopts a mostly extended conformation and binds to the proteinase in a reverse orientation (i.e., opposite to substrates) (Fig. S4). Anophelin interacts with both the active site region (through its C terminus) and the exosite I of the enzyme, establishing the specific contacts briefly described below (Fig. 3B and C and Fig. S4).
Anophelin Residues D32-F45 Block the Exosite I of Thrombin.
The N-terminal portion of anophelin spanning residues AY34 to AS40 binds across the exosite I of thrombin (Fig. 3B). This region displays a pseudohelical conformation mostly stabilized by a polar contact between the carboxylate of the central residue AE38 and the nitrogen amide of AY34 rather than main chain-to-main chain interactions. This particular arrangement allows the aliphatic part of the AE38 side chain to engage in Van der Waals interactions with the phenolic ring of the critical exosite I residue, TY76. The interaction of anophelin with the positively charged exosite I of thrombin is also strengthened by hydrophobic contacts of the well-conserved AL41 with the pocket formed by the side chains of TF34, TL65, TR67, TY76, and TI82. Specific binding is also determined by additional hydrogen bonds (e.g., the carboxamide group of thrombin’s TQ38 interacts with both the carbonyl oxygen of AS40 and the main-chain nitrogen atom of AS42), whereas a trapped water molecule bridges the AS42 carbonyl oxygen with both TT74 O and the TR67 guanidinium group.
The following portion of the inhibitor, connecting the exosite I- and the active site-contacting regions, associates closely with the proteinase; residues AN46 to AA48 form a short, distorted β-strand that pairs with the N terminus of the TE39-TS48 strand of thrombin. This extended conformation allows multiple water-mediated contacts of AT47 with proteinase residues TW141 and TN143. Furthermore, the side-chain carboxylate of AE43 accepts a hydrogen bond from the main-chain nitrogen of TY76. The phenolic side chain of AF45 is fully inserted in a shallow thrombin pocket, and it engages particularly in a stacking interaction with the recessed TF34, a central docking site for multiple thrombin interactors [e.g., protease-activated receptor 1 (PAR1) (31) and hirudin (32)] (Fig. S5).
Strictly Conserved AD50-AR53 Tetrapeptide of Anophelin Runs Across Thrombin's Active Site Cleft in a Reverse Direction to Substrates.
The most relevant interactions established between anophelin and thrombin are on the segment stretching from AD50 to AR53. A central role is played by the carboxylate of AD50, which accepts hydrogen bonds from the side chains of the catalytic residues TS195 and TH57 (NE2) and the main-chain nitrogen of TG193 (Fig. 3C). These interactions result in disruption of the critical hydrogen bond between the side chains of TS195 and TH57 (NE2 displaced by 0.74 Å and OG displaced by 1.36 Å on 180° rotation in relation to free thrombin) (30), with the concomitant disturbance of the characteristic serine proteinase catalytic triad charge–relay system. Accordingly, mutation of this residue to alanine essentially abolished binding to immobilized thrombin (Table 3 and Fig. S3) and stabilization as measured by differential scanning fluorimetry (DSF) (Fig. 2A and Table S1). Furthermore, the AD50A mutant did not display significant anticoagulant activity or inhibited γ-thrombin (Tables 1 and 2 and Fig. S2). Even the conservative replacements of AD50 by glutamate or asparagine led to a dramatic decrease in inhibition efficiency both in terms of TT prolongation and inhibition kinetics (Tables 1, 2, and 3).
As suggested by mutational studies (see above), the experimental complex structure revealed that thrombin’s S1 pocket is occupied by AR53. However, the reverse-binding mode of anophelin imposes significant differences in the interactions of this residue with the proteinase compared with substrates/substrate-like inhibitors (Fig. S5A). The NH1 atom of the AR53 guanidinium group donates hydrogen bonds to TD189 OD2 and the carbonyl oxygen of TG219, whereas the NH2 nitrogen engages in a water-mediated hydrogen bond with TF227 O (Fig. 3C). Furthermore, the AR53 carbonyl oxygen is hydrogen-bonded to the main-chain nitrogen of TG219. There is also a unique water-mediated interaction between AR53 NE and the catalytic TS195 OG. The extent and quality of these interactions fully corroborate the results of the mutagenesis studies described above.
The S2 hydrophobic pocket of thrombin is partially occupied by AP51, which slots between TH57 and TW60D, and by AG52, which establishes Van der Waals contacts with TL99 and TW215. The position of this strictly conserved glycine, which is further stabilized by hydrogen bonds between its main-chain nitrogen atom and the carboxylate of AD50 and between its carboxyl oxygen and the main-chain N of TG216, forces AP51 toward the 60-loop (Fig. S5A). An additional interaction that exploits thrombin’s characteristic residues is made by the side chain of the gate-keeping TE192, which flips to accept two hydrogen bonds from AD50 N and AR54 NE. AnophelinAa R54A and R54N mutants, the latter mimicking the primary sequence of anophelinAg, are likely unable to establish these interactions to TE192 and therefore, display relatively high Ki values. Unsurprisingly, this effect is even more dramatic for the reversal-of-charge mutant R54E (see above). The following AL55 occupies the aryl binding site (S4) of the proteinase, making important Van der Waals interactions with TL99, TI174, and TW215.
The AL55-AG58 residues of anophelinAa make a sharp turn to leave the active site cleft of the proteinase. In this manner, the C-terminal region of the inhibitor runs parallel to and shields from bulk solvent the side chain of AR54, and in addition, it engages in several mostly polar interactions with thrombin. The side chains of AE57 and AK60 form salt bridges to the oppositely charged TK224 and TE146, respectively, whereas the guanidinium group of TR221A hydrogen bonds to the carbonyl oxygen of AE57. However, these solvent-exposed polar contacts seem to only marginally contribute to complex stability, because these residues are not conserved in anophelins from Old World mosquitoes.
C-Terminal Segment of Anophelin Is Sufficient for Tight Thrombin Inhibition.
In the two independent crystal structures of the human α-thrombin·anophelin complex, only the C-terminal portion of the inhibitor, comprising residues AE32 to AP61, was fully defined by electron density. To further verify that this segment was sufficient for thrombin inhibition, the corresponding synthetic peptide (anophelin32–61) was thoroughly characterized. The anticoagulant and antiamidolytic activities of anophelin32–61 were only three- and ninefold lower than the activites of full-length anophelinAa (Tables 1 and 2). The C-terminal inhibitor fragment also bound immobilized α-thrombin with an affinity comparable with the affinity of the full-length form (Fig. 2B and Table 3), and it had similar effects on proteinase stabilization (Fig. 2A and Table S1).
However, the 31-residue-long N-terminal segment (anophelin1–31) neither prolonged TT nor inhibited α- or γ-thrombin in vitro (Tables 1 and 2 and Fig. S2). Additionally, no interaction of anophelin1–31 with α-thrombin was detectable either by DSF assays or SPR analyses (Fig. 2A, Table 3, Fig. S3, and Table S1).
Discussion
We have performed a thorough structural and functional analysis of anophelin, a unique anticoagulant from the malaria vector, disclosing an unprecedented molecular mechanism of thrombin inhibition. Studies of the salivary gland transcriptomes of hematophagous animals have revealed that anophelins are exclusively found in Anopheles mosquitoes (1, 2, 33, 34), being absent from the related culicine species, from which they diverged ∼150 Mya (35). Despite significant sequence divergence between New and Old World species, consistent with the evolutionary separation of the two anopheline branches ∼95 Mya (35), thrombin-inhibiting activity is retained in anophelin homologs from A. albimanus and A. gambiae (6, 7, 9). Here, we show that all known family I77 members are effective thrombin-targeting anticoagulants, and we identify the critical residues responsible for the inhibitory activity of anophelins.
Mechanism of Thrombin Targeting by I77 Family Inhibitors.
The C-terminal moiety of anophelin establishes a multitude of interactions with the exosite I and the active site of thrombin (Fig. 3A and Fig. S5C). In contrast to a previous suggestion (9), the N-terminal hirudin-like motif of anophelin (residues AG7 to AD15 in anophelinAa) is not involved in exosite I binding. Instead, this important recognition surface of thrombin is occupied by the acidic AE32-AF45 segment. Similar to other thrombin complexes with substrates, cofactors, and inhibitors (13, 14, 20–23) and despite complementary electrostatic surface potentials, nonpolar interactions dominate at this interface.
Pivotal to the inhibition mechanism of I77 family members is the unexpected reverse-binding mode to thrombin (Figs. 3A and 4). Curiously, this binding mechanism is distantly reminiscent of the interaction of X-linked inhibitor of apoptosis with the apoptotic caspases 3 and 7 and the contact between the pro- and catalytic domains of papain-like cysteine proteinases, such as cathepsins B and L (36–38). In contrast to the intrinsically disordered anophelins, the extensive contacts between the folded scaffold of X-linked inhibitor of apoptosis and the target proteinases are essential for placing an 8-residue-long segment across the active site of the enzymes, where it establishes a limited number of interactions. Contrary to anophelin, there are no interactions with the catalytic residues, the primed subsites, or even the S1 and S2 specificity subsites. In the case of procathepsins, the C-terminal prodomain residues pass across the substrate-binding cleft in a reverse orientation (39, 40). However, also in this case, binding is dependent on the presentation of these residues by a highly structured moiety, and even limited N-terminal truncation of the prodomain results in a dramatic decrease of inhibitory potency (38). Anophelin displays a considerably more extensive interaction with the active site cleft of the target proteinase. The most important contribution to the binding energy results from the insertion of the AD50-AR54 segment into the active site cleft of the enzyme, with AR53 occupying thrombin’s acidic S1 specificity pocket. Indeed, single-point mutations to alanine convert a tight binding into a poor competitive inhibitor (D50A) or abolish inhibitory activity altogether (R53A) (Table 2). In particular, the multiple interactions established by the AD50 carboxylate with active site residues TH57 and TS195 significantly account to the binding efficiency as indicated by the D50A mutant (ΔΔG°WT-D50A ∼ −9,600 cal·mol−1).
Fig. 4.

Schematic representation of the unique mechanism of thrombin recognition by family I77 inhibitors compared with substrates and other inhibitors. The thrombin molecule is represented as an orange ellipse, with exosites in blue and the active site in red. (A) The natural substrate PAR1 binds to the active site, and the region C terminus to the scissile bond interacts with the exosite I of the proteinase. (B) Thrombin-activated coagulation factors V and VIII bind in a canonical way to the active site of the enzyme, interacting with both exosites through the region upstream to the cleavable bond. (C) The natural anticoagulant from Hirudo medicinalis, hirudin, interacts with the exosite I of thrombin through its C-terminal acidic segment and blocks the active site of the enzyme with its N terminus. (D) MEROPS family I77 inhibitors from the Anopheles mosquitoes bind to thrombin in a reverse orientation relative to natural substrates: the N-terminal portion of anophelin recognizes the exosite I of the enzyme, whereas the C-terminal acidic segment binds to the active site region of the proteinase.
The N-terminal portion of the inhibitor (AA1-AD31) does not interact with thrombin in the two independent experimental structures reported here, although there is no impairment arising from the crystal packing. In addition, deletion of this segment only marginally reduced anophelin’s inhibitory potency in vitro and had essentially no effect in binding to immobilized thrombin. Finally, the isolated N-terminal peptide showed no effect on TT, did not impair thrombin activity against chromogenic substrates, and did not bind to the immobilized proteinase. Altogether, these findings exclude a functional role of anophelin’s N-terminal moiety in its antithrombin activity. Several not mutually exclusive explanations can be put forward to explain the striking conservation of this region across I77 family members. In vivo, under high flow conditions, this negatively charged segment might help direct the inhibitor to active thrombin. Alternatively, in thrombin-bound anophelin, this exposed polypeptide might interact with other hitherto unidentified element(s) of the hemostatic system.
Role of Flexibility in Thrombin⋅Anophelin Complex Formation.
As indicated by the CD analysis, anophelin is essentially disordered in solution, and it adopts a definite 3D structure only in complex with the target proteinase (Fig. S1), similar to what was previously observed for IA3, an aspartic proteinase inhibitor from Saccharomyces cerevisiae and the cysteine proteinase-targeting calpastatin (41–43). However, recent evidence indicates that thrombin itself is more flexible than previously thought and in particular, that exosite I is in dynamic equilibrium in the free proteinase (44). These findings indicate that formation of a stable complex between thrombin and anophelin deviates significantly from the classical lock-and-key model, and it involves simultaneous stabilization of their 3D structures. Complex formation, in turn, can only occur because the extent and quality of interactions between the two moieties revealed in the current crystal structures exceed the entropy loss caused by reduced degrees of freedom of both thrombin and anophelin. This feature needs to be considered for the design of novel antithrombotics, and it is likely also relevant for other members of the family of trypsin-like serine proteinases.
Comparison with Other Thrombin Inhibitors.
Although I77 inhibitors display an original mechanism of serine proteinase inhibition, several structural aspects of the interaction are reminiscent of those aspects previously observed in other thrombin inhibitors. Despite the reverse direction of the respective polypeptide chains, some relevant anophelin interactions resemble those interactions established by the canonical I14 family inhibitor, hirudin, with the regulatory exosite I (19, 32, 45). In particular, residue AE43 is topologically equivalent to HE57 in hirudin, also establishing a direct polar interaction with the proteinase (Fig. S5C). In addition, the phenolic side chain of AF45 is nearly superposable to the side chain of hirudin HF56, therefore preserving the edge-on stacking interaction with TF34 (19). Also substantiating the importance of this interaction, similar contacts are established not only by the recently reported cleavable inhibitor variegin (25) (VF20) but also in thrombin complexes with two critical substrates, PAR1 (31) (PY52) and factor V (14) (hFVF668).
Regarding the interactions made by the conserved AD50-AR54 segment, mechanistically similar contacts have been observed for both noncanonical inhibitors of family I2, ornithodorin and boophilin (20, 23), and I14 family members (19, 24, 32, 45). Notably, the interaction observed between the S1-occupying arginine residue NH1 atom and both TD189 OD2 and TG219 O is preserved in boophilin (23) and hemadin (24) (Fig. S5 A and B).
Anophelin as a Model for Other Cysteine-Less Thrombin Inhibitors.
It is noteworthy that several thrombin inhibitors isolated from hematophagous animals share with anophelin important features, such as the preponderance of acidic residues and a complete lack of stabilizing disulfide linkages, but they are, nevertheless, not cleaved by the target proteinase. This is, for instance, the case of a potent inhibitor isolated from the tsetse fly, Glossina morsitans morsitans (46, 47). Inspection of the amino acid sequence of the tsetse inhibitor (TTI; family I76 in the MEROPS database) reveals that the C-terminal end, TTIP28-TTIL32, is closely related to the active site-targeting region of anophelin (numbers for mature TTI) (47) (Fig. 1), and therefore, it could bind thrombin essentially as revealed in the current structures. Loss of favorable electrostatic interactions with the S1 pocket because of the replacement of residue AR53 by TTII30 might be compensated by more extensive contacts with exosite I. In this regard, it is noteworthy that recombinant and synthetic TTIs were much less potent thrombin inhibitors than the natural product (47) (Ki values of 150 nM and 584 fM, respectively). This discrepancy has been attributed to a nonidentified posttranslational modification of natural TTI. Sulfation of tyrosine residues at positions 9 and 12 of mature TTI, as predicted by Sulfinator (48), seems to be the likely modification responsible for enhanced affinity of the natural TTI to the procoagulant proteinase. The positive effect of tyrosine sulfation on hirudin binding to thrombin has been well-established (45).
Implications for the Design of Antithrombotics.
To date, hirudin is the only natural proteinaceous thrombin inhibitor approved for clinical use. Because of its relatively large size and narrow therapeutic window, considerable efforts have been made to simplify hirudin’s scaffold. Briefly, two major paths have been followed, both of which graft the exosite I-binding C-terminal tail of hirudin to distinct active site-targeting moieties (45). In these hirudomimetics, either reverse binding to the active site is preserved at the expense of a long connecting linker or a substrate-like binding moiety is used, requiring nonpeptidic linkages to the shorter linker to circumvent degradation by thrombin. The molecular mechanism of anophelin, here reported, provides an elegant way to bridge exosite I and parallel active site-binding moieties with the shortest linker described to date, yielding highly specific and potent inhibition, while assuring resistance to proteolysis by thrombin.
Methods
Anophelin Production.
A synthetic gene coding for mature A. albimanus anophelin (anophelinAa) was generated by PCR-based gene assembly (49) and cloned into pTYB11 (New England BioLabs) in fusion with an N-terminal intein tag. Expression constructs of anophelinAa point mutants and the truncated anophelin1–31 form were generated by site-directed mutagenesis. Similar constructs coding for anophelin homologs were synthesized at GenScript.
Escherichia coli ER2566 cells (New England BioLabs) transformed with pTYB11-anophelin plasmids were grown in LB medium supplemented with ampicillin to OD600 = 0.5, and expression was induced with 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). After overnight growth at 18 °C, cells were lysed by sonication in 20 mM Tris⋅HCl (pH 8.5), 500 mM NaCl, and 1 mM EDTA (buffer A). Clarified protein extracts were loaded onto chitin-agarose columns (New England BioLabs) preequilibrated with buffer A and eluted with buffer A supplemented with 50 mM DTT. Anophelin-containing fractions were concentrated and further purified on a HiPrep 16/60 Sephacryl S-100 column (GE Healthcare) preequilibrated with 20 mM Tris⋅HCl (pH 8.0) and 150 mM NaCl. Protein authenticity was verified by N-terminal sequencing.
A peptide corresponding to the AE32-AP61 sequence of anophelinAa (anophelin32–61) was obtained from GenScript.
CD Spectroscopy.
Far-UV region (190–260 nm) spectra were recorded in a 1.0-mm-path quartz cuvette at 20 °C with a Peltier temperature-controlled cell holder-equipped Jasco J-815 spectropolarimeter. Secondary structure content was estimated using DichroWeb (50).
MS Analysis.
MALDI TOF/TOF MS analyses were performed using a 4700 Proteomics Analyzer (AB SCIEX) in the linear positive mode. Samples were desalted and concentrated in C18 chromatographic columns (ZipTip; Millipore) according to the manufacturer’s protocol and eluted in the presence of appropriate MALDI matrices.
Thrombin Inhibition Assays.
The inhibition of the amidolytic activity of titrated (51) bovine α-thrombin (GE Healthcare) was followed spectrophotometrically using Tos-Gly-Pro-Arg-p-nitroanilide (Roche) as chromogenic substrate. The inhibition constants (Ki) of all anophelin variants were determined according to a tight-binding or competitive inhibitor model. Tight-binding inhibitors bind reversibly to and inhibit their target enzyme with a binding energy much greater than substrates (52). Therefore, an intermediate degree of inhibition is observed when the concentration of inhibitor is comparable with the concentration of enzyme necessary to observe a measurable reaction rate. As a consequence, a significant fraction of the total inhibitor is bound to the enzyme, and the concentration of free inhibitor is not the same as the concentration of total inhibitor, as is assumed for classical inhibitors. Experimentally, substrate depletion in tight-binding inhibition is accounted for by the Morrison equation (53) (Eq. 1), where v is the inhibited steady-state velocity, v0 is the enzyme velocity with no inhibitor, [E] is the concentration of enzyme, [I] is the concentration of inhibitor, KM is the Michaelis constant for substrate S (7,721 nM), [S] is the concentration of substrate, and Ki is the inhibition constant (Eq. 1):
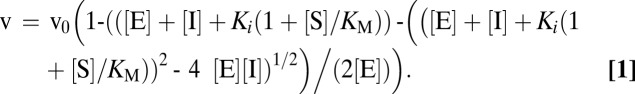 |
Tight-binding assays were performed using 0.5 nM bovine α-thrombin, 100 μM substrate, and increasing concentrations (0–80 nM) of inhibitor. The inhibition constant Ki was determined by fitting the inhibited steady-state velocity data to the Morrison equation (Eq. 1) using GraphPad Prism 5.0 (GraphPad Software).
By contrast, the binding energy of competitive inhibitors is comparable with the binding energy of substrates, and therefore, the experimental concentration range of the inhibitor is within a few orders of magnitude of the concentration of the substrate and much larger than the concentration of the enzyme. Competitive inhibition assays were performed using 0.5 or 1 nM bovine α-thrombin and increasing concentrations (0–100 μM) of substrate in the presence of inhibitor (0–500 nM). The inhibition constant Ki was determined by fitting the observed initial velocity data to the competitive inhibition model (Eq. 2) using GraphPad Prism 5.0 (GraphPad Software), where v is the initial rate of the reaction, vmax is the maximum velocity of the reaction, KM is the Michaelis constant for substrate S (7,721 nM), [S] is the concentration of substrate, and Ki is the inhibition constant (Eq. 2):
All reactions were carried out at 37 °C in 50 mM Tris⋅HCl (pH 8.0), 100 mM NaCl, and 1 mg/mL BSA in 96-well microtiter plates. Reaction progress was monitored at 405 nm for 1–2 h on a Synergy2 Multimode microplate reader (Biotek).
Dose–response curves (percentage of thrombin inhibition at 120 min as a logarithmic function of the concentration of inhibitor) were used to determine the IC50 values using GraphPad Prism 5.0 (GraphPad Software).
The inhibition of the amidolytic activity of human γ-thrombin (Haematologic Technologies) was measured as described above for bovine α-thrombin. The final concentrations of substrate, inhibitor, and γ-thrombin were 95 µM, from 0 to 5 µM, and 0.5 nM, respectively. For each inhibitor, at least two independent experiments with duplicate reactions were performed together with control reactions in the absence of enzyme. For all curves, the goodness of fitting parameter R2 was between 0.964 and 0.998.
DSF Assays.
The thermal stability of thrombin·anophelin complexes was assessed by following SYPRO Orange (Invitrogen) fluorescence at 585 nm (excitation = 545 nm) on an iQ5 Real-Time PCR System (BioRad) as a function of temperature (20–95 °C in 0.5 °C/15-s steps); 20 pmol human α-thrombin (Haematologic Technologies) were mixed with anophelin variants (0–100 pmol) in a 20 μL reaction volume and incubated on ice for 15 min before SYPRO Orange addition to a final concentration of 5× . Duplicate reactions were performed, and controls included no enzyme and no dye. The melting curves were analyzed using CFX Manager (BioRad), and the melting temperature (Tm) was calculated as the inflection point of the curve.
SPR.
SPR experiments using CM5 chips were performed at 25 °C on a Biacore 3000 (GE Healthcare) with 10 mM Hepes (pH 7.4), 150 mM NaCl, 3 mM EDTA, and 0.005% (vol/vol) P20. Kinetic experiments were performed on flow cells with different amine-coupled human α-thrombin densities and using at least five different analyte concentrations covering three orders of magnitude. D-Phe-L-ProL-Arg-chloromethyl ketone-inhibited bovine trypsin (Roche) was immobilized to the reference channel as control for nonspecific binding events and bulk refractive index effects. The association and dissociation phases lasted 3 min each, and surface regeneration was achieved with a 5-µL pulse of 10 mM sodium acetate (pH 4.5) and 2 M MgCl2. Kinetic parameters (ka and kd) and equilibrium dissociation constants (KD) were determined by nonlinear fitting of the sensorgrams to a 1:1 interaction model (Langmuir fitting) using BIAevaluation 4.1.1.
TT Assays.
The anticoagulant activity of anophelin mutants was determined by measuring their ability to prolong clotting of human plasma. Human plasma (800 µL) was mixed with 200 µL recombinant anophelin solutions [0–10 μM final concentration in 20 mM Tris⋅HCl (pH 8.0) and 100 mM NaCl], and TT was measured at BM Análises Clínicas following standard protocols.
Crystallization of Human α-Thrombin·Anophelin Complexes.
Human α-thrombin was mixed with a fivefold molar excess of anophelinAa, incubated on ice for 1 h, and concentrated by ultrafiltration. Crystals grew at 20 °C using the sitting-drop method at the High Throughput Crystallization Laboratory of the European Molecular Biology Laboratory (Grenoble, France). Crystals obtained using 100 mM Hepes (pH 7.5) and 1.4 M trisodium citrate belong to space group P3112, whereas crystals grown using 100 mM sodium acetate (pH 4.5) and 3 M NaCl belong to space group H32.
Data Collection and Processing.
Crystals cryoprotected in mother liquor supplemented with 10–15% glycerol were flash-cooled in liquid nitrogen, and diffraction data were collected at beam lines ID23-EH1 (54) (space group P3112; wavelength = 0.9792 Å) or ID29 (55) (space group H32; wavelength = 0.9763 Å) of the European Synchrotron Radiation Facility (ESRF; Grenoble, France). Data were processed with XDS (56) and scaled using XSCALE (57).
Structure Determination and Refinement.
The structure of the human α-thrombin·anophelin complex (space group P3112) was solved by molecular replacement with Phaser (58) using the coordinates of free human α-thrombin (Protein Data Bank ID code 3U69) (30, 59). The refined model was used as the search model to solve the structure of the other crystal form (space group H32). Alternating cycles of model building with Coot (60) and refinement with PHENIX (61) were performed until model completion. Both complex structures had good stereochemistry, with 100% and 99.0% of the residues in the allowed regions of the Ramachandran plot for the high- and low-resolution models, respectively.
Supplementary Material
Acknowledgments
We acknowledge the ESRF for provision of synchrotron radiation facilities and thank the ESRF staff for help with data collection. Reinhard Mentele (MPI für Biochemie, Martinsried) is acknowledged for N-terminal sequencing of recombinant anophelin, and Hugo Osório (Instituto de Patologia e Imunologia Molecular da Universidade do Porto, Porto) is acknowledged for MS analysis. Transnational Access to the High Throughput Crystallization Laboratory of the European Molecular Biology Laboratory Grenoble Outstation was supported by the European Community-Seventh Framework Program (FP7/2007-2013) Grant Protein Production Platform (PCUBE Agreement No. 227764). This work was funded in part by Spanish Ministry of Science and Innovation Grant SAF2010-15668, by Fundo Europeu de Desenvolvimento Regional (FEDER) funds through the Operational Competitiveness Programme (COMPETE), by national funds through Fundação para a Ciência e a Tecnologia (Portugal) under projects FCOMP-01-0124-FEDER-007076 (PTDC/BIA-PRO/70627/2006) and FCOMP-01-0124-FEDER-022718 (PEst-C/SAU/LA0002/2011), and postdoctoral fellowship SFR/BPD/46722/2008 (to A.C.F.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The crystallographic atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4E05 and 4E06).
See Author Summary on page 21191 (volume 109, number 52).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1211614109/-/DCSupplemental.
References
- 1.Valenzuela JG, Francischetti IMB, Pham VM, Garfield MK, Ribeiro JMC. Exploring the salivary gland transcriptome and proteome of the Anopheles stephensi mosquito. Insect Biochem Mol Biol. 2003;33(7):717–732. doi: 10.1016/s0965-1748(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 2.Calvo E, Pham VM, Marinotti O, Andersen JF, Ribeiro JM. The salivary gland transcriptome of the neotropical malaria vector Anopheles darlingi reveals accelerated evolution of genes relevant to hematophagy. BMC Genomics. 2009;10:57. doi: 10.1186/1471-2164-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calvo E, Dao A, Pham VM, Ribeiro JMC. An insight into the sialome of Anopheles funestus reveals an emerging pattern in anopheline salivary protein families. Insect Biochem Mol Biol. 2007;37(2):164–175. doi: 10.1016/j.ibmb.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization . World Malaria Report. Geneva: World Health Organization; 2010. [Google Scholar]
- 5.Rawlings ND, Barrett AJ, Bateman A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40(Database issue):D343–D350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valenzuela JG, Francischetti IM, Ribeiro JM. Purification, cloning, and synthesis of a novel salivary anti-thrombin from the mosquito Anopheles albimanus. Biochemistry. 1999;38(34):11209–11215. doi: 10.1021/bi990761i. [DOI] [PubMed] [Google Scholar]
- 7.Francischetti IM, Valenzuela JG, Ribeiro JM. Anophelin: Kinetics and mechanism of thrombin inhibition. Biochemistry. 1999;38(50):16678–16685. doi: 10.1021/bi991231p. [DOI] [PubMed] [Google Scholar]
- 8.Arcà B, et al. An updated catalogue of salivary gland transcripts in the adult female mosquito, Anopheles gambiae. J Exp Biol. 2005;208(Pt 20):3971–3986. doi: 10.1242/jeb.01849. [DOI] [PubMed] [Google Scholar]
- 9.Ronca R, et al. The Anopheles gambiae cE5, a tight- and fast-binding thrombin inhibitor with post-transcriptionally regulated salivary-restricted expression. Insect Biochem Mol Biol. 2012;42(9):610–620. doi: 10.1016/j.ibmb.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixit R, et al. Salivary gland transcriptome analysis during Plasmodium infection in malaria vector Anopheles stephensi. Int J Infect Dis. 2009;13(5):636–646. doi: 10.1016/j.ijid.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 11.Das S, et al. Transcriptomic and functional analysis of the Anopheles gambiae salivary gland in relation to blood feeding. BMC Genomics. 2010;11:566. doi: 10.1186/1471-2164-11-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stubbs MT, Bode W. A player of many parts: The spotlight falls on thrombin’s structure. Thromb Res. 1993;69(1):1–58. doi: 10.1016/0049-3848(93)90002-6. [DOI] [PubMed] [Google Scholar]
- 13.Fuentes-Prior P, et al. Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature. 2000;404(6777):518–525. doi: 10.1038/35006683. [DOI] [PubMed] [Google Scholar]
- 14.Corral-Rodríguez MA, Bock PE, Hernández-Carvajal E, Gutiérrez-Gallego R, Fuentes-Prior P. Structural basis of thrombin-mediated factor V activation: The Glu666-Glu672 sequence is critical for processing at the heavy chain-B domain junction. Blood. 2011;117(26):7164–7173. doi: 10.1182/blood-2010-10-315309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsiang M, et al. Functional mapping of the surface residues of human thrombin. J Biol Chem. 1995;270(28):16854–16863. doi: 10.1074/jbc.270.28.16854. [DOI] [PubMed] [Google Scholar]
- 16.Myles T, Yun TH, Leung LLK. Structural requirements for the activation of human factor VIII by thrombin. Blood. 2002;100(8):2820–2826. doi: 10.1182/blood-2002-03-0843. [DOI] [PubMed] [Google Scholar]
- 17.Dumas JJ, Kumar R, Seehra J, Somers WS, Mosyak L. Crystal structure of the GpIbalpha-thrombin complex essential for platelet aggregation. Science. 2003;301(5630):222–226. doi: 10.1126/science.1083917. [DOI] [PubMed] [Google Scholar]
- 18.Celikel R, et al. Modulation of α-thrombin function by distinct interactions with platelet glycoprotein Ibalpha. Science. 2003;301(5630):218–221. doi: 10.1126/science.1084183. [DOI] [PubMed] [Google Scholar]
- 19.Rydel TJ, et al. The structure of a complex of recombinant hirudin and human α-thrombin. Science. 1990;249(4966):277–280. doi: 10.1126/science.2374926. [DOI] [PubMed] [Google Scholar]
- 20.van de Locht A, et al. The ornithodorin-thrombin crystal structure, a key to the TAP enigma? EMBO J. 1996;15(22):6011–6017. [PMC free article] [PubMed] [Google Scholar]
- 21.van de Locht A, et al. Two heads are better than one: Crystal structure of the insect derived double domain Kazal inhibitor rhodniin in complex with thrombin. EMBO J. 1995;14(21):5149–5157. doi: 10.1002/j.1460-2075.1995.tb00199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuentes-Prior P, et al. Structure of the thrombin complex with triabin, a lipocalin-like exosite-binding inhibitor derived from a triatomine bug. Proc Natl Acad Sci USA. 1997;94(22):11845–11850. doi: 10.1073/pnas.94.22.11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macedo-Ribeiro S, et al. Isolation, cloning and structural characterisation of boophilin, a multifunctional Kunitz-type proteinase inhibitor from the cattle tick. PLos One. 2008;3:e1624. doi: 10.1371/journal.pone.0001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richardson JL, et al. Crystal structure of the human α-thrombin-haemadin complex: An exosite II-binding inhibitor. EMBO J. 2000;19(21):5650–5660. doi: 10.1093/emboj/19.21.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koh CY, et al. Crystal structure of thrombin in complex with S-variegin: Insights of a novel mechanism of inhibition and design of tunable thrombin inhibitors. PLoS One. 2011;6(10):e26367. doi: 10.1371/journal.pone.0026367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang JY. The structures and proteolytic specificities of autolysed human thrombin. Biochem J. 1986;240(3):797–802. doi: 10.1042/bj2400797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corral-Rodríguez MA, Macedo-Ribeiro S, Barbosa Pereira PJ, Fuentes-Prior P. Tick-derived Kunitz-type inhibitors as antihemostatic factors. Insect Biochem Mol Biol. 2009;39(9):579–595. doi: 10.1016/j.ibmb.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Bode W, Huber R. Structural basis of the endoproteinase-protein inhibitor interaction. Biochim Biophys Acta. 2000;1477(1–2):241–252. doi: 10.1016/s0167-4838(99)00276-9. [DOI] [PubMed] [Google Scholar]
- 29.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27(2):157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 30.Figueiredo AC, et al. Rational design and characterization of D-Phe-Pro-D-Arg-derived direct thrombin inhibitors. PLoS One. 2012;7(3):e34354. doi: 10.1371/journal.pone.0034354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gandhi PS, Chen Z, Di Cera E. Crystal structure of thrombin bound to the uncleaved extracellular fragment of PAR1. J Biol Chem. 2010;285(20):15393–15398. doi: 10.1074/jbc.M110.115337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu CC, Brustad E, Liu W, Schultz PG. Crystal structure of a biosynthetic sulfo-hirudin complexed to thrombin. J Am Chem Soc. 2007;129(35):10648–10649. doi: 10.1021/ja0735002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Francischetti IM, Valenzuela JG, Pham VM, Garfield MK, Ribeiro JM. Toward a catalog for the transcripts and proteins (sialome) from the salivary gland of the malaria vector Anopheles gambiae. J Exp Biol. 2002;205(Pt 16):2429–2451. doi: 10.1242/jeb.205.16.2429. [DOI] [PubMed] [Google Scholar]
- 34.Ribeiro JMC, Mans BJ, Arcà B. An insight into the sialome of blood-feeding Nematocera. Insect Biochem Mol Biol. 2010;40(11):767–784. doi: 10.1016/j.ibmb.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krzywinski J, Grushko OG, Besansky NJ. Analysis of the complete mitochondrial DNA from Anopheles funestus: An improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution. Mol Phylogenet Evol. 2006;39(2):417–423. doi: 10.1016/j.ympev.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 36.Riedl SJ, et al. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104(5):791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 37.Chai J, et al. Structural basis of caspase-7 inhibition by XIAP. Cell. 2001;104(5):769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 38.Carmona E, et al. Potency and selectivity of the cathepsin L propeptide as an inhibitor of cysteine proteases. Biochemistry. 1996;35(25):8149–8157. doi: 10.1021/bi952736s. [DOI] [PubMed] [Google Scholar]
- 39.Coulombe R, et al. Structure of human procathepsin L reveals the molecular basis of inhibition by the prosegment. EMBO J. 1996;15(20):5492–5503. [PMC free article] [PubMed] [Google Scholar]
- 40.Cygler M, et al. Structure of rat procathepsin B: Model for inhibition of cysteine protease activity by the proregion. Structure. 1996;4(4):405–416. doi: 10.1016/s0969-2126(96)00046-9. [DOI] [PubMed] [Google Scholar]
- 41.Li M, et al. The aspartic proteinase from Saccharomyces cerevisiae folds its own inhibitor into a helix. Nat Struct Biol. 2000;7(2):113–117. doi: 10.1038/72378. [DOI] [PubMed] [Google Scholar]
- 42.Moldoveanu T, Gehring K, Green DR. Concerted multi-pronged attack by calpastatin to occlude the catalytic cleft of heterodimeric calpains. Nature. 2008;456(7220):404–408. doi: 10.1038/nature07353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanna RA, Campbell RL, Davies PL. Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature. 2008;456(7220):409–412. doi: 10.1038/nature07451. [DOI] [PubMed] [Google Scholar]
- 44.Lechtenberg BC, Johnson DJD, Freund SMV, Huntington JA. NMR resonance assignments of thrombin reveal the conformational and dynamic effects of ligation. Proc Natl Acad Sci USA. 2010;107(32):14087–14092. doi: 10.1073/pnas.1005255107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corral-Rodríguez MA, Macedo-Ribeiro S, Pereira PJB, Fuentes-Prior P. Leech-derived thrombin inhibitors: From structures to mechanisms to clinical applications. J Med Chem. 2010;53(10):3847–3861. doi: 10.1021/jm901743x. [DOI] [PubMed] [Google Scholar]
- 46.Cappello M, et al. Isolation and characterization of the tsetse thrombin inhibitor: A potent antithrombotic peptide from the saliva of Glossina morsitans morsitans. Am J Trop Med Hyg. 1996;54(5):475–480. doi: 10.4269/ajtmh.1996.54.475. [DOI] [PubMed] [Google Scholar]
- 47.Cappello M, et al. Tsetse thrombin inhibitor: Bloodmeal-induced expression of an anticoagulant in salivary glands and gut tissue of Glossina morsitans morsitans. Proc Natl Acad Sci USA. 1998;95(24):14290–14295. doi: 10.1073/pnas.95.24.14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monigatti F, Gasteiger E, Bairoch A, Jung E. The Sulfinator: Predicting tyrosine sulfation sites in protein sequences. Bioinformatics. 2002;18(5):769–770. doi: 10.1093/bioinformatics/18.5.769. [DOI] [PubMed] [Google Scholar]
- 49.Stemmer WPC, Crameri A, Ha KD, Brennan TM, Heyneker HL. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene. 1995;164(1):49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]
- 50.Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers. 2008;89(5):392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 51.Chase T, Jr, Shaw E. p-Nitrophenyl-p’-guanidinobenzoate HCl: A new active site titrant for trypsin. Biochem Biophys Res Commun. 1967;29(4):508–514. doi: 10.1016/0006-291x(67)90513-x. [DOI] [PubMed] [Google Scholar]
- 52.Szedlacsek SE, Duggleby RG. Kinetics of slow and tight-binding inhibitors. Methods Enzymol. 1995;249:144–180. doi: 10.1016/0076-6879(95)49034-5. [DOI] [PubMed] [Google Scholar]
- 53.Williams JW, Morrison JF. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 1979;63:437–467. doi: 10.1016/0076-6879(79)63019-7. [DOI] [PubMed] [Google Scholar]
- 54.Nurizzo D, et al. The ID23-1 structural biology beamline at the ESRF. J Synchrotron Radiat. 2006;13(Pt 3):227–238. doi: 10.1107/S0909049506004341. [DOI] [PubMed] [Google Scholar]
- 55.de Sanctis D, et al. ID29: A high-intensity highly automated ESRF beamline for macromolecular crystallography experiments exploiting anomalous scattering. J Synchrotron Radiat. 2012;19(Pt 3):455–461. doi: 10.1107/S0909049512009715. [DOI] [PubMed] [Google Scholar]
- 56.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carvalho Figueiredo A, Clement CC, Philipp M, Barbosa Pereira PJ. Crystallization and preliminary crystallographic characterization of three peptidic inhibitors in complex with α-thrombin. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67(Pt 1):54–58. doi: 10.1107/S1744309110043472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 61.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 4):352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arcá B, et al. Trapping cDNAs encoding secreted proteins from the salivary glands of the malaria vector Anopheles gambiae. Proc Natl Acad Sci USA. 1999;96(4):1516–1521. doi: 10.1073/pnas.96.4.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mongin E, Louis C, Holt RA, Birney E, Collins FH. The Anopheles gambiae genome: An update. Trends Parasitol. 2004;20(2):49–52. doi: 10.1016/j.pt.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 64.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 65.Bond CS, Schüttelkopf AW. ALINE: A WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 5):510–512. doi: 10.1107/S0907444909007835. [DOI] [PubMed] [Google Scholar]
- 66.Stierand K, Rarey M. Drawing the PDB: Protein−ligand complexes in two dimensions. ACS Med Chem Lett. 2010;1(9):540–545. doi: 10.1021/ml100164p. [DOI] [PMC free article] [PubMed] [Google Scholar]
