

Abstract
A novel competitive enzyme-linked immunosorbent assay (cELISA) was developed and evaluated for detection of antibodies against Francisella tularensis in humans. The assay is based on the ability of serum antibodies to inhibit the binding of monoclonal antibodies (MAbs) directed against F. tularensis lipopolysaccharide antigens. The assay was evaluated using serum samples of tularemia patients, inactivated F. tularensis-immunized rabbits, and F. tularensis-infected mice. Antibodies against F. tularensis were successfully detected in serum samples of tularemia patients as well as the immunized and infected animals. The cELISA method was compared to indirect ELISA (iELISA) and the commonly used microagglutination test (MA) using serum samples of 19 tularemia patients and 50 healthy individuals. The sensitivity and specificity of cELISA were 93.9 and 96.1%, respectively, in comparison to the iELISA. MA was less sensitive than cELISA with a sensitivity and specificity of only 81.8 and 98.0%, respectively. A high degree of correlation (R2 = 0.8226) was observed between cELISA and iELISA results. The novel cELISA developed in this study appears to be highly sensitive and specific for serodiagnosis of human tularemia. The potential of the MAb-based cELISA to be used in both human and animal samples emphasizes its usefulness for serological survey of tularemia among multiple animal species.
INTRODUCTION
Tularemia is a highly infectious zoonotic disease caused by an intracellular Gram-negative bacterium, Francisella tularensis. It was first reported in North America in 1911 during plague studies of rodents (1); subsequently, both human and animal infections were identified in Japan, as well as in European countries and the former Soviet Union (2). Tularemia exists primarily as two clinically relevant strains, the highly virulent type A (F. tularensis subsp. tularensis) found predominantly in North America and the less virulent type B (F. tularensis subsp. holarctica) found in the northern hemisphere (3, 4). Transmission to humans is mostly associated with handling of infected animals, arthropod bites, ingestion of contaminated water or food, and inhalation of infective aerosols (2). The clinical manifestation of the disease in humans ranges from skin ulcers to life-threatening pneumonia (3). Clinical signs and the course of the infection vary among species. For example, rodents and hares generally die rapidly after being infected without mounting an antibody response, whereas other animal species, such as cats, dogs, and cattle, are relatively resistant to infection (5).
To date, tularemia outbreaks, both sporadic cases and epidemiological surveillance data, have been reported (6–8); however, little is known about the prevalence rate in Asia. Currently, pathogen isolation, molecular detection, and serology are the most commonly used methods for the diagnosis of tularemia (9). However, a high risk of laboratory infection associated with isolation of the organism and lack of a well-evaluated standardized PCR protocol make these techniques difficult to apply for routine diagnosis of large numbers of samples (10–12). For these reasons, serological assays are the best choice for surveillance of tularemia in humans and animals.
The most commonly utilized serological assays for tularemia are microagglutination (MA), enzyme-linked immunosorbent assay (ELISA), and Western blotting (WB) (13, 14). In patients with tularemia, antibodies appear approximately 2 to 3 weeks after infection and may be detected several years after recovery (15–17). Monitoring of antibody titers in serum during acute and convalescent phases is thus necessary to identify tularemia infection. MA seems to be an appropriate test because it is easy and applicable to various animal species (17, 18); however, it is not applicable to hemolyzed serum, and the sensitivity is relatively low (14), particularly for serum with lower antibody titers. Furthermore, cross-reaction with other bacterial species makes this assay difficult to use for examination of serum suspected to have antibodies against F. tularensis (19, 20). Indirect ELISA (iELISA) is appropriate for seroepidemiological studies because the test is relatively sensitive (10, 13, 14); however, iELISA requires enzyme-conjugated secondary antibodies against immunoglobulins of respective animal species. For seroepidemiological surveillance of many wild animals, it is almost impossible to prepare antibodies that are specifically directed against immunoglobulins of each animal species. Although the combination of WB and iELISA is often used for confirmatory serodiagnosis (14), it is difficult for use with a large number of samples. Therefore, there is a need for a high-throughput assay that is specific and sensitive in detecting antibodies against F. tularensis. Monoclonal antibody (MAb)-based competitive ELISA (cELISA) appears to be ideal because it is able to overcome the problems associated with the currently available tests. Therefore, we attempted to develop cELISA for detection of antibodies against F. tularensis in serum of humans and animals.
MATERIALS AND METHODS
Serum samples.
A total of 84 human serum samples were used in the present study. Twenty serum samples from 15 patients with confirmed tularemia and 5 healthy subjects were obtained by H. Fujita, Ohara Research Laboratories, Fukushima, Japan. Fourteen serum samples were obtained from four patients at several other hospitals in Japan. Patient serum samples were obtained as early as day 1after onset of tularemia symptoms (Table 1). All patients were diagnosed with tularemia by a significant rise of MA or tube agglutination titer. A total of 45 serum samples of healthy donors were also obtained from several hospitals in Japan. The identity of the patients was not disclosed to us and was derived from various contributors. The studies in human subjects were approved by the research and ethical committees of the National Institute of Infectious Diseases (NIID), Tokyo, Japan, and written informed consent was obtained from all participants.
Table 1.
Antibodies against F. tularensis in patients determined by cELISA, iELISA, and MAa
| Patient | Blood collection (days after onset of tularemia) | Test |
||
|---|---|---|---|---|
| cELISA (% inhibition) | iELISA (OD) | MA (titer) | ||
| P1 | 11 | 25.8 | 0.60 | <10 |
| 36 | 79.7 | 1.91 | 320 | |
| P2 | 12 | 54.1 | 1.84 | 20 |
| 37 | 74.5 | 2.65 | 320 | |
| P3 | 37 | 84.9 | 2.99 | >1,280 |
| 83 | 80.4 | 3.07 | >1,280 | |
| P4 | 1 | 9.9 | 0.27 | <10 |
| 13 | 43.4 | 0.79 | 40 | |
| P5 | 9 | 47.9 | 1.01 | <10 |
| 25 | 77.3 | 2.15 | 80 | |
| P6 | 80 | 78.2 | 2.54 | 320 |
| P7 | 21 | 82.8 | 2.57 | 640 |
| P8 | ND | 42.7 | 1.02 | 80 |
| P9 | 87 | 75.8 | 2.66 | 320 |
| P10 | 13 | 43.2 | 0.89 | <10 |
| P11 | 59 | 41.1 | 1.18 | 160 |
| P12 | ND | 36.1 | 0.79 | <10 |
| P13 | 8 | 79.2 | 2.49 | 320 |
| P14 | 78 | 54.8 | 2.44 | >1,280 |
| P15 | ND | 59.7 | 1.64 | >1,280 |
| P16 | 42 | 71.0 | 2.15 | 640 |
| 89 | 53.4 | 1.68 | 320 | |
| P17 | 13 | 42.0 | 2.84 | <10 |
| 241 | −1.5 | 1.31 | 40 | |
| P18 | 16 | 83.0 | 2.36 | 40 |
| 23 | 94.2 | 2.86 | 160 | |
| 30 | 90.3 | 2.83 | 160 | |
| 59 | 86.1 | 3.34 | 80 | |
| 185 | 68.7 | 2.42 | 40 | |
| P19 | 16 | 66.6 | 0.95 | 40 |
| 23 | 65.3 | 1.19 | 160 | |
| 30 | 66.2 | 1.18 | 160 | |
| 59 | 79.0 | 1.62 | 160 | |
| 185 | 60.2 | 1.01 | 40 | |
A total of 34 serum samples of 19 patients were used. Sera of patients P1 to P5, P16, and P17 are paired samples obtained at the indicated days after the onset of tularemia symptoms. Patients P18 and P19 represent the five sets of samples obtained for several days after symptom onset. ND, not documented. Shaded numbers are values below the cutoff level in each test.
Anti-F. tularensis serum samples were obtained by immunizing rabbits or mice with formalin-inactivated F. tularensis subsp. tularensis or subsp. holarctica suspension as follows. To prepare immunized serum samples, specific-pathogen-free 10-week-old female Kbl:JW rabbits (Kitayama Rabesu Co., Nagano, Japan) were inoculated subcutaneously with formalin-inactivated F. tularensis subsp. tularensis (38 strain) or subsp. holarctica (Yama strain, a Japanese isolate) (400 μg of protein/rabbit) suspended in TiterMax Gold (KIEL Lab, Norcross, GA). The protein concentration of F. tularensis whole cells was determined by a Bradford protein assay (Bio-Rad). The rabbits were inoculated again with 400 μg of the protein together with the adjuvant 4, 6, and 8 weeks after the first injection. A final booster was injected intravenously 2 weeks after the fourth injection with formalin-inactivated bacteria (50 μg of protein/rabbit) in phosphate-buffered saline (PBS). Similarly, specific-pathogen-free 6-week-old female BALB/c mice (SLC, Shizuoka, Japan) were immunized twice with formalin-inactivated F. tularensis subsp. holarctica (Yama) or Francisella novicida (U112; 100 μg of protein/mouse) suspended in Titer Max Gold 4 weeks apart. At 2 weeks after the second inoculation, mice were boosted by intravenous injection with the formalin-inactivated bacteria (50 μg of protein/mouse) in PBS. Serum samples of mice that had recovered from experimental infection with attenuated F. tularensis subsp. tularensis (Schu strain) were also included. Eight-week-old female BALB/c mice were infected intraperitoneally with 6.2 × 106 CFU bacterial suspension in saline, and the blood was collected at 6 days postinfection. These animal experiments were approved by the Animal Care and Use Committee of NIID. Serum samples of rabbits immunized with formalin-inactivated Brucella abortus, Brucella canis, Brucella melitensis, Brucella suis, Yersinia enterocolitica, Yersinia pestis, and Yersinia pseudotuberculosis were gifts from Koichi Imaoka of our department.
Bacteria and purification of LPS.
F. tularensis subsp. holarctica, NVF1 strain, which was isolated from a hare in 2009, was grown at 37°C for 72 h on chocolate agar (II) plates (Becton Dickinson, Tokyo, Japan). The bacteria were harvested into saline, and the suspension was adjusted to an optical density at 600 nm (OD600) of 1.2. Lipopolysaccharide (LPS) was extracted using an LPS extraction kit (iNtRON Biotechnology, Kyungki-Do, Korea) according to the manufacturer's protocol after mixing the bacterial suspension with extraction buffer, followed by incubation at 65°C for 10 min. The dried LPS pellets were dissolved in 10 mM Tris-HCl buffer (pH 8.0) at a concentration of 10 μg/μl and stored at 4°C until use. Live bacteria were handled in a biosafety level 3 laboratory at the NIID.
cELISA.
A 96-well flat-bottom microtiter plate (Greiner Bio-One, Frickenhausen, Germany) was coated with LPS antigen in carbonate-bicarbonate buffer (pH 9.6; 2.5 μg/50 μl/well) at 37°C overnight. The wells were rinsed thrice with PBS containing 0.1% Tween 20 (MP Biomedicals, Illkirch, France) (PBST) to remove unbound antigens and were blocked with PBST containing 3% (wt/vol) skim milk (150 μl/well) at 37°C for 1 h. For all subsequent steps, PBST containing 1% (wt/vol) skim milk was used as dilution buffer. After three washes with PBST, 50 μl of diluted sample serum was added to the antigen coated wells in duplicate, and the plates were incubated at 37°C for 90 min. Pooled patient sera (n = 10) and pooled healthy human sera (n = 5) were also added as positive and negative controls during each test. After the wells were washed three times with PBST, biotin-labeled anti-LPS MAb (M14B11) (21) (50 μl/well, 1:5,000 dilution) was added, and the plates were further incubated at 37°C for 60 min. The biotin labeling of M14B11 (isotype IgG2a; 3.4 mg of IgG/ml and a biotin/IgG coupling ratio of 4/67) was performed at a commercial laboratory (T. K. Craft, Maebashi, Japan). After three washes, streptavidin-peroxidase (Thermo Scientific, Rockford, IL) (50 μl/well, 1:5,000 dilution) was added to each well, and the plates were incubated at 37°C for 60 min. After three washing steps, 100 μl of 3,3′,5,5′-tetramethylbenzidine (TMB) enzyme substrate (SureBlue Reserve, TMB microwell peroxidase substrate; KPL, Gaithersburg, MD) was added to each well, and the plates were incubated at 37°C for 30 min. Finally, 100 μl of stop solution (1 N HCl) was added, and the OD values were measured at 450 nm using an iMark microplate reader (Bio-Rad, Hercules, CA). The percent inhibition was calculated using the following formula: {1 − [(ODsample − ODbackground)/(ODMAb − ODbackground)]} × 100, where ODsample and ODMAb are the absorbances observed in the presence and in the absence of samples, respectively, and ODbackground was obtained in the absence of sample or labeled MAb.
MA test.
Portions (25 μl) of 2-fold serial dilutions of serum were mixed with an equal volume of formalin-inactivated F. tularensis subsp. holarctica (Yama) whole-cell suspension (OD560 = 1.0) in a 96-well round-bottom microtiter plate (IWAKI, Tokyo, Japan). The reactions in the plates were observed 18 h after incubation at 37°C for agglutination. Agglutination titers were expressed as reciprocals of the highest serum dilution showing agglutination with the antigen. Agglutination at dilutions of 1:10 or higher were considered MA positive.
iELISA.
The LPS solution was diluted 1:800 in carbonate-bicarbonate buffer (pH 9.6). Ninety-six-well microtiter plates (Greiner Bio-One) were coated with 50 μl of antigen at 37°C overnight. The wells were washed with PBST and blocked with 150 μl of PBST containing 3% (wt/vol) skim milk. After another washing step, 50 μl of patient serum samples, diluted 1:500 in PBST containing 1% (wt/vol) skim milk, were added, followed by incubation at 37°C for 1 h. After the plates were washed three times, 50 μl of horseradish peroxidase (HRP)-conjugated goat anti-human immunoglobulin G (IgG) (ICN Pharmaceuticals, Cappel, OH), diluted 1:8,000 in PBST containing 1% (wt/vol) skim milk, was added, followed by incubation at 37°C for 1 h. Colorimetric development and measurement of the OD values was performed as described above in the cELISA protocol.
WB.
Western blot (WB) analyses were performed as described previously (7) using whole bacterial cell lysates and purified LPS as antigens. After SDS-PAGE, antigens were transferred to a polyvinylidene difluoride membrane (Immobilon; Millipore Corp., Bedford, MA), and the membranes were saturated with PBST containing 3% (wt/vol) skim milk for 1 h. After being washed with PBST, the membranes were incubated with samples diluted 1:1,000 with PBST containing 1% (wt/vol) skim milk for 1 h. After three washes with PBST for 5 min, the membranes were incubated with HRP-conjugated goat anti-mouse IgG (H+L; Zymed Laboratories, Inc., CA) or HRP-conjugated goat anti-human IgG (ICN Pharmaceuticals) at a dilution of 1:8,000 for 1 h. The antigens were visualized by soaking the membranes in 3,3′-diaminobenzidine tetrahydrochloride (Wako Pure Chemicals, Osaka, Japan) and hydrogen peroxide visualization solution. Samples were considered to contain specific antibodies when the typical LPS ladder-like banding pattern was observed.
Statistical analysis.
Diagnostic efficiency of the assay in terms of sensitivity and specificity were determined initially by receiver operating characteristics (ROC) and two-graph-ROC (TG-ROC) curves using StatFlex software (Artech Co., Ltd., Osaka, Japan) (22, 23). An optimal cutoff value was estimated by comparing a range of sensitivity and specificity values for a range of cutoff values. The relationship between results for patients and healthy humans determined by cELISA and iELISA were evaluated by linear regression analysis using Microsoft Excel software for Windows.
RESULTS
Target antigens recognized by MAb and antibodies induced by infection.
WB was performed to determine antigens predominantly recognized by serum samples of infected humans and animals (Fig. 1). Regardless of whether LPS or whole-cell antigen was targeted, typical LPS ladder-like banding patterns were observed in the serum samples of patients with tularemia and mice that had recovered from experimental infection. These reactions were similar to that of the MAb M14B11 recognizing LPS. No band was obtained from healthy human serum samples.
Fig 1.
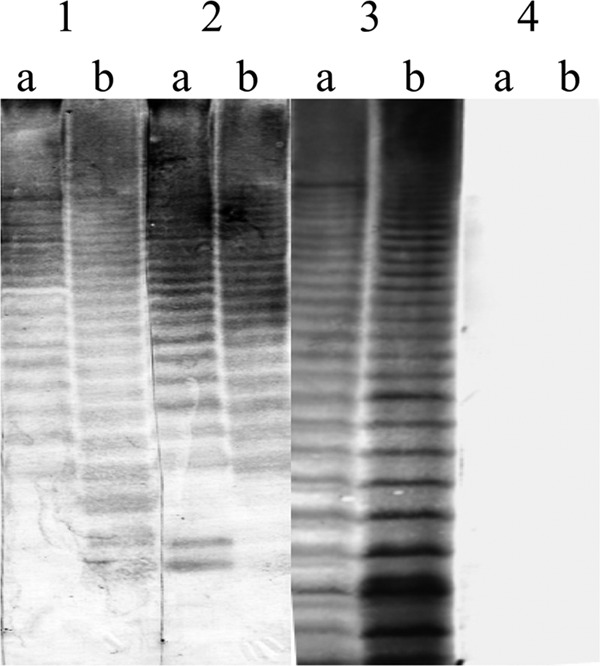
Western blot analysis of target antigens recognized by MAb and antibodies induced by infection. MAb M14B11 (lane 1) and sera from an F. tularensis-infected mouse (lane 2) and a patient with tularemia (lane 3) were reacted with F. tularensis whole-cell lysate (a) or purified LPS antigens (b). Sera obtained from healthy human donors (lane 4) were also included.
Development of cELISA.
Optimization of LPS concentration and dilution of antibodies were performed by checkerboard titration (data not shown). The composition of blocking and dilution buffers and incubation times for serum samples were also determined by preliminary experiments (data not shown). After optimization of the test conditions, samples in which the presence or absence of F. tularensis antibodies was known were subjected to cELISA; the representative results are shown in Fig. 2. The reaction of MAb to LPS was inhibited by serum samples of patients with tularemia in a dose-dependent manner (Fig. 2a). Serum samples of hyperimmune rabbits and mice infected with F. tularensis also inhibited MAb reaction to LPS (Fig. 2b and c). When the serum samples of three tularemia patients were tested, all serum samples were shown to inhibit MAb reactions at dilutions up to 1:1,000. No significant inhibition was observed in serum samples of healthy individuals (Fig. 2a).
Fig 2.

Inhibition of binding of MAb targeting the F. tularensis LPS by sera of humans, rabbits, and mice positive for F. tularensis antibodies in MA and iELISA. (a) Serial dilutions of three MA-positive (tularemia 1, 2, and 3) and three MA-negative (healthy 1, 2, and 3) human serum samples were examined for their ability to inhibit MAb binding. (b) Serum samples of four rabbits were subjected to cELISA. Two rabbits (immunized 1 and 2) were immunized with F. tularensis subsp. holarctica and F. tularensis subsp. tularensis, respectively, whereas two rabbits (normal 1 and 2) were unimmunized. (c) Serum obtained from a mouse infected with attenuated F. tularensis subsp. tularensis (Ftt infected), a mouse immunized with formalin-fixed F. tularensis subsp. holarctica (Fth immunized), and a mouse immunized with formalin-fixed F. novicida (Fn immunized) were subjected to cELISA. Plasma and serum of a normal mouse were also tested as negative controls. The error bars in each figure indicate standard deviations from four-well replications for each serum sample.
Serum samples of rabbits immunized with F. tularensis subsp. holarctica and subsp. tularensis exhibited 100 and 88% inhibition of MAb reaction, respectively, at a dilution of 1:125 and thereafter exhibited decreasing percent inhibitions with increasing serum dilutions (Fig. 2b). Thus, the level of inhibition was directly proportional to the amount of antibody in the samples. No inhibition of MAb reaction was observed with normal serum obtained from two different rabbits.
Furthermore, serum samples obtained from convalescent mice experimentally infected with attenuated F. tularensis subsp. tularensis and mice immunized with formalin-fixed F. tularensis subsp. holarctica exhibited clear inhibition of MAb reaction at dilutions up to 1:1,000, whereas the sera of mice immunized with F. novicida exhibited only slight inhibition (ca. 11%) at a dilution of 1:125. The plasma or sera of normal mice did not exhibit inhibition of MAb reaction at any of the dilutions tested (Fig. 2c).
Inhibition of MAb reaction was consistently achieved by both patient sera and immune and infected animal sera even at higher dilutions, whereas inhibition caused by normal serum never exceeded 20%, even at lower dilutions. Immune serum diluted 1:125 exhibited the highest levels of inhibition, whereas normal serum at this dilution exhibited the least inhibition. We also determined whether cELISA designed for F. tularensis cross-reacted with other bacterial species, particularly, Brucella spp. and Yersinia spp. None of the rabbit immune serum samples against Brucella abortus, Brucella canis, Brucella melitensis, Brucella suis, Yersinia enterocolitica, Yersinia pestis, and Yersinia pseudotuberculosis exhibited significant inhibition (<10%) at a dilution of 1:125 (Fig. 3).
Fig 3.

Inhibition of binding of F. tularensis LPS-specific MAb by rabbit antisera against various bacterial species. Serial dilutions of the rabbit antisera against B. abortus, B. canis, B. melitensis, B. suis, Y. enterocolitica, Y. pestis, and Y. pseudotuberculosis were negative by cELISA, whereas antisera against F. tularensis were positive, with clear inhibition of MAb binding. Error bars indicate the standard deviations from four-well replications for each serum sample.
Determination of cutoff values for cELISA and iELISA.
cELISA was performed to detect specific antibodies against F. tularensis in 34 serum samples collected from 19 patients and 50 healthy individuals. The percent inhibition values for each serum sample at a dilution of 1:100 were plotted (Fig. 4a), and ROC and TG-ROC curves were plotted (Fig. 4b and c). The area under the ROC curve was 0.98559, indicating that the assay had an excellent ability to discriminate between patients and healthy donors (Fig. 4b). The TG-ROC analysis initially identified the cutoff value for discrimination of patients and healthy donors to be 25.8% inhibition (Fig. 4c). With this value, cELISA had a sensitivity of 91.1% and a specificity of 100%. To increase the sensitivity of the assay without decreasing the specificity, the cutoff value was decreased to 8.5% inhibition (Fig. 4c). As a result, the sensitivity increased from 91.1 to 97.0%. Antibodies against F. tularensis were successfully detected in samples of patients with tularemia at a cutoff value of 8.5% inhibition, except for one serum sample. The cutoff value and diagnostic performance of iELISA were determined similarly. As a result, iELISA was considered positive at a cutoff OD of 0.61 at which the sensitivity and the specificity were 94.1 and 98.0%, respectively (data not shown).
Fig 4.
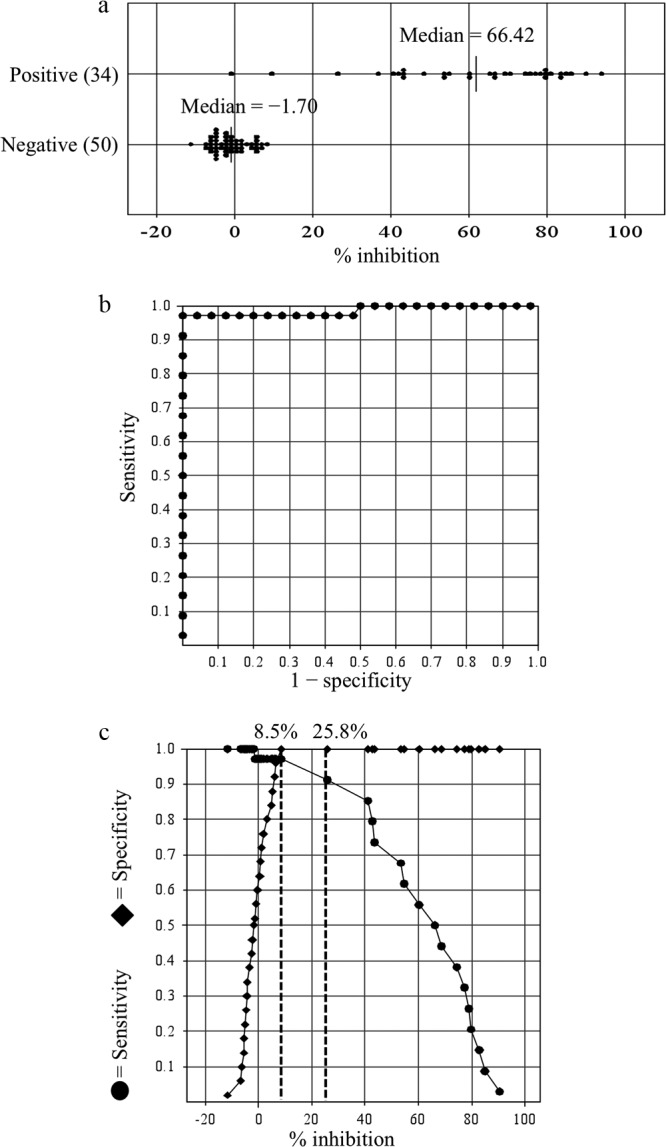
Distribution of percent inhibition values and ROC and TG-ROC analyses of cELISA specific for F. tularensis LPS. (a) The percent inhibition value of each serum sample at a dilution of 1:100 was plotted for serum samples obtained from patients with tularemia and healthy donors. (b and c) The ROC curve (b) and two-graph-ROC (TG-ROC) curve (c) were analyzed and drawn using Stat Flex software. The area under the ROC curve is 0.98559, which indicates the test has a good probability of distinguishing between patients with tularemia and healthy individuals (see panel b). The graphs show the relationship between the sensitivity and specificity of cELISA for each cutoff percent inhibition. The dashed vertical lines indicate the cutoff values at 8.5 and 25.8% inhibition, respectively (see panel c). The most optimal sensitivity (97.0%) and specificity (100%) are obtained when the cutoff value is set at 8.5% inhibition.
Sensitivity and specificity of cELISA, iELISA, and MA.
cELISA, iELISA, and MA were performed simultaneously on 84 human serum samples. The results for the 34 serum samples collected from 19 patients are summarized in Table 1. Only one serum sample was found to be false negative by cELISA, whereas the number of false-negative samples by iELISA and MA were 2 and 6, respectively (Table 1). Similarly, among the 50 healthy human serum samples, only one sample was found to be false-positive by iELISA, and no sample was found to be false positive by cELISA and MA (data not shown). In comparison to cELISA, the sensitivity and specificity of MA were 81.8 and 98.0%, respectively (Table 2). In addition, when cELISA was compared to iELISA, the relative sensitivity and specificity of cELISA were 93.9 and 96.1%, respectively (Table 3).
Table 2.
Sensitivity and specificity of MA relative to cELISAa
| cELISA | MA (no. of samples) |
||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 27 | 6 | 33 |
| Negative | 1 | 50 | 51 |
| Total | 28 | 56 | 84 |
Relative sensitivity = 27 of 33 (81.8%); relative specificity = 50 of 51 (98%). Positive, human serum with a positive MA titer with agglutination at dilutions of ≥1:10; negative, human serum with no agglutination in MA.
Table 3.
Sensitivity and specificity of cELISA relative to iELISAa
| iELISA | cELISA |
||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 31 | 2 | 33 |
| Negative | 2 | 49 | 51 |
| Total | 33 | 51 | 84 |
Relative sensitivity = 31 of 33 (93.9%); relative specificity = 49 of 51 (96.1%). Positive, human serum with an iELISA cutoff OD of >0.61; negative, human serum with an iELISA cutoff OD of ≤0.61.
Correlation between cELISA and iELISA.
The results of cELISA and iELISA using the 84 human serum samples were analyzed to determine whether the two assays were statistically correlated. Linear regression analysis showed a significant linear correlation between cELISA percent inhibition values and OD values determined by iELISA (R2 = 0.82, r = 0.91) (Fig. 5).
Fig 5.

Correlation between cELISA and iELISA for detection of F. tularensis-specific antibodies. A scatter plot of the percent inhibition obtained by cELISA and OD values obtained by iELISA for serum samples of patient (n = 34) and healthy human donor (n = 50). Linear regression analysis showed that a significant linear correlation was observed between cELISA and iELISA with a correlation coefficient of 0.91.
Persistence of F. tularensis-specific antibodies in patients.
Two patients were evaluated by cELISA and iELISA to determine the level and persistence of disease-specific antibodies between 19 to 188 days after exposure to F. tularensis (Fig. 6). The antibodies were detected on day 19, and thereafter their levels increased or remained constant until 62 days after exposure. However, antibody levels in the sera of both patients decreased moderately at 188 days. cELISA and iELISA demonstrated similar patterns of antibody persistence in both patients.
Fig 6.

Kinetics of antibody levels in patients with tularemia over a 6-month period. F. tularensis-specific antibodies were measured by cELISA and iELISA in two patients with tularemia (P18 and P19, Table 1) at the indicated days after exposure to F. tularensis. The percent inhibition values in cELISA and OD values in iELISA are shown on the left and right sides of the y axis, respectively.
DISCUSSION
Human tularemia is diagnosed on the basis of clinical findings and laboratory tests that include serological and molecular methods (11, 13, 14, 17). However, variations in sensitivity and specificity of different assays during surveillance studies sometimes lead to misdiagnosis of the disease. The cELISA developed here is a simple, specific, and sensitive test that is applicable to both humans and animals for detection of antibodies against F. tularensis.
Our cELISA is based on the measurement of competition between the test sample and MAb for LPS, the major antigen of F. tularensis (14, 24, 25). Similar assays using MAb against LPS have been described for Brucella species (26, 27). The typical ladder-like bands observed on WB demonstrated that both MAb and F. tularensis-infected human and mice serum contained antibodies predominantly recognizing the LPS antigen (Fig. 1). In cELISA, the human and animal serum samples that were positive by MA and iELISA clearly inhibited MAb binding to LPS (Fig. 2). Serum samples of tularemia patients with MA titers of 1:160 (tularemia 1 and 2 in Fig. 2a) and 1:40 (tularemia 3 in Fig. 2a) positively inhibited MAb binding up to a dilution of 1:1,000, whereas the serum samples of healthy donors did not. This result shows that cELISA is useful for detecting F. tularensis antibodies and is more sensitive than MA.
In previous studies, the possible difference in antigenic structure of LPS between F. tularensis subsp. tularensis and subsp. holarctica were suggested (28). In the present study, we used anti-LPS MAb (M14B11) (21), which recognized both F. tularensis subsp. tularensis and subsp. holarctica in the cELISA. Furthermore, antibodies against both F. tularensis subsp. tularensis and subsp. holarctica in rabbit and mouse sera were detected by cELISA (Fig. 2b and c). Although we could not test convalescent human patients sera infected with F. tularensis subsp. tularensis due to unavailability in Japan, the result in the present study indicated that the cELISA is capable to detect antibodies induced after both F. tularensis subsp. tularensis and subsp. holarctica infections. On the other hand, when serum sample of a mouse immunized with F. novicida were tested, 11% inhibition was observed (Fig. 2c). This slight inhibition may be due to the common antigenic epitopes shared by F. novicida and F. tularensis since they are genetically closely related (29). In addition, no serum samples of rabbits immunized with Brucella spp. and Yersinia spp. interfered with the specific binding of MAb to LPS (Fig. 3). These results indicate that MAb-based cELISA is highly specific for F. tularensis without any cross-reactions with other microorganisms.
The results of cELISA of serum samples of tularemia patients and healthy donors were evaluated. ROC analysis showed that cELISA had sensitivities of 91.1 and 97.0% at cutoff values of 25.8% inhibition and 8.5% inhibition, respectively (Fig. 4). Two serum samples of patients at day 1 (P4) and day 11 (P1) after onset of symptoms were negative by cELISA at a cutoff value of 25.8%. These sera were also negative by iELISA and MA (Table 1), and thus it was conceivable that these acute-phase sera did not contain detectable levels of antibodies against F. tularensis. However, when the cutoff value was decreased to 8.5% inhibition, these two acute-phase sera became positive, indicating that cELISA with a cutoff value of 8.5% is more sensitive in detecting antibodies against F. tularensis than iELISA and MA. In contrast, serum sample of the patient (P17) obtained 8 months (241 days) after recovery was negative even at a cutoff value of 8.5% inhibition, although the serum was positive by iELISA and MA. The paired serum of the same patient (P17) obtained 2 weeks after disease onset was clearly positive by both cELISA and iELISA, but negative by MA. These results might suggest that LPS antibodies recognizing the specific epitope of the MAb M14B11 in patients did not persist for several months, whereas the antibodies recognizing the other epitopes on LPS persisted for more than 8 months. The testing of large numbers of serum samples from both acute and convalescent phases of tularemia patients might have improved the precise interpretation of data. For the 50 healthy human serum samples, cELISA exhibited 100% specificity at both cutoff values of 25.8 and 8.5% inhibition (Fig. 4c). Therefore, the cutoff value of 8.5% inhibition was considered to be suitable for sensitive and specific detection of F. tularensis antibodies in human serum samples based on the ROC analysis.
The sensitivity of cELISA and iELISA were comparable, whereas MA was less sensitive in detecting F. tularensis antibodies (Tables 2 and 3). All of these results were comparable to those of previously reported cELISA methods that detected antibodies against F. tularensis outer membrane protein antigens with a sensitivity and specificity of 95.7 and 96%, respectively (30). However, it is noteworthy that LPS is a more suitable antigen for detecting an antibody response elicited in the early phase after F. tularensis infection as well as antibodies that persist for several years (10, 24).
In the present study, we also found, in agreement with a previous report, that antibodies against F. tularensis persist for months after infection (16). Although we could not test how long the antibodies persist, a moderately decreasing antibody level was observed after 6 months (Fig. 6), and this pattern was the same in serum samples of two different patients with comparable antibody levels in cELISA and iELISA. Indeed, the correlation between cELISA and iELISA was high (R2 = 0.8226) (Fig. 5). These data suggest that the results obtained for detection of antibodies against F. tularensis by the novel cELISA are reliable and consistent with those obtained by iELISA.
Our cELISA method based on the inhibition of binding of MAb specific to the F. tularensis LPS is highly sensitive and specific. This method is well suited as a routine, confirmatory laboratory test for tularemia in humans. It can overcome the problems associated with conventional serological assays such as low sensitivity and specificity in MA or requirements of species-specific secondary antibodies in iELISA or indirect immunofluorescence assay. Therefore, our MAb-based cELISA is expected to be advantageous and useful for serosurveillance of various wild animals such as bears, wild boars, fox, raccoon dogs, and even birds, except for the highly susceptible animals such as some species of rodents or lagomorphs since they die before producing antibodies to F. tularensis. A seroepidemiological study of various wild animals with this novel cELISA is ongoing. We believe this cELISA method will help facilitate the surveillance of tularemia in humans and wild animals, leading to better understanding of the ecology and epidemiology of tularemia.
ACKNOWLEDGMENTS
We thank H. Fujita, Ohara Research Laboratories, Ohara General Hospital, Fukushima, Japan, for providing serum samples of patients and healthy blood donors. We also thank K. Imaoka (Department of Veterinary Science, NIID) for providing rabbit sera immunized with Brucella spp. and Yersinia spp.
This study was supported in part by Health and Labor Science Research Grants for Research on Emerging and Re-Emerging Infectious Diseases from the Ministry of Health, Labor, and Welfare in Japan (H22-Shinkou-Ippan-010). N.S. was supported by a Tokyu Foundation Scholarship for Inbound Students.
Footnotes
Published ahead of print 31 October 2012
REFERENCES
- 1. McCoy GW, CC. 1912. Bacterium tularense, the cause of a plaguelike disease of rodents. Public Health Bull. 53:17–23 [Google Scholar]
- 2. Ellis J, Oyston PC, Green M, Titball RW. 2002. Tularemia. Clin. Microbiol. Rev. 15:631–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Layton M, Lillibridge SR, McDade JE, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Tonat K. 2001. Tularemia as a biological weapon: medical and public health management. JAMA 285:2763–2773 [DOI] [PubMed] [Google Scholar]
- 4. Oyston PC, Sjostedt A, Titball RW. 2004. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2:967–978 [DOI] [PubMed] [Google Scholar]
- 5. Hopla CE. 1974. The ecology of tularemia. Adv. Vet. Sci. Comp. Med. 18:25–53 [PubMed] [Google Scholar]
- 6. Hansen CM, Vogler AJ, Keim P, Wagner DM, Hueffer K. 2011. Tularemia in Alaska, 1938–2010. Acta Vet. Scand. 53:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hotta A, Tanabayashi K, Yamamoto Y, Fujita O, Uda A, Mizoguchi T, Yamada A. 2012. Seroprevalence of tularemia in wild bears and hares in Japan. Zoonoses Public Health 59:89–95 [DOI] [PubMed] [Google Scholar]
- 8. Maurin M, Pelloux I, Brion JP, Del Bano JN, Picard A. 2011. Human tularemia in France, 2006–2010. Clin. Infect. Dis. 53:e133–141 [DOI] [PubMed] [Google Scholar]
- 9. World Health Organization Epidemic and Pandemic Alert and Response 2007. WHO guidelines on tularemia. World Health Organization, Geneva, Switzerland [Google Scholar]
- 10. Eliasson H, Olcen P, Sjostedt A, Jurstrand M, Back E, Andersson S. 2008. Kinetics of the immune response associated with tularemia: comparison of an enzyme-linked immunosorbent assay, a tube agglutination test, and a novel whole-blood lymphocyte stimulation test. Clin. Vaccine Immunol. 15:1238–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Splettstoesser WD, Tomaso H, Al Dahouk S, Neubauer H, Schuff-Werner P. 2005. Diagnostic procedures in tularaemia with special focus on molecular and immunological techniques. J. Vet. Med. B Infect. Dis. Vet. Public Health 52:249–261 [DOI] [PubMed] [Google Scholar]
- 12. Versage JL, Severin DD, Chu MC, Petersen JM. 2003. Development of a multitarget real-time TaqMan PCR assay for enhanced detection of Francisella tularensis in complex specimens. J. Clin. Microbiol. 41:5492–5499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Porsch-Ozcurumez M, Kischel N, Priebe H, Splettstosser W, Finke EJ, Grunow R. 2004. Comparison of enzyme-linked immunosorbent assay, Western blotting, microagglutination, indirect immunofluorescence assay, and flow cytometry for serological diagnosis of tularemia. Clin. Diagn. Lab. Immunol. 11:1008–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmitt P, Splettstosser W, Porsch-Ozcurumez M, Finke EJ, Grunow R. 2005. A novel screening ELISA and a confirmatory Western blot useful for diagnosis and epidemiological studies of tularemia. Epidemiol. Infect. 133:759–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bevanger L, Maeland JA, Kvan AI. 1994. Comparative analysis of antibodies to Francisella tularensis antigens during the acute phase of tularemia and eight years later. Clin. Diagn. Lab. Immunol. 1:238–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koskela P, Salminen A. 1985. Humoral immunity against Francisella tularensis after natural infection. J. Clin. Microbiol. 22:973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sato T, Fujita H, Ohara Y, Homma M. 1990. Microagglutination test for early and specific serodiagnosis of tularemia. J. Clin. Microbiol. 28:2372–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown SL, McKinney FT, Klein GC, Jones WL. 1980. Evaluation of a safranin-O-stained antigen microagglutination test for francisella tularensis antibodies. J. Clin. Microbiol. 11:146–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Behan KA, Klein GC. 1982. Reduction of Brucella species and Francisella tularensis cross-reacting agglutinins by dithiothreitol. J. Clin. Microbiol. 16:756–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bevanger L, Maeland JA, Naess AI. 1988. Agglutinins and antibodies to Francisella tularensis outer membrane antigens in the early diagnosis of disease during an outbreak of tularemia. J. Clin. Microbiol. 26:433–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hotta A, Uda A, Fujita O, Tanabayashi K, Yamada A. 2007. Preparation of monoclonal antibodies for detection and identification of Francisella tularensis. Clin. Vaccine Immunol. 14:81–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greiner M, Sohr D, Gobel P. 1995. A modified ROC analysis for the selection of cutoff values and the definition of intermediate results of serodiagnostic tests. J. Immunol. Methods 185:123–132 [DOI] [PubMed] [Google Scholar]
- 23. Zweig MH, Campbell G. 1993. Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin. Chem. 39:561–577 [PubMed] [Google Scholar]
- 24. Carlsson HE, Lindberg AA, Lindberg G, Hederstedt B, Karlsson KA, Agell BO. 1979. Enzyme-linked immunosorbent assay for immunological diagnosis of human tularemia. J. Clin. Microbiol. 10:615–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Viljanen MK, Nurmi T, Salminen A. 1983. Enzyme-linked immunosorbent assay (ELISA) with bacterial sonicate antigen for IgM, IgA, and IgG antibodies to Francisella tularensis: comparison with bacterial agglutination test and ELISA with lipopolysaccharide antigen. J. Infect. Dis. 148:715–720 [DOI] [PubMed] [Google Scholar]
- 26. Meegan J, Field C, Sidor I, Romano T, Casinghino S, Smith CR, Kashinsky L, Fair PA, Bossart G, Wells R, Dunn JL. 2010. Development, validation, and utilization of a competitive enzyme-linked immunosorbent assay for the detection of antibodies against Brucella species in marine mammals. J. Vet. Diagn. Invest. 22:856–862 [DOI] [PubMed] [Google Scholar]
- 27. Weynants V, Gilson D, Cloeckaert A, Denoel PA, Tibor A, Thiange P, Limet JN, Letesson JJ. 1996. Characterization of a monoclonal antibody specific for Brucella smooth lipopolysaccharide and development of a competitive enzyme-linked immunosorbent assay to improve the serological diagnosis of brucellosis. Clin. Diagn. Lab. Immunol. 3:309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gunn JS, Ernst RK. 2007. The structure and function of Francisella lipopolysaccharide. Ann. N. Y. Acad. Sci. 1105:202–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Broekhuijsen M, Larsson P, Johansson A, Bystrom M, Eriksson U, Larsson E, Prior RG, Sjostedt A, Titball RW, Forsman M. 2003. Genome-wide DNA microarray analysis of Francisella tularensis strains demonstrates extensive genetic conservation within the species but identifies regions that are unique to the highly virulent F. tularensis subsp. tularensis. J. Clin. Microbiol. 41:2924–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bevanger L, Maeland JA, Naess AI. 1989. Competitive enzyme immunoassay for antibodies to a 43,000-molecular-weight Francisella tularensis outer membrane protein for the diagnosis of tularemia. J. Clin. Microbiol. 27:922–926 [DOI] [PMC free article] [PubMed] [Google Scholar]