

Abstract
Calcium (Ca2+)-mediated signaling events in fungal pathogens such as Cryptococcus neoformans are central to physiological processes, including those that mediate stress responses and promote virulence. The Cch1-Mid1 channel (CMC) represents the only high-affinity Ca2+ channel in the plasma membrane of fungal cells; consequently, cryptococci cannot survive in low-Ca2+ environments in the absence of CMC. Previous electrophysiological characterization revealed that Cch1, the predicted channel pore, and Mid1, a binding partner of Cch1, function as a store-operated Ca2+-selective channel gated by depletion of endoplasmic reticulum (ER) Ca2+ stores. Cryptococci lacking CMC did not survive ER stress, indicating its critical role in restoring Ca2+ homeostasis. Despite the requirement for Mid1 in promoting Ca2+ influx via Cch1, identification of the role of Mid1 remains elusive. Here we show that the C-terminal tail of Mid1 is a modulatory region that impinges on Cch1 channel activity directly and mediates the trafficking of Mid1 to the plasma membrane. This region consists of the last 24 residues of Mid1, and the functional expression of Mid1 in a human embryonic cell line (HEK293) and in C. neoformans is dependent on this domain. Substitutions of arginine (R619A) or cysteine (C621A) in the modulatory region failed to target Mid1 to the plasma membrane and prevented CMC activity. Interestingly, loss of a predicted protein kinase C (PKC)-phosphorylated serine residue (S605A) had no effect on Mid1 trafficking but did alter the kinetics of Cch1 channel activity. Thus, establishment of Ca2+ homeostasis in C. neoformans is dependent on a modulatory domain of Mid1.
INTRODUCTION
Cryptococcus neoformans and other fungi express the Cch1-Mid1 channel (CMC channel) in their plasma membrane (1–7). C. neoformans is a fungal pathogen that causes life-threatening disease primarily in patients with a compromised immune system (8, 9). The survival of C. neoformans in low Ca2+ environments requires the activity of CMC (3–5). The notion of the role of CMC as the only high-affinity Ca2+ channel in the plasma membrane is supported by genetic analysis demonstrating that strains of C. neoformans lacking CCH1 or MID1 display growth sensitivity under conditions of limiting extracellular, free [Ca2+] (3, 4). Store-operated Ca2+ (SOC) entry in eukaryotes involves the influx of Ca2+ across the plasma membrane in response to the depletion of Ca2+ from endoplasmic reticulum (ER) stores (10–14). A prolonged depletion of Ca2+ due to stresses inflicted on the ER can result in cell death unless the Ca2+ is restored to homeostatic levels (11, 13, 14).
The recent electrophysiological characterization of CMC revealed that the channel is gated by the depletion of intracellular ER Ca2+ stores, indicating that CMC functions primarily to replenish these stores and reestablish Ca2+ homeostasis (2). This notion was supported by evidence from recordings of inward Ca2+ currents that were specifically activated upon the depletion of intracellular ER Ca2+ stores with 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid, cesium salt (BAPTA-AM; a Ca2+ chelator), or thapsigargin (inhibitor of the SERCA Ca2+-ATPase) (2). Cch1 was also found to be highly permeable with respect to to both Ca2+ and Ba2+ ions, while La3+ specifically blocked Cch1-mediated Ca2+ currents (2). Ca2+ movement through Cch1 was not voltage dependent, suggesting that CMC is not gated by voltage. This was consistent with the lack of the highly conserved voltage sensor, a hallmark of voltage-gated channels, within the Cch1 protein sequence (3, 15–17). Collectively, these studies indicated that CMC displayed some features intrinsic to SOC channels (10–12, 14, 18).
The survival of C. neoforomans under conditions of ER stress was dependent on the expression of CMC, further indicating that Cch1 and its partner Mid1 play a critical role in the restoration of Ca2+ homeostasis (2). Similarly, yeast (Saccharomyces cerevisiae) survival under conditions of ER stress is supported by an active CMC that likely promotes Ca2+ influx during secretory Ca2+ depletion, consistent with the electrophysiological evidence (5, 19, 20). The activation of Cch1-mediated Ca2+ currents is dependent on the coexpression of Mid1, indicating that Mid1 is an essential component of CMC (2). This is consistent with earlier genetic data that demonstrated identical phenotypic defects of strains lacking either MID1 or CCH1 (10, 11, 21, 22). Unlike other accessory proteins of channel pores, Mid1 does not mediate the trafficking of Cch1 to the plasma membrane, but Mid1 expression in yeast appears to be stabilized by Cch1 (4, 23). Although it has been shown that Mid1 in S. cerevisiae (ScMid1) can operate as a stretch-activated nonselective cation channel under certain conditions, we found that CnMid1 does not have any independent channel activity under conditions of Ca2+ depletion, suggesting that its primary role, although elusive, is to activate Cch1 (2, 24).
In this study, we sought to resolve the contribution of Mid1 to the role of CMC in C. neoformans through the analysis of Mid1-truncated mutants and site-directed mutagenesis of Mid1. Microscopy, biochemical, and electrophysiological analysis revealed that Mid1 contains a modulatory region in its C-terminal tail that is required for establishing Ca2+ homeostasis. This region consists of the last 24 amino acids of Mid1 and functions as a modulatory region that affects Cch1 channel activity directly and mediates the trafficking of Mid1 to the plasma membrane. The modulatory region may serve as a likely point of contact between Cch1 and Mid1. This report demonstrates that restoring Ca2+ homeostasis via CMC in C. neoformans is dependent on the fundamental role of a modulatory region of Mid1.
MATERIALS AND METHODS
Cell culture and reagents.
All C. neoformans var. grubii strains (H99 MATa serotype A) and S. cerevisiae strains (W303 MATa, mid1Δ) were recovered from 15% glycerol stocks at −80°C prior to use in these experiments. The mid1Δ, cch1Δ, and mid1Δ cch1Δ mutant C. neoformans strains were constructed as previously described (2–4, 25). Strains were maintained on YPD (1% yeast extract, 2% peptone, and 2% dextrose) medium. Cells of C. neoformans were cultured in YPD medium at 30°C for 24 h. Cells from the HEK293 human embryonic kidney cell line were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% calf serum and antibiotics (penicillin-streptomycin [PEN-STREP]) in a 5% CO2 incubator at 37°C. HEK293 cells were purchased from ATCC (CRL-1573) (2). Where indicated, a cell-impermeative form of 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid, cesium salt (BAPTA; Invitrogen/Molecular Probes, Carlsbad, CA), was added to YPD medium (26).
Plasmids and transfection.
The cDNA of C. neoformans MID1 (CnMID1) was cloned into the pcDNA3.1/CT-GFP-TOPO expression plasmid, under the control of the cytomegalovirus (CMV) promoter, using conventional molecular biological techniques as previously described (2). Briefly, the MID1 amplicon was generated from cDNA synthesized by a SuperScript III first-strand synthesis system for reverse transcriptase PCR (RT-PCR) (Invitrogen) using total RNA as the template with the following primers: MID1-cDNA-F and MID1-cDNA-R (Table 1). RNA was isolated from a culture (∼5 × 107 cells) of a wild-type strain of C. neoformans var. grubii (H99, serotype A). Cells were lysed with acid-washed glass beads, and total RNA was isolated and purified according to the manufacturer's instructions (RNeasy kit; Qiagen). Following blue-white colony selection, positive transformants were isolated and sequenced. To assess the localization of the Mid1 truncated mutants in C. neoformans, fusion products for red fluorescent proteins (DsRed) expressed from the H99 C. neoformans actin promoter using primers listed in Table 1 were constructed (27). The DsRed was provided by J. Heitman (28). Construction of the Mid1 amino acid point mutations was performed by the substitution of R619, C621, and S605 amino acids to alanine by PCR using the reverse primers MID1-R619A, MID1-C621A, and MID1-S605A along with the forward primer MID1-cDNA-F (Table 1). The MID1 truncated mutants were constructed using the reverse primers MID1-R-CA-124, MID1-R-CB-91, and MID1-R-CC-24 and primer 1 as the forward primer (Table 1). The CnMID1 cDNA was also subcloned into a yeast expression vector (p424ADH; ATCC catalog no. 87373) and transformed into a mid1Δ deletion strain of S. cerevisiae.
Table 1.
Primers used in this study
| Primer no. | Primer designationb | Primer sequence |
|---|---|---|
| Primers for amplification of MID1 cDNA | ||
| 1 | MID1-cDNA-F | ATGCCAGCGAGAGAGGTGTA |
| 2 | MID1-cDNA-R | CTATCCGTTACACCATCTAT |
| Primers for construction of MID1 point mutations | ||
| 3 | MID1-F-point mutations | ATGCCAGCGAGAGAGGTGTATTTCAAA |
| 4 | MID1-R-R619A | TTCCGTTACACCATGCATTTCCCCAGCGATCTTGTGCGGT |
| 5 | MID1-R-C621A | TTCCGTTAGCCCATCTATTTCCCCAGCGATCTTGTGCGGT |
| 6 | MID1-R-S605A | TTCCGTTACACCATCTATTTCCCCAGCGATCTTGTGCGGTGACCCCTTGCTCTGCGGCCCGTC |
| Primers for construction of MID1 truncated mutantsa | ||
| 7 | MID1-R-CA-124 | GAAGCGTCTCTGATCGGTTTTGAACCGA |
| 8 | MID1-R-CB-91 | GTTTATGGCGCATATAGAGTTGTCCAAA |
| 9 | MID1-R-CC-24 | GCCGGCACAACCAGTCTCTGTAAGCAGA |
The forward primer used to make the MID1 truncated mutants was primer 1.
R indicates a reverse primer (3′ to 5′ direction), and F indicates a forward primer (5′ to 3′ direction).
HEK293 cells were counted and trypsinized 24 h before transfection. Approximately 1.25 × 105 cells in 1 ml of complete medium were plated per well such that the cell density represented ∼50% to 80% confluence on the day of transfection. Transient transfection of HEK293 cells with MID1 (cDNA)-green fluorescent protein (GFP) plasmid DNA or Mid1 truncated mutant or MID1 single-amino-acid mutant DNA was performed with Lipofectamine 2000 and Plus reagents according to the manufacturer's instructions (Invitrogen) (2). The cDNA of CCH1 was also expressed in HEK293 cells as previously described (2). Approximately 1 μg plasmid DNA was added to 100 μl of Opti-MEM I reduced-serum media and mixed with 1.75 ml of Lipofectamine 2000, and the mixture was incubated for 30 min at room temperature. The mixture was added directly to HEK293 cells that had been grown in a culture dish and maintained in DMEM supplemented with 4 mM l-glutamine–10% fetal bovine serum. HEK293 cells were maintained at 37°C with 5% CO2 18 to 24 h posttransfection prior to assaying cells for transgene expression (2, 25).
Sensitivity spot assays.
For spot assays, C. neoformans strains and S. cerevisiae strains were cultured in YPD medium overnight at 30°C. Cultures were pelleted and resuspended in sterile H2O. Serially diluted cells (106, 105, 104, 103, 102) were added to YPD agar plates supplemented with 1 mM BAPTA (2).
Protein analysis.
Biotinylation of HEK293 cells expressing Mid1-GFP or the Mid1 truncated GFP mutants or the Mid1 point GFP mutants was performed according to the protocol described in the manual for a Pierce cell surface biotin protein isolation kit (Thermo Scientific) and as described previously (2). HEK293 cells that had been transfected with Mid1-GFP or the Mid1-GFP truncated or point mutants were biotinylated according to the specifications outlined in the kit. Biotinylated protein samples were separated on a 10% SDS-PAGE gel and transferred to polyvinylidene difluoride (PVDF) membranes using semidry transfer (Bio-Rad). Western blotting for Mid1 was performed using a rabbit polyclonal antibody to GFP (Abcam, Cambridge, MA) (1:5,000) as the primary antibody followed by detection with a goat polyclonal antibody to rabbit immunoglobulin G (IgG) (H&L horseradish peroxidase [HRP]; Abcam, Cambridge, MA) (1:5,000).
Confocal microscopy.
Cells of C. neoformans were grown to mid-log phase in YPD overnight at 30°C. Cells were fixed in 3% formaldehyde for 1 h at 30°C and washed twice, and spheroplasts were obtained by digesting cell walls with 40 mg/ml lysing enzyme from Trichoderma harzianum (Amersham) in 1 M sorbitol–10 mM sodium citrate (pH 5.8) for 3 h at 30°C. Cells were then washed in 1 M sorbitol–10 mM sodium citrate buffer, diluted in phosphate-buffered saline (PBS), and dried on microscope slides. Cells were then incubated with a peptide antibody raised against Cch1 (Antibodies Incorporated, Davis, CA) (1:500 dilution) plus 1 mg/ml bovine serum albumin (Sigma) at 4°C overnight. The specificity of the primary peptide antibody for Cch1 was confirmed in previous publications (2, 4). Cells were subsequently washed extensively in PBS and then incubated for 1 h with fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Abcam, Cambridge, MA) (1:1,000). To examine the localization of Mid1 and Mid1 truncated mutants in HEK293 cells, cells expressing plasmids containing Mid1-GFP mutant fusion proteins were fixed in 4% paraformaldehyde for 20 min at room temperature, washed in PBS, and subsequently visualized with a confocal microscope. Immunofluorescence analysis of Cch1 was performed in HEK293 cells that had been fixed in 4% paraformaldehyde and permeabilized. A primary peptide antibody against the C terminus of Cch1 (Antibodies Incorporated, Davis, CA) was added to HEK cells at a dilution of 1:500 as previously done (2, 4, 25). Cch1 was visualized with a secondary antibody (1:1,000) conjugated with Texas Red (Abcam) (2, 4, 25). Fluorescence was examined using a Carl Zeiss LSM-5 inverted confocal microscope. Mid1 and Cch1 were examined with a laser line (488 nm and a 568 nm) and a 40× objective. The images were scanned and captured at a resolution of 1,024 by 1,024 pixels.
Patch clamp measurements.
Experiments were performed using conventional whole-cell and single-channel patch clamp techniques (29, 30). In order to measure currents through Cch1, HEK cells expressing Cch1 and Mid1 were visually selected based on bright GFP fluorescence. Currents were stimulated by passive Ca2+ store depletion with a pipette solution containing 10 μM BAPTA-AM or by activation with thapsigargin (100 μM). Recording electrodes were pulled with a vertical puller (HEKA Instruments Inc., Bellmore, NY) from borosilicate glass capillaries, coated with Sylgard, fire polished, and filled with a solution containing either BAPTA-AM or thapsigargin plus 130 mM Cs-positive (Cs+) methane sulfonate, 5 mM MgCl2, 500 μM Mg-ATP, and 20 mM HEPES (pH 7.2) with CsOH. Recording electrodes had a tip resistance of ∼ 5 MΩ when placed in an external solution containing 2, 5, or 10 mM CaCl2 or BaCl2 plus 140 mM NMDG (N-methyl-d-glucamine) (or 140 mM Na+ methane sulfonate in divalent-free solution and free of NMDG), 10 mM glucose, and 10 mM HEPES (pH 7.4) with NaOH. Whole-cell currents were measured by voltage ramps (−120 to +60 mV, lasting 200 ms) from a holding potential of −60 mV. All voltages have been corrected for liquid junction potentials. Currents were filtered at 2 kHz with a four-pole Bessel filter (Dagan) contained in the Dagan amplifier and sampled at 5 kHz. The amplifier was interfaced with a Digidata 1322A digitizer (Axon Instruments, Foster City, CA) in order to digitize data. All data were corrected for leak currents. For single-channel measurements, the outside-out patch configuration was used. Both the extracellular and pipette solutions used for single-channel recordings were the same as those used in whole-cell experiments. Single-channel records were obtained at voltages of −60 mV and −80 mV and sampled at 25 kHz. Currents were analyzed with pCLAMP 9.0 software (Axon) and graphed with SigmaPlot 8.0 software on an IBM 3-GHz computer.
RESULTS
A low, free Ca2+ assay identified the C terminus of Mid1 as a regulatory region required for CMC activity.
In order to determine the functional activity of Mid1 in C. neoformans, truncated mutants of Mid1 that lacked regions of the C terminus and N terminus were created. We reasoned that since Mid1 is an integral membrane protein, both of the predicted cytosolic ends of the Mid1 protein represented viable regions for association with Cch1 (2, 19, 20). Three C-terminal-truncated mutants were created: (i) a Mid1-CA-124 mutant (lacking 124 amino acids); (ii) a Mid1-CB-91 mutant (lacking 91 amino acids); and (iii) a Mid1-CC-24 mutant (lacking 24 amino acids) (Fig. 1A). Three N-terminal-truncated mutants of Mid1 were also created, but despite several attempts, expression of these mutants in HEK293 cells or in C. neoformans was not detected (data not shown). These results indicated that the N-terminal region of the Mid1 protein might be required for stability of the Mid1 polypeptide.
Fig 1.

Activity of the Cch1 channel in C. neoformans is dependent on a C-terminal modulatory region of the subunit, Mid1. (A) Three truncated mutants of Mid1, each lacking a portion of the C terminus, were constructed. Mid1-CA-124 lacked 124 amino acids (aa), Mid1-CB-91 lacked 91 amino acids, and Mid1-CC-24 lacked 24 amino acids. Each mutant was constitutively expressed in the mid1Δ deletion background of C. neoformans under the control of the actin promoter (27). (B) Sensitivity spot assays were used to identify regions of the Mid1 protein critical for Cch1 activity. The Mid1-CC-24 truncated mutant did not rescue the sensitivity of the mid1Δ deletion strain on low, free [Ca2+] medium, suggesting that this region is critical for Cch1 activity. (C) A wild-type yeast (Saccharomyces cerevisiae) strain was transformed with full-length CnMid1. Two representative transformants expressing CnMid1 did not restore growth of the Scmid1Δ deletion strain on low, free [Ca2+] medium. (B and C) Serially diluted cells (106, 105, 104, 103, 102) from wild-type and mid1Δ deletion strains of C. neoformans and yeast were added to YPD agar plates supplemented with BAPTA (cell impermeative), and spot assays were performed as explained above.
It is known that Mid1 is an essential partner and coregulator of Cch1 in C. neoformans, yeast, and other fungi (1–6) Accordingly, a strain of C. neoformans lacking Mid1 displays growth sensitivity under conditions of limited extracellular Ca2+ (∼100 nM)—a similar phenotype was reported for the cch1Δ deletion strain (2, 3, 5). Thus, we used a simple functional assay based on this phenotype to resolve whether the channel activity of Cch1 was dependent on the C-terminal and/or N-terminal regions of the Mid1 protein. To do this, we monitored whether the expression of the Mid1 truncated mutants in the mid1Δ deletion background could rescue the mid1Δ deletion strain sensitivity in the presence of low, free Ca2+ medium. For this assay, growth of C. neoformans and yeast strains on agar plates with YPD media (where the free [Ca2+] = ∼0.140 mM) and growth on YPD medium plus BAPTA, a Ca2+ chelator (where the free [Ca2+] = ∼100 nM), were compared (Fig. 1B and C) (26).
The assays revealed that a mid1Δ deletion strain of C. neoformans expressing a Mid1-CC-24 truncated mutant (lacking the last 24 amino acids) displayed a significant sensitivity to an extracellular environment that was low in free Ca2+, suggesting that CMC activity was compromised (Fig. 1B). A similar result was observed for the mid1Δ deletion strain expressing the Mid1-CA-124 mutant or the Mid1-CB-91 mutant (data not shown). However, the expression of full-length Mid1 in the mid1Δ strain did indeed rescue the growth sensitivity of the mid1Δ deletion strain in low, free Ca2++, suggesting that the assay worked as predicted (Fig. 1B). These results suggested that CMC activity was dependent on the C-terminal tail (the last 24 residues) of Mid1. Interestingly, the expression of CnMid1 full-length cDNA in yeast (S. cerevisiae) did not rescue the mid1Δ deletion strain sensitivity of yeast, suggesting significant differences in the amino acid sequences of CnMid1 and ScMid1 and/or a lack of accessory proteins in yeast that are required for Cch1 channel activity in C. neoformans (Fig. 1C).
Multiple alignment of the primary structures of Mid1 reveals a high degree of similarity in the latter half of Mid1 but not within the modulatory region.
A protein structure for Mid1 has not yet been resolved; however, previous results have determined that Mid1 forms an oligomeric structure that exists primarily as an integral plasma membrane protein and specifically associates with Cch1 (2, 5). In silico analysis suggested that Mid1 may have 2 to 4 predicted transmembrane regions and that both the C-terminal and N-terminal ends likely face the cytoplasm. Interestingly, similar analyses revealed that the N-terminal region of Mid1 lacks a predicted signal sequence that is often present in plasma membrane-bound proteins. This is consistent with previous observations of ScMid1 characteristics (20, 31).
Analysis of the Mid1 amino acid sequence alignments revealed some degree of similarity in the region closest to the C terminus; however, very little similarity was observed within the modulatory region (last 24 residues) (see Fig. S1 in the supplemental material) (32, 33). The conservation of 11 of the 12 cysteine residues within the latter half of Mid1 was striking, and it indicated a potential role for this group of cysteines in complex formation (see Fig. S1 in the supplemental material [black filled triangles]). Another noteworthy feature of the Mid1 alignment was the presence of two conserved heme-regulatory cysteine-proline (CP) motifs (CP dipeptide) at position 924 and position 932 (see Fig. S1 in the supplemental material [black filled ovals]). This motif has been implicated in mediating cell signaling events that in some cases involved a Ca2+-dependent Slo1 potassium (BK) channel that operated via the binding of heme to the CP motifs (21, 34–38).
Mid1 trafficking to the plasma membrane is dependent on a modulatory region within the C-terminal end.
To further explore the mechanism of Mid1, confocal microscopy was used to examine the Mid1-CC-24 truncated mutant protein in vivo. To do this, a Mid1-CC-24–DsRed fusion protein was constructed and expressed in C. neoformans. As a control, we examined the mid1Δ deletion strain of C. neoformans expressing the Mid1-CC-24–DsRed fusion protein by light microscopy and found that these cells appeared physically unaltered, as expected (Fig. 2A, differential interference contrast [DIC]). Confocal microscopy was then used to examine the expression of the Mid1-CC-24–DsRed fusion protein. Unexpectedly, the Mid1-CC-24 truncated mutant protein was not observed on the cell surface of cryptococci as is typically observed for both Mid1 and Cch1 full-length proteins (Fig. 2B, C, and D). Indeed, it has been previously shown that Cch1 and Mid1 colocalize and reside in the plasma membrane as a complex (2, 4, 5). Instead, the Mid1-CC-24–DsRed fusion protein appeared to be largely cytosolic, indicating that Mid1 could not localize to the plasma membrane of C. neoformans independently of the 24 amino acids in its C-terminal region (Fig. 2B).
Fig 2.
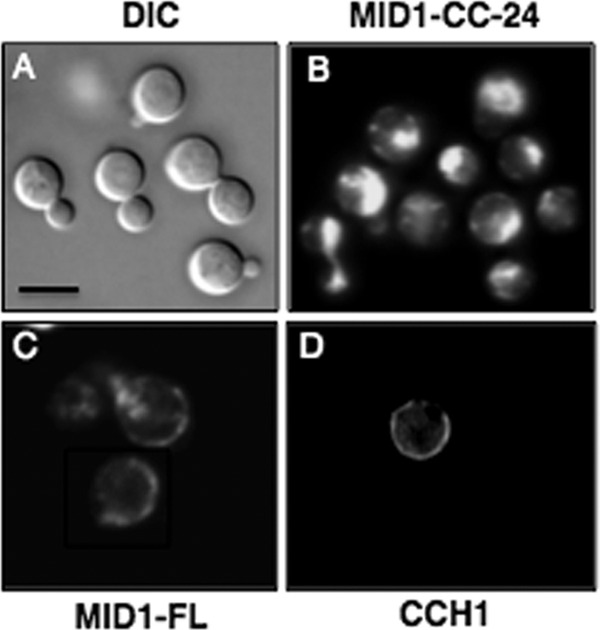
Loss of the modulatory region of Mid1 alters the localization pattern of Mid1 in C. neoformans. (A) Mid1-CC-24–DsRed fusion protein was constructed by tagging the Mid1-CC-24 truncated mutant with DsRed (28) and visualized by DIC and confocal microscopy. (A) A DIC image of cryptococci expressing the Mid1-CC-24–DsRed fusion protein revealed bright, spherical cells of C. neoformans. (B and C) Interestingly, Mid1-CC-24–DsRed appeared to be mislocalized. This was in stark contrast to the full-length Mid1 (Mid1-FL) dsRed fusion protein, which is predominately localized to the cell surface of C. neoformans, consistent with its plasma membrane distribution as previously reported (2). (D) Immunofluorescence (Texas Red) of Cch1 revealed a surface distribution in C. neoformans similar to that of Mid1, confirming that, like Cch1, Mid1 is primarily localized to the plasma membrane in cells of C. neoformans. A primary peptide antibody to the C terminus of Cch1 and a Texas Red-conjugated secondary antibody were used to visualize Cch1 as reported previously (2, 4). The specificity of the primary peptide antibody to Cch1 was confirmed in previous publications (2, 4). Scale bars represent 10 μM.
We further examined this region of Mid1 more closely in HEK293 (human embryonic kidney) cells. The HEK293 cell line was chosen because it represents an excellent expression system for characterizing channel proteins and because we previously successfully expressed the Cch1-Mid1 channel complex in HEK293 cells and demonstrated by patch clamp analysis that Cch1 is activated by the depletion of intracellular Ca2+ stores (2). Our aim in this study was to determine whether the C-terminal region of Mid1 directly affected Cch1 channel activity. Thus, prior to examining the channel activity of the Cch1 pore upon heterologous coexpression with the truncated mutants of Mid1 in HEK293 cells, it was necessary to determine whether the Mid1 truncated mutants remained targeted to the plasma membrane. To do this, a GFP fusion protein of each Mid1 truncated mutant was made and expressed in HEK293 cells (Fig. 3). Biotin labeling of surface proteins in HEK293 cells expressing a full-length Mid1 protein followed by Western blotting revealed two specific bands corresponding to full-length Mid1 (Fig. 3A, Mid1-FL). The lower band (∼100 kDa) is representative of the monomer form of Mid1, whereas the higher band (∼ 200 kDa) may suggest that Mid1 forms a complex (Fig. 3A). This property of Mid1 has been previously reported, and it was further demonstrated that the complex might form via disulfide bonding (31).
Fig 3.
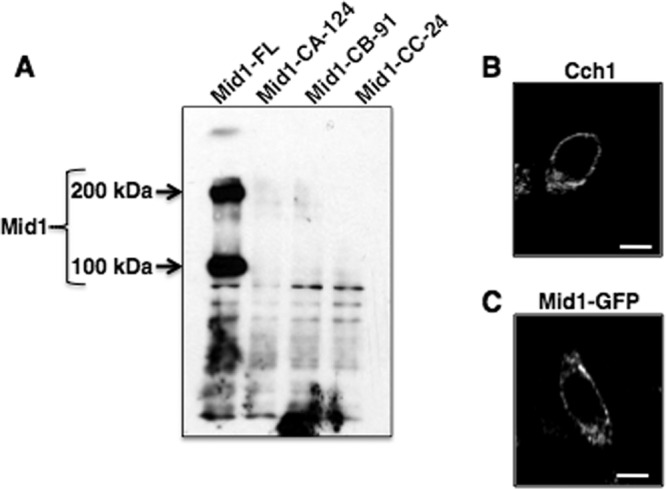
The expression of Mid1 on the surface of HEK293 cells is dependent on the C-terminal modulatory region. (A) Western blot analysis of surface biotinylation of HEK293 cells expressing full-length Mid1-GFP fusion protein revealed two distinct bands corresponding to Mid1 (monomer, ∼ 100 kDa; complex, ∼ 200 kDa [indicated by arrows]). In contrast, Mid1 polypeptides were not detected in Western blots of biotinylated HEK293 cells expressing Mid1 truncation mutants (Mid1-CA-124, Mid1-CB-91, or Mid1-CC-24). The Mid1-CA-124 mutant lacked 124 residues, Mid1-CB-91 lacked 91 residues, and Mid1-CC-24 lacked 24 residues. Western blotting for Mid1 was performed using a commercial rabbit polyclonal antibody to GFP (1:5,000) as the primary antibody (Abcam, Cambridge, MA) followed by detection with a goat polyclonal antibody to rabbit immunoglobulin G (IgG) (1:5,000). (B and C) Surface expression of Cch1 is independent of Mid1. Full-length Mid1 was tagged with a C-terminal GFP expressed in HEK293 cells and visualized by confocal microscopy. Immunofluorescence (Texas Red) of Cch1 (not tagged) in HEK293 cells showed surface localization similar to that of Mid1. A primary peptide antibody to the C terminus of Cch1 and a Texas Red-conjugated secondary antibody were used to visualize Cch1 (2, 4). Scale bars represent 10 μM.
In striking contrast, the protein bands corresponding to Mid1 were not detected in HEK293 cells expressing the three Mid1 C-terminal-truncated mutants (Fig. 3A). This suggested that these mutants (Mid1-CA-124, Mid1-CB-91, and Mid1-CC-24) were not expressed on the surface of HEK293 cells, further indicating that Mid1 could not localize to the surface of HEK293 cells without an intact C terminus as observed in C. neoformans. Consistent with this analysis, Ca2+ channel currents from HEK293 cells expressing Cch1-GFP and the Mid1-GPF truncated mutants were not detected by conventional patch clamp techniques (data not shown). Confocal microscopy revealed that Cch1 and Mid1 were targeted to the surface of HEK293 cells independently of each other; thus, the absence of CMC activity upon expression of the Mid1 truncated mutants was not due to a defect in Cch1 trafficking (Fig. 3B and C).
These results were further corroborated by confocal microscopy of HEK293 cells expressing the Mid1 truncated mutant-GFP fusion proteins (Fig. 4). There is little doubt that polypeptides corresponding to the Mid1 truncation mutants were indeed synthesized and expressed in HEK293 cells, since strong fluorescence signals corresponding to the Mid1 truncated mutant-GFP fusion proteins were clearly detected (Fig. 4B, C, and D). However, the Mid1 truncated mutant proteins appeared mislocalized in HEK293 cells, since the mutants were no longer observed on the surface of HEK293 cells (Fig. 4B, C, and D). The localization patterns of the Mid1 truncation mutants in HEK293 cells were strikingly different from those of the full-length Mid1 protein, which displayed a cell surface distribution in HEK293 cells (Fig. 4A). These results supported the biotin labeling experiment data and further indicated that the last 24 amino acid residues in the C-terminal tail of Mid1 are critical for targeting Mid1 to the plasma membrane in HEK293 cells.
Fig 4.
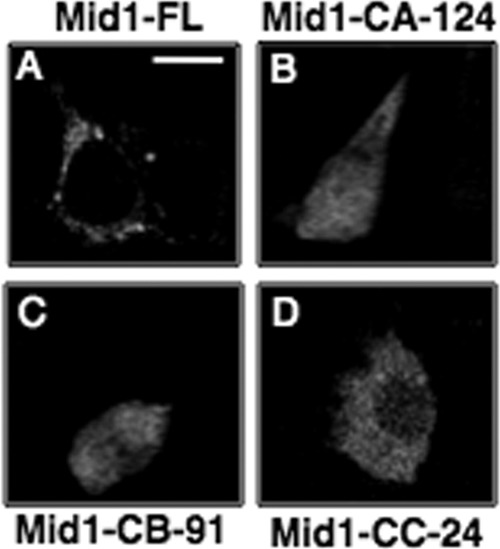
C-terminal-truncated mutants of Mid1 are mislocalized in HEK293 cells. Confocal microscopy was used to examine the localization of three truncated mutants of Mid1 in HEK293 cells. (A) Full-length Mid1-GFP under the control of the CMV promoter was expressed in HEK293 and observed on the cell surface. (B, C, and D) However, expression of the Mid1 truncated mutants lacking specified regions of the C terminus (Mid1-CA-124, Mid1-CB-91, and Mid1-CC-24) displayed a diffuse and predominately cytosolic localization pattern unlike that of full-length Mid1. The Mid1-CA-124 mutant lacked 124 residues, Mid1-CB-91 lacked 91 residues, and Mid1-CC-24 lacked 24 residues.
Key amino acids in the C-terminal region of Mid1 promote CMC activity.
Among the 24 amino acids within the C-terminal modulatory domain of Mid1, 3 amino acids were identified as potentially relevant residues. The charged amino acid (arginine; R619) and the only cysteine (C621) residue in this region mediating disulfide bond formation were selected for substitution experiments because of their key roles in bridging protein complexes. In addition, a predicted protein kinase C (PKC)-phosphorylated serine residue (S605) was selected because of its potential role in Ca2+ signaling. To establish the role of these residues in the molecular mechanism of Mid1, cysteine (C621A), arginine (R619A), and a serine residue (S605A) were replaced with alanine (Fig. 5A). Single GFP fusion proteins of the Mid1 point mutations were constructed and examined in HEK293 cells. Interestingly, confocal microscopy revealed an altered localization pattern for all the Mid1 point mutations, with the exception of the Mid1 serine mutant (S605A) (Fig. 5B). In this case, HEK293 cells expressing the Mid1-S605A–GFP fusion revealed a localization pattern of the Mid1-S605A mutant that was similar to the expression of full-length, unaltered Mid1-GFP (Fig. 5B). In contrast, the most striking result occurred in HEK293 cells expressing the Mid1-R619A–GFP mutant protein, where a strongly diffuse localization pattern was observed (Fig. 5B). The Mid1-C621A–GFP mutant also appeared mislocalized, as determined by its presence throughout regions of the cytosol (Fig. 5B). This expression pattern was not consistent with the more uniform expression of Mid1-GFP along the surface of HEK293 cells (Fig. 5B).
Fig 5.
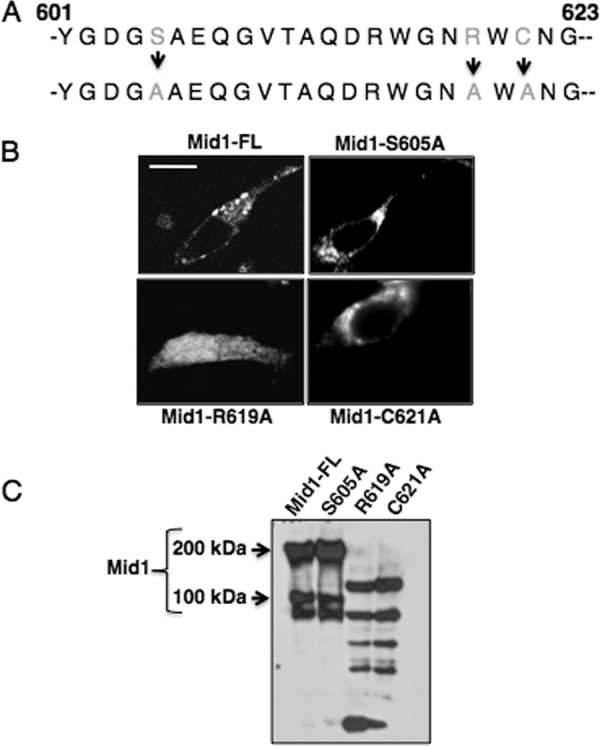
Arginine and cysteine residues of the modulatory region of Mid1 are critical for its localization. (A) Three key residues in the C-terminal modulatory region of Mid1 were altered. A predicted PKC-phosphorylation serine residue (Mid1-S605A), an arginine residue (Mid1-R619A), and a cysteine residue (Mid1-C621A) were substituted with alanine residues by PCR and confirmed by DNA sequencing. (B) GFP fusion proteins of the Mid1 point mutations were constructed and expressed in HEK293 cells under the control of the CMV promoter. Confocal microscopy revealed that the R619A Mid1 mutant displayed a diffuse localization pattern whereas the C621A mutant appeared to be localized in distinct patches throughout the cell. In contrast, the S605A mutant displayed a localization pattern similar to that of Mid1, suggesting predominant plasma membrane localization. (C) Biotin labeling of the HEK293 cells expressing the Mid1 point mutations further supported the notion of the absence of the R619A and C621A mutants from the cell surface and confirmed the unaltered targeting of S605A to the surface of HEK293 cells. Western blotting was performed using a commercial rabbit polyclonal antibody to GFP (1:5,000) as the primary antibody (Abcam, Cambridge, MA) followed by detection with a goat polyclonal antibody to rabbit immunoglobulin G (IgG) (1:5,000) (2).
To confirm the surface expression of the Mid1 serine mutant observed by confocal microscopy, HEK293 cells expressing Mid1-S605A–GFP, Mid1-R619A–GFP, Mid1-C621A–GFP, or Mid1-GFP were biotin labeled (Fig. 5C). Western blot analysis revealed that the polypeptides corresponding to full-length, unaltered Mid1 were detected in HEK293 expressing Mid1-S605A–GFP or Mid1-GFP (Fig. 5C), suggesting that the serine residue within the C-terminal modulatory region was not required for trafficking of Mid1 to the cell surface. However, Mid1 protein bands were not detected in biotin-labeled HEK293 cells expressing Mid1-R619A–GFP or Mid1-C621A–GFP mutants, indicating that both residues were required for the surface localization of Mid1 (Fig. 5C).
A predicted phosphorylation site in the modulatory domain of Mid1 alters the kinetics of CMC-mediated Ca2+ influx.
Our results suggested that the serine residue in the C-terminal region of Mid1 was not involved in the trafficking of Mid1. However, since this residue is a predicted PKC-phosphorylation site and the only serine residue within this region, we questioned whether it might have a direct role in mediating Ca2+ influx via CMC. To examine this, the Mid1-S605A mutant was expressed in the mid1Δ deletion strain of C. neoformans under the control of a constitutive, actin promoter (27). Reverse transcriptase PCR analysis revealed robust expression of the Mid1-S605A transcript in C. neoformans (Fig. 6A). Interestingly, expression of the Mid1-S605A mutant in C. neoformans rescued the sensitivity associated with the mid1Δ deletion strain on low, free Ca2+ media, suggesting that the lack of a S605 residue did not impair CMC-mediated Ca2+ influx (Fig. 6B). However, patch clamp analysis of Ca2+ channel currents from HEK293 cells expressing Cch1-GFP and Mid1-S605A–GFP revealed differences in the kinetics of CMC (Fig. 6C and D). Similar to previous electrophysiological characterization of CMC by ramp protocols, the depletion of ER Ca2+ stores by BAPTA-AM led to the development of robust, time-dependent Cch1-mediated Ca2+ currents (Fig. 6C) (2). We previously demonstrated that these inward Ca2+ currents activated specifically by Ca2+ store depletion were not detected in HEK293 cells expressing either Cch1 or Mid1 alone or an empty plasmid, indicating that Cch1 activity required Mid1 and, importantly, that the measured Ca2+ currents were not endogenous to HEK293 cells (2). The coexpression of Cch1 and Mid1-S605A in HEK293 cells resulted in Cch1-mediated Ca2+ currents that displayed a more rapid activation and a significantly slower inactivation; however, current amplitude levels were largely unchanged (Fig. 6D). These differences in CMC kinetics suggest that Mid1 may have a direct role in regulating the activity of the Cch1channel pore.
Fig 6.

A predicted phosphorylation site in the modulatory domain of Mid1 alters the kinetics of CMC activity. (A) The Mid1-S605A point mutation was expressed in the mid1Δ deletion strain of C. neoformans. Transcript analysis by reverse transcriptase PCR analysis revealed a robust expression of a transcript corresponding to the Mid1-S605A point mutation in C. neoformans. The S605A mutant was constitutively expressed under the control of the actin promoter. (B) Sensitivity assays demonstrated that the strain of C. neoformans expressing Mid1-S605A rescued the sensitivity of the mid1Δ strain on low-Ca2+ media (YPD plus 1 mM BAPTA; [Ca2+] = ∼100 nM), suggesting that serine 605 was not required for CMC activity. Strains H99, mid1Δ, and mid1Δ::S605A were grown overnight in YPD, serially diluted (104, 103, 102, 101), and spotted onto agar plates of YPD alone or YPD plus 1 mM BAPTA. (C and D) Representative traces of a time course of whole-cell Ca2+ currents through the Cch1 channel. Ca2+ currents were measured upon the depletion of Ca2+ stores by the addition of 10 μM BAPTA-AM in the patch pipette from HEK293 cells expressing Cch1 and full-length Mid1 (C) or expressing Cch1 and Mid1-S605A (D).
DISCUSSION
The first mechanistic study of Cch1 by electrophysiological characterization revealed that the Cch1-Mid1 channel complex is a Ca2+-selective store-operated channel that is gated by the depletion of intracellular Ca2+ (2). This conclusion is largely based on the direct measurement of Cch1-mediated inward Ca2+ currents activated by the passive depletion of Ca2+ stores during ER stress (2). The influx of Ca2+ coupled to the Ca2+-depleted state of the ER strongly suggested that Cch1 functions primarily to replenish these stores and reestablish Ca2+ homeostasis. This activation of the channel pore is dependent on Mid1, but how Mid1 contributes to this activity is not known.
We identified a modulatory region that consisted of 24 amino acids in the C terminus of Mid1 that is critical for Mid1 trafficking and CMC activity. A strain of C. neoformans expressing a mutant of Mid1 lacking this region was not viable on low, free Ca2+ medium, indicating that Cch1 could not mediate the influx of Ca2+ from the extracellular environment in the absence of the Mid1 modulatory domain. This is consistent with the role of CMC as the only high-affinity Ca2+ channel in the plasma membrane of C. neoformans. We found that the C-terminal tail mediated the trafficking of Mid1 to the cell surface; however, Mid1 did not affect the surface expression of the channel pore (Cch1), since we have shown that Cch1 is expressed in the plasma membrane independently of Mid1 (2). The inability of the Mid1 C-terminal tail mutant to activate Cch1 was likely due to its inability to associate with Cch1, since the Mid1 mutant was no longer targeted to the plasma membrane. Interestingly, a similar observation was made in S. cerevisiae, indicating some similarity in the functional mechanism of Mid1 (39). However, although the C-terminal modulatory region of Mid1 in C. neoformans appeared to be fundamentally important for Mid1 trafficking, as observed for yeast, it is surprising that very little similarity was observed in the amino acid sequence within this region among fungi. This finding argues against the presence of a conserved targeting sequence within the modulatory domain of Mid1. In the case of C. neoformans, Mid1 trafficking could be the result of palmitoylation—a posttranslational modification of the cysteine within the modulatory region (C621). This thioester linkage of a 16-carbon palmitate lipid to a cytosolic-exposed cysteine residue could promote anchoring of Mid1 to the plasma membrane (40). Indeed, many regulatory subunits of ion channels have been identified as being palmitoylated (41). The lipid modification of channel pore proteins and/or channel subunits can promote both the surface expression and regulation of ion channels (41). In C. neoformans, it has been shown that palmitoylation regulates different aspects of cytoplasmic proteins and G proteins (9, 22). In the case of Mid1, lipid modification of C621 could provide a viable mechanism for membrane association and this would support the essential function of C621 in the modulatory domain during Mid1 trafficking; however, more work is needed to confirm this notion. Biochemical analysis has revealed that Mid1 forms an oligomeric structure likely mediated by disulfide bonds. The 11 conserved cysteine residues within the C-terminal half of Mid1 likely play a role in Mid1 complex formation. Our results indicated that substitution of the only cysteine residue (C621) within the modulatory domain of Mid1 prevented the targeting of Mid1 to the plasma membrane and consequently precluded the functional activity of CMC. Nevertheless, it is not clear whether the C621 residue also mediates the formation of the Mid1 oligomeric complex.
Further analysis of Mid1 identified a key arginine residue within the modulatory domain that was crucial for Mid1 function. It is known that polar and charged amino acids primarily cover the surface of proteins and usually associate with each other. Positively charged amino acids form salt bridges with charged residues, and this noncovalent interaction is crucial because it provides stability to the folded protein conformation (42, 43). Thus, it is likely that the arginine residue is responsible for maintaining a stable conformation of the modulatory region of Mid1.
Although not part of the modulatory region, two CP dipeptides in the C-terminal region of Mid1 are highly conserved among fungi. It has been shown in eukaryotes that heme binds to proteins such as transcription factors and mitogen-activated protein (MAP) kinases through CP motifs (34, 35, 37). In addition to the role of heme in the transport and storage of oxygen, heme also regulates several other molecular and cellular processes involved in oxygen sensing (34). In yeast, heme binds to the HapI transcription factor via CP motifs and subsequently mediates expression of genes induced by heme and oxygen (21, 37, 38). Interestingly, heme has been shown to bind and inhibit Ca2+-dependent Slo1 BK (potassium) channels in mammalian cells, suggesting that heme can act as an acute signaling molecule, with ion channels as its target (36). In the case of C. neoformans, it is conceivable that certain stress conditions might promote the signaling activity of heme, which could then bind the CP motifs in Mid1 and possibly contribute to further regulation of CMC activity.
It is not surprising that Ca2+ currents mediated by Cch1 were not detected upon the coexpression of Cch1 and the Mid1 C-terminal-tail-truncated mutants, since it has been shown that the channel pore of CMC cannot conduct Ca2+ currents independently of Mid1 (2). It is intriguing, however, that the loss of a serine residue (S605) within the modulatory region of Mid1 altered the kinetics of the Cch1 channel pore but did not prevent Cch1-mediated Ca2+ influx in C. neoformans or in HEK293 cells. Patch clamp analysis revealed a significantly faster activation of channel currents upon depletion of ER Ca2+ with BAPTA-AM and a slower inactivation of the channel; however, Ca2+ current amplitude levels remained the same. These results suggest that the modulatory region of Mid1 may directly control the activity of the Cch1 pore. This notion is consistent with the functional mechanisms of some ion channels found in mammalian cells. For example, the beta-subunit of voltage-gated calcium channels has been shown to regulate biophysical properties of high-voltage-activated Ca2+ channels in addition to regulating their own functional expression (44).
In summary, Ca2+ homeostasis in C. neoformans is established through the action of the Cch1-Mid1 channel complex whose activity is dependent on a crucial set of amino acids in the C-terminal tail of Mid1. This nonconserved modulatory region is essential for Mid1 trafficking and for CMC activity.
Supplementary Material
ACKNOWLEDGMENTS
We are especially grateful to K. W. Cunningham for invaluable discussions, to E. Blumwald for critical assessment of the manuscript, and to S. Y. Park for technical assistance.
This work was supported by Public Health Service grants from the National Institutes of Health (AI054477 and AI078848) awarded to A.G.
Footnotes
Published ahead of print 21 November 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00130-12.
REFERENCES
- 1. Hallen HE, Trail E. 2008. The L-type calcium ion channel Cch1 affects ascospore discharge and mycelial growth in the filamentous fungus Gibberella zeae (anamorph Fusarium graminearum). Eukaryot. Cell 7:415–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hong MP, Vu K, Bautos J, Gelli A. 2010. Cch1 restores intracellular Ca2+ in fungal cells during endoplasmic reticulum stress. J. Biol. Chem. 285:10951–10958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu M, Du P, Heinrich G, Cox GM, Gelli A. 2006. Cch1 mediates calcium entry in Cryptococcus neoformans and is essential in low-calcium environments. Eukaryot. Cell 5:1788–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu M, Gelli A. 2008. Elongation factor 3, EF3, associates with the calcium channel Cch1 and targets Cch1 to the plasma membrane in Cryptococcus neoformans. Eukaryot. Cell 7:1118–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Locke EG, Bonilla M, Liang L, Takita Y, Cunningham KW. 2000. A homolog of voltage-gated Ca2+ channels is stimulated by depletion of secretory Ca2+ in yeast. Mol. Cell. Biol. 20:6686–6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Paidhungat M, Garrett S. 1997. A homologue of mammalian voltage-gated calcium channels mediates yeast pheromone-stiumulated calcium uptake and exacerbates the cdc1(Ts) growth defect. Mol. Cell. Biol. 17:6339–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zelter A, Bencina M, Bowman BJ, Yarden O, Read ND. 2004. A comparative genomic analysis of the calcium signaling machinery in Neurospora crassa, Magnaporthe grisea, and Saccharomyces cerevisiae. Fungal Genet. Biol. 41:827–841 [DOI] [PubMed] [Google Scholar]
- 8. Casadevall A, Perfect JR. 1998. Cryptococcus neoformans. ASM Press, Washington, DC [Google Scholar]
- 9. Mitchell TG, Perfect JR. 1995. Cryptococcsis in the ear of AIDS—100 years after the discovery of Cryptococcus neoformans. Clin. Microbiol. Rev. 8:515–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Rao A. 2006. A mutation in Orai causes immune deficiency by abrogating CRAC channel function. Nature 441:179–185 [DOI] [PubMed] [Google Scholar]
- 11. Hogan PG, Rao A. 2007. Dissecting ICRAC, a store-operated calcium current. TRENDS Biochem. Sci. 32:235–245 [DOI] [PubMed] [Google Scholar]
- 12. Parekh AB, Putney JW., Jr 2005. Store-operated calcium channels. Physiol. Rev. 85:757–810 [DOI] [PubMed] [Google Scholar]
- 13. Putney JW., Jr 1986. A model for receptor-regulated calcium entry. Cell Calcium 7:1–12 [DOI] [PubMed] [Google Scholar]
- 14. Putney JW., Jr 1990. Capacitative calcium entry revisited. Cell Calcium 11:611–624 [DOI] [PubMed] [Google Scholar]
- 15. Catterall WA. 2000. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16:521–555 [DOI] [PubMed] [Google Scholar]
- 16. Durrell SR, Shrivastava IH, Guy HR. 2004. Models of the structure and voltage-gating mechanism of the shaker K+ channel. Biophys. J. 87:2116–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Long SB, Campbell EB, MacKinnon R. 2005. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 309:903–908 [DOI] [PubMed] [Google Scholar]
- 18. Bakowski D, Parekh AB. 2007. Voltage-dependent Ba2+ permeation through store operated CRAC channels: implications for channel selectivity. Cell Calcium 42:333–339 [DOI] [PubMed] [Google Scholar]
- 19. Bonilla M, Cunningham KW. 2003. Mitogen-activated protein kinase stimulation of Ca2+ signaling is required for survival of endoplasmic reticulum stress in yeast. Mol. Biol. Cell 14:4296–4305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bonilla M, Nastase KK, Cunningham KW. 2002. Essential role of calcineurin in response to endoplasmic reticulum stress. EMBO J. 21:2343–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lan C, Lee HC, Tang S, Zhang L. 2004. A novel mode of chaperone action: heme activation of HapI by enhanced association of Hsp90 with the repressed Hsp70-HapI complex. J. Biol. Chem. 279:27607–27612 [DOI] [PubMed] [Google Scholar]
- 22. Leach MD, Brown AJP. 2012. Post-translational modifications of proteins in the pathobiology of medically relevant fungi. Eukaryot. Cell 11:98–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin DC, Kim H, Mackin NA, Maldonado-Baez L, Evangelista CC, Beaudry VG, Dudgeon DD, Naiman DQ, Erdman SE, Cunningham KW. 2011. New regulators of a high-affinity Ca2+ influx system revealed through a genome-wide screen in yeast. J. Biol. Chem. 286:10744–10754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanzaki M, Nagasawa M, Kojima I, Sato C, Naruse K, Sokabe M, Ida H. 1999. Molecular identification of a eukaryotic, stretch-activated nonselective cation channel. Science 285:882–886 [DOI] [PubMed] [Google Scholar]
- 25. Vu K, Bautos JM, Hong MP, Gelli A. 2009. The functional expression of toxic genes: lessons learned from molecular cloning of CCH1, a high-affinity Ca2+ channel. Anal. Biochem. 393:234–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsien RY. 1980. New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis and properties of prototype structures. Biochemistry 19:2396–2403 [DOI] [PubMed] [Google Scholar]
- 27. Gorlach JM, McDade HC, Perfect JR, Cox GM. 2002. Antisense repression in Cryptococcus neoformans as a laboratory tool and potential antifungal strategy. Microbiology 148:213–219 [DOI] [PubMed] [Google Scholar]
- 28. Idnurm A, Giles SS, Perfect JR, Heitman J. 2007. Peroxisome function regulates growth on glucose in the Basidiomycete fungus Cryptococcus neoformans. Eukaryot. Cell 6:60–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. 1981. Improved patch-clamp techniques for high-resolution current recordings from cells and cell-free membrane patches. Pflugers Arch. 391:85–100 [DOI] [PubMed] [Google Scholar]
- 30. Hille B. 1992. Ionic channels of excitable membranes, 2nd ed. Sinauer Associates Inc., Sunderland, MA [Google Scholar]
- 31. Maruoka T, et al. 2002. Essential hydrophilic carboxyl-terminal regions including cysteine residues of the yeast stretch-activated calcium-permeable channel Mid1. J. Biol. Chem. 277:11645–11652 [DOI] [PubMed] [Google Scholar]
- 32. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 33. Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment and analysis workbench. Bioinformatics 25:1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mense S, Zhang L. 2006. Heme: a versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 16:681–692 [DOI] [PubMed] [Google Scholar]
- 35. Ogawa K, et al. 2001. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 20:2835–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tang XD, Xu R, Reynolds MF, Garcia ML, Heinemann SH, Hoshi T. 2003. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature 425:531–535 [DOI] [PubMed] [Google Scholar]
- 37. Zhang L, Guarente L. 1995. Heme binds to a short sequence that serves a regulatory function in diverse proteins. EMBO J. 14:313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang L, Hach A. 1999. Molecular mechanism of heme signaling in yeast: the transcription activator HapI serves as the key mediator. Cell. Mol. Life Sci. 56:415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yoshimura H, Tada T, Ida H. 2004. Subcellular localization and oligomeric structure of the yeast putative stretch-activated Ca2+ channel component Mid1. Exp. Cell Res. 293:185–195 [DOI] [PubMed] [Google Scholar]
- 40. Bijlmakers MJ, Marsh M. 2003. The on-off story of protein palmitoylation. Trends Cell Biol. 13:32–42 [DOI] [PubMed] [Google Scholar]
- 41. Shipston MJ. 2011. Ion channel regulation by protein palmitoylation. J. Biol. Chem. 286:8709–8716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gromiha MM, Oobatake M, Kono H, Uedaira H, Sarai A. 1999. Relationship between amino acid properties and protein stability: buried mutations. J. Protein Chem. 18:565–578 [DOI] [PubMed] [Google Scholar]
- 43. Strub C, Alies C, Lougarre A, Ladurantie C, Czaplicki J, Fournier D. 2004. Mutation of exposed hydrophobic amino acids to arginine to increase protein stability. BMC Biochem. 5:9 doi:10.1186/1471-2091-5-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suh BC, Kim DI, Falkenburger BH, Hille B. 2012. Membrane-localized β-subunits alter the PIP2 regulation of high-voltage activated Ca2+ channels. Proc. Natl. Acad. Sci. U. S. A. 109:3161–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brand A, Shanks S, Duncan VMS, Yang M, MacKenzie K, Gow NA. 2007. Hyphal orientation of Candia albicans is regulated by a calcium-dependent mechanism. Curr. Biol. 17:347–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fischer MN, Schnell N, Chattaway J, Davies P, Dixon G, Saunders D. 1997. The Saccharomyces cerevisiae CCH1 gene is involved in calcium influx and mating. FEBS Lett. 419:259–262 [DOI] [PubMed] [Google Scholar]
- 47. Iida H, Nakamura N, Ono T, Okumura MS, Anraku Y. 1994. MID1, a novel Saccharomyces cerevisiae gene encoding a plasma membrane protein, is required for Ca2+ influx and mating. Mol. Cell. Biol. 14:8259–8271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nichols CB, Ferreyra J, Ballou ER, Alsphaugh A. 2009. Subcellular localization directs signaling specificity of the Cryptococcus neoformans Ras1 protein. Eukaryot. Cell 8:181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.