

Abstract
The extensive use and misuse of antibiotics in medicine result in the emergence of multidrug-resistant bacteria, creating an urgent need for the development of new chemotherapeutic agents. Nowadays, antimicrobial peptides are widely recognized as a class of promising candidates with activity against multidrug-resistant bacteria. NK-18 is a truncated peptide derived from NK-Lysin, an effector of cytotoxic T cells and natural killer cells. In this study, we studied the antibacterial mechanism of action of NK-18. The results revealed that NK-18 has potent antibacterial activity against Escherichia coli and Staphylococcus aureus. According to our findings, NK-18 is membrane active and its target of action is not only the bacterial membrane but also the DNA in the cytoplasm. The double targets of NK-18 make it difficult for bacteria to generate resistance, which may present a new strategy to defend against multidrug-resistant bacteria and provide a new lead in the design of potent antimicrobial peptides with therapeutic application in the presence of increasing resistance to conventional antibiotics.
INTRODUCTION
In recent decades, the misuse of conventional antibiotics has resulted in the emergence of many multidrug-resistant strains (1–3). With the increasing severity of this phenomenon, such strains have become a serious menace to human health and quality of life. Therefore, development of a new class of antibiotics with mechanisms of action different from those of conventional antibiotics is becoming more and more urgent and critical. The outbreak of superbugs in several countries in the world in 2011 emphasized once again the need to search for and develop new antimicrobial agents or resources. Nowadays, it is widely recognized that antimicrobial peptides (AMPs) could play a promising role in the fight against multidrug-resistant strains. AMPs are considered a new class of antibiotic with characteristics including an ability to kill target cells rapidly, an unusually broad spectrum of activity, and activity against some of the more serious antibiotic-resistant pathogens in the clinic. Furthermore, the selection of mutants resistant to AMPs in vitro is relatively difficult (4–6).
AMPs are widespread in nature, including microorganisms, insects, invertebrates, amphibians, plants, birds, and mammals (7, 8). They comprise a wide range of short, cationic peptides which constitute the first line of innate immune defense against infectious agents (9–12). To date, more than 1,800 AMPs have been purified from a wide range of organisms (13) or chemically synthesized on the basis of the sequences of purified peptides. NK-18 is a derivative of a mammalian protein, NK-lysin. It derives from the core region of NK-lysin (residues 39 to 56) and possesses potent antitumor activity against prostate and bladder tumors with a membrane-active mode of action (14). The amino acid sequence of NK-18 is KILRGVCKKIMRTFLRRI-NH2. Compared with NK-lysin, NK-18 has the potential to be used clinically because of its shorter amino acid sequence and lower cost of chemosynthesis.
In the present study, the antibacterial activity of NK-18 was studied by the 2-fold dilution method and the radial diffusion assay. In order to get a better understanding of its antibacterial mechanism, the outer membrane (OM)/inner membrane (IM) permeability assay and confocal laser scanning microscopy assay were employed to examine the effect of NK-18 on the integrity of the bacterial membrane. The effect of NK-18 on the morphology of bacteria was observed under a scanning electron microscope (SEM). To investigate the interaction between NK-18 and DNA, a DNA retardation assay and a DNA absorption assay, as well as atomic force microscopy (AFM) imaging, were employed in this study. Furthermore, in order to exclude the influence of other nonlipid components on the exhibition of NK-18 activity, we also examined the effect of NK-18 on large unilamellar liposomes (LUVs), composed of egg yolk l-α-phosphatidylethanolamine (EYPE) and egg yolk l-α-phosphatidyl-dl-glycerol (EYPG), as a model membrane.
MATERIALS AND METHODS
Peptide synthesis and purification.
NK-18 was synthesized by a stepwise solid-phase method using N-9-fluorenylmethoxycarbonyl (Fmoc) chemistry, as reported previously (15). Fluorescein isothiocyanate (FITC)-labeled NK-18 was synthesized as described by Wender et al. (16). All the peptides were purified on a Sephadex gel column by reverse-phase high-pressure liquid chromatography (HPLC; Waters) using a μ-Bondapak C18 column (19 mm by 300 mm) with gradient elution of 20% to 75% CH3CN-H2O with 0.1% trifluoroacetic acid at a flow rate of 8 ml/min. The atomic masses of these peptides were confirmed by electrospray ionization mass spectrometry (ESI-MS).
Bacteria and reagents.
The bacterial strains used in this study were obtained from the Institute of Microbiology, Lanzhou University. Escherichia coli (CMCC44102) and Staphylococcus aureus (CMCC26003) were cultured in LB broth. Prior to assays, bacteria were grown overnight to the stationary phase at 37°C in 5 ml LB broth. A 1/50 dilution of the overnight culture was suspended in 5 ml fresh LB broth and grown for an additional few hours at 37°C to obtain a mid-log-phase culture, and then the optical density (OD) was determined using a spectrophotometer to obtain a certain cell population. 1-N-Phenylnaphthylamine (NPN) was purchased from J&K Scientific Ltd. o-Nitrophenyl-β-d-galactoside (ONPG) was obtained from the Beyotime Institute of Biotechnology (Shanghai, China). EYPE and EYPG were purchased from Avanti Polar. Plasmid DNA (pBR322) was purchased from Fermentas. Propidium iodide (PI) was purchased from Invitrogen.
Radial diffusion assay.
The antibacterial activity of NK-18 was evaluated by a modification of the sensitive radial diffusion assay described by Takemura et al. (17). Briefly, the bacteria were cultured as described above until the optical density at 600 nm (OD600) of an aliquot reached 0.5. One milliliter of the bacteria was added to 100 ml of previously autoclaved, warm LB agar. After rapid dispersion of the bacteria, the agar was poured into an agar plate to form a layer approximately 5 mm deep and was punched with a 3-mm-diameter gel punch to make evenly spaced wells. Following the addition of 20 μl with different concentrations of NK-18 to each well, the plates were incubated at 37°C for 18 to 24 h. The size of the clear zone surrounding each well was then measured. Twenty microliters of sterile water was also added as a control.
MIC determinations.
The MICs were measured using methods described previously (18) and by the CLSI (formerly NCCLS) broth microdilution method (19), with some modification. Briefly, for MIC determinations, 2-fold serial dilutions of NK-18 were prepared in LB medium. One hundred-microliter aliquots of the bacterial suspension were dispensed into each well of a 96-well microtiter plate (Costar 3599; Corning), and then 100 μl of peptide solution was added. Phosphate-buffered saline (PBS) buffer was used as a negative control. The antibacterial activity of NK-18 was evaluated from the turbidity visible in each well after 18 h of incubation at 37°C. The MICs were expressed as the minimum concentration of each sample required for the visible inhibition of growth. All MIC determinations were made in triplicate, and if the results were within 2 doubling dilutions of each other, the highest reading was recorded for analysis.
Killing kinetics.
Bacterial suspensions were prepared as described above. The killing kinetics assay was performed according to the method described previously, with a little modification (20). Bacteria were incubated with different concentrations of peptides determined by the MIC assay. The viable count was monitored for up to 24 h. Aliquots were taken at defined intervals and appropriately diluted in phosphate saline buffer (pH 7.4), and then 100 μl of each of the dilutions was plated in triplicate on LB agar. The plates were incubated at 37°C, and the numbers of CFU were counted after 24 h.
Flow cytometric analysis.
The integrity of the bacterial membrane after NK-18 treatment was determined by FACScan analysis via nuclear staining with PI as described by Jang et al. (21). The test strains in the mid-log phase (OD600 = 0.5) were mixed with different concentrations of NK-18 and then incubated for 60 min at 37°C. The peptide-treated cells were stained with PI solution (20 μg/ml) for 25 min at room temperature in the dark. After incubation, the unbound dye was removed via excessive washing with PBS. Flow cytometric measurements were performed on a flow cytometer (Beckman Coulter) with computer-assisted evaluation of the data (CellQuest software).
OM permeability.
Permeation of the E. coli OM by NK-18 was evaluated using the hydrophobic NPN fluorescent probe as described before, with a little modification (22). E. coli cultures with an OD600 value of 0.5 ± 0.02 were centrifuged for 10 min at 1,000 × g, and the cells were suspended in a half volume of 5 mM HEPES (pH 7.2). One hundred microliters E. coli and 50 μl NK-18 were mixed with 50 μl NPN (final concentration, 10 μM). The final concentrations of NK-18 were 12.5 μg/ml, 25 μg/ml, 50 μg/ml, and 100 μg/ml. A control was carried out with 0.5% NaCl alone. An increase in fluorescence due to partitioning of NPN into the OM was recorded as a function of time until no further increase in intensity was observed. The excitation and emission wavelengths were set at 350 and 420 nm, respectively. Each assay was performed at least three times by a multimode reader (Tecan Infinite M200 Pro).
IM permeability.
Permeation of the E. coli IM was determined by measuring the release of cytoplasmic β-galactosidase activity from E. coli into the culture medium using ONPG as the substrate (23). E. coli cells cultured with LB broth containing 5% lactose were harvested, washed, resuspended in 0.5% NaCl solution to get an absorbance of 1.2 at 420 nm, and then resuspended in a half volume of 0.5% NaCl solution. The concentrations of peptide were the same as those described in the previous section. One hundred microliters of E. coli and 90 μl of the peptide were mixed with 10 μl of ONPG (30 mM) in a 96-well plate. A negative control was carried out with 0.5% NaCl, and a positive control was carried out with Triton X-100. The production of o-nitrophenol over time was determined by monitoring the changes in absorbance at 420 nm using a Tecan Infinite M200 Pro multireader.
Examination of bacterial membrane damage by SEM.
The test strain of E. coli was grown to mid-logarithmic phase in LB broth. A suspension of E. coli (∼105 CFU/ml) was incubated with NK-18 (100 μg/ml) at 37°C for 2 h and then centrifuged for 5 min at 10,000 rpm. The resulting pellet was gently washed with PBS. One milliliter of 3% glutaraldehyde solution was subsequently added into the tube to fix these cells. After fixation with glutaraldehyde, the precipitations were impregnated in 2.5% tannic acid (Sigma) for 2 days. Counterfixation in 2% osmium tetroxide (Sigma) for 1 h was followed by dehydration in ethanol and drying in a freeze-drying device (JFD-310; JEOL, Japan). Cells were coated with gold and analyzed by scanning electron microscope (JSM-6380Lv; Japan).
Calcein release from LUVs.
In order to assess the ability of NK-18 to cause leakage of liposomal content, LUVs composed of an EYPE-EYPG (7:3, wt/wt) bacterial mimic membrane were used. LUVs were prepared by dissolving the required amounts of dry lipids in chloroform (24, 25). The solvents were removed by rotary evaporation to form a thin lipid film. After being dried under vacuum overnight, the lipid was hydrated in dye buffer solution (70 mM calcein, 10 mM Tris, 150 mM NaCl, 1 mM EDTA, pH 7.4) (18). The suspensions were frozen in liquid nitrogen, thawed for 10 cycles, and then successively extruded through polycarbonate filters (100-nm-pore-size filter, 21 times) by an Avanti miniextruder. Untrapped calcein was removed by gel filtration chromatography on a Sephadex G-50 column (10 mm by 150 mm). The release of calcein from the LUVs with or without NK-18 was monitored on a Tecan Infinite M200 Pro plate reader by measuring the fluorescence intensity at an excitation wavelength of 490 nm and an emission wavelength of 520 nm. For determination of 100% dye release, 2% Triton X-100 in Tris buffer was added to dissolve the vesicles. The percentage of dye leakage caused by the peptides was calculated as follows: 100 × [(F − F0)/(Ft − F0)], where F is the fluorescence intensity after addition of the peptide and F0 and Ft are fluorescence intensities without the peptides and with Triton X-100, respectively.
TEM.
LUVs for transmission electron microscopy (TEM) analysis were prepared as described before but were hydrated with Tris-EDTA buffer (10 mM Tris, 150 mM NaCl, 1 mM EDTA, pH 7.4). LUVs in the presence or absence of 100 μg/ml of NK-18 were deposited on carbon-coated copper grids and dried naturally. The grids were negative stained with (2%, wt/vol) phosphotungstic acid for 1 min. Then, the additional phosphotungstic acid was soaked up by filter paper and electron micrographs were recorded at nominal magnification (×20,000 to ×40,000) with an accelerating voltage of 120 kV (JEM-1230; Japan).
Confocal laser scanning microscopy.
E. coli cells in mid-log phase were prepared and washed with PBS buffer. After incubation with 100 μg/ml FITC-labeled NK-18 at 37°C for 1 h, the cells were washed with the same buffer three times and immobilized on a glass slide. The accumulation of the FITC-labeled peptides in the bacteria was observed with a confocal laser scanning microscope (Leica SP2; Germany).
DNA binding assay.
Gel retardation experiments were performed for NK-18 as described previously (26). Briefly, 300 ng of the plasmid DNA (pBR322) was mixed with increasing amounts of peptide in 30 μl of buffer (10 mM Tris-HCl, 1 mM EDTA buffer, pH 8.0). Reaction mixtures were incubated at room temperature for 30 min. Subsequently, native loading buffer was added (10% Ficoll 400, 10 mM Tris-HCl, pH 7.5, 50 mM EDTA, 0.25% bromophenol blue, 0.25% xylene cyanol), and a 20-μl aliquot was subjected to 1.5% agarose gel electrophoresis. The migration of DNA was detected by the fluorescence of ethidium bromide (Bio-Rad).
Absorption spectroscopy.
Extraction of genomic DNA from E. coli was performed according to a procedure described previously, with a little modification (27). The purity and concentration of the extracted DNA were evaluated by use of a Nanodrop 2000 spectrophotometer (Thermo Scientific), as described previously (28, 29). DNA and NK-18 were dissolved in 10 mM Tris-HCl buffer (pH 8.0) and then mixed to obtain various samples of DNA and NK-18 with a constant DNA concentration (82.0 ng/μl) and increasing NK-18 concentrations (0, 10, 50, 100, and 200 μg/ml). Reaction mixtures were incubated for 30 min. The absorbance of the mixed solutions was measured over a range of wavelengths from 220 to 320 nm (Nanodrop 2000 spectrophotometer). All measurements were performed at room temperature.
AFM imaging.
DNA and NK-18 were dissolved in 10 mM Tris-HCl buffer (pH 8.0) and then mixed for 30 min at room temperature to obtain a DNA–NK-18 complex with 92.8 ng/μl of DNA and 100 μg/ml of NK-18. Prior to AFM measurements, 50 μl of the suspension was deposited onto a freshly cleaved mica substrate. Samples were imaged after complete water evaporation. Experiments were performed with a multimode nanoscope atomic force microscope (5400; Agilent) operating in contact mode (29, 30). All images were recorded in air at room temperature at a scan speed of 0.8 Hz.
RESULTS
Antimicrobial activity of NK-18.
To investigate the antimicrobial activity of NK-18, it was synthesized by stepwise solid-phase assay, purified by HPLC, and confirmed by ESI-MS. The primary sequence of NK-18 is KILRGVCKKIMRTFLRRI-NH2. The antimicrobial activities of NK-18 against E. coli and S. aureus were screened by radial diffusion assay and determined by the broth microdilution method. The zones of inhibition corresponded to the antibacterial effect of NK-18 after incubation at 37°C for 18 to 24 h. As shown in Fig. 1, NK-18 showed remarkable antibacterial activity. It could induce significantly larger zones of clearance than the control group with sterile water. Furthermore, the diameter of the inhibition zone produced by NK-18 increased with an increase in the peptide concentration, which indicates that the antimicrobial activity of NK-18 was concentration dependent. According to the broth microdilution method, NK-18 displayed very impressive antimicrobial activity against both E. coli and S. aureus. The MICs of NK-18 against E. coli and S. aureus were 12.5 μg/ml and 6.25 μg/ml, respectively.
Fig 1.

NK-18 exhibits antimicrobial activity against different bacterial strains. The direct antimicrobial activity of NK-18 (0, 5, 10, 20, 30, and 40 μg/well) against E. coli and S. aureus was determined in a radial diffusion assay. NK-18 gave rise to clear, bacterium-free zones around the wells of both E. coli and S. aureus.
Time-killing kinetics of NK-18.
Although the MIC assay could reflect the antimicrobial activity of NK-18, it was determined by the overnight effect and the detailed information about NK-18 that killing rates might be obscured in this assay. In order to understand the detail of the NK-18 action, time-killing kinetic assays were performed against E. coli and S. aureus. As shown in Fig. 2, NK-18 acted at different rates against E. coli and S. aureus. For the targeted bacterium S. aureus, NK-18 showed a very rapid rate of killing that was about 3 times log killing and 7 times log killing at 2.5× MIC and 5× MIC, respectively, within 5 min of treatment. NK-18 acted a little more slowly against E. coli than against S. aureus.
Fig 2.

Bacterial killing kinetics of NK-18 at different concentrations against S. aureus (A) and E. coli (B). Controls were samples treated with PBS.
PI uptake assay.
In order to characterize the effects of NK-18 on the integrity of E. coli membranes, PI, a DNA-staining fluorescent probe, was employed. Figure 3 showed the results of the intracellular PI measurements. The majority of the E. coli cells were fluorescently labeled after 60 min of incubation with NK-18, thereby indicating that NK-18 induced the influx of PI into the cells. That is, NK-18 may destabilize or disturb the integrity of the bacterial membrane and induce the uptake of PI.
Fig 3.
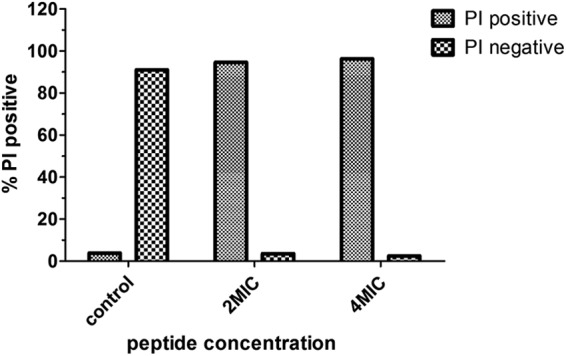
Effect of NK-18 on integrity of E. coli membrane. Bacteria (OD600 = 0.5) were incubated with NK-18 at 25 μg/ml and 50 μg/ml for 60 min. The percentage of PI-fluorescent cells after treatment is shown. The results of one of two tests are presented.
OM and IM permeability.
In order to further confirm the effect of NK-18 on the integrity of the membrane of the bacteria, OM and IM permeability was measured. OM permeability was detected by NPN, which is a hydrophobic fluorescent probe. The fluorescence of NPN is weak in an aqueous environment and strong in a hydrophobic environment, e.g., the hydrophobic core of the bacterial membrane (31). As shown in Fig. 4, NK-18 could induce NPN incorporation into the hydrophobic core of the bacterial cell membrane in a dose-dependent manner, and the relative fluorescence increased to a maximum in 5 min. Apart from this, the integrity of the inner membrane of E. coli was also evaluated by ONPG assay. As we know, cytoplasmic β-galactosidase can be released only as a consequence of an alteration/compromise in the inner membrane of E. coli. β-Galactosidase could react with ONPG and yield ortho-nitrophenol, which is absorbed at 420 nm (32). As shown in Fig. 5, after treatment with different concentrations of NK-18, β-galactosidase was released from E. coli cells with a lag time of about 5 min and reached steady state at 90 min. Meanwhile, the release of β-galactosidase was dose dependent.
Fig 4.

Uptake of 1-N-phenylnaphthylamine (measured as fluorescence intensity in arbitrary units) by E. coli with addition of different concentrations of NK-18. Data are the means ± standard errors of the means (n = 3).
Fig 5.

Release of cytoplasmic β-galactosidase activity (measured from the absorbance at 420 nm) of E. coli cells treated by NK-18. C−, bacterial cells treated with 0.5% NaCl; C+, bacterial cells treated with 2% Triton X-100. Data are the means ± standard errors of the means (n = 3).
Examination of bacterial membrane change by SEM.
In order to gain a more direct insight into the interaction between NK-18 and E. coli, the morphological changes in the bacterial membrane were also examined by SEM in order to confirm the results obtained earlier. As shown in Fig. 6, untreated E. coli cells had a normal smooth surface (Fig. 6A); in contrast, cells treated with NK-18 for 1 h showed clear morphological changes. The changes caused by NK-18 appeared to involve the formation of blebs on the cell surface, and the surface seemed to have been disrupted (Fig. 6B). The SEM observations provided direct morphological evidence of the strong influence of NK-18 on the bacterial membrane.
Fig 6.

Effect of NK-18 on the bacterial membrane of E. coli by scanning electron microscopy. A typical example of untreated E. coli (A) shows a normal smooth surface, while treatment with 100 μg/ml NK-18 reveals an incomplete pattern and bubbling on the outer face (B).
Peptide-lipid interaction using a model of a liposome mimicking the bacterial membrane.
Because the bacterial membrane is complicated and it is sometimes hard to investigate the interaction between AMPs and the bacterial membrane under the influence of different factors, most studies of the interaction of cationic AMPs with membranes have been performed using the model of a liposome with compositions chosen to reflect those of the bacterial cytoplasmic membrane (33). To determine whether the antimicrobial activity of NK-18 depends on its capacity to permeate bacterial membranes, its ability to induce calcein leakage from negatively charged EYPE-EYPG (7:3, wt/wt) LUVs mimicking the composition of the E. coli membrane was also measured. For this study, we employed LUVs encapsulating calcein at a concentration of 70 mM, a concentration at which the fluorescence of calcein is self-quenched. If the peptide is able to break the liposome membrane after treatment, the LUVs encapsulating the calcein will burst and release calcein into the aqueous environment. Meanwhile, the dye starts fluorescing and thus the fluorescence values increase. The fluorescence intensity curves for calcein release induced by NK-18 are shown in Fig. 7. NK-18 induced a sharp increase in the leakage of calcein, and this ability was concentration dependent. At concentrations of 6.25 μg/ml and 12.5 μg/ml, the fluorescence intensity induced by NK-18 was similar to that for the positive control (Fig. 7, left). Furthermore, NK-18 interacted with LUVs as soon as it was added to the specimen and caused the maximum leakage of calcein dye of up to 90% at a concentration of 6.25 μg/ml (Fig. 7, right).
Fig 7.

Release of the fluorescent dye calcein from LUVs composed of EYPE-EYPG (7:3) induced by NK-18. (Left) Change in fluorescence intensity of samples treated with NK-18 for 10 min; (right) concentration-dependent leakage of calcein from LUVs measured after 1 min, 5 min, and 10 min of incubation with peptide at different concentrations.
Knowing that the peptides are acting on the surface of the bacterial membrane, TEM studies were conducted with NK-18 and LUVs mimicking bacterial membranes. TEM studies were conducted with NK-18 at a concentration of 100 μg/ml to monitor the morphological changes of LUVs, and the results showed surface alterations of the liposome. As shown in Fig. 8, the membranes of untreated LUVs were smooth and intact. On the contrary, the surface of LUVs treated with NK-18 changed thoroughly. The membrane was incomplete, and the edge of it was vague. This was strong evidence that NK-18 could act against the model liposome mimicking a bacterial membrane, and this result was consistent with that of the calcein leakage assay.
Fig 8.

Transmission electron microscopy images of LUVs mimicking a bacterial membrane. (A) Untreated liposomes; (B) liposomes treated with 100 μg/ml NK-18 for 3 min.
Internalization of FITC-labeled NK-18 in E. coli cells.
For determination of the site of action of NK-18, FITC-labeled NK-18 (100 μg/ml) was incubated with log-phase E. coli cells for 1 h, and their localization was visualized by confocal laser scanning microscopy. On the basis of the observed phenomenon, the localization of this peptide was almost in the cytoplasm of the bacterial cells (Fig. 9). Figure 9 illustrates that FITC-labeled NK-18 could penetrate the cell membrane of E. coli and accumulate in the cytoplasm.
Fig 9.

Localization of FITC-labeled peptide in E. coli cells by confocal laser scanning microscopy. (A to C) Untreated E. coli cells; (D to F) cells treated with FITC-labeled NK-18. For each treatment, fluorescence (A and D) and differential inference contrast (B and E) images, as well as merged images (C and F), are presented.
Interaction between NK-18 and plasmid and genomic DNA of E. coli.
Although NK-18 showed activity against both model liposomes and the bacterial membrane, as described above, we still could not conclude that the membrane was the only target against which NK-18 exerted its antibacterial activity on the basis of the detection of FITC-labeled NK-18 in bacteria. Therefore, the DNA-interacting ability of NK-18 was also studied by monitoring the electrophoretic mobility of plasmid DNA on an agarose gel. Different concentrations of NK-18 were mixed with a fixed amount of plasmid DNA, after which the mixture was electrophoresed on an agarose gel. Figure 10 indicates that NK-18 could interact with plasmid DNA and retard its migration in the gels in a concentration-dependent manner. At a peptide concentration of 12 μg/ml, some of the plasmid DNA was still able to migrate into the gel as nontreated DNA, whereas at a concentration of 16 μg/ml, almost all of the DNA remained at the origin. At higher concentrations, complete retardation of DNA migration was observed, showing that the DNA was aggregated by NK-18. To compare its mechanism of action with that of a membrane-active antimicrobial peptide, the same experiment was performed with magainin 2. In contrast, magainin 2 did not show any DNA-interacting ability even at concentrations up to 128 μg/ml, a result that was absolutely different from that for NK-18. These results show that NK-18 has intrinsic DNA binding ability.
Fig 10.

DNA binding activity of NK-18. Binding was assayed by determination of the inhibitory effect of peptides on the migration of DNA bands. Various amounts of peptides were incubated with 300 ng of plasmid DNA at room temperature for 30 min, and the reaction mixtures were applied to a 1.5% agarose gel. The gel was visualized after ethidium bromide staining and UV irradiation. The numbers below the lanes represent the concentration (in μg/ml) of NK-18 or magainin 2. Lanes C, control consisting of plasmid DNA only. The results shown are representative of three experiments.
Now that we showed that NK-18 could interact with the plasmid DNA of bacteria and retard DNA migration in an agarose gel, the relationship between this peptide and the genomic DNA of E. coli was also evaluated by electronic absorption spectroscopy. As shown in Fig. 11, in the absence of NK-18, the genomic DNA exhibited a fluorescence emission maximum at 257 nm, and addition of NK-18 caused the fluorescence intensity to decrease and the emission maximum to shift from 257 to 261 nm, showing that NK-18 could bind with the genomic DNA of E. coli. The results of this experiment are in agreement with those of our gel retardation assay.
Fig 11.
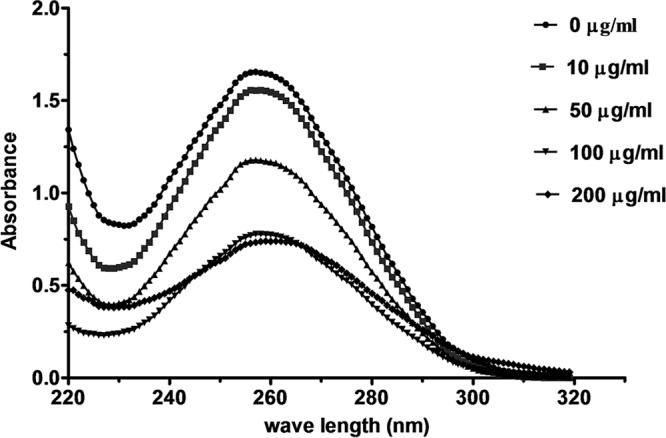
UV spectra of E. coli genomic DNA in the presence of different concentrations of NK-18. A fixed concentration (82.0 ng/μl) of E. coli genomic DNA was treated with increasing amounts of NK-18 for 30 min. UV spectra were measured over a range of wavelengths from 220 to 320 nm. All measurements were recorded at room temperature.
AFM imaging.
The gel retardation and fluorescence spectra data presented above indicate that NK-18 interacts with DNA. To understand the interaction between DNA and the peptide more directly, the genomic DNA of E. coli was immobilized and AFM imaging was employed. Representative AFM images of free DNA and DNA after treatment are shown in Fig. 12. The images show that in the absence of peptide, DNA appears as circles. However, most geometric structures of DNA were influenced in the presence of NK-18 and were accompanied by an increased height of the peaks. This phenomenon indicates that most of the DNA molecules could interact with NK-18, which proved the results of the gel retardation assay and absorption spectroscopy.
Fig 12.

AFM images of genomic DNA of E. coli in the absence or presence of NK-18. (A) DNA in the absence of NK-18; (B) DNA treated with 100 μg/ml of NK-18 for 30 min.
DISCUSSION
AMPs are common in nature as one of the primary defense or attack strategies. Nowadays, with the emergency of the burst in the prevalence of multidrug-resistant bacteria, AMPs have attracted great interest for their potential antimicrobial activity and different mechanisms of action compared with those of conventional antibiotics. Although extensive research has been conducted (34–37), the exact mode of action of antimicrobial peptides has been controversial, until now. It is generally accepted that AMPs produce effects on bacterial function via changes in cell wall synthesis and composition, inhibition of proteases and/or enzyme activity, inhibition of nucleic acid and/or protein synthesis, binding to DNA, and transmembrane pore formation (38, 39).
NK-18 is a derivative of the mammalian natural killer cell effector NK-lysin. It has a much shorter sequence than NK-lysin and has more desirable features that make it useful clinically. NK-18 exhibits antitumor activity through a membrane-active mode of action which is superior to that of conventional chemotherapeutic agents (14). However, the effects and the mode of action of NK-18 against bacteria are unknown. With the increase in the incidence of multidrug-resistant bacteria, the development of membrane-active antibiotics should be a new strategy. Therefore, the aim of this study was to investigate the antimicrobial activity and to clarify the antimicrobial mechanism of action of NK-18 with the purpose of developing a novel antibiotic template because of the current urgent need. Our results showed that NK-18 could inhibit the growth of E. coli and S. aureus in a dose-dependent manner. It could induce PI, a membrane-impermeant fluorescent dye, entering into E. coli cells and binding with DNA. To our knowledge, the cell membrane of Gram-positive bacteria is absolutely distinct (Medical Microbiology, Digital Proteus). In order to further verify the effect of NK-18 on the membrane of bacteria, its ability to permeate the E. coli OM and IM was examined. Our results showed that the integrity of both the outer membrane and the inner membrane was disturbed after treatment with NK-18. Finally, the morphological changes to the bacteria detected by SEM provide evidence that NK-18 has an effect on the E. coli cell membrane. To exclude the possibility that nonlipid components influence the activity of NK-18 on the membrane and further confirm that NK-18 has the ability to affect the microbial plasma membrane, we investigated the effect of NK-18 on the integrity of artificial liposome LUVs, which were composed of EYPE and EYPG (7:3, wt/wt), as a model for the bacterial plasma membrane. The results showed that NK-18 could disrupt the integrity of LUVs, which implies that NK-18 is active against both the bacterial plasma membrane and a model of a bacterial membrane.
In addition to these results, we also found by confocal laser scanning microscopy that FITC-labeled NK-18 was visible on the cell membrane and in the cytoplasm of E. coli. This result indicates that NK-18 can penetrate both the cell wall and cell membrane and accumulate inside bacterial cells. This result may imply that the peptides not only were associated with the plasma membrane but also were internalized. This prompted us to look for some intracellular targeting mode of action for this peptide. With this aim, we compared the relative affinity of NK-18 to bind plasmid DNA. In accordance with our confocal microscopy data, the DNA binding affinity of NK-18 was observed in a DNA binding assay and was reflected as retardation in the movement of plasmid DNA. For magainin 2, an antimicrobial peptide studied more thoroughly, no retardation was observed even at a concentration of 128 μg/ml, which was in accordance with the findings of studies carried out by Park et al. (40). Absorption spectroscopy and AFM were then employed to investigate the interaction between NK-18 and bacterial genomic DNA. That analysis showed that NK-18 not only could induce a change in the maximum absorption intensity but also could shift the maximum absorption wavelength (from 257 nm to 261 nm) of the genome DNA of bacteria.
So, taking all the data into account, we provide a certain understanding of the whole mechanism of antibacterial activity of NK-18. It could reasonably be assumed that the cationic, hydrophilic characteristic of NK-18 first initiates an electrostatic interaction with the negatively charged components of the membrane of microbes (41, 42). The peptide then leads to permeabilization and disturbance of the outer and inner membranes, as well as the cytoplasmic membrane of bacteria, allowing the free exchange of intra- and extracellular ions and resulting in the leakage of cytoplasmic components. The changes in membrane permeability further reflect the transport of the peptide across the membrane. Because DNA appeared as a polyion with considerable negative charges, alkalescent amino acids (e.g., Lys and Arg) in NK-18 could spontaneously bind to the phosphate fragments of DNA through electrostatic interaction. So, after the perturbation and transfer to the cytoplasm, NK-18 then interacts with its intracellular target DNA. As the barrier function of the cell membrane has been disrupted and the cellular functions (such as the DNA repair function) are also inhibited owing to binding with the peptide, the bacteria are killed in a rapid manner without the exploitation of a specific receptor. As with any new class of antimicrobial therapeutics, one key point is whether resistance can be provoked. The fact that NK-18 has double targets, the cell membrane and intracellular DNA, means that the chances of resistance are slim, as this would require the complete alteration of the cell membrane or bypassing of several biochemical pathways (43, 44).
In conclusion, our results reveal that NK-18 can effectively kill bacteria by disturbing the bacterial membrane and binding to DNA in a rapid manner. These are not affected by known mechanisms of resistance. Although ongoing work on other properties of NK-18 will be necessary to obtain a systematic understanding of its application in the future, knowledge of the mechanism of action of NK-18 could no doubt provide a new lead in the design of potent antimicrobial peptides with therapeutic application in this time of increasing resistance to conventional antibiotics.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (grants 90813012, 20932003, 21272102, 31200584, 21272108, and 81202400) and the Key National S&T Program Major New Drug Development of the Ministry of Science and Technology (grant 2012ZX09504001-003).
Footnotes
Published ahead of print 22 October 2012
REFERENCES
- 1. Alanis AJ. 2005. Resistance to antibiotics: are we in the post-antibiotic era? Arch. Med. Res. 36:697–705 [DOI] [PubMed] [Google Scholar]
- 2. Heymann DL. 2006. Resistance to anti-infective drugs and the threat to public health. Cell 124:671–675 [DOI] [PubMed] [Google Scholar]
- 3. Straand J, Gradmann C, Lindbæk M, Simonsen GS. 2008. Antibiotic development and resistance, p 200–211 In Kris H. (ed), International encyclopedia of public health. Academic Press, Oxford, United Kingdom [Google Scholar]
- 4. Chen Y, Guarnieri MT, Vasil AI, Vasil ML, Mant CT, Hodges RS. 2007. Role of peptide hydrophobicity in the mechanism of action of {alpha}-helical antimicrobial peptides. Antimicrob. Agents Chemother. 51:1398–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kraus D, Peschel A. 2006. Molecular mechanisms of bacterial resistance to antimicrobial peptides, antimicrobial peptides and human disease, p 231–250 In Shafer WM. (ed), vol 306 Springer, Berlin, Germany: [DOI] [PubMed] [Google Scholar]
- 6. Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395 [DOI] [PubMed] [Google Scholar]
- 7. Jenssen H, Hamill P, Hancock REW. 2006. Peptide antimicrobial agents. Clin. Microbiol. Rev. 19:491–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nicolas P, Mor A. 1995. Peptides as weapons against microorganisms in the chemical defense system of vertebrates. Annu. Rev. Microbiol. 49:277–304 [DOI] [PubMed] [Google Scholar]
- 9. Boman HG. 1995. Peptide antibiotics and their role in innate immunity. Annu. Rev. Immunol. 13:61–92 [DOI] [PubMed] [Google Scholar]
- 10. Hancock REW, Diamond G. 2000. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 8:402–410 [DOI] [PubMed] [Google Scholar]
- 11. Oppenheim JJ, Biragyn A, Kwak LW, Yang D. 2003. Roles of antimicrobial peptides such as defensins in innate and adaptive immunity. Ann. Rheum. Dis. 62(Suppl. 2):ii17–ii21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang K, Yan J, Liu X, Zhang J, Chen R, Zhang B, Dang W, Zhang W, Kai M, Song J, Wang R. 2011. Novel cytotoxicity exhibition mode of polybia-CP, a novel antimicrobial peptide from the venom of the social wasp Polybia paulista. Toxicology 288:27–33 [DOI] [PubMed] [Google Scholar]
- 13. Wang Z, Wang G. 2004. APD: the antimicrobial peptide database. Nucleic Acids Res. 32:D590–D592 doi:10.1093/nar/gkh025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yan J-X, Wang K-R, Chen R, Song J-J, Zhang B-Z, Dang W, Zhang W, Wang R. 2012. Membrane active antitumor activity of NK-18, a mammalian NK-lysin-derived cationic antimicrobial peptide. Biochimie 94:184–191 [DOI] [PubMed] [Google Scholar]
- 15. Fields GB, Noble RL. 1990. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 35:161–214 [DOI] [PubMed] [Google Scholar]
- 16. Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. 2000. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc. Natl. Acad. Sci. U. S. A. 97:13003–13008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takemura H, Kaku M, Kohno S, Hirakata Y, Tanaka H, Yoshida R, Tomono K, Koga H, Wada A, Hirayama T, Kamihira S. 1996. Evaluation of susceptibility of gram-positive and -negative bacteria to human defensins by using radial diffusion assay. Antimicrob. Agents Chemother. 40:2280–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Joshi S, Bisht GS, Rawat DS, Kumar A, Kumar R, Maiti S, Pasha S. 2010. Interaction studies of novel cell selective antimicrobial peptides with model membranes and E. coli ATCC 11775. Biochim. Biophys. Acta 1798:1864–1875 [DOI] [PubMed] [Google Scholar]
- 19. CLSI 2006. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard M7-A7. CLSI, Wayne, PA [Google Scholar]
- 20. Pag U, Oedenkoven M, Papo N, Oren Z, Shai Y, Sahl H-G. 2004. In vitro activity and mode of action of diastereomeric antimicrobial peptides against bacterial clinical isolates. J. Antimicrob. Chemother. 53:230–239 [DOI] [PubMed] [Google Scholar]
- 21. Jang WS, Kim HK, Lee KY, Kim SA, Han YS, Lee IH. 2006. Antifungal activity of synthetic peptide derived from halocidin, antimicrobial peptide from the tunicate, Halocynthia aurantium. FEBS Lett. 580:1490–1496 [DOI] [PubMed] [Google Scholar]
- 22. Wang B, Navath RS, Menjoge AR, Balakrishnan B, Bellair R, Dai H, Romero R, Kannan S, Kannan RM. 2010. Inhibition of bacterial growth and intramniotic infection in a guinea pig model of chorioamnionitis using PAMAM dendrimers. Int. J. Pharm. 395:298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Je J-Y, Kim S-K. 2006. Chitosan derivatives killed bacteria by disrupting the outer and inner membrane. J. Agric. Food Chem. 54:6629–6633 [DOI] [PubMed] [Google Scholar]
- 24. Papo N, Oren Z, Pag U, Sahl H-G, Shai Y. 2002. The consequence of sequence alteration of an amphipathic α-helical antimicrobial peptide and its diastereomers. J. Biol. Chem. 277:33913–33921 [DOI] [PubMed] [Google Scholar]
- 25. Zhu WL, Song YM, Park Y, Park KH, Yang S-T, Kim JI, Park I-S, Hahm K-S, Shin SY. 2007. Substitution of the leucine zipper sequence in melittin with peptoid residues affects self-association, cell selectivity, and mode of action. Biochim. Biophys. Acta 1768:1506–1517 [DOI] [PubMed] [Google Scholar]
- 26. Beckloff N, Laube D, Castro T, Furgang D, Park S, Perlin D, Clements D, Tang H, Scott RW, Tew GN, Diamond G. 2007. Activity of an antimicrobial peptide mimetic against planktonic and biofilm cultures of oral pathogens. Antimicrob. Agents Chemother. 51:4125–4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Soolingen D, Hermans PW, de Haas PE, Soll DR, van Embden JD. 1991. Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J. Clin. Microbiol. 29:2578–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Amagliani G, Giammarini C, Omiccioli E, Brandi G, Magnani M. 2007. Detection of Listeria monocytogenes using a commercial PCR kit and different DNA extraction methods. Food Control 18:1137–1142 [Google Scholar]
- 29. Tang Y-L, Shi Y-H, Zhao W, Hao G, Le G-W. 2009. Interaction of MDpep9, a novel antimicrobial peptide from Chinese traditional edible larvae of housefly, with Escherichia coli genomic DNA. Food Chem. 115:867–872 [Google Scholar]
- 30. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang Y-H, Homey B, Cao W, Wang Y-H, Su B, Nestle FO, Zal T, Mellman I, Schroder J-M, Liu Y-J, Gilliet M. 2007. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449:564–569 [DOI] [PubMed] [Google Scholar]
- 31. Loh B, Grant C, Hancock RE. 1984. Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 26:546–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Falla T, Hancock R. 1997. Improved activity of a synthetic indolicidin analog. Antimicrob. Agents Chemother. 41:771–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Glukhov E, Stark M, Burrows LL, Deber CM. 2005. Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J. Biol. Chem. 280:33960–33967 [DOI] [PubMed] [Google Scholar]
- 34. Henzler Wildman KA, Lee DK, Ramamoorthy A. 2003. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 42:6545–6558 [DOI] [PubMed] [Google Scholar]
- 35. Oren Z, Shai Y. 1998. Mode of action of linear amphipathic α-helical antimicrobial peptides. Pept. Sci. 47:451–463 [DOI] [PubMed] [Google Scholar]
- 36. Wang K, Yan J, Chen R, Dang W, Zhang B, Zhang W, Song J, Wang R. 2012. Membrane-active action mode of polybia-CP, a novel antimicrobial peptide isolated from the venom of Polybia paulista. Antimicrob. Agents Chemother. 56:3318–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu M, Maier E, Benz R, Hancock RE. 1999. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 38:7235–7242 [DOI] [PubMed] [Google Scholar]
- 38. Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238–250 [DOI] [PubMed] [Google Scholar]
- 39. Kawasaki H, Koyama T, Conlon JM, Yamakura F, Iwamuro S. 2008. Antimicrobial action of histone H2B in Escherichia coli: evidence for membrane translocation and DNA-binding of a histone H2B fragment after proteolytic cleavage by outer membrane proteinase T. Biochimie 90:1693–1702 [DOI] [PubMed] [Google Scholar]
- 40. Park CB, Kim HS, Kim SC. 1998. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 244:253–257 [DOI] [PubMed] [Google Scholar]
- 41. Hancock REW. 1984. Alterations in outer membrane permeability. Annu. Rev. Microbiol. 38:237–264 [DOI] [PubMed] [Google Scholar]
- 42. Matsuzaki K. 1999. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1462:1–10 [DOI] [PubMed] [Google Scholar]
- 43. Cho JH, Kim SC. 2010. Non-membrane targets of antimicrobial peptides: novel therapeutic opportunities? In Wang G. (ed), Antimicrobial peptides: discovery, design and novel therapeutic strategies. CABI Publishing, Wallingford, United Kingdom [Google Scholar]
- 44. Jang SA, Kim H, Lee JY, Shin JR, Kim DJ, Cho JH, Kim SC. 2012. Mechanism of action and specificity of antimicrobial peptides designed based on buforin IIb. Peptides 34:283–289 [DOI] [PubMed] [Google Scholar]