

Abstract
Acute lung injury (ALI) is a devastating disease characterized by pulmonary edema. Removal of edema from the air spaces is a critical function of the epithelial sodium channel (ENaC) in ALI. The molecular mechanisms behind resolution of pulmonary edema are incompletely understood. MicroRNA’s (miRNA) are crucial gene regulators and are dysregulated in various diseases including ALI. Recent studies suggest that microRNA-16 (miR-16) targets serotonin transporter (SERT) involved in the serotonin (5-HT) transmitter system. Alterations in serotonin levels have been reported in various pulmonary diseases. However, the role of miR-16 on its target SERT, and ENaC, a key ion channel involved in the resolution of pulmonary edema, have not been studied. In the present study, the expression patterns of miR-16, SERT, ENaC and serotonin were investigated in mice exposed to room air and hyperoxia. The effects of miR-16 overexpression on ENaC, SERT, TGF-β and Nedd4 in human alveolar epithelial cells were analyzed. miR-16 and ENaC were downregulated in mice exposed to hyperoxia. miR-16 downregulation in mouse lung was correlated with an increase in SERT expression and pulmonary edema. Overexpression of miR-16 in human alveolar epithelial cells (A549) suppressed SERT and increased ENaCβ levels when compared to control-vector transfected cells. In addition, miR-16 over expression suppressed TGFβ release, a critical inhibitor of ENaC. Interestingly Nedd4, a negative regulator of ENaC remained unaltered in miR-16 over expressed A549 cells when compared to controls. Taken together, our data suggests that miR-16 upregulates ENaC, a major sodium channel involved in resolution of pulmonary edema in ALI.
Keywords: Acute lung injury, Hyperoxia, Pulmonary edema, Ion transport, Ion channels
1. Introduction
Acute lung injury (ALI) is a devastating disease characterized by flooding of alveolar spaces with a protein-rich fluid that impairs pulmonary gas exchange, leading to arterial hypoxemia and respiratory failure [1]. Hyperoxic acute lung injury is a well-established animal model to study human ALI and acute respiratory distress syndrome (ARDS) [2, 3]. Alveolar epithelial injury is a major contributor to alveolar flooding, because the epithelial barrier is much less permeable under normal conditions than the endothelial barrier [4]. Injury to alveolar epithelial cells can also disrupt normal epithelial fluid transport, impairing the removal of edema fluid from the alveolar space [1, 5]. Earlier reports have shown that reduced alveolar fluid clearance is a characteristic feature of ALI. However, these factors for this impair in epithelial fluid transport have not been well studied. The removal of lung edema fluid from the airspaces occurs via an active transport-dependent sodium concentration gradient across the distal lung epithelium [6, 7].
Epithelial sodium channel (ENaC) is the main force to drive sodium ions for transepithelial reabsorption and for formation of ion gradients to drive fluid out of alveolar spaces. ENaC is composed of α, β, and γ subunits. Several studies have found that ENaC is the major determinant of alveolar fluid clearance across the alveolar epithelium. Earlier studies have demonstrated that serotonin (5-HT) significantly inhibits amiloride-sensitive sodium transport across rat and human lung alveolar epithelial cell monolayers via a receptor-independent inhibition of ENaC activity [8]. As 5-HT also significantly inhibits the amiloride-sensitive fraction of the alveolar fluid clearance in mice, 5-HT can be considered as an endogenous inhibitor of ENaC. Increased levels of 5-HT have been reported in various pulmonary diseases, and inappropriate function of the 5-HT transmitter system further aggravates the disease condition. Therefore, any molecule that modulates the 5-HT transmitter system and upregulates EnaC show considerable therapeutic promise.
Recently, microRNAs (miRNAs) emerged as a major class of gene expression regulators implicated in deregulation of ion channel genes leading to channelopathies [9]. The consequence of miRNAs in fine-tuning gene expression shows that changes in the abundance of a single miRNA can affect the levels of expression of several different functional proteins [10, 11]. Thus, it is possible that a single dysregulated miRNA can push cells into an injured or inflammatory state. Recently, microRNA-16 (miR-16) has been reported to selectively target the serotonin transporter (SERT) involved in the 5-HT transmitter system [12]. However, miRNAs have never been studied in regulation of amiloride sensitive channels in ALI and role of miR-16 on its targets has not yet been elucidated. In this study, we investigated the role of miR-16 on ENaC expression and its target SERT in human lung alveolar epithelial cells. The results indicate that miR-16 modulates ENaC protein and SERT expression in alveolar epithelial cells. In addition, miR-16 suppressed the expression of transforming growth factor β (TGFβ), an inhibitor of ENaC and a critical mediator of ALI [13]. miR-16 mediated upregulation of ENaC is independent of Nedd4 (E3 ubiquitin protein ligase) pathway. Targeting SERT by miR-16 can be considered as a novel therapeutic approach to modulate ENaC expression and restore alveolar fluid balance in acute lung injury.
2. Materials and methods
2.1 Cell culture and transfection
Human alveolar epithelial cells (A549) derived from human lung adenocarcinoma were obtained from American Type Culture Collection (ATCC, Manassas, VA), and were maintained in complete Dulbecco’s modified Eagle medium (Invitrogen, Grand Island, NY) as per vendor instructions. Constructs expressing miR-16 and control vector were purchased from Add gene (Cambridge MA) and plasmid isolation was performed using miniprep plasmid isolation kits (Qiagen, Valencia, CA). Briefly, 2×105 cells were suspended in complete DMEM and seeded in a 6 well plate. At 70% confluency, cells were transfected with miR-16 plasmid or control-vector using Lipofectamine (Invitrogen). Complete DMEM was added to the A549 cells, 4 h post-transfection. Serotonin, at final concentration of 1 or 2 mM, was added to the respective wells.
2.2 Reagents and antibodies
Antibodies directed to ENaC α, β, γ and SERT were obtained from Alpha Diagnostics Int. (San Antonio, TX). Antibodies against ß-actin and GAPDH were obtained from Cell signaling technology Inc. (Boston, MA). Serotonin hydrochloride and HRP-conjugated secondary antibodies were obtained from Sigma (St. Louis, MO).
2.3 Mice
The Animal Care and Use Committee at the University of South Florida approved all animal procedures. C57BL/6J mice were purchased from Harlan Laboratories (Indianapolis, IN). In all experiments, mice aged 7–9 wks, were exposed to 100% oxygen as described earlier [14].
2.4 Quantitative RT-PCR
Total mRNA was isolated using the Trizol method [15]. Equal amounts of RNA were reverse transcribed into cDNA using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) and cDNA was amplified using qPCR performed on a Biorad CFX96 real-time PCR detection system as described earlier [16]. All primers were purchased from Integrated DNA Technologies (Coralville, IA) and were used at final concentrations of 100 nM. All experiments were performed in triplicate. Primers used were: mouse SERT (forward): 5′TCGCCCAGGACAACATCACCTGGAC3′, (reverse):5′TATGTGATGAAAAGGAGGCTGG3′;mouse18S(forward):5′ACCTGGTTGATCCTGCCAGT AG3′,(reverse):5′TTAATGAGCCATTCGCAGTTTC-3′;humanSERT(forward):5′ CAGCGTGTGAAGATGGAGAAG3′, (reverse): 5′TGGGATAGAGTGCCGTGTGT3′;human GADPH (forward):5′CGGAGTCAACGGATTTGGTCGTAT3′, (reverse): 5′ AGCCTTCTCCATGGTGGTGAAGAC3′.
2.5 Isolation of mouse RNA and quantification of miR-16
Total RNA enriched with small RNAs was extracted from mouse lung homogenates, using mirVana™ miRNA isolation kit (Ambion, Foster City, CA). miR-16 levels were measured using the TaqMan microRNA reverse transcription kit (Applied Biosystems, Carlsbad, CA).
2.6 Western blot
Cells were lysed with RIPA buffer and protein concentrations were determined with a Bicinchronoic Acid Assay kit (Pierce, Rockford, IL). Protein lysates were prepared for SDS-PAGE by adding 1/4 volume of 4× SDS-PAGE sample buffer (100 mM Tris-Cl, pH 6.8, 200 mM DTT, 4% SDS, 0.2% bromophenol blue, 20% glycerol) and the mixture was heated at 95°C for 5 min. Thirty micrograms of the protein was separated via SDS-PAGE and transferred onto a PVDF membrane by electrophoresis (Immobilon P; Millipore, Bedford, MA). After blocking with TBS Tween buffer (10 Mm Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% Tween-20) containing 5% nonfat dry milk for 1 hr, the membranes were incubated overnight at 4°C with blocking solution containing the indicated antibody (diluted 1:1,000–1:2,000). Membranes were washed and incubated with a suitable HRP-conjugated secondary antibody (Cell Signaling Technology, Danvers, MA) as mentioned previously [15]. HRP activity was detected using an ECL kit according to the manufacturer’s instructions (Pierce, Rockford, IL). Densitometric analysis of the immunoblots are added in the supplemental section.
2.7 Quantification of serotonin and TGFβ by ELISA
Serotonin levels in mouse lung homogenates were analyzed using ELISA as per the manufacturer’s instructions (GenWay Biotech Inc., San Diego, CA). TGFβ concentrations in cell supernatants were measured using the human TGFβ ELISA kit (R&D Systems, Minneapolis, MN).
2.8 Statistical analysis
All experiments were performed in triplicate. A paired t-test was used to determine the statistical significance. A p-value of p<0.05 was accepted as statistically significant.
3. Results
3.1 Elevated lung serotonin and increased SERT expression were observed in hyperoxia exposed mice
It was shown that serotonin decreases alveolar epithelial fluid transport via a direct inhibition of epithelial sodium channel [8]. Many studies suggest that defects in alveolar epithelial fluid transport can lead to pulmonary edema [5]. High plasma serotonin levels are implicated in pulmonary disorders such as primary pulmonary hypertension and pulmonary fibrosis [17]. However, serotonin levels have never been reported in hyperoxia-induced acute lung injury. To test the hypothesis that high serotonin levels may contribute to hyperoxia-induced ALI, we analyzed lung serotonin levels in mice under room air (normoxic) and hyperoxic (100% O2) conditions. We found that exposure to hyperoxia causes a significant increase in lung serotonin levels when compared to room air controls (Fig. 1A). The increase in serotonin levels after hyperoxia exposure was highly significant (p<0.001) when compared with room air controls. To assess the functional damage that occurs in ALI in terms of lung edema, we monitored the wet to dry ratio of lung tissue in mice exposed to hyperoxia and room air. The edematous lung damage caused by hyperoxia was highly significant (p<0.001) compared with room air controls (Fig. 1B). These results clearly show that hyperoxia-induced lung edema is associated with elevated lung serotonin levels.
Figure 1.
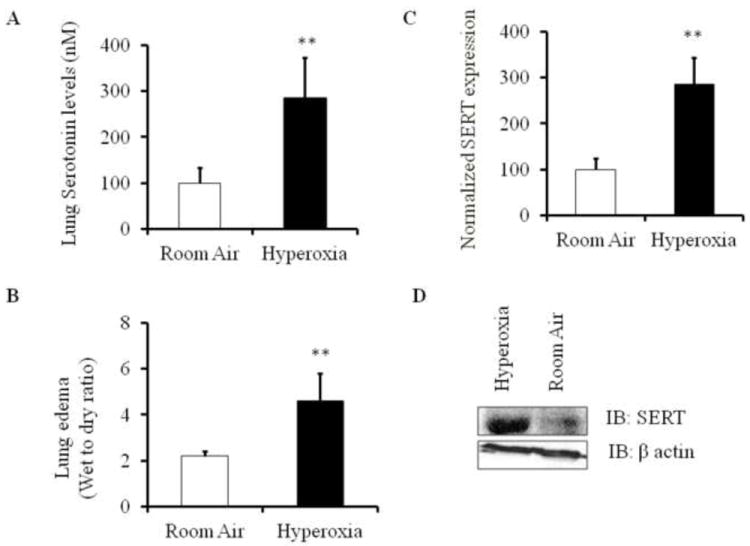
Elevated serotonin and SERT expression in mice exposed to hyperoxia (100% O2). A) Lung serotonin levels in mice exposed to room air and hyperoxic conditions. Lung homogenates were obtained from mice and subjected to capture ELISA to assess serotonin concentrations. B) Lung edema was measured using wet to dry ratio of mouse lungs exposed to hyperoxia and room air. C) Quantitative RT-PCR analysis to measure SERT mRNA expression in lung homogenates of mice exposed to hyperoxia and their room air controls. SERT mRNA was measured using reverse transcription and qPCR with SERT primers. 18S rRNA was used as internal control D) Relative SERT protein expression in mice lung homogenates was measured using Western blot analysis. **P<0.001 compared with room air controls.
Polymorphisms in SERT expression and function can lead to various lung diseases like pulmonary arterial hypertension and chronic obstructive lung disease. As per our knowledge, role of SERT in ALI has not yet been elucidated. To determine whether SERT expression is dysregulated in ALI, we measured SERT mRNA and protein expression levels in mice exposed to hyperoxia and room air (Fig. 1C and Fig. 1D, S2). SERT mRNA expression and protein levels were induced in mouse lungs exposed to hyperoxia when compared to normoxic controls.
3.2 miR-16 levels were downregulated in mice exposed to hyperoxia
Genetic factors that regulate SERT in acute lung injury are unknown. Recent studies suggest that SERT is a known direct target for miR-16[12]. However, the role of miR-16 in hyperoxic ALI has not been studied. To test the hypothesis that miR-16 may contribute to dysregulated SERT expression in hyperoxia-induced ALI, we analyzed lung miR-16 levels after hyperoxia exposure. We found that exposure to hyperoxia caused a significant decrease in lung miR-16 levels (Fig. 2A) showing that increased SERT levels are associated with suppressed lung miR-16 levels in hyperoxia induced ALI.
Figure 2.
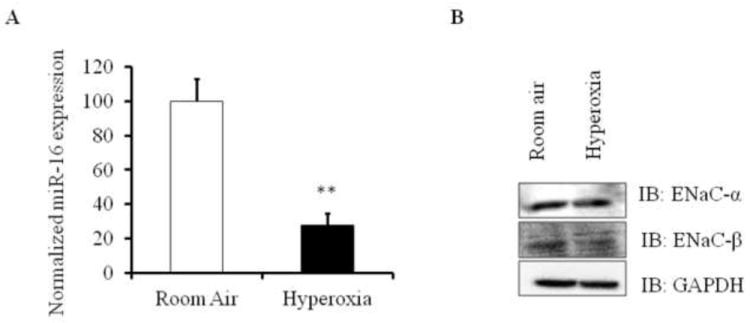
Expression levels of miR-16 and ENaC in hyperoxia induced ALI mouse models. A) miR-16 levels were analyzed by qRT-PCR in lung homogenates of mice exposed to hyperoxia and room air. Total RNA from the lung was isolated, reverse transcribed and analyzed for miR-16 using miR-16 specific assay primers and SYBR green method. miR-16 expression was normalized to 18S. B) ENaC-α and ENaC-β protein levels in mouse lung homogenates exposed to normoxia and hyperoxia were measured by western blot analysis. Lung homogenates (n=4) were subjected to immunoblotting with the indicated antibodies. **P<0.001 compared with room air controls.
3.3 Effect of hyperoxia on lung epithelial sodium channel (ENaC) expression levels
Elevated serotonin decreases alveolar epithelial fluid transport via a direct inhibition of the epithelial sodium channel [8]. Active vectorial Na+ transport through the distal lung epithelium is essential to maintain a physiological lung fluid balance [18]. Vectorial fluid and ion transport are altered in ALI. Clinical data and experimental studies provided a potential role of ENaC expression in fluid clearance and active ion transport [19]. We therefore analyzed the levels of ENaC expression in hyperoxia induced ALI mice. ENaC protein expression in mouse lung homogenates was analyzed by western blot (Fig. 2B, S3). When mice were exposed to hyperoxic conditions, there was a significant decrease in lung ENaCβ subunit compared to normoxic controls. ENaCα subunit expression was slightly decreased in hyperoxia induced ALI mice when compared to normoxic controls. Our results suggest that hyperoxia alters ENaCα expression and decreases ENaCβ expression.
3.4 Over expression of miR-16 down regulates SERT in the presence of serotonin in A549 cells
We next evaluated if overexpression of miR-16 can down regulate SERT in the presence of serotonin. To test this hypothesis, A549 cells were cultured in complete DMEM for 24 h and were treated with different concentrations of serotonin. Cells treated with 1 or 2 mM serotonin, showed significant increase in SERT mRNA levels by 10-40 fold (Fig. 3A). Surprisingly, cells treated with 4 mM serotonin did not show an increase in SERT mRNA, which may be due to a feedback inhibition of these transporters at higher serotonin concentrations. We next transfected A549 cells with miR-16 or control-vector and then treated cells with 1 or 2 mM serotonin. SERT mRNA and protein expression were analyzed by qRT-PCR and western blot analysis respectively. Down regulation of SERT mRNA and protein was observed in cells containing miR-16 treated with 1 or 2 mM serotonin (Fig. 3B and Fig. 3C, S4). These results demonstrate that miR-16 targets SERT and downregulates its expression in A549 cells post-serotonin treatment.
Figure 3.
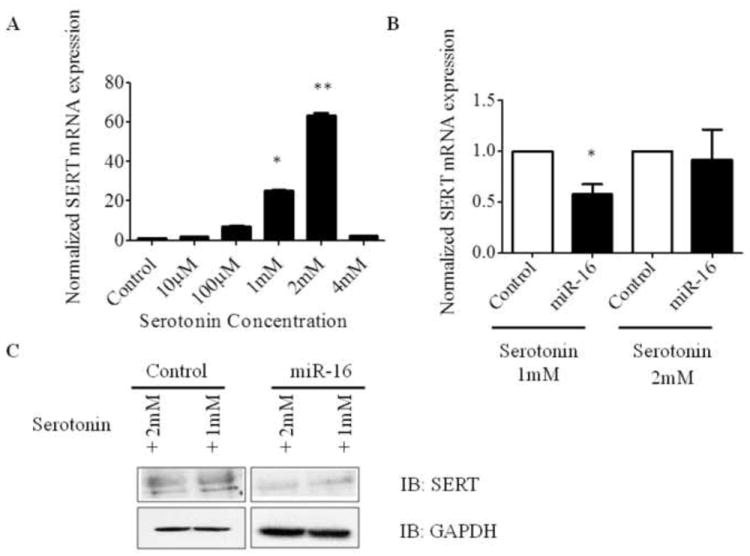
Over expression of miR-16 downregulated SERT expression in A549 cells in vitro. A) Serotonin transporter expression varied dose dependently with the addition of different concentrations of serotonin. A549 cells were treated with increasing concentrations of serotonin and were subjected to qRT-PCR to assess SERT mRNA expression levels. SERT mRNA expression was normalized to GAPDH. B) qRT-PCR analysis of SERT in miR-16 overexpressed A549 cells post-treated with serotonin. A549 cells were transfected with miR-16; 4 h post-transfection, cells were treated with either 1 or 2 mM serotonin, and 24 h later, RNA was isolated and subjected to qRT-PCR analysis. All experiments were repeated at least 3 independent times. C) SERT protein expression was analyzed in miR-16 overexpressed A549 cells post-treated with serotonin. A549 cells were transfected with miR-16 or control plasmid and post-treated with serotonin for 24 h. Cell lysates were subjected to immunoblotting with the indicated antibodies. *P<0.01 and **P<0.001 compared with control.
3.5 Overexpression of miR-16 induces epithelial sodium channel-β expression in alveolar epithelial cells
A majority of patients with increased permeability and edema in ALI have impaired alveolar epithelial fluid transport associated with dysregulation of epithelial sodium channel expression [5, 6]. It has been reported that higher serotonin levels can decrease alveolar epithelial fluid transport by inhibition of the epithelial sodium channel [8]. Since, our data showed miR-16 mediated modulation of SERT in epithelial cells, we hypothesized that miR-16 may regulate ENaC expression in epithelial cells. To test this hypothesis, A549 cells were transfected with miR-16 or control plasmid, and 24 h post-transfection and post-serotonin treatment, the cells were assayed for ENaC expression. The effect of miR-16 on different ENaC subunits in A549 cells was examined by western blot analysis (Fig. 4A, S5, S6). Transfection of miR-16 into A549 cells resulted in a significant increase of ENaC-β subunit when compared to control. However, ENaC-γ expression was unaltered in miR-16 transfected, post-serotonin treated A549 cells when compared to controls. ENaC-α expression was decreased in miR-16 transfected cells, which may be due to a significant increase in ENaC-β subunits. These two subunits complement each other to form a functional epithelial sodium channel. These results demonstrate that miR-16 has the ability to induce ENaC expression in human alveolar epithelial cells.
Figure 4.
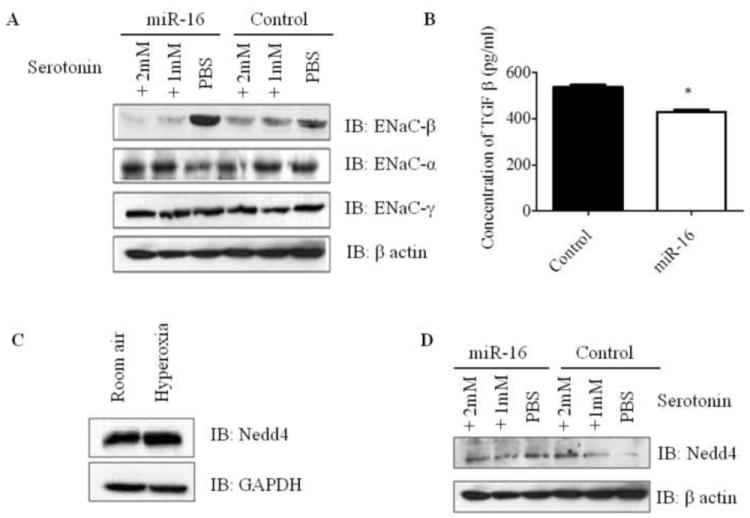
miR-16 overexpression in A549 cells modulates ENaC and TGF-β expression via Nedd4 independent pathway. A) Immunoblot analysis of different subunits of ENaC in miR-16 and control plasmid transfected A549 cells. A549 cells transfected with miR-16 or control plasmid were post-treated with or without serotonin and protein lysates were analyzed for ENaC-α, -β and -γ by immunoblot. B) TGF-β concentration in cell supernatants was analyzed by capture ELISA. Human alveolar epithelial cells were transfected with miR-16 or control plasmid and 24 h post-transfection, the supernatants were subjected to ELISA. C) Mice were exposed to hyperoxia and room air, and lung homogenates were analyzed for Nedd4 levels by immunoblot. D) Nedd4 immunoblot in A549 cells transfected with miR-16 or control plasmid in the presence or absence of serotonin treatment post-transfection. *P<0.01 compared with control.
3.6 miR-16 alters TGFβ levels in human alveolar epithelial cells in vitro
TGFβ has been shown to be a critical mediator of ALI [1]. TGFβ is known to decrease ENaC expression and alveolar sodium transport in epithelial cells [13]. Since our results showed that miR-16 modulates ENaC expression, we next evaluated the effect of miR-16 on TGFβ levels in A549 cells. To check whether miR-16 can modulate TGFβ expression, TGFβ levels were analyzed in culture supernatants from A549 cells transfected with miR-16 or control plasmid. There was a significant reduction of TGFβ levels in miR-16 transfected A549 cells compared to controls (Fig. 4B). Increased ENaC-β expression as observed earlier in miR-16 transfected human alveolar epithelial cells (Fig. 4A) may thus be associated with a significant decrease in TGFβ levels.
3.7 miR-16 mediated ENaC upregulation is independent of Nedd4
Target scan revealed several targets for miR-16. One among them is Nedd4 binding partner 1 (N4BP1)[20]. It has been recently reported that N4BP1 acts as an expressed interactor and mono-ubiquitinylation substrate of Nedd4. Nedd4 has been implicated in the regulation of fluid and electrolyte homeostasis by controlling the surface abundance of ENaC subunits [21]. We checked the levels of Nedd4 in lungs of mice exposed to hyperoxia and normoxia. Nedd4 was upregulated in mice exposed to hyperoxia compared to their room air controls (Fig. 4C, S6). This showed an association with decreased ENaC subunits in mice exposed to hyperoxia. To further determine if miR-16-mediated ENaC expression is through Nedd4 modulation, we monitored the expression levels of Nedd4 in miR-16 overexpressed A549 cells and controls post-treated with 1 or 2 mM serotonin. Levels of Nedd4 were slightly reduced in miR-16 transfected A549 cells post-treated with 2 mM serotonin when compared to controls (Fig. 4D, S6).Our data suggest that miR-16 mediated upregulation of ENaC is independent of Nedd4.
4. Discussion
In this study we have shown that miR-16 suppression in ALI is correlated with increased serotonin levels. Serotonin (5-HT) significantly inhibits amiloride-sensitive sodium transport across rat and human lung alveolar epithelial cell monolayers via a receptor-independent inhibition of epithelial sodium channel (ENaC) expression [8]. It is known that SERT is a key player for 5-HT mediated effects in alveolar epithelial transport. Therefore, inhibitors of SERT offer considerable therapeutic promise. miR-16 is known to target SERT and overexpression of miR-16 in A549 cells downregulated its target SERT and upregulated EnaCβ in the presence of serotonin. For the first time, a biological explanation for an increase in lung SERT levels, during ALI by means of downregulation of miR-16 (S1) and effects of overexpression of miR-16 on ENaC is provided in this study.
Previous studies have shown that TGF-β a critical mediator of ALI, significantly decreased ENaC-α mRNA and protein expression [13]. The inhibitory effect of TGF-β1 on sodium uptake and ENaC-α expression in rat alveolar type II (ATII) cells was mediated by activation of the MAPK, ERK1/2 pathway. Serotonin is also known to induce TGFβ [22] and increased TGFβ levels are known to suppress ENaC expression. To determine whether miR-16 mediated effect of ENaC upregulation is due to alteration of TGF-β, we checked the levels of TGF-β in miR-16 overexpressed alveolar epithelial cells. Our study shows that increased ENaC expression by miR-16 is associated with a significant decrease in TGF-β levels. Therefore miR-16 mediated ENaC expression is further dependent on TGF-β downregulation.
The importance of Nedd4-2 in regulating ENaC expression and distal lung fluid absorption near term and at birth was also recently demonstrated by artificially removing Nedd4-2 using specific siRNA [23]. In that report, it was demonstrated that reducing Nedd4 expression led to an increased lung fluid absorption rate, and thus it was suggested that Nedd4 plays an important role in regulating ENaC expression and lung fluid absorption at birth. However, factors that modulate Nedd4 expression in ALI are not clear. Since miR-16 is a known target for N4BP1, a Nedd4 binding partner, we checked the levels of Nedd4 in miR-16 overexpressed A549 cells. Our results show that overexpression of miR-16 did not significantly suppress Nedd4 in epithelial cells. However, a slight decrease in Nedd4 levels was observed in miR-16 overexpressed cells post-treated with 2 mM serotonin. Therefore, miR-16 modulation of ENaC, which we observed in our study, may be independent of the NEDD4 pathway.
In conclusion, we have provided evidence that overexpressed miR-16 is an endogenous inhibitor of SERT and we also provided the functional effects of miR-16 on ENaC upregulation, which is a key protein involved in alveolar fluid clearance in ALI. Based on our results, we propose that exogenous miR-16 provides a therapeutic strategy to induce ENaC expression by SERT suppression and may have use as a therapeutic to treat ALI and other diseases involving pulmonary edema.
Supplementary Material
Highlights.
We indentified a novel role for miR-16 in regulating epithelial sodium channel in ALI
Decreased miR-16 levels in ALI were correlated with increased SERT and pulmonary edema
Over expression of miR-16 upregulated EnaC and suppressed SERT, TGFβ in epithelial cells
Acknowledgments
This work was funded by the American Heart Association National Scientist Development Grant 09SDG2260957 and National Institutes of Health R01 HL105932 to NK and the Joy McCann Culverhouse Endowment to the Division of Allergy and Immunology. Authors thank Dr. Brenda Flam for critical reading and editing this manuscript.
Abbreviations
acute lung injury
- miR-16
microRNA-16
- ENaC
epithelial sodium channel
- SERT
serotonin transporter
- TGFβ
transforming growth factor β
- 5-HT
serotonin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pittet JF, Griffiths MJ, Geiser T, Kaminski N, Dalton SL, Huang X, Brown LA, Gotwals PJ, Koteliansky VE, Matthay MA, Sheppard D. TGF-beta is a critical mediator of acute lung injury. J Clin Invest. 2001;107:1537–1544. doi: 10.1172/JCI11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhandari V, Choo-Wing R, Lee CG, Zhu Z, Nedrelow JH, Chupp GL, Zhang X, Matthay MA, Ware LB, Homer RJ, Lee PJ, Geick A, de Fougerolles AR, Elias JA. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat Med. 2006;12:1286–1293. doi: 10.1038/nm1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kolliputi N, Waxman AB. IL-6 cytoprotection in hyperoxic acute lung injury occurs via PI3K/Akt-mediated bax phosphorylation. Am J Physiol Lung Cell Mol Physiol. 2009;297:L6–16. doi: 10.1152/ajplung.90381.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waxman AB, Kolliputi N. IL-6 protects against hyperoxia-induced mitochondrial damage via bcl-2-induced bak interactions with mitofusins. Am J Respir Cell Mol Biol. 2009;41:385–396. doi: 10.1165/rcmb.2008-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berthiaume Y, Matthay MA. Alveolar edema fluid clearance and acute lung injury. Respir Physiol Neurobiol. 2007;159:350–359. doi: 10.1016/j.resp.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82:569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- 7.Geiser T, Atabai K, Jarreau PH, Ware LB, Pugin J, Matthay MA. Pulmonary edema fluid from patients with acute lung injury augments in vitro alveolar epithelial repair by an IL-1beta-dependent mechanism. Am J Respir Crit Care Med. 2001;163:1384–1388. doi: 10.1164/ajrccm.163.6.2006131. [DOI] [PubMed] [Google Scholar]
- 8.Goolaerts A, Roux J, Ganter MT, Shlyonsky V, Chraibi A, Stephane R, Mies F, Matthay MA, Naeije R, Sariban-Sohraby S, Howard M, Pittet JF. Serotonin decreases alveolar epithelial fluid transport via a direct inhibition of the epithelial sodium channel. Am J Respir Cell Mol Biol. 2010;43:99–108. doi: 10.1165/rcmb.2008-0472OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elvira-Matelot E, Jeunemaitre X, Hadchouel J. Regulation of ion transport by microRNAs. Curr Opin Nephrol Hypertens. 2011;20:541–546. doi: 10.1097/MNH.0b013e328348b4aa. [DOI] [PubMed] [Google Scholar]
- 10.Malumbres M. miRNAs versus oncogenes: The power of social networking. Mol Syst Biol. 2012;8:569. doi: 10.1038/msb.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdellatif M. Differential expression of microRNAs in different disease states. Circ Res. 2012;110:638–650. doi: 10.1161/CIRCRESAHA.111.247437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baudry A, Mouillet-Richard S, Schneider B, Launay JM, Kellermann O. miR-16 targets the serotonin transporter: A new facet for adaptive responses to antidepressants. Science. 2010;329:1537–1541. doi: 10.1126/science.1193692. [DOI] [PubMed] [Google Scholar]
- 13.Frank J, Roux J, Kawakatsu H, Su G, Dagenais A, Berthiaume Y, Howard M, Canessa CM, Fang X, Sheppard D, Matthay MA, Pittet JF. Transforming growth factor-beta1 decreases expression of the epithelial sodium channel alphaENaC and alveolar epithelial vectorial sodium and fluid transport via an ERK1/2-dependent mechanism. J Biol Chem. 2003;278:43939–43950. doi: 10.1074/jbc.M304882200. [DOI] [PubMed] [Google Scholar]
- 14.Kolliputi N, Galam L, Tamarapu Parthasarathy P, Tipparaju SM, Lockey RF. NALP-3 inflammasome silencing attenuates ceramide-induced transepithelial permeability. J Cell Physiol. 2012;227:3310–3316. doi: 10.1002/jcp.24026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolliputi N, Shaik RS, Waxman AB. The inflammasome mediates hyperoxia-induced alveolar cell permeability. J Immunol. 2010;184:5819–5826. doi: 10.4049/jimmunol.0902766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aljubran SA, Cox R, Jr, Tamarapu Parthasarathy P, Kollongod Ramanathan G, Rajanbabu V, Bao H, Mohapatra SM, Lockey R, Kolliputi N. Enhancer of zeste homolog 2 induces pulmonary artery smooth muscle cell proliferation. PLoS One. 2012;7:e37712. doi: 10.1371/journal.pone.0037712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egermayer P, Town GI, Peacock AJ. Role of serotonin in the pathogenesis of acute and chronic pulmonary hypertension. Thorax. 1999;54:161–168. doi: 10.1136/thx.54.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eaton DC, Chen J, Ramosevac S, Matalon S, Jain L. Regulation of na+ channels in lung alveolar type II epithelial cells. Proc Am Thorac Soc. 2004;1:10–16. doi: 10.1513/pats.2306008. [DOI] [PubMed] [Google Scholar]
- 19.Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol. 2009;71:403–423. doi: 10.1146/annurev.physiol.010908.163250. [DOI] [PubMed] [Google Scholar]
- 20.Oberst A, Malatesta M, Aqeilan RI, Rossi M, Salomoni P, Murillas R, Sharma P, Kuehn MR, Oren M, Croce CM, Bernassola F, Melino G. The Nedd4-binding partner 1 (N4BP1) protein is an inhibitor of the E3 ligase itch. Proc Natl Acad Sci U S A. 2007;104:11280–11285. doi: 10.1073/pnas.0701773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dinudom A, Harvey KF, Komwatana P, Young JA, Kumar S, Cook DI. Nedd4 mediates control of an epithelial na+ channel in salivary duct cells by cytosolic na+ Proc Natl Acad Sci U S A. 1998;95:7169–7173. doi: 10.1073/pnas.95.12.7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grewal JS, Mukhin YV, Garnovskaya MN, Raymond JR, Greene EL. Serotonin 5-HT2A receptor induces TGF-beta1 expression in mesangial cells via ERK: Proliferative and fibrotic signals. Am J Physiol. 1999;276:F922–30. doi: 10.1152/ajprenal.1999.276.6.F922. [DOI] [PubMed] [Google Scholar]
- 23.Li T, Koshy S, Folkesson HG. IL-1beta-induced cortisol stimulates lung fluid absorption in fetal guinea pigs via SGK-mediated Nedd4-2 inhibition. Am J Physiol Lung Cell Mol Physiol. 2009;296:L527–33. doi: 10.1152/ajplung.90506.2008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.