

Abstract
Carboxyethylarginine synthase is the first dedicated enzyme of clavam biosynthesis in Streptomyces clavuligerus and is present in two isoforms encoded by two separate genes. When grown on a liquid soy medium, strains with ceaS1 deleted showed only a mild reduction of clavam biosynthesis, while disruption of ceaS2 abolished all clavam biosynthesis. Creation of an in-frame ceaS2 deletion mutant to avoid polarity did not restore clavam production, nor did creation of a site-directed mutant altered only in a single amino acid residue important for activity. Reverse transcriptase PCR analyses of these mutants indicated that the failure to produce clavam metabolites could be traced to reduced or abolished transcription of ceaS1 in the ceaS2 mutants, despite the location of ceaS1 on a replicon completely separate from that of ceaS2. Western analyses further showed that the CeaS1 protein (as well as the CeaS2 protein) was absent from the ceaS2 mutants. Complementation experiments were able to restore clavam production partially, but only by virtue of restoring CeaS2 production. CeaS1 was still absent from the complemented strains. While this dependence of CeaS1 production on the expression of ceaS2 from its native chromosomal location was seen in all of the ceaS2 mutants, the effect was limited to growth in liquid medium. When the same mutants were grown on solid soy medium, clavam production was restored and CeaS1 was produced, albeit at low levels compared to the wild type.
INTRODUCTION
Clavulanic acid is a powerful β-lactamase inhibitor used in conjunction with conventional β-lactam antibiotics to combat diseases caused by antibiotic-resistant pathogens. It is produced industrially in fermentations of Streptomyces clavuligerus. Due to its clinical value (1), the biochemical and genetic aspects of clavulanic acid biosynthesis in S. clavuligerus have been subjected to intensive research in recent years. It is now known that the biosynthetic pathway to clavulanic acid is partly shared with the pathway for structurally related 5S clavam metabolites that are also produced by S. clavuligerus, up to the level of clavaminic acid (2). The pathway branches at this point and goes on to form either clavulanic acid or 5S clavams (Fig. 1A). The enzymes involved in the early shared steps of the pathway have been well characterized (3), although the later stages remain to be clarified.
Fig 1.

Production of clavam metabolites. (A) The biosynthetic pathway to clavaminic acid, the branch point intermediate leading to production of clavulanic acid and 5S clavams. Gene designations are shown in italics. (B) Part of the cephamycin-clavulanic acid gene cluster. Cephamycin-specific genes are shown in white, and clavulanic acid-specific genes are shown in gray. Locations of restriction endonuclease cleavage sites relevant to descriptions in the text are shown.
From their first discovery, it was known that there are two isozymes of clavaminic acid synthase (CAS), one of the early pathway enzymes, in S. clavuligerus (4, 5). The two isozymes are encoded by two cas genes, cas1 and cas2, and disruption of either copy causes only partial loss of clavulanic acid production, while disruption of both genes causes a complete loss. cas2 lies near one end of the chromosome within the clavulanic acid gene cluster, which includes all of the genes associated with the production of clavulanic acid (Fig. 1B), whereas cas1 is located several megabases away in the clavam gene cluster. The gene pah2, encoding proclavaminic acid synthase (PAH), another of the early enzymes, lies adjacent to cas2 in the clavulanic acid gene cluster. When disruption of pah2 caused only partial loss of clavulanic acid production (6), this suggested that there must also be two pah genes. The bls2 gene, which encodes β-lactam synthetase, follows this same paradigm, in that disruption of bls2 caused only partial loss of clavulanic acid production, leading to prediction of a second bls gene, but the ceaS2 gene stood apart. ceaS2 encodes carboxyethylarginine synthase (CEAS), the first enzyme of the pathway. Initial studies showed that disruption of ceaS2 with an apramycin resistance (apr) gene to generate a ceaS2::apr mutant caused complete loss of production of all clavam metabolites, suggesting that there might be only one ceaS gene. However, trace amounts of clavulanic acid (2 to 5% of wild-type levels) were produced by ceaS2::apr mutants on rare occasions (6). An early report by Perez Redondo et al. (7) also indicated that ceaS2 mutants could produce clavulanic acid under certain growth conditions, but we could not reproduce this effect when using our ceaS2 mutants. In all of our recent studies, ceaS2 mutants were completely blocked in production of all clavam metabolites. This led us to conclude that initial findings of clavam production by ceaS2 mutants were uncertain and perhaps attributable to limitations of early bioassay and high-performance liquid chromatography (HPLC) techniques. However, the eventual discovery of the paralogue gene cluster located on a giant linear plasmid (8–10) proved that there are second copies of all early genes, including a second ceaS gene. The second copies of these early genes were designated ceaS1, bls1, and pah1, to correlate with the previously named cas1 gene and to distinguish them from the ceaS2, bls2, pah2, and cas2 genes of the clavulanic acid gene cluster. The genes of the clavam and paralogue clusters are associated with the production of the 5S clavam metabolites, while the clavulanic acid cluster is involved with the production of clavulanic acid. The two processes are regulated independently of one another (11–16), although the shared early steps in the pathway mean that there is some cross talk between clavulanic acid and 5S clavam biosynthesis. The ceaS1 gene is highly similar to ceaS2 (66% identity and 80% similarity at the amino acid level), but deletion of the ceaS1 gene caused only a minor decrease in clavulanic acid production (10), which left open the question of why ceaS2 mutants typically do not produce any clavam metabolites.
Investigation of the blocked phenotype of the ceas2::apr mutant was complicated by the fact that ceaS2 is the lead gene of an operon encompassing all of the early genes of the clavulanic acid cluster (Fig. 1B) (17). To prevent polarity from influencing expression of downstream genes, a second ceaS2 mutant was created in which the disrupting apr gene was replaced by a simple frameshift (FS) (6). However, the phenotype of the resulting ceaS2-FS mutant was the same as that of the ceaS2::apr mutant. In this study, we investigated further the basis for the severe effect of ceaS2 disruption on clavam metabolite production in S. clavuligerus.
MATERIALS AND METHODS
Bacterial strains, plasmid and cosmid vectors, and culture conditions.
Bacterial strains and the plasmid and cosmid vectors used in this study are listed in Table 1 (18–20). S. clavuligerus strains were maintained on International Streptomyces Project-4 (ISP-4) agar plates as described previously (21) and stored in 20% glycerol at −80°C as spore stocks. Escherichia coli strains were grown and maintained on Lennox broth (LB) medium at 37°C. Plasmid-containing strains were selected with appropriate antibiotics as described previously (10). Fermentation studies were performed by inoculating Trypticase soy broth plus 1% soluble starch (TSBS) with S. clavuligerus spore stocks. After 40 h of incubation at 28°C on a rotary shaker at 280 rpm, these seed cultures were used to inoculate 2% (vol/vol) soy (5) or starch asparagine (SA) medium (17), and incubation was continued for 72 to 96 h, when metabolite production was maximal in wild-type cultures. All production cultures were staged using 100 ml of medium in 500-ml flasks, since production of 5S clavam metabolites was found to be particularly sensitive to medium-to-flask volume ratios.
Table 1.
Bacterial strains, plasmids, and cosmids used in this study
| Strain, plasmid, or cosmid | Description | Reference or source |
|---|---|---|
| E. coli strains | ||
| BL21(DE3) | Host for T7 promoter-driven protein expression | Stratagene |
| BW25115/pIJ790 | Host for Redirect PCR targeting system, produces λ Red proteins from temperature-sensitive plasmid pIJ790 | 24 |
| DH5α | General cloning host | Gibco BRL |
| DH5α(BT340) | Produces FLP recombinase from temperature-sensitive plasmid BT340 | 24 |
| ET12567/pUZ8002 | Methylation-deficient host, expresses transfer functions from pUZ8002 | 22 |
| S. clavuligerus strains | ||
| NRRL 3585 | Wild-type strain producing cephamycin C, clavulanic acid, and 5S clavams | Northern Regional Research Laboratory, Peoria, IL |
| ceaS2::apr | ceaS2 insertional disruption mutant | 6 |
| ceaS2-FS | ceaS2 frameshift mutant | 10 |
| ΔceaS1::apr | Deletion mutant with ceaS1 replaced by apr cassette | 10 |
| ΔceaS1::apr/ ceaS2-FS | ceaS1/ceaS2 double mutant | 10 |
| ΔceaS2::apr | Deletion mutant with ceaS2 replaced by apr cassette | This study |
| ΔceaS2-IF | ceaS2 in-frame deletion mutant | This study |
| ΔceaS2-c5 | ceaS2 in-frame deletion mutant complemented with pSET152-c5 | This study |
| ΔceaS2-c11.6 | ceaS2 in-frame deletion mutant complemented with pSET152-c11.6 | This study |
| E57WceaS2 | ceaS2 site-directed mutant with codon 57 changed from glutamate to tryptophan | This study |
| E57WceaS2-c5 | ceaS2 E57W mutant complemented with pSET152-c5 | This study |
| E57WceaS2-c11.6 | ceaS2 E57W mutant complemented with pSET152-c11.6 | This study |
| Plasmids | ||
| pBB5.3A | pUC119 carrying 5-kb DNA insert with pcbR and ceaS2 | 16 |
| pET-9c | Protein expression vector | Novagen |
| pET-24b | Protein expression vector | Novagen |
| pET-9c-ceaS1 | pET-9c derivative carrying ceaS1 | This study |
| pET-24a-ceaS2 | pET-24a derivative carrying ceaS2 | C. Schofield, Oxford University |
| pIJ773 | Plasmid carrying template for Redirect apr cassette | 24 |
| pSET152 | E. coli-Streptomyces shuttle plasmid, integrates in Streptomyces | Northern Regional Research Laboratory |
| pSET152-ceaS1 | pSET152 derivative carrying promoter region of ceaS1 coupled to promoterless egfp gene | 16 |
| pSET152-c5 | pSET152 derivative carrying 5-kb insert with ceaS2 and upstream regions | This study |
| pSET152-c11.6 | pSET152 derivative carrying 11.6-kb insert extending from mid-pcbC through cad | This study |
| pUC119 | General cloning vector | 18 |
| pUWL-KS | E. coli-Streptomyces shuttle plasmid | 19 |
| pUWL-oriT | Derivative of pUWL-KS carrying oriT | 20 |
| pUWL-ΔceaS2::apr | Derivative of pUWL-KS carrying 5.5-kb insert encompassing ΔceaS2::apr mutation | This study |
| Cosmids | ||
| pWE15 | General purpose cosmid vector | Promega |
| 12B8 | pWE15-derived cosmid carrying ceaS2 and downstream region | 8 |
| 12B8-AP | 12B8 derivative with ceaS2 replaced by apr gene cassette | This study |
Metabolite production on solid medium was tested using plates containing 20 ml of soy medium solidified with agar (2%, wt/vol) and spread with ∼108 spores. Sterile cellophane disks (22) were placed on the inoculated plates, and another 108 spores was spread on each cellophane disk. This double inoculation technique was necessitated by the insoluble nature of the soy medium nutrients. Inoculated plates were incubated at 28°C for 9 days.
DNA manipulation, PCR, and sequencing.
Standard procedures were used to manipulate plasmid DNA purified from E. coli (23). PCRs were performed using the Expand high-fidelity PCR system (Roche) according to the manufacturer's protocol. Oligonucleotide primers are listed in Table S1 of the supplemental material and were purchased from Integrated DNA Technologies (IDT). DNA sequence analysis was carried out by the Molecular Biology Service Unit, University of Alberta. The BLAST program (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used for performing sequence alignments and similarity searches.
Construction of ΔceaS2::apr, ΔceaS2 in-frame (IF), and E57WceaS2 mutants.
ΔceaS2::apr mutants were prepared by using the Redirect PCR-targeted mutagenesis procedure (24) as described in previous studies (10). Primers ceaS2-Redirect For and ceaS2-Redirect Rev were used to amplify the apr cassette to give a PCR product with sequences on each end targeting it to the ceaS2 gene. The PCR product was transformed into E. coli BW25115/pIJ790 carrying cosmid 12B8, wherein the ceaS2 gene was deleted and replaced by the apr cassette to generate mutant cosmid 12B8-AP. Cosmid 12B8-AP was then conjugated into wild-type S. clavuligerus, and ΔceaS2::apr mutant strains were selected based on the presence of apr and confirmed by Southern analysis.
ΔceaS2-IF mutants were prepared by cloning a 5.5-kb KpnI DNA fragment carrying the apr cassette and regions flanking the deleted ceaS2 gene from cosmid 12B8-AP into pUWL-KS to create pUWL-ΔceaS2::apr. pUWL-ΔceaS2::apr was then transformed into E. coli DH5α(BT340), and transformants were grown at 42°C to induce FLP recombinase and excise the disruption cassette, leaving in its place an 81-bp scar sequence. The resulting plasmid, pUWL-ΔceaS2-IF, was transformed into ΔceaS2::apr protoplasts, where homologous recombination generated the ΔceaS2-IF mutants.
E57WceaS2 mutants were created by PCR using plasmid pBB5.3A, which carries an ∼5-kb DNA fragment extending from a Sau3A site inside pcbC to a BglII site inside bls2 (Fig. 1B) (16), as the template, along with primers Mutate ceaS2-For and Mutate ceaS2-Rev to amplify a 580-bp fragment from the 5′ end and upstream of ceaS2. The amplified fragment encompassed naturally occurring NcoI and SacII sites and introduced a GA-to-TG mutation to change the glutamate 57 residue of CeaS2 to tryptophan. The fragment, digested with NcoI and SacII, was used to replace the corresponding wild-type fragment in pBB5.3A, and then the E57W mutant version of the entire 5-kb DNA fragment was subcloned into pUWL-oriT and conjugated into the ceaS2::apr mutant. Thiostrepton-resistant exconjugants were sporulated in the presence of thiostrepton and then again in its absence. Resulting spores were replica plated to identify thiostrepton-sensitive, apramycin-sensitive E57WceaS2 mutants. The identity of the mutants was verified by sequencing PCR products amplified from genomic DNA using primers ceaS2-RT-for and ceaS2-RT-rev.
Several independent isolates of each ceaS2 mutant type were examined. All isolates sporulated and showed growth characteristics on solid and liquid media that were indistinguishable from wild-type strains. Metabolite production patterns were consistent for all mutants, so a representative strain of each type was chosen for further studies.
Complementation of the ΔceaS2-IF mutation in S. clavuligerus.
The ∼5-kb insert of S. clavuligerus DNA that encompasses ceaS2 was released from pBB5.3A as a BamHI-XbaI fragment and cloned into pSET152 to create pSET152-c5. Similarly, an ∼11.6-kb EcoRI fragment that extends from upstream of ceaS2 to beyond the end of cad (Fig. 1B) was cloned into pSET152 to generate pSET152-c11.6. The plasmids were introduced into ΔceaS2-IF and E57WceaS2 mutants by conjugation, and complemented strains were selected based on the presence of apr.
Overproduction of CeaS1 and CeaS2.
Plasmid pET-24a-ceaS2 was generously provided by C. Schofield (Oxford University). Plasmid pET-9c-ceaS1 was prepared by amplifying ceaS1 by PCR using primers ceas1-exp-fwd and ceaS1-exp-rev. The amplified fragment was subcloned into pTOPO2.1, sequenced to verify fidelity, and then transferred to pET9c. The two expression plasmids were transformed into E. coli BL21(DE3), and when freshly grown cultures reached an optical density at 600 nm of approximately 0.6, isopropyl-β-d-thiogalactoside (IPTG) was added to 0.5 mM to induce protein production, and cultures were incubated at 37°C for a further 4 h. Bacterial cells were harvested, resuspended in 50 mM Tris-HCl (pH 7.4), 2 mM EDTA, 0.1% Triton X-100, and lysozyme at 0.15 mg/ml, disrupted by sonication, and centrifuged for 10 min at 14,000 × g to sediment insoluble material.
RNA isolation and RT-PCR.
Total RNAs were isolated from S. clavuligerus strains grown in soy medium for 48 h by using the modified Kirby procedure (22). Preliminary studies indicated that ceaS1 and ceaS2 transcript levels in wild-type cultures were greatest at this time point. Reverse transcriptase PCR (RT-PCR) analysis of RNA was carried out using SuperScript III (SSIII) reverse transcriptase (Invitrogen) to generate cDNA. RT reaction mixtures were set up according to the manufacturer's protocol but with the following changes: 1 μg of total RNA, 5% dimethyl sulfoxide (DMSO), and 0.5 μl of SSIII (200 U/μl) were included in each 10-μl reaction mixture, and reaction mixtures were incubated at 55°C for 1 h. Each 50-μl PCR mixture contained 10 μl of RT product, 5% DMSO, and 25 pmol of forward and reverse primers. PCR conditions were as follows: initial denaturation at 94°C for 2 min, followed by 28 cycles of denaturation at 94°C for 45 s, annealing at 62°C for 45 s, and elongation at 72°C for 45 s. Negative controls (no RT) were carried out with each RNA preparation to confirm the absence of contaminating chromosomal DNA.
Western blot analysis.
Cell extracts (CFEs) were prepared from 5-ml aliquots of 48-h S. clavuligerus cultures grown in soy or SA medium, after supplementing with Complete protease inhibitor (Roche) and chilling to 4°C. Cultures were harvested by centrifugation, resuspended in 1 ml of lysis buffer (50 mM Tris-HCl [pH 7.4] containing Complete protease inhibitor), and disrupted by sonication. Cell debris was removed by centrifugation, and clarified cell lysates were stored at −80°C. Protein concentrations were determined using a Coomassie dye binding protein assay (Bio-Rad) with bovine gamma globulin as the standard.
CFEs were prepared from cultures grown on solid medium by scraping cell material from cellophane disks on the surface of soy agar plate cultures into lysis buffer, 1 ml per plate, and disrupting by sonication as described above.
Samples of total CFE protein were separated using 8% SDS-PAGE. Resolved proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Immobilon-P; Amersham) in transfer buffer (25 mM Tris, 192 mM glycine, 10% methanol) and using a mini-Trans-Blot apparatus (Bio-Rad). The enhanced chemiluminescent Western blotting system (ECL-Plus; Pierce) was used to determine protein production according to the manufacturer's instructions. As a cautionary note, CeaS proteins are very sensitive to proteolysis, and so blocking and hybridization solutions containing skim milk were prepared in small quantities immediately before use. Primary anti-CeaS2 antibodies were prepared by standard procedures (25) as described previously (26). Both primary antibody and horseradish peroxidase-linked anti-rabbit IgG secondary antibody (from donkey; Amersham) were used at a dilution of 1/15,000.
EGFP expression and confocal microscopy.
pSET-ceaS1 (16), which carries the promoter region of ceaS1 coupled to a promoterless egfp gene, was conjugated into wild-type and ΔceaS2-IF mutant strains of S. clavuligerus with selection for apr. Confocal microscopy was carried out using a Leica DM IRB inverted microscope to detect enhanced green fluorescent protein (EGFP) production under conditions described previously (16), except samples were harvested after 48 h of growth in soy medium. An argon laser with 50 to 52% attenuation was used to provide excitation at 488 nm, and EGFP excitation was detected between 500 nm and 520 nm.
HPLC analyses.
HPLC was used to measure clavulanic acid and 5S clavam metabolites in S. clavuligerus cultures grown in soy or SA medium. Supernatants from 72- and 96-h cultures were derivatized with imidazole and analyzed using a Phenomenex Bondclone C18 column (100 by 800 mm, 10 μm; Phenomenex, Torrance, CA) under previously described conditions (17).
Clavam metabolites produced during growth on solid soy medium were assessed by cutting 4-cm by 4-cm squares of agar from plate cultures after the cell material had been harvested for production of CFE. Agar squares were frozen at −80°C, thawed slowly, and pressed to release fluid. The resulting exudates (25-μl samples) were derivatized with imidazole and analyzed by HPLC as described above for culture supernatants.
RESULTS
Production of ΔceaS2::apr and ΔceaS2-IF mutants.
Disruption of the ceaS2 gene in S. clavuligerus gave ceaS2::apr mutants that routinely produced no clavam metabolites, and ceaS2 frameshift mutants showed the same nonproducer phenotype (6, 10). Although the ceaS2-FS mutants were unmarked, translational termination of the out-of-frame 3′ end of the gene would occur 334 bp upstream of the normal stop codon, and so transcription of downstream genes could still be affected. Furthermore, both the ceaS2::apr and ceaS2-FS mutants could potentially produce truncated or C-terminally mistranslated CeaS2 proteins. Since the CeaS protein is known to exist as a tetramer (a dimer of dimers) (27), the defective Ceas2 subunits might interact with active CeaS1 subunits to affect activity.
To address these possibilities, two more ceaS2 mutants were produced using PCR-targeted mutagenesis (24). In the mutant ΔceaS2::apr, the entire ceaS2 gene was replaced by an apr gene cassette, and in mutant ΔceaS2-IF, the apr cassette was removed from ΔceaS2::apr by FLP recombinase to leave an 81-bp unmarked, in-frame scar in place of the ceaS2 gene. Both mutants were verified by Southern analysis and then analyzed along with wild-type S. clavuligerus and earlier versions of ceaS2 mutants for the ability to produce clavulanic acid and 5S clavams. HPLC analysis of culture supernatants showed that all of the ceaS2 mutant types had similar phenotypes, and the newly created ΔceaS2::apr and ΔceaS2-IF mutants, like the earlier versions, produced no detectable clavulanic acid or 5S clavams when grown in soy medium, whereas the wild-type strain produced all of the expected clavam metabolites (Fig. 2).
Fig 2.

Production of clavam metabolites by S. clavuligerus wild-type and ceaS2 mutant cultures. Culture supernatants from 96-h soy cultures were derivatized with imidazole to detect clavam metabolites and analyzed by HPLC. AC, alanylclavam; C2C, clavam-2-carboxylate; 2HMC, 2-hydroxymethylclavam; CA, clavulanic acid.
Effect of ΔceaS2-IF mutation on ceaS1 and bls2 transcription.
RT-PCR was carried out to investigate if the ΔceaS2-IF mutant showed altered transcription of either ceaS1 or downstream genes compared to the wild type. Total RNAs were isolated from S. clavuligerus wild type, ΔceaS2-IF, and ΔceaS1::apr mutants grown for 48 h in soy medium. Reverse primers (see Table S1 in the supplemental material) were used to generate cDNA, and specific primer pairs (see Table S1) were then used to determine if the genes of interest were transcribed in the mutant strains. When RNA from wild-type S. clavuligerus was subjected to RT-PCR, transcripts of ceaS1, ceaS2, bls1, and bls2 were all observed (Fig. 3). When RNA from the ΔceaS1::apr mutant was analyzed, only ceaS2 and bls2 transcripts were observed, consistent with previous studies showing that ceaS-bls gene pairs are cotranscribed in polycistronic transcripts (17). However, when RNA from the ΔceaS2-IF mutant was analyzed, no ceaS2 transcripts were seen, as expected, but bls1 or bls2 transcripts were also not detected, and only a faint band from ceaS1 was observed. This indicated that transcription of ceaS1 and bls1 was severely impaired or absent in the ΔceaS2-IF mutant, and also that transcription of bls2 was blocked despite the supposedly nonpolar character of the mutation. All RNA samples showed similar transcript levels for hrdB, which served as a control for RNA quality and quantity (Fig. 3).
Fig 3.
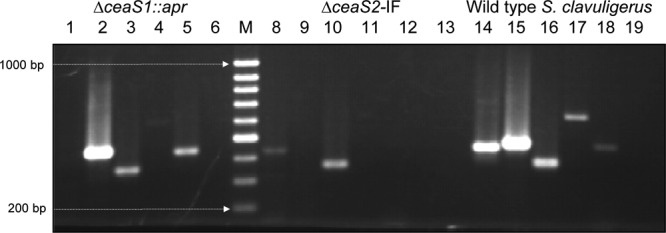
Assessment of transcript levels by RT-PCR. Total RNA was isolated from wild-type S. clavuligerus, ΔceaS2-IF, and ΔceaS1::apr mutants grown on liquid soy medium for 48 h. PCRs were carried out for 28 cycles, and transcripts examined were the following: ceaS1 (lanes 1, 8, and 14), ceaS2 (lanes 2, 9, and 15), hrdB (lanes 3, 10, and 16), bls1 (lanes 4, 11, and 17), and bls2 (lanes 5, 12, and 18). Lane M, GeneRuler 100-bp DNA ladder. Lanes 6, 13, and 19 represent negative controls for each RNA sample used, in which no reverse transcription was carried out.
Promoter activity analysis with egfp.
To confirm that ceaS1 transcription was affected in a ΔceaS2-IF mutant, promoter activity of ceaS1 was examined using EGFP as a reporter. pSET-ceaS1 carrying the promoter region of ceaS1 coupled to egfp was conjugated into wild-type and ΔceaS2-IF mutant strains of S. clavuligerus. The resulting reporter strains were grown in soy medium for 48 h, and samples were analyzed by confocal microscopy. Fluorescence was observed for wild-type S. clavuligerus carrying pSET-ceaS1 but not for the ΔceaS2-IF strain carrying the same reporter construct (Fig. 4).
Fig 4.

Detection of ceaS1 promoter activity in wild-type S. clavuligerus and ΔceaS2-IF mutant cultures by using EGFP as a reporter. (A) Mycelia of wild-type S. clavuligerus carrying the pSET-p-ceaS1 reporter construct. (B) Mycelia of ΔceaS2-IF carrying pSET-ceaS1. Both reporter strains were grown in liquid soy medium for 48 h and then analyzed by confocal microscopy. Fluorescence images are shown on the left. Differential interference contrast (DIC) images are shown on the right.
Production of CeaS1 and CeaS2 in ceaS2 mutants.
To gain further evidence that mutation of ceaS2 affects production of CeaS1, Western analyses were carried out using CFEs from the ceaS2 mutant strains. Recombinant CeaS1 and CeaS2 proteins were overproduced in E. coli and recovered from inclusion bodies for use as protein markers. Although only anti-CeaS2 antibody was available, Western analysis showed that the antibodies reacted with both CeaS1 and CeaS2 (Fig. 5). In addition, despite the similar molecular masses of CeaS1 and CeaS2 (59 and 61 kDA, respectively), the two proteins could be resolved from each other by prolonged SDS-PAGE (Fig. 5).
Fig 5.
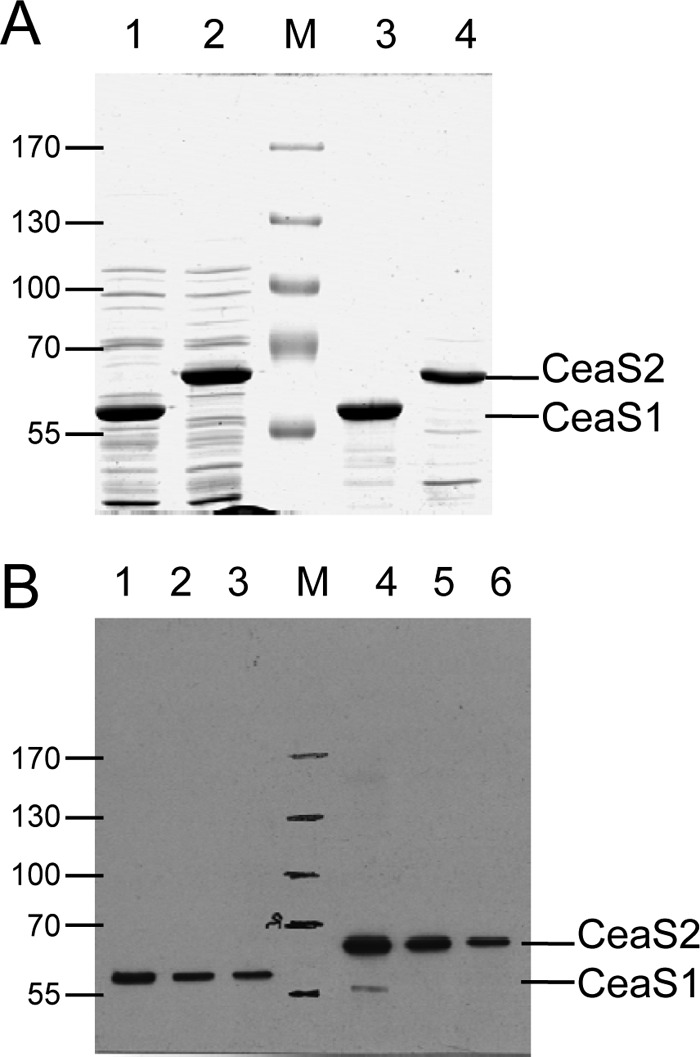
Western analysis of recombinant CeaS1 and CeaS2 proteins. Broken cell suspensions of E. coli expressing ceaS1 and ceaS2 were separated into soluble and insoluble fractions by centrifugation and then analyzed by electrophoresis on 8% SDS-PAGE gels. (A) Coomassie-stained gel, Lanes 1 and 3, soluble (8.5 μg of protein) and insoluble (2.5 μg of protein) CeaS1; lanes 2 and 4, soluble (8.5 μg of protein) and insoluble (2.5 μg of protein) CeaS2. (B) Western analysis of insoluble CeaS1 (lanes 1 to 3, 50, 25, and 12.5 ng of protein, respectively) and CeaS2 (lanes 4 to 6, 50, 25, and 12.5 ng of protein, respectively) samples used in panel A.
Previous studies showed that expression of ceaS1 and ceas2 is nutritionally regulated such that both ceaS1 and ceaS2 are expressed in soy medium but only ceaS2 is expressed in SA medium (16). Western analyses confirmed these observations by showing that CeaS2 protein was present in CFEs from wild-type S. clavuligerus grown on either soy or SA medium, while CeaS1 protein was almost undetectable in CFEs from SA-grown cultures (Fig. 6). Cross-reacting bands of unknown identity were observed between CeaS2 and CeaS1 and beneath Ceas1. These bands appeared to be degradation products of CeaS2, even though protease inhibitors were routinely included when preparing CFEs to prevent protein degradation.
Fig 6.
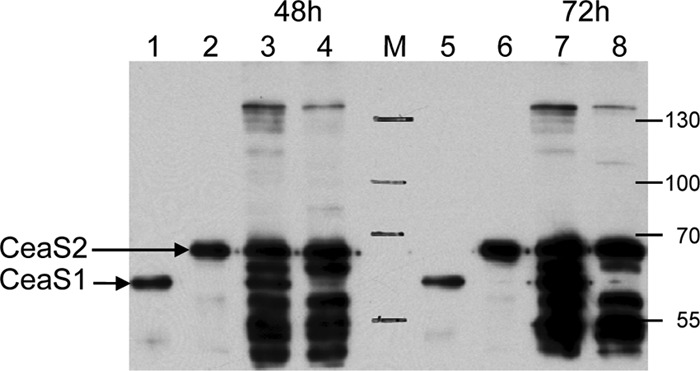
Western analysis of CeaS proteins from cell extracts of S. clavuligerus grown on soy or SA medium. Samples were separated by electrophoresis on 8% SDS-PAGE gels and transferred to PVDF membranes for analysis. Lanes 1 and 5, CeaS1 standard (25 ng of protein); lanes 2 and 6, Ceas2 standard (25 ng of protein); lanes 3 and 7, soy medium-grown S. clavuligerus CFE (200 ng of protein); lanes 4 and 8, SA medium-grown S. clavuligerus CFE (100 ng of protein); M, molecular weight marker proteins.
CFEs of wild-type S. clavuligerus and ceaS2::apr, ceaS2-FS, ΔceaS2::apr, ΔceaS2-IF, ΔceaS1::apr, and ΔceaS1::apr ceaS2-FS mutants were prepared from cultures grown in soy medium and subjected to Western analysis. Protein bands corresponding to CeaS1 and CeaS2 were seen in the CFE of wild-type S. clavuligerus, and protein bands due to CeaS2 were absent from all of the ceaS2 mutants (Fig. 7). However, protein bands representing CeaS1 were also absent from these mutants, indicating that neither CeaS1 nor CeaS2 protein was produced in the ceaS2 mutants. In contrast, CeaS2 was observed in CFE from the ΔceaS1::apr mutant. As expected, neither Ceas1 nor CeaS2 was seen in the ceaS1/ceaS2 double mutants. The cross-reacting bands seen in the wild type CFE are missing from the various ceaS2 mutants and from the ceaS1 ceaS2 double mutant, which supports the contention that they are degradation products that arise from CeaS2.
Fig 7.
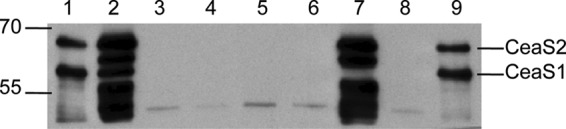
Western analysis of CeaS proteins from the wild type and mutant strains of S. clavuligerus. Cell extracts were isolated from soy medium-grown cultures of wild-type S. clavuligerus (lane 2), ceaS2::apr (lane 3), ceaS2-FS (lane 4), ΔceaS2::apr (lane 5), ΔceaS2 (lane 6), ΔceaS1::apr (lane7), and ΔceaS1::apr/ceaS2-FS (lane 8). Lanes 1 and 9 contained 25 ng each of CeaS1 and CeaS2 standard proteins. Cell extract proteins (200-ng amounts) were separated by electrophoresis on 8% SDS-PAGE gels and transferred to PVDF membranes for analysis.
Creation of an E57WceaS2 site-directed mutant.
All four types of ceaS2 mutants produced neither clavam metabolites nor CeaS1 protein. However, residual polarity affecting expression of downstream genes in the ceaS2 operon was still evident, even in the ΔceaS2-IF mutant. Furthermore, it was unclear whether the ceaS2 mutants failed to produce CeaS1 because of the physical disturbance at the ceaS2 locus caused by the mutations or if active CeaS2 protein was required for ceaS1 expression. To gain further insights, a fifth type of ceaS2 mutant was created in which the ceaS2 gene was modified by a site-directed mutation of 2 bp, with GA changed to TG, such that the glutamate codon at amino acid 57 became a tryptophan codon. Structural analyses of CeaS2 have suggested that the glutamate residue at position 57 (E57) is intimately involved in the catalytic function of the protein (27), and so the E57W mutant CeaS2 protein was expected to be defective in CeaS2 activity but otherwise unchanged from the wild type. A cloned copy of the E57W mutant version of ceaS2 was conjugated into a ceaS2::apr mutant, whereupon the apr-disrupted ceaS2 was replaced by E57WceaS2. Upon cultivation in soy medium, culture filtrates of the E57W mutants, like all of the previous ceas2 mutants, showed a complete inability to produce clavam metabolites (Fig. 8A).
Fig 8.

Analysis of clavam metabolite and CeaS1 and CeaS2 production in complemented ceaS2 mutants. (A) Culture supernatants from 96-h soy cultures were derivatized with imidazole to detect clavam metabolites and then analyzed by HPLC. AC, alanylclavam; C2C, clavam-2-carboxylate; 2HMC, 2-hydroxymethylclavam; CA, clavulanic acid. (B) Cell extra proteins (200-ng amounts) from wild-type, ΔceaS2 and E57WceaS2 mutant and complemented mutant cultures were separated by electrophoresis on 8% SDS-PAGE gels and transferred to PVDF membranes for analysis. Lane 1, ΔceaS2; lane 2, ΔceaS2-c5; lane 3, ΔceaS2-c11.6; lane 4, 25-ng amounts of CeaS1 and CeaS2 standard proteins; lane 5, wild type; lane 6, E57WceaS2; lane 7, E57WceaS2c5; lane 8, E57WceaS2-c11.6.
Complementation of S. clavuligerus ΔceaS2-IF and E57Wceas2 mutants with ceaS2.
Complementation experiments to restore clavam production to the ΔceaS2-IF and E57Wceas2 mutant strains were carried out using two different complementation constructs. Both constructs were based on the integrative vector, pSET152, and both employed the native ceaS2 promoter. In the first instance, an ∼5-kb Sau3A-BglII DNA fragment was used, which encompassed the ceaS2 gene extending upstream into pcbC and downstream into bls2 (Fig. 1B). This construct, pSET152-c5, was introduced into the ΔceaS2-IF and E57WceaS2 mutant strains by conjugation, and exconjugants were cultured in soy medium. Culture filtrates were analyzed by HPLC and showed small amounts of clavam metabolite production in the complemented ΔceaS2-IF strain and somewhat greater complementation in the E57WceaS2 strain, but production was still well below wild-type levels (Fig. 8A). A second complementation construct, pSET152-c11.6, was prepared in which an ∼11.6-kb EcoRI DNA fragment, extending from 183 bp upstream of ceaS2 to beyond the end of cad (Fig. 1B) and including all of the genes in the ceaS2 operon was inserted in pSET152. When this larger complementation construct was inserted into the ΔceaS2-IF and E57WceaS2 mutants, clavulanic acid and 5S clavam production were again partially restored, but only to levels similar to those seen with the 5-kb complementing construct (Fig. 8A).
When CFEs of the complemented strains ΔceaS2-c5, ΔceaS2-c11.6, E57WceaS2-c5, and E57WceaS2-c11.6 were examined by Western analysis, protein bands corresponding to CeaS2 were observed in all of them. The amounts of CeaS2 produced varied, and the level of Ceas2 protein did not necessarily reflect the level of clavam metabolites produced. However, and more significantly, the CeaS1 protein band was still absent in all of these complemented strains (Fig. 8B).
Clavam metabolite production by ceaS2 mutants on solid medium.
None of the ceaS2 mutants produced clavam metabolites, and none expressed ceaS1 even when they were complemented with an intact ceaS2 gene. Since no conditions were encountered under which ceaS1 was expressed and clavam metabolites produced in the absence of CeaS2, the question of whether CeaS1 is functionally equivalent to CeaS2 remained unanswered. Therefore, we looked for cultural conditions that would allow the ceaS2 mutant strains to express ceaS1 and produce clavam metabolites. We knew from previous studies that expression of ceaS1 and ceaS2 is subject to different nutritional regulation and that production of 5S clavam metabolites is very sensitive to changes in growth conditions, such as the culture medium-to-flask volume ratio and shaker speed (unpublished observations). Despite testing various growth conditions and media (including the GSPG medium originally reported by Perez-Redondo et al. as supporting clavulanic acid production by ceaS2 mutants [7]), no conditions supporting clavam metabolite production in liquid medium by ceaS2 mutants were identified. Surprisingly, however, if the ceaS2 mutants were grown in solid culture on the surface of soy medium-based agar plates, all of the mutants regained at least some ability to produce clavam metabolites (Fig. 9A). Furthermore, when CFEs were prepared from cell material growing on cellophane disks on the surface of the plates, Western analyses showed that CeaS1 was now clearly present (Fig. 9B), although at low levels compared to the wild type. This indicated that at least some transcription of ceaS1 takes place in the absence of CeaS2, providing that cells are grown on solid medium.
Fig 9.

Analysis of clavam metabolite and CeaS1 and CeaS2 production in wild-type and mutant strains of S. clavuligerus grown on solid soy medium. (A) Freeze-and-squeeze exudates from 9-day-old solid soy medium-grown cultures were derivatized with imidazole to detect clavam metabolites and then analyzed by HPLC. AC, alanylclavam; C2C, clavam-2-carboxylate; 2HMC, 2-hydroxymethylclavam; CA, clavulanic acid. (B) Cell extract proteins (200-ng amounts) from wild-type and various ceaS2 mutant cultures harvested after 9 days growth on solid soy medium were separated by electrophoresis on 8% SDS-PAGE gels and transferred to PVDF membranes for analysis. Lanes 1 and 8, CeaS1 and CeaS2 standards (25 ng of each protein); lane 2, wild-type S. clavuligerus; lane 3, ceaS2::apr; lane 4, ceaS2-FS; lane 3, ΔceaS2::apr; lane 6, ΔceaS2; lane 7, E57WceaS2.
DISCUSSION
S. clavuligerus ceaS2::apr and ceaS2-FS mutants typically showed a complete inability to produce clavam metabolites, which was unexpected given that they contain intact ceaS1 genes that should be able to compensate for the loss of CeaS2. Since ceaS2 is transcribed together with bls2, pah2, and cas2, a ΔceaS2-IF mutant was constructed to ensure that polar effects on downstream genes were not contributing to this defect (17). Like the other ceaS2 mutants (6, 10), the ΔceaS2-IF mutant produced no clavam metabolites, but RT-PCR analyses indicated that transcription of the downstream bls2 gene was still blocked despite the mutation being unmarked and in frame.
Although polarity persisted in ΔceaS2-IF mutants, this alone could not explain why clavam production was so severely affected in ceaS2 mutants. Even if disruption of ceaS2 silenced the entire ceaS2 operon, the redundant ceaS1 operon was still present and so a reduced level, rather than a total loss of clavam production, would be expected. As an alternative, we considered whether CeaS1 might not be functionally equivalent to CeaS2. However, RT-PCR, eGFP, and Western analyses all clearly showed that expression of ceaS1 was severely depressed or abolished in ceaS2 mutants, making the failure of these mutants to produce CeaS1 a more likely reason for the defect in clavam production, rather than any differences in activities between the two proteins.
Interestingly, when the E57W site-directed mutant version of ceaS2 was generated to produce full-length but enzymatically inactive CeaS2 protein, clavam metabolite and CeaS1 production were still completely blocked, suggesting that active Ceas2 protein must be required for ceaS1 expression. The inability to detect the E57W mutant form of CeaS2 by Western analysis further suggested either that the mutation in the protein rendered it so unstable that all remnants of it escaped detection by Western analysis or that active CeaS2 protein is also required for efficient ceaS2 expression.
Despite their locations on separate replicons within the cell (ceaS2 resides on the chromosome, while ceaS1 is located on a giant linear plasmid), cross-regulation must tie the expression of ceaS1 to ceaS2. In this regard, complementation studies showed that CeaS1 production was not restored in the complemented ceaS2 mutant strains, even though both CeaS2 and some clavam metabolite production were observed. The failure of complementation to restore expression of ceaS1 suggested that there might be some type of a positional effect, such that the location from which the ceaS2 gene is expressed, as well as the resulting gene product itself, are critical in order to support efficient ceaS1 expression. But whether this might involve a topological effect related to supercoiling, or possibly involve sense or antisense noncoding RNA, or the involvement of small effector molecules, is not clear at present. Alternatively, if mutation of ceaS2 at its native location on the chromosome somehow negatively impacts expression of genes further downstream from cad in the clavulanic acid gene cluster, this could account for some of the complementation results.
Expression of developmental genes associated with fruiting body formation in Myxococcus xanthus requires activator proteins that bind to DNA elements similar to enhancer sequences from eukaryotes to activate transcription from their promoters (28, 29). While analysis of genome sequence information has not indicated the presence of such enhancer binding-type activator proteins in Streptomyces spp., perhaps active CeaS2 plays such a role, but how these effects would be transmitted to ceaS1, residing as it does on a separate replicon within the cell, remains unclear.
In previous studies, Kyung et al. showed that expression of genes encoding both enzymes and transcriptional regulators involved in antibiotic biosynthesis were spatially and temporally regulated in S. clavuligerus (30). Prokaryotes are typified by a lack of subcellular compartments, but if the enzymes of clavulanic acid and 5S clavam biosynthesis associate into multienzyme complexes as they are produced and reside in separate cellular locations, as has been described for bacillaene production in Bacillus subtilis (31), this may provide an opportunity for higher levels of regulation and organization more similar to those seen in eukaryotes.
Finally, the ability to obtain clavam metabolite production when ceaS2 mutants are grown on solid medium demonstrated that Ceas1 protein is functional in its own right and can support clavam metabolite production. While the specific production of Ceas1 in ceaS2 mutants was low compared to the production levels for CeaS1 and CeaS2 in the wild type on solid medium, ceaS1 was nonetheless expressed and clavams were produced in the absence of any CeaS2 protein. This also implied that there must be two separate pathways to achieve ceaS1 expression, a CeaS2-dependent pathway that is functional during growth in both liquid and solid medium and a Ceas2-independent pathway seen only during growth on solid medium. This Ceas2-independent pathway may respond to signals reflecting the physical environment, such as osmolarity, concentrations of volatile metabolites, or oxygen tension, in the same way that such signals may affect other processes, such as sporulation of Streptomyces spp. on solid versus liquid media. Previous studies have shown that an atypical two-component regulatory system specifically controls the production of the 5S clavam metabolites, and so growth conditions may influence ceaS1 expression through interaction with this system (12).
Given the complexity of the process, a model that attempts to incorporate all of these possible explanations to account for the cross-regulation of ceaS1 expression by CeaS2 would be overly speculative at this stage and must await clarification through further studies. Since sequencing of the S. clavuligerus genome sequence has recently been completed (9, 32), microarray-type analyses comparing transcription patterns of a ΔceaS2-IF mutant grown in liquid and solid medium may help to identify additional genes that are involved and provide greater insights into this complex regulatory system.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Natural Sciences and Engineering Research Council of Canada (Discovery grant 2824-06 to S.E.J.), by a graduate student scholarship from the Alberta Heritage Foundation for Medical Research to K.T., and by the Department of Biological Sciences, University of Alberta.
We have no conflicts of interest to declare.
Footnotes
Published ahead of print 26 October 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02600-12.
REFERENCES
- 1. Demain AL. 2000. Small bugs, big business: the economic power of the microbe. Biotechnol. Adv. 18:499–514 [DOI] [PubMed] [Google Scholar]
- 2. Egan LA, Busby RW, IwataReuyl D, Townsend CA. 1997. Probable role of clavaminic acid as the terminal intermediate in the common pathway to clavulanic acid and the antipodal clavam metabolites. J. Am. Chem. Soc. 119:2348–2355 [Google Scholar]
- 3. Jensen SE, Paradkar AS. 1999. Biosynthesis and molecular genetics of clavulanic acid. Antonie Van Leeuwenhoek 75:125–133 [DOI] [PubMed] [Google Scholar]
- 4. Marsh EN, Chang MD, Townsend CA. 1992. Two isozymes of clavaminate synthase central to clavulanic acid formation: cloning and sequencing of both genes from Streptomyces clavuligerus. Biochemistry 31:12648–12657 [DOI] [PubMed] [Google Scholar]
- 5. Salowe SP, Marsh EN, Townsend CA. 1990. Purification and characterization of clavaminate synthase from Streptomyces clavuligerus: an unusual oxidative enzyme in natural product biosynthesis. Biochemistry 29:6499–6508 [DOI] [PubMed] [Google Scholar]
- 6. Jensen SE, Elder KJ, Aidoo KA, Paradkar AS. 2000. Enzymes catalyzing the early steps of clavulanic acid biosynthesis are encoded by two sets of paralogous genes in Streptomyces clavuligerus. Antimicrob. Agents Chemother. 44:720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perez-Redondo R, Rodriguez-Garcia A, Martin JF, Liras P. 1999. Deletion of the pyc gene blocks clavulanic acid biosynthesis except in glycerol-containing medium: evidence for two different genes in formation of the C3 unit. J. Bacteriol. 181:6922–6928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jensen SE, Wong A, Griffin A, Barton B. 2004. Streptomyces clavuligerus has a second copy of the proclavaminate amidinohydrolase gene. Antimicrob. Agents Chemother. 48:514–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Medema MH, Trefzer A, Kovalchuk A, van den Berg M, Muller U, Heijne W, Wu L, Alam MT, Ronning CM, Nierman WC, Bovenberg RA, Breitling R, Takano E. 2010. The sequence of a 1.8-Mb bacterial linear plasmid reveals a rich evolutionary reservoir of secondary metabolic pathways. Genome Biol. Evol. 2:212–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tahlan K, Park HU, Wong A, Beatty PH, Jensen SE. 2004. Two sets of paralogous genes encode the enzymes involved in the early stages of clavulanic acid and clavam metabolite biosynthesis in Streptomyces clavuligerus. Antimicrob. Agents Chemother. 48:930–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alexander DC, Jensen SE. 1998. Investigation of the Streptomyces clavuligerus cephamycin C gene cluster and its regulation by the CcaR protein. J. Bacteriol. 180:4068–4079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kwong T, Zelyas NJ, Cai H, Tahlan K, Wong A, Jensen SE. 2012. 5S clavam biosynthesis is controlled by an atypical two-component regulatory system in Streptomyces clavuligerus. Antimicrob. Agents Chemother. 56:4845–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paradkar AS, Aidoo KA, Jensen SE. 1998. A pathway-specific transcriptional activator regulates late steps of clavulanic acid biosynthesis in Streptomyces clavuligerus. Mol. Microbiol. 27:831–843 [DOI] [PubMed] [Google Scholar]
- 14. Perez-Llarena FJ, Liras P, Rodriguez-Garcia A, Martin JF. 1997. A regulatory gene (ccaR) required for cephamycin and clavulanic acid production in Streptomyces clavuligerus: amplification results in overproduction of both β-lactam compounds. J. Bacteriol. 179:2053–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perez-Redondo R, Rodriguez-Garcia A, Martin JF, Liras P. 1998. The claR gene of Streptomyces clavuligerus, encoding a LysR-type regulatory protein controlling clavulanic acid biosynthesis, is linked to the clavulanate-9-aldehyde reductase (car) gene. Gene 211:311–321 [DOI] [PubMed] [Google Scholar]
- 16. Tahlan K, Anders C, Jensen SE. 2004. The paralogous pairs of genes involved in clavulanic acid and clavam metabolite biosynthesis are differently regulated in Streptomyces clavuligerus. J. Bacteriol. 186:6286–6297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Paradkar AS, Jensen SE. 1995. Functional analysis of the gene encoding the clavaminate synthase 2 isoenzyme involved in clavulanic acid biosynthesis in Streptomyces clavuligerus. J. Bacteriol. 177:1307–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vieira J, Messing J. 1987. Production of single stranded plasmid DNA. Methods Enzymol. 153:3–11 [DOI] [PubMed] [Google Scholar]
- 19. Wehmeier UF. 1995. New multifunctional Escherichia coli-Streptomyces shuttle vectors allowing blue-white screening on XGal plates. Gene 165:149–150 [DOI] [PubMed] [Google Scholar]
- 20. Zelyas N, Tahlan K, Jensen SE. 2009. Use of the native flp gene to generate in-frame unmarked mutations in Streptomyces spp. Gene 443:48–54 [DOI] [PubMed] [Google Scholar]
- 21. Jensen SE, Paradkar AS, Mosher RH, Anders C, Beatty PH, Brumlik MJ, Griffin A, Barton B. 2004. Five additional genes are involved in clavulanic acid biosynthesis in Streptomyces clavuligerus. Antimicrob. Agents Chemother. 48:192–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. John Innes Foundation, Norwich, England [Google Scholar]
- 23. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 24. Gust B, Challis GL, Fowler K, Kieser T, Chater KF. 2003. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U. S. A. 100:1541–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harlow E, Lane D. 1988. Antibodies: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 26. Paradkar AS, Aidoo KA, Wong A, Jensen SE. 1996. Molecular analysis of a β-lactam resistance gene encoded within the cephamycin gene cluster of Streptomyces clavuligerus. J. Bacteriol. 178:6266–6274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Caines MEC, Elkins JM, Hewitson KS, Schofield CJ. 2004. Crystal structure and mechanistic implications of N2-(2-carboxyethyl)arginine synthase, the first enzyme in the clavulanic acid biosynthesis pathway. J. Biol. Chem. 279:5685–5692 [DOI] [PubMed] [Google Scholar]
- 28. Buck M, Gallegos MT, Studholme DJ, Guo Y, Gralla JD. 2000. The bacterial enhancer-dependent σ54 (σN) transcription factor. J. Bacteriol. 182:4129–4136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jelsbak L, Givskov M, Kaiser D. 2005. Enhancer-binding proteins with a forkhead-associated domain and the σ54 regulon in Myxococcus xanthus fruiting body development. Proc. Natl. Acad. Sci. U. S. A. 102:3010–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kyung YS, Hu W, Sherman DS. 2001. Analysis of temporal and spatial expression of the CcaR regulatory element in the cephamycin C biosynthetic pathway using green fluorescent protein. Mol. Microbiol. 40:530–541 [DOI] [PubMed] [Google Scholar]
- 31. Straight PD, Fischbach MA, Walsh CT, Rudner DZ, Kolter R. 2007. A singular enzymatic megacomplex from Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 104:305–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Song JY, Jeong H, Yu DS, Fischbach MA, Park HS, Kim JJ, Seo JS, Jensen SE, Oh TK, Lee KJ, Kim JF. 2010. Draft genome sequence of Streptomyces clavuligerus NRRL 3585, a producer of diverse secondary metabolites. J. Bacteriol. 192:6317–6318 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.