

Abstract
In many bacteria, the second messenger cyclic AMP (cAMP) interacts with the transcription factor cAMP receptor protein (CRP), forming active cAMP-CRP complexes that can control a multitude of cellular activities, including expanded carbon source utilization, stress response pathways, and virulence. Here, we assessed the role of cAMP-CRP as a regulator of stress resistance and virulence in uropathogenic Escherichia coli (UPEC), the principal cause of urinary tract infections worldwide. Deletion of genes encoding either CRP or CyaA, the enzyme responsible for cAMP synthesis, attenuates the ability of UPEC to colonize the bladder in a mouse infection model, dependent on intact innate host defenses. UPEC mutants lacking cAMP-CRP grow normally in the presence of glucose but are unable to utilize alternate carbon sources like amino acids, the primary nutrients available to UPEC within the urinary tract. Relative to the wild-type UPEC isolate, the cyaA and crp deletion mutants are sensitive to nitrosative stress and the superoxide generator methyl viologen but remarkably resistant to hydrogen peroxide (H2O2) and acid stress. In the mutant strains, H2O2 resistance correlates with elevated catalase activity attributable in part to enhanced translation of the alternate sigma factor RpoS. Acid resistance was promoted by both RpoS-independent and RpoS-dependent mechanisms, including expression of the RpoS-regulated DNA-binding ferritin-like protein Dps. We conclude that balanced input from many cAMP-CRP-responsive elements, including RpoS, is critical to the ability of UPEC to handle the nutrient limitations and severe environmental stresses present within the mammalian urinary tract.
INTRODUCTION
Under homeostatic conditions, the mammalian urinary tract is maintained as a sterile environment through the production of antimicrobial peptides and other toxic compounds, the bulk flow of urine, innate immune cell surveillance mechanisms, and nutrient limitations (1–5). However, select microbial pathogens are capable of colonizing and infecting this normally inhospitable niche. Uropathogenic Escherichia coli (UPEC) is the major cause of urinary tract infections (UTI) worldwide, affecting millions and requiring billions of dollars for diagnosis and treatment annually (6). To overcome host defenses and effectively colonize the urinary tract, UPEC employs a variety of virulence factors and stress resistance mechanisms, including adhesive and motility organelles that mediate attachment to and invasion of host cells, toxins that disarm innate immune responses, and multiple iron-scavenging proteins (1, 7–9). The ability to sense and prioritize the use of limited carbon sources within the nutrient-poor environment of the urinary tract is also likely critical to the success of UPEC, but our understanding of the impact that bacterial metabolic pathways have on the establishment and progression of a UTI is incomplete.
Within the urinary tract, UPEC relies largely on the catabolism of small peptides and amino acids for survival and growth (4). UPEC strains that are defective in peptide and carbohydrate transport systems, the tricarboxylic acid (TCA) cycle, and gluconeogenesis are unable to effectively colonize the urinary tract (10, 11). Of note, many UPEC isolates are able to utilize the gluconeogenic amino acid d-serine, which is typically present in urine (12, 13). d-Serine not only provides substrates for the TCA cycle and gluconeogenesis but also serves as an environmental cue that can regulate UPEC virulence. Interplay between bacterial metabolism and virulence is also evident by analysis of the QseBC two-component regulatory system, which was recently shown to modulate carbon flux through key metabolic pathways as well as the expression of UPEC-associated virulence factors like type 1 pili and flagella (14, 15). It is likely that numerous other systems help coordinate the expression of virulence and stress resistance factors with the ability of UPEC to sense and respond to the various carbon sources encountered within the host.
Although E. coli strains are generalists with the capacity to metabolize myriad metabolites, they preferentially utilize glucose as a primary carbon source. Transitioning into glucose-limiting conditions triggers the activation of the adenylate cyclase CyaA, producing high levels of the second messenger molecule cyclic AMP (cAMP) (16). Binding of cAMP to the transcription factor cAMP receptor protein (CRP) forms the active cAMP-CRP complex, which directly regulates expression of genes necessary for utilization of alternative carbon sources. However, not all genes that are regulated by cAMP-CRP function in bacterial metabolism (17, 18). Disruption of cAMP signaling within the prominent pathogens Vibrio cholerae, Mycobacterium tuberculosis, Salmonella enterica serovar Typhimurium, and Pseudomonas aeruginosa attenuates virulence through the misregulation of key virulence genes (18–22). Within pathogenic subsets of E. coli, the cAMP-CRP complex has been shown to modulate the expression of type 1 pili, major facilitators of bacterial colonization of the bladder mucosa (23). However, the cumulative effects of cAMP-CRP on the virulence potential of UPEC within the urinary tract remain to be elucidated.
Here, we report that the deletion of genes encoding either CyaA or CRP within the UPEC reference strain UTI89 does not affect growth in the presence of glucose but mutants with these deletions are unable to utilize amino acids as the sole carbon source. Furthermore, these mutants are significantly attenuated in the ability to colonize the bladders of mice, dependent upon the presence of intact innate host defenses. In broth culture assays, the cyaA and crp mutants are both sensitive to reactive nitrogen species and superoxide radicals generated by methyl viologen but highly resistant to hydrogen peroxide (H2O2) and acid stress. Resistance of the mutants to H2O2 and acid stress is in part attributable to increased translation of the alternate sigma factor RpoS (σS) and RpoS-regulated gene products that include catalases and the DNA-binding, iron storage protein Dps. In total, these data indicate that balanced input from cAMP-CRP is critical to the ability of UPEC to catabolize amino acids and appropriately handle harsh environmental stresses, characteristics that are pertinent to bacterial fitness and survival within the urinary tract.
MATERIALS AND METHODS
Bacterial strains and plasmids.
All bacterial strains and plasmids are listed in Table 1. The human cystitis isolate UTI89 has been described previously (24, 25). Expression constructs were made using standard molecular biology techniques with the plasmid pRR48 (26). Where indicated, gene expression from the Ptac promoter in the pRR48 backbone was induced by addition of 0.5 or 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Primers used to construct all plasmids are indicated in Table 2, along with primers used to verify each clone by sequencing. Antibiotics (50 μg/ml kanamycin, 20 μg/ml chloramphenicol, or 100 μg/ml ampicillin) were added to plates and growth media to select for mutants and to maintain plasmids when necessary. Targeted gene knockouts were generated in UTI89 using the lambda Red-mediated linear transformation system (27, 28). Briefly, an antibiotic resistance cassette was amplified from pKD3, pKD4, or the template strain TT23691 with 40-bp overhangs specific to sites at the 5′ and 3′ ends of each target gene. PCR products were introduced via electroporation into UTI89 carrying pKM208, which encodes an IPTG-inducible lambda Red recombinase. Knockouts were confirmed by PCR using primers listed in Table 2.
Table 1.
Bacterial strains and plasmids
| Strain or plasmid | Description | Source |
|---|---|---|
| Strains | ||
| UTI89 | UPEC strain (cystitis isolate, O18:K1:H7) | 25 |
| UTI89ΔcyaA | UTI89cyaA::Clmr (pKD3) | This study |
| UTI89Δcrp | UTI89crp::Clmr (pKD3) | This study |
| UTI89Δdps | UTI89dps::Kanr (TT23691) | This study |
| UTI89ΔfimH | UTI89fimH::Clmr (pKD3) | 83 |
| UTI89ΔotsBA | UTI89otsBA::Kanr (pKD4) | This study |
| UTI89ΔrpoS | UTI89rpoS::Kanr (TT23691) | This study |
| UTI89ΔcyaAΔotsBA | UTI89cyaA::Clmr (pKD3) otsBA::Kanr (pDK4) | This study |
| UTI89ΔcrpΔrpoS | UTI89crp::Clmr (pKD3) rpoS::Kanr (TT23691) | This study |
| UTI89ΔcrpΔdps | UTI89crp::Clmr (pKD3) dps::Kanr (TT23691) | This study |
| TT23691 | Strain with Kanr cassette flanked by universal primer sites | 84 |
| Plasmids | ||
| pRR48 | Ampr cloning plasmid containing an IPTG-inducible Ptac promoter upstream of the MCS | 26 |
| pKM208 | Ampr plasmid; encodes IPTG-inducible lambda red recombinase | 27 |
| pKD3 | Template plasmid for gene disruption; contains FRTa-flanked Clmr cassette | 28 |
| pKD4 | Template plasmid for gene disruption; contains FRT-flanked Kanr cassette | 28 |
| pcrp | Ampr plasmid; crp (from UTI89) cloned into PstI, KpnI sites of pRR48 | This study |
| pcyaA | Ampr plasmid; cyaA (from UTI89) cloned into PstI, KpnI sites of pRR48 | This study |
| pdps | Ampr plasmid; IPTG-inducible expression of E. coli Dps | 55 |
| prpoS | Ampr plasmid; rpoS (from UTI89) cloned into PstI, HindIII sites of pRR48 | This study |
FRT, FLP recombination target.
Table 2.
Primer sequences
| Primer | Sequence |
|---|---|
| crp KOa | |
| Forward | GCGCATGGTGCTTGGCAAACCGCAAACAGACCCGACTCTCTGTGTAGGCTGGAGCTGCTTCG |
| Reverse | CGCGCTACCAGGTAACGCGCCACTCTGACGGGATTAACGACATATGAATATCCTCCTTAG |
| crp KO confirmation | |
| Forward | GTATGCAAAGGACGCCACAT |
| Reverse | TTCGCCAAGCATTAACCCAA |
| cyaA KO | |
| Forward | GCGGAATCACAGTCATGACGGGTAGCAAATCAGGCGATACTGTGTAGGCTGGAGCTGCTTCG |
| Reverse | TACTGCTGCAACAGCGGCGCGTCATGCTCCTGATTGGCAGCATATGAATATCCTCCTTAG |
| cyaA KO confirmation | |
| Forward | AACCAGGCGCGAAAAGTGGT |
| Reverse | CTGAAAGGCGACGAGTGGAT |
| otsBA KO | |
| Forward | ATGTCTGTAAAGCGCGTTCTGCGCAACACAATAAGAAATGTGTAGGCTGGAGCTGCTTCG |
| Reverse | CTACGCAAGCTTAGGAAAGGTAGCAACTTTATCGCGCTGCCATATGAATATCCTCCTTAG |
| otsBA KO confirmation | |
| Forward | AGCGAAACGCACTGTCTGAT |
| Reverse | TTGCCTACGGTGAGTTAAGC |
| dps KO | |
| Forward | TTATTCGATGTTAGACTCGATAAACCACAGGAATTTATCCAGGTCGCGAGCACCAAACACCCCCCAAAACC |
| Reverse | GTGATAGGAACAGCCAGAATAGCGGAACACATAGCTGGTGCTATACTTAGCACACAACCACACCACACCAC |
| dps KO confirmation | |
| Forward | GATAGCAGATGGATGCACTA |
| Reverse | TGACAGTACGCAAAGAGAGC |
| rpoS KO | |
| Forward | CCAGCCTCGCTTGAGACTGGCCTTTCTGACAGATGCTTACCACCAAACACCCCCCAAAACC |
| Reverse | TGCCGCAGCGATAAATCGGCGGAACCAGGCTTTTGCTTGACACACAACCACACCACACCAC |
| rpoS KO confirmation | |
| Forward | AATGATGATTGCCGAATGTGACGCTG |
| Reverse | GCATTGTGTCGTTATGGGCGTAGG |
| pcrp | |
| Forward | CCCCC CTGCAG ATGGTGCTTGGCAAACCGCA |
| Reverse | CCCCC GGTACC TTAACGAGTGCCGTAAACGA |
| pcyaA | |
| Forward | CCCCC CTGCAG TTGTACCTCTATATTGAGAC |
| Reverse | CCCCC GGTACC TCACGAAAAATACTGCTGCA |
| prpoS | |
| Forward | CATTC CTGCAG ATGTTCCGTCAAGGGATCA |
| Reverse | AGTGC AAGCTT TTATTCGCGGAACAGCGCT |
| pRR48 sequencing primer | |
| Forward | CTGCTGAAGAGTACTTTGG |
| Reverse | CCAAAGCTGAAGACATCCAG |
KO, knockout.
Mouse infections.
Seven- to 9-week-old female CBA/J or C3H/HeJ mice (Jackson Laboratory) were used in accordance with IACUC-approved protocols as previously described (29). Mice were anesthetized using isoflurane inhalation and inoculated via transurethral catheterization with 50 μl of a bacterial suspension containing approximately 1 × 107 bacteria. For these noncompetitive infection assays, UTI89 and isogenic knockout mutants were grown statically for 24 h in Luria-Bertani (LB) broth, pelleted by spinning at 10,000 × g for 8 min, and resuspended in phosphate-buffered saline (PBS) prior to inoculation. Bladders were recovered at 6 h or 3 days postinoculation, weighed, and homogenized in 1 ml PBS containing 0.025% Triton X-100. Homogenates were serially diluted and plated on LB agar plates to determine the number of bacteria per gram of tissue. Mouse experiments were repeated at least twice, and the total combined data from at least 11 animals are presented.
Growth assays.
Bacteria were grown from frozen stocks at 37°C with shaking overnight in 5 ml of LB broth, 100 mM morpholineethanesulfonic acid (MES)-buffered LB broth (MES-LB broth; pH 5.0), or modified M9 minimal medium (6 g/liter Na2HPO4, 3 g/liter KH2PO4, 1 g/liter NH4Cl, 0.5 g/liter NaCl, 1 mM MgSO4, 0.1 mM CaCl2, 0.1% glucose, 0.0025% nicotinic acid, 16.5 μg/ml thiamine, and 0.2% casein amino acids). Bacteria were then diluted 1:100 into the appropriate corresponding medium with or without additives as indicated. Growth of quadruplicate 200-μl samples in shaking 100-well honeycomb plates was assessed at 37°C using a Bioscreen C instrument (Growth Curves USA). Stocks of methyl viologen (MV) (also known as paraquat), acidified sodium nitrite (ASN), trehalose, and H2O2 were prepared fresh prior to addition to LB or MES-LB broth cultures. Where indicated, IPTG was added to cultures to induce high-level expression of recombinant proteins from pdps or prpoS. MacConkey agar and other reagents used in these assays were obtained from Sigma-Aldrich.
Western blots.
UTI89, UTI89ΔcyaA, and UTI89Δcrp were diluted 1:50 from overnight cultures into fresh LB broth and grown with shaking at 37°C until an optical density at 600 nm (OD600) of 0.4 was reached. One milliliter of each culture was pelleted, resuspended in 200 μl B-PER lysis reagent (Thermo Scientific) containing 1 mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail (Roche), and incubated for 15 min at room temperature. Protein concentrations within the lysates were determined using the BCA reagent system (Pierce), and equivalent protein amounts were resolved by SDS-PAGE and transferred to an Immobilon PVDF-FL membrane (Millipore). Blots were probed using anti-RpoS (Neoclone) and anti-E. coli antibodies (Biodesign International) and visualized using enhanced chemiluminescence as previously described (30).
pH stress resistance assays.
Bacterial strains from overnight cultures were diluted 1:100 in fresh LB broth and grown with shaking at 37°C for 3 h. LB broth containing 100 μg/ml ampicillin and 0.5 mM IPTG was used for strains carrying plasmids pRR48, pdps, or prpoS. Strains were subjected to acid stress (pH 3.5) by the addition of concentrated HCl for 30 min. Bacteria in 1 ml of culture were pelleted at 14,000 × g for 5 min and washed in PBS. Surviving bacteria were enumerated by plating serial dilutions on LB agar.
Catalase assays.
Overnight bacterial cultures were diluted 1:100 in LB broth and grown with shaking at 37°C to an OD600 of 1.0. UTI89/prpoS was grown in broth containing 1 mM IPTG. Bacteria in 1 ml of culture were pelleted, resuspended in 200 μl B-PER lysis reagent (Thermo Scientific), and incubated at room temperature for 15 min. Catalase activity present in the lysates was determined using a Fluoro Catalase kit (Cell Technology) and a Synergy HT multidetection microplate reader (BioTek Instruments, Inc.) according to instructions from the manufacturer.
Trehalose analysis.
Chemicals and reagents were purchased from Sigma-Aldrich, except for MSTFA [N-methyl-N-(trimethylsilyl) trifluoroacetamide], which was purchased from Thermo Scientific, and methoxyamine hydrochloride, which was purchased from MP Biomedicals. Bacterial cultures were grown in modified M9 medium to an OD600 of 1.0, pelleted by centrifugation, and frozen. Pellets were suspended in 5 ml of boiling 75% ethanol (EtOH) (aqueous), vortexed, and then incubated at 90°C for 5 min. Cell debris was removed by centrifugation at 5,000 × g for 3 min. Supernatants were transferred to new tubes and dried in vacuo. Gas chromatography-mass spectrometry (GC-MS) analysis was performed using a Waters GCT Premier mass spectrometer fitted with an Agilent 6890 gas chromatograph and a Gerstel MPS2 autosampler. Dried samples were suspended in 40 μl of a pyridine solution containing 40 mg/ml O-methoxylamine hydrochloride and incubated for 1 h at 30°C. Twenty microliters of each sample was transferred to an autosampler vial and incubated with MSTFA for 30 min at 37°C with shaking. One microliter of sample was injected into the inlet at a 75:1 split ratio. The injector temperature was held at 250°C. The gas chromatograph was obtained using an initial temperature of 95°C for 1 min followed by a 40°C/min ramp up to 110°C, with a hold time of 2 min. This was followed by a second 5°C/min ramp up to 250°C and then a third ramp up to 350°C, with a final hold time of 3 min. A 30-m Restek Rxi-5 MS column with a 5-m-long guard column was employed for analysis. Data were collected using MassLynx 4.1 software. To determine trehalose concentrations specifically, an external calibration curve was developed by performing a 2-fold dilution series starting at 10 μg/μl trehalose in pyridine. This series was analyzed to determine the linear range of analysis, the upper and lower limits of detection and quantitation, and the fragment ion to be utilized for analysis. For quantification, the fragment ion of 331 m/z was monitored and the linear range for analysis was determined to be from 100 to 1,000 μg/μl.
Metal stress assay.
UTI89/pRR48, UTI89Δdps/pRR48, and UTI89Δdps/pdps were grown with shaking overnight at 37°C in LB broth containing 100 μg/ml ampicillin. Nine hundred microliters of each overnight culture was added to a sterile microcentrifuge tube, followed by 100 μl of 0.5 M CuSO4. Tubes were then incubated for 15 min at room temperature, and surviving bacteria were pelleted at 14,000 × g for 5 min, washed in PBS, and enumerated by plating serial dilutions on LB agar.
Statistics.
Results from in vivo mouse assays were analyzed by Mann-Whitney two-tailed t tests. Results from the catalase and survival assays were analyzed using two-tailed unpaired t tests. Data analysis was performed using Prism 5.0c (GraphPad Software, Inc.). P values of less than 0.05 are considered significant.
RESULTS
cAMP-CRP is necessary for lactose and amino acid catabolism by UPEC.
Carbon catabolite repression—the preferential use of a carbon source like glucose instead of other secondary carbon sources—is regulated by the generation of cAMP-CRP (16). In the classic example, decreased glucose levels result in enhanced production of cAMP-CRP, which in turn activates the expression of genes needed to catabolize alternate carbon sources like lactose. Using MacConkey agar plates, we established that wild-type UTI89 could consume lactose in the absence of glucose, creating lactic acid and causing the pH indicator neutral red present in the agar to produce a pink color (Fig. 1A). In contrast, the isogenic cyaA and crp deletion mutants UTI89ΔcyaA and UTI89Δcrp could not utilize lactose and instead had to ferment available peptone, producing basic ammonia and turning the pH indicator yellow. The ability of the ΔcyaA and Δcrp mutants to use lactose in these assays was restored by complementation with plasmids pcyaA and pcrp, respectively (data not shown).
Fig 1.

Impaired use of alternative carbon sources by UTI89ΔcyaA and UTI89Δcrp. (A) MacConkey agar plate streaked with UTI89, UTI89ΔcyaA, and UTI89Δcrp. Growth of UTI89, UTI89ΔcyaA, UTI89Δcrp, and complemented mutants in modified M9 medium (B) and modified M9 medium lacking glucose (C). Mutant strains were complemented by uninduced expression of cyaA or crp from a Ptac promoter. Strains transformed with the empty vector pRR48 served as controls. Each curve reflects the means of results from a single experiment and is representative of at least three independent experiments performed in quadruplicate.
Within the urinary tract, UPEC cells are mostly dependent on the catabolism of small peptides and amino acids (10, 12, 13). In modified M9 media containing both glucose and amino acids, wild-type UTI89, UTI89ΔcyaA, and UTI89Δcrp grew with similar kinetics (Fig. 1B). However, in modified M9 media containing only amino acids as a carbon source, the ΔcyaA and Δcrp mutants failed to grow unless appropriately complemented with plasmid pcyaA or pcrp (Fig. 1C). The inability of UTI89ΔcyaA and UTI89Δcrp to catabolize amino acids and other secondary carbon sources such as lactose may affect the fitness of these mutants within the urinary tract.
CRP and CyaA promote UPEC colonization of the bladder.
To assess the contribution of cAMP-CRP to UPEC pathogenicity, wild-type UTI89, UTI89ΔcyaA, and UTI89Δcrp were individually inoculated via transurethral catheterization into adult female CBA/J mice. In comparison with wild-type UTI89, significantly reduced numbers of both the ΔcyaA and Δcrp mutants were recovered from bladders at 6 h and 3 days postinoculation (Fig. 2A and B). Interestingly, differences between wild-type UTI89 and the ΔcyaA and Δcrp mutants were blunted in C3H/HeJ mice (Fig. 2C). Due to defects in Toll-like receptor 4 (TLR4) and possibly other host factors, C3H/HeJ mice have attenuated inflammatory responses and are consequently hypersensitive to UTIs (31–36). In total, these data indicate that cAMP-CRP is critical to the fitness of UPEC within the urinary tract of immunocompetent animals, probably due to regulatory effects of cAMP-CRP on factors that control bacterial resistance to stresses generated by stimulation of host inflammatory cascades. These results prompted us to investigate further potential interplay between cAMP-CRP and stress response mechanisms in UPEC.
Fig 2.

UPEC requires functional cAMP-CRP for virulence in the murine urinary tract. Adult female CBA/J mice (A, B) or C3H/HeJ mice (C) were infected via catheterization with 1 × 107 CFU of wild-type UTI89 or isogenic mutants lacking cyaA or crp. Graphs show bacterial titers present in the bladder at 6 h (A) and 3 days (B, C) postinoculation. Bars indicate median values for each group; n ≥ 11 mice. P values were determined using Mann-Whitney U tests.
UTI89ΔcyaA and UTI89Δcrp are sensitive to nitrosative stress and methyl viologen but resistant to H2O2.
Key stresses encountered by UPEC during the course of a UTI include damage elicited by reactive oxygen and nitrogen radicals that can be produced by both host and bacterial cells (37–42). To test the sensitivity of UTI89ΔcyaA and UTI89Δcrp to nitrosative and oxidative stresses, we utilized acidified sodium nitrite (ASN) and the superoxide generator methyl viologen (MV), respectively (Fig. 3). In these assays, the addition of sodium nitrite to MES-LB broth (pH 5.0) to create ASN results in the production of nitrous acid, NO, and other reactive nitrogen intermediates (43). In MES-LB broth, the ΔcyaA and Δcrp mutants grew like the wild-type strain, but in the presence of ASN growth of the mutants was markedly attenuated (Fig. 3A and B). Likewise, the addition of MV to LB broth severely impeded growth of both UTI89ΔcyaA and UTI89Δcrp (Fig. 3D and E). Of note, the mutant cultures did not attain the same optical density as the wild-type pathogen when grown with shaking in nutrient-rich LB broth, but in stationary cultures the mutant and wild-type strains reached equivalent bacterial titers, consistent with results using laboratory K-12 strains (reference 44 and data not shown). While MV impaired growth of UTI89ΔcyaA and UTI89Δcrp, the same mutants grew remarkably better than the wild-type strain in the presence of H2O2 (Fig. 3C). This effect was observed in both MES-LB and regular LB broth cultures, though results were more variable in the latter. Complementation of UTI89ΔcyaA and UTI89Δcrp with plasmids pcyaA and pcrp, respectively, caused the mutant strains to behave like wild-type UTI89, rendering them sensitive to H2O2 and resistant to ASN and MV (Fig. 4 and data not shown).
Fig 3.

UTI89ΔcyaA and UTI89Δcrp are sensitive to nitrosative stress and methyl viologen but resistant to H2O2. Growth of UTI89, UTI89ΔcyaA, and UTI89Δcrp in MES-LB broth (A), MES-LB broth + 1 mM ASN (B), MES-LB broth + 1 mM H2O2 (C), LB broth (D), and LB broth containing 1 mM MV (E). Growth curves show the means of results from a single experiment and are representative of at least three independent experiments carried out in quadruplicate.
Fig 4.

Complementation of UTI89ΔcyaA and UTI89Δcrp in the presence of nitrosative and oxidative stresses. Graphs show growth of UTI89 versus UTI89ΔcyaA (A to C) and UTI89Δcrp (D to F) in MES-LB (A, D), MES-LB ± 1 mM ASN (B, E), or MES-LB ± 1 mM H2O2 (C, F), all without added IPTG. Strains carried pcyaA, pcrp, or the control plasmid pRR48, as indicated. Growth curves show the means of results from a single experiment and are representative of at least three independent experiments carried out in quadruplicate.
H2O2 resistance correlates with elevated RpoS expression and catalase activity in the absence of cAMP-CRP.
cAMP-CRP represses the transcription of the alternate sigma factor RpoS, a master regulator of the general stress response in E. coli (45). In enterohemorrhagic E. coli and laboratory K-12 mutant strains that lack cAMP-CRP, RpoS levels are abnormally increased during the exponential growth phase (44, 46, 47). By Western blot analyses, we observed a similar phenomenon, with RpoS levels in UTI89ΔcyaA and UTI89Δcrp notably increased relative to those of the wild-type strain during exponential growth in broth culture (Fig. 5A). RpoS regulates the expression of various genes that enable bacteria to deal with multiple environmental stresses, including reactive oxygen species like H2O2 (45, 48). IPTG-induced expression of RpoS from the plasmid prpoS rescued growth of wild-type UTI89 in the presence of H2O2, phenocopying the H2O2 resistance seen with the ΔcyaA and Δcrp mutants (Fig. 5B and C). The high-level resistance of UTI89/prpoS, UTI89ΔcyaA, and UTI89Δcrp to H2O2 correlated with increased catalase activity in these strains (Fig. 5D). These results are in line with previous work showing that RpoS can stimulate expression of stress-responsive catalase genes necessary for the detoxification of H2O2 (48, 49).
Fig 5.

H2O2 resistance correlates with increased RpoS expression and catalase activity in UTI89ΔcyaA and UTI89Δcrp. (A) Western blot of RpoS (σS) in UTI89, UTI89ΔcyaA, and UTI89Δcrp after growth to mid-exponential phase in LB broth. Relative levels of RpoS normalized to loading control (Ctrl) are indicated. (B, C) Curves show growth of UTI89 and UTI89Δcrp carrying empty vector pRR48 or prpoS, as indicated, in MES-LB ± 1 mM H2O2. (D) Graph of catalase activity in UTI89, UTI89ΔcyaA, UTI89Δcrp, and UTI89/prpoS following growth to stationary phase in LB broth + 1 mM IPTG. Data are expressed relative to wild-type UTI89 as the means ± standard errors of three independent experiments carried out in triplicate. (E) Levels of trehalose present in UTI89ΔfimH, UTI89ΔotsBA, UTI89ΔcyaA, and UTI89Δcrp following growth to stationary phase (OD600 = 1.0). The ΔfimH mutant carries the same chloramphenicol resistance cassette as the ΔcyaA and Δcrp mutants and served as the control. (F, G) Graphs show growth of UTI89 and its mutant derivatives (ΔcyaA, Δcrp, and Δdps mutants) carrying pRR48 or pdps, as indicated, in MES-LB ± 1 mM H2O2. Each growth curve (B, C, F, and G) shows the means of results from a single experiment and is representative of at least three independent experiments carried out in quadruplicate. Dps and RpoS expression in these assays was induced by addition of 0.5 mM IPTG. (H) Survival of UTI89Δdps complemented with empty vector pRR48 or pdps following 15 min of exposure to 0.05 M CuSO4. Results were obtained without addition of IPTG and are presented relative to those of wild-type UTI89/pRR48. The indicated P values were determined using two-tailed unpaired t tests.
While these data argue that enhanced expression of one or more RpoS-regulated catalases promotes high-level resistance of UTI89ΔcyaA and UTI89Δcrp toH2O2, it is feasible that other RpoS-regulated genes also contribute to the resistance phenotype of these mutants. To explore this possibility, we investigated two additional loci—otsBA and dps—known to be regulated by RpoS and previously linked with oxidative stress resistance. The first, otsBA, encodes two enzymes used to catalyze the biosynthesis of the disaccharide trehalose, a universal stress protectant produced in abundance by many prokaryotic and eukaryotic organisms (50). In yeast, trehalose protects against oxidative stress caused by H2O2 (51, 52). We found that UTI89 mutants lacking cAMP-CRP generate sizeable amounts of trehalose relative to an isogenic ΔotsAB mutant or a control mutant strain missing an unrelated gene (ΔfimH) (Fig. 5E). However, the addition of exogenous trehalose (up to 3,783 μg/ml) failed to rescue growth of wild-type UTI89 in broth cultures containing 1 mM H2O2, and the double deletion mutant UTI89ΔcyaAΔostBA was as resistant to H2O2 as UTI89ΔcyaA (data not shown). These results indicate that while trehalose levels are greatly elevated in bacteria lacking cAMP-CRP, this phenomenon is likely not essential to the heightened H2O2 resistance associated with UTI89ΔcyaA and UTI89Δcrp.
We next examined Dps, an abundant RpoS- and cAMP-CRP-regulated stationary-phase protein that can protect E. coli cells from multiple stresses, including oxidants (44, 46, 53–55). Dps can bind and shield DNA and also has ferritin-like properties, enabling it to sequester and oxidize ferrous ions while detoxifying H2O2 in the process (54). In consideration of this information, we hypothesized that forced expression of recombinant Dps would render wild-type UTI89 more resistant to H2O2, potentially mimicking UTI89ΔcyaA and UTI89Δcrp. However, IPTG-induced expression of Dps from plasmid pdps had no effect on the growth of either wild-type UTI89 or a Δdps mutant in the presence or absence of H2O2 (Fig. 5F and G). Induced expression of Dps did promote survival of UTI89Δdps in a metal (CuSO4) stress resistance assay, confirming the functionality of the pdps plasmid (Fig. 5H). In total, these data indicate that increased Dps expression is surprisingly ineffective at promoting H2O2 resistance in UTI89.
Acid stress resistance of UTI89ΔcyaA and UTI89Δcrp is linked with increased RpoS and Dps expression.
Within the urinary tract, UPEC will likely come across pH extremes, both within the urine and within host epithelial cells and infiltrating phagocytes (56, 57). In laboratory E. coli K-12 strains, cAMP-CRP, RpoS, and Dps can mediate acid stress resistance (58–60). Potential involvement of cAMP-CRP as a regulator of acid stress resistance in UTI89 was assessed using survival assays. Following a 30-min exposure of exponential-growth-phase cultures to acidic (pH 3.5) conditions, we found that the ΔcyaA and Δcrp mutants had a significant survival advantage over wild-type UTI89 (Fig. 6). In these assays, IPTG-induced expression of recombinant RpoS or Dps was sufficient to enhance survival of the wild-type strain to levels observed with UTI89ΔcyaA and UTI89Δcrp. To address whether or not RpoS or Dps is necessary for acid resistance, we constructed the mutant strains UTI89ΔrpoS, UTI89Δdps, UTI89ΔdpsΔcrp, and UTI89ΔrpoSΔcrp. In agreement with results observed with other E. coli strains (61–63), we found that UTI89ΔrpoS is highly sensitive to acid stress (Fig. 6). This sensitivity was reduced nearly 10,000-fold when crp was deleted along with rpoS. However, the ΔrpoS Δcrp mutant was still more sensitive than wild-type UTI89 and much more sensitive than UTI89 lacking only crp. In contrast, deletion of dps had only modest effects on the acid resistance of either UTI89 or UTI89Δcrp. Together, these data indicate that the acid stress resistance of UTI89 mutants lacking cAMP-CRP is likely attributable in part to increased cellular levels of RpoS and RpoS-regulated factors like Dps, in addition to other cAMP-CRP-repressible gene products.
Fig 6.
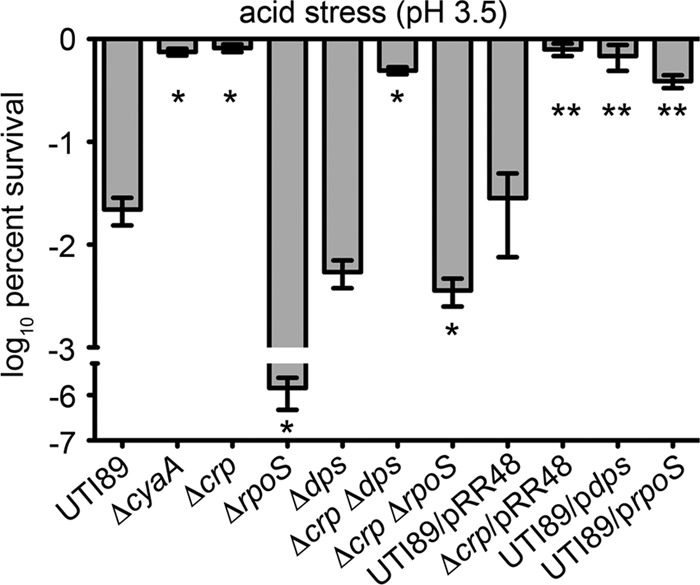
Expression of RpoS or Dps enables wild-type UTI89 to survive low-pH stress at levels similar to those of the ΔcyaA and Δcrp mutants. After reaching mid-logarithmic growth phase in LB broth, UTI89 and the UTI89ΔcyaA, UTI89Δcrp, UTI89Δdps, UTI89ΔrpoS, UTI89ΔcrpΔdps, and UTI89ΔcrpΔrpoS mutants (± pRR48, pdps, and prpoS, as indicated) were exposed to acid (pH 3.5) stress for 30 min. Following washes in PBS, numbers of surviving bacteria were determined by dilution plating. Plasmid-containing strains were grown in the presence of 0.5 mM IPTG prior to challenge with pH stress. Data are expressed as the means ± standard deviations of three independent experiments. P values of <0.05 are indicated by one asterisk (*) for comparison with UTI89 and by two asterisks (**) for comparison with UTI89/pRR48, as determined by two-tailed unpaired t tests.
DISCUSSION
The misregulation of carbon flux through metabolic pathways can restrict niche availability and alter the virulence potential of E. coli and other bacterial species (4, 18, 64–66). cAMP-CRP—a central regulator of carbon metabolism—has been implicated as an important facilitator of host colonization and virulence in many bacterial pathogens, including the uropathogen Proteus mirabilis (18, 67). This is not entirely unexpected given the known capacity of cAMP-CRP to modulate far-ranging activities in addition to metabolism (16). Among these is the ability to influence key stress response pathways such as those controlled by OxyR and RpoS (44, 45, 68). Results presented here demonstrate that cAMP-CRP is also critical to the ability of UPEC to effectively colonize the urinary tract.
Earlier work indicated that E. coli strains that are deficient in the production of cAMP-CRP express more type 1 pili (23). These filamentous adhesive organelles promote bacterial colonization of the bladder, suggesting that the defects observed with the ΔcyaA and Δcrp mutants in our in vivo assays may be attributable to misregulation of type 1 pilus expression. However, this possibility is countered by recent work showing that elevated levels of type 1 pilus expression actually enhance the ability of UPEC to colonize and persist within the bladder (69). Consequently, we conclude that the inability of the ΔcyaA and Δcrp mutants to effectively colonize the bladder is mostly due to the effects of diminished cAMP-CRP levels on factors other than type 1 pili.
In our in vitro assays, deletion of either cyaA or crp increased the ability of UPEC to withstand levels of H2O2 that prevent growth of the wild-type strain. The ΔcyaA and Δcrp mutants were also substantially more resistant to acid stress. UPEC likely comes across similar stresses during the course of a UTI, but any increase in stress resistance afforded by the deletion of cyaA or crp is apparently countered and surpassed in vivo by detrimental effects on other systems. For example, the ΔcyaA and Δcrp mutants are highly sensitive to nitrosative stress and the superoxide generator MV. Both reactive nitrogen and reactive oxygen species like superoxide are abundantly produced in response to a UTI and could compromise the fitness of mutants lacking cAMP-CRP (5, 38, 39, 41, 70). An inability to utilize alternate carbon sources like amino acids may also limit successful colonization of the urinary tract by the ΔcyaA and Δcrp mutants, as peptides and amino acids are a primary energy source utilized by UPEC during a UTI (10, 12, 13). Furthermore, the massive upregulation of trehalose production by UTI89ΔcyaA and UTI89Δcrp, while potentially offering a degree of protection under some stressful conditions, may exact a high fitness cost within the nutrient-poor confines of the urinary tract.
Results obtained using C3H/HeJ mice suggest that innate host defenses, and not nutrient availability per se, are the primary factors that restrict UTI89ΔcyaA and UTI89Δcrp from effectively colonizing the urinary tract. C3H/HeJ mice are hyporesponsive to lipopolysaccharide and are therefore unable to mount full-on TLR4-dependent inflammatory responses (32–36, 71, 72). Specific defects associated with C3H/HeJ mice include poor expression of chemokines and greatly reduced infiltration of the bladder mucosa by neutrophils in response to UTI (35, 73, 74). In our assays, wild-type UTI89 and the ΔcyaA and Δcrp mutants colonized C3H/HeJ mice much better than immunocompetent CBA/J animals, although the bladder-associated titers of the mutant strains were more variable within C3H/HeJ mice (Fig. 2). Significantly, the clear differences in bladder titers observed between wild-type UTI89 and the ΔcyaA and Δcrp mutants in CBA/J mice were markedly diminished in the C3H/HeJ strain, probably because C3H/HeJ mice present the mutants with a less inflammatory and therefore less stressful environment.
Cumulatively, our data indicate that the effects of cAMP-CRP on multiple metabolic and stress response pathways must be balanced in order for UPEC to effectively colonize the urinary tract. This likely involves input from many cAMP-CRP-responsive regulatory factors, including the alternate sigma factor RpoS. In vitro, we found that UPEC mutants lacking cAMP-CRP have elevated levels of RpoS expression, in line with results obtained using other E. coli strains (44, 46, 47, 75, 76). The high-level resistance of UTI89ΔcyaA and UTI89Δcrp to H2O2 could be phenocopied by inducing the expression of RpoS in the wild-type pathogen. Other researchers have reported that a laboratory E. coli K-12 mutant lacking cAMP-CRP is also highly resistant to H2O2 (44). In this case, it was suggested that resistance of the mutant to H2O2 was partially attributable to increased RpoS-dependent expression of the DNA binding, ferritin-like protein Dps. We found that IPTG-induced expression of recombinant Dps is not sufficient to rescue growth of wild-type UTI89 in the presence of H2O2, suggesting that other, as-yet-undefined factor(s) acting downstream of RpoS mediate H2O2 resistance in this pathogen. Chief among the candidate gene products that may promote H2O2 resistance are the RpoS-inducible catalases (48, 49), which by inference appear to be upregulated in UTI89ΔcyaA and UTI89Δcrp (Fig. 5D). Interestingly, in our assays, the UTI89ΔcyaA mutant consistently had higher levels of catalase activity than the Δcrp mutant, suggesting that cAMP generated by CyaA may boost catalase activity in part via CRP-independent mechanisms.
As with the H2O2 sensitivity assays, induced expression of recombinant RpoS increased the acid resistance of wild-type UTI89 to levels observed with the ΔcyaA and Δcrp mutants (Fig. 6). In this case, overexpression of recombinant Dps gave similar results, suggesting that enhanced production of RpoS in the absence of cAMP-CRP promotes acid stress resistance in UPEC via transcriptional effects on dps. However, deletion of dps only slightly decreases the acid resistance of either UTI89 or the Δcrp mutant. In contrast, deletion of rpoS greatly increased the acid sensitivity of UTI89 and, to a far lesser extent, UTI89Δcrp. These observations indicate that acid resistance in UPEC does not require Dps, implying the possible involvement of other RpoS-regulated pH stress-responsive genes such as asr, gadA, and gadBC (46, 59). Furthermore, since UTI89ΔrpoS is much more sensitive to acid stress than the ΔcrpΔrpoS double-knockout mutant, we conclude that the absence of cAMP-CRP promotes acid resistance in UPEC via both RpoS-dependent and RpoS-independent mechanisms. The number of cAMP-CRP-repressible genes that could contribute to the observed acid resistance phenotypes independent of RpoS is potentially high (77, 78).
At first glance, the high-level resistance of UTI89ΔcyaA and UTI89Δcrp to H2O2 seems at odds with the increased sensitivity of these mutants to MV. While H2O2 and MV both generate oxidative stress, there are appreciable differences in their reactivities, duration of activity, and side effects that may differentially influence their toxicity (79). Methyl viologen is a superoxide generator that reduces diatomic oxygen to form superoxide. Oxidized MV can then be reduced by cellular electron donors, creating a redox cycle that consumes reducing equivalents like NADPH while continually producing superoxide molecules (80). In comparison, the oxidizing effects of H2O2 are not regenerated. Within E. coli, superoxide dismutase (Sod) enzymes convert superoxide into oxygen and H2O2. In turn, catalases convert H2O2 into innocuous diatomic oxygen and water. Decreased repression of RpoS within the ΔcyaA and Δcrp mutants results in increased catalase activity, as reported here (Fig. 5D), and may also stimulate the expression of cAMP-CRP- and RpoS-regulated enzymes like SodB and SodC (81, 82). Consequently, mutants lacking cAMP-CRP are likely better equipped to detoxify both superoxide and H2O2, which in turn may drive the redox cycle centered around MV so that reducing equivalents needed by the bacteria are consumed at a rate that disrupts essential cellular processes. The exact mechanisms by which the ΔcyaA and Δcrp mutants differentially deal with MV and H2O2 require further investigation. However, the opposing effects of these two oxidants on cAMP-CRP-dependent bacterial growth and fitness highlight the complex interplay that is possible between cAMP-CRP and the myriad metabolic and stress response systems that can contribute to the pathogenicity of UPEC.
ACKNOWLEDGMENTS
We are grateful to James Cox in the Metabolomics Facility of the University of Utah for help with quantifying trehalose levels, and we thank James Imlay for providing plasmid pdps and Sandy Parkinson for pRR48. We are also grateful to Rich Kulesus for providing UTI89ΔrpoS.
This work was supported by grants AI095647, AI090369, and AI088086 from the National Institute of Allergy and Infectious Diseases. J.P.N. was supported by NIH Genetics Training Grant T32-GM007464.
Footnotes
Published ahead of print 31 October 2012
REFERENCES
- 1. Wiles TJ, Kulesus RR, Mulvey MA. 2008. Origins and virulence mechanisms of uropathogenic Escherichia coli. Exp. Mol. Pathol. 85:11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zasloff M. 2007. Antimicrobial peptides, innate immunity, and the normally sterile urinary tract. J. Am. Soc. Nephrol. 18:2810–2816 [DOI] [PubMed] [Google Scholar]
- 3. Gawel D, Seed PC. 2011. Urinary tract infection drives genome instability in uropathogenic Escherichia coli and necessitates translesion synthesis DNA polymerase IV for virulence. Virulence 2:222–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alteri CJ, Mobley HL. 2012. Escherichia coli physiology and metabolism dictates adaptation to diverse host microenvironments. Curr. Opin. Microbiol. 15:3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li B, Smith P, Horvath DJ, Jr, Romesberg FE, Justice SS. 2010. SOS regulatory elements are essential for UPEC pathogenesis. Microbes Infect. 12:662–668 [DOI] [PubMed] [Google Scholar]
- 6. Foxman B. 2010. The epidemiology of urinary tract infection. Nat. Rev. Urol. 7:653–660 [DOI] [PubMed] [Google Scholar]
- 7. Sivick KE, Mobley HL. 2010. Waging war against uropathogenic Escherichia coli: winning back the urinary tract. Infect. Immun. 78:568–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hunstad DA, Justice SS. 2010. Intracellular lifestyles and immune evasion strategies of uropathogenic Escherichia coli. Annu. Rev. Microbiol. 64:203–221 [DOI] [PubMed] [Google Scholar]
- 9. Dhakal BK, Kulesus RR, Mulvey MA. 2008. Mechanisms and consequences of bladder cell invasion by uropathogenic Escherichia coli. Eur. J. Clin. Invest. 38(S2):2–11 [DOI] [PubMed] [Google Scholar]
- 10. Alteri CJ, Smith SN, Mobley HL. 2009. Fitness of Escherichia coli during urinary tract infection requires gluconeogenesis and the TCA cycle. PLoS Pathog. 5:e1000448 doi:10.1371/journal.ppat.1000448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Martinez-Jehanne V, Pichon C, du Merle L, Poupel O, Cayet N, Bouchier C, Le Bouguenec C. 2012. Role of the Vpe carbohydrate permease in Escherichia coli urovirulence and fitness in vivo. Infect. Immun. 80:2655–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anfora AT, Haugen BJ, Roesch P, Redford P, Welch RA. 2007. Roles of serine accumulation and catabolism in the colonization of the murine urinary tract by Escherichia coli CFT073. Infect. Immun. 75:5298–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roesch PL, Redford P, Batchelet S, Moritz RL, Pellett S, Haugen BJ, Blattner FR, Welch RA. 2003. Uropathogenic Escherichia coli use d-serine deaminase to modulate infection of the murine urinary tract. Mol. Microbiol. 49:55–67 [DOI] [PubMed] [Google Scholar]
- 14. Hadjifrangiskou M, Kostakioti M, Chen SL, Henderson JP, Greene SE, Hultgren SJ. 2011. A central metabolic circuit controlled by QseC in pathogenic Escherichia coli. Mol. Microbiol. 80:1516–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kostakioti M, Hadjifrangiskou M, Pinkner JS, Hultgren SJ. 2009. QseC-mediated dephosphorylation of QseB is required for expression of genes associated with virulence in uropathogenic Escherichia coli. Mol. Microbiol. 73:1020–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gorke B, Stulke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6:613–624 [DOI] [PubMed] [Google Scholar]
- 17. Poncet S, Milohanic E, Maze A, Nait Abdallah J, Ake F, Larribe M, Deghmane AE, Taha MK, Dozot M, De Bolle X, Letesson JJ, Deutscher J. 2009. Correlations between carbon metabolism and virulence in bacteria. Contrib. Microbiol. 16:88–102 [DOI] [PubMed] [Google Scholar]
- 18. McDonough KA, Rodriguez A. 2012. The myriad roles of cyclic AMP in microbial pathogens: from signal to sword. Nat. Rev. Microbiol. 10:27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nielsen AT, Dolganov NA, Rasmussen T, Otto G, Miller MC, Felt SA, Torreilles S, Schoolnik GK. 2010. A bistable switch and anatomical site control Vibrio cholerae virulence gene expression in the intestine. PLoS Pathog. 6:e1001102 doi:10.1371/journal.ppat.1001102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rickman L, Scott C, Hunt DM, Hutchinson T, Menendez MC, Whalan R, Hinds J, Colston MJ, Green J, Buxton RS. 2005. A member of the cAMP receptor protein family of transcription regulators in Mycobacterium tuberculosis is required for virulence in mice and controls transcription of the rpfA gene coding for a resuscitation promoting factor. Mol. Microbiol. 56:1274–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Curtiss R, III, Kelly SM. 1987. Salmonella typhimurium deletion mutants lacking adenylate cyclase and cyclic AMP receptor protein are avirulent and immunogenic. Infect. Immun. 55:3035–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smith RS, Wolfgang MC, Lory S. 2004. An adenylate cyclase-controlled signaling network regulates Pseudomonas aeruginosa virulence in a mouse model of acute pneumonia. Infect. Immun. 72:1677–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muller CM, Aberg A, Straseviciene J, Emody L, Uhlin BE, Balsalobre C. 2009. Type 1 fimbriae, a colonization factor of uropathogenic Escherichia coli, are controlled by the metabolic sensor CRP-cAMP. PLoS Pathog. 5:e1000303 doi:10.1371/journal.ppat.1000303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen SL, Hung CS, Xu J, Reigstad CS, Magrini V, Sabo A, Blasiar D, Bieri T, Meyer RR, Ozersky P, Armstrong JR, Fulton RS, Latreille JP, Spieth J, Hooton TM, Mardis ER, Hultgren SJ, Gordon JI. 2006. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc. Natl. Acad. Sci. U. S. A. 103:5977–5982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mulvey MA, Schilling JD, Hultgren SJ. 2001. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect. Immun. 69:4572–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou Q, Ames P, Parkinson JS. 2009. Mutational analyses of HAMP helices suggest a dynamic bundle model of input-output signalling in chemoreceptors. Mol. Microbiol. 73:801–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murphy KC, Campellone KG. 2003. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 4:11 doi:10.1186/1471-2199-4-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blango MG, Mulvey MA. 2010. Persistence of uropathogenic Escherichia coli in the face of multiple antibiotics. Antimicrob. Agents Chemother. 54:1855–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eto DS, Gordon HB, Dhakal BK, Jones TA, Mulvey MA. 2008. Clathrin, AP-2, and the NPXY-binding subset of alternate endocytic adaptors facilitate FimH-mediated bacterial invasion of host cells. Cell. Microbiol. 10:2553–2567 [DOI] [PubMed] [Google Scholar]
- 31. Hopkins W, Gendron-Fitzpatrick A, McCarthy DO, Haine JE, Uehling DT. 1996. Lipopolysaccharide-responder and nonresponder C3H mouse strains are equally susceptible to an induced Escherichia coli urinary tract infection. Infect. Immun. 64:1369–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hagberg L, Hull R, Hull S, McGhee JR, Michalek SM, Svanborg Eden C. 1984. Difference in susceptibility to gram-negative urinary tract infection between C3H/HeJ and C3H/HeN mice. Infect. Immun. 46:839–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Suhs KA, Marthaler BR, Welch RA, Hopkins WJ. 2011. Lack of association between the Tlr4 (Lpsd/Lpsd) genotype and increased susceptibility to Escherichia coli bladder infections in female C3H/HeJ mice. mBio 2:e00094–11 doi:10.1128/mBio.00094-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hopkins WJ, Elkahwaji J, Kendziorski C, Moser AR, Briggs PM, Suhs KA. 2009. Quantitative trait loci associated with susceptibility to bladder and kidney infections induced by Escherichia coli in female C3H/HeJ mice. J. Infect. Dis. 199:355–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haraoka M, Hang L, Frendéus B, Godaly G, Burdick M, Strieter R, Svanborg C. 1999. Neutrophil recruitment and resistance to urinary tract infection. J. Infect. Dis. 180:1220–1229 [DOI] [PubMed] [Google Scholar]
- 36. Schilling JD, Martin SM, Hung CS, Lorenz RG, Hultgren SJ. 2003. Toll-like receptor 4 on stromal and hematopoietic cells mediates innate resistance to uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 100:4203–4208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aubron C, Glodt J, Matar C, Huet O, Borderie D, Dobrindt U, Duranteau J, Denamur E, Conti M, Bouvet O. 2012. Variation in endogenous oxidative stress in Escherichia coli natural isolates during growth in urine. BMC Microbiol. 12:120 doi:10.1186/1471-2180-12-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bower JM, Gordon-Raagas HB, Mulvey MA. 2009. Conditioning of uropathogenic Escherichia coli for enhanced colonization of host. Infect. Immun. 77:2104–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johnson JR, Clabots C, Rosen H. 2006. Effect of inactivation of the global oxidative stress regulator oxyR on the colonization ability of Escherichia coli O1:K1:H7 in a mouse model of ascending urinary tract infection. Infect. Immun. 74:461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Korshunov S, Imlay JA. 2006. Detection and quantification of superoxide formed within the periplasm of Escherichia coli. J. Bacteriol. 188:6326–6334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lundberg JO, Ehren I, Jansson O, Adolfsson J, Lundberg JM, Weitzberg E, Alving K, Wiklund NP. 1996. Elevated nitric oxide in the urinary bladder in infectious and noninfectious cystitis. Urology 48:700–702 [DOI] [PubMed] [Google Scholar]
- 42. Poljakovic M, Svensson ML, Svanborg C, Johansson K, Larsson B, Persson K. 2001. Escherichia coli-induced inducible nitric oxide synthase and cyclooxygenase expression in the mouse bladder and kidney. Kidney Int. 59:893–904 [DOI] [PubMed] [Google Scholar]
- 43. Woolford G, Casselden RJ, Walters CL. 1972. Gaseous products of the interaction of sodium nitrite with porcine skeletal muscle. Biochem. J. 130:82P–83P [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barth E, Gora KV, Gebendorfer KM, Settele F, Jakob U, Winter J. 2009. Interplay of cellular cAMP levels, {sigma}S activity and oxidative stress resistance in Escherichia coli. Microbiology 155:1680–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hengge-Aronis R. 2002. Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol. Mol. Biol. Rev. 66:373–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jeong KC, Baumler DJ, Kaspar CW. 2006. dps expression in Escherichia coli O157:H7 requires an extended −10 region and is affected by the cAMP receptor protein. Biochim. Biophys. Acta 1759:51–59 [DOI] [PubMed] [Google Scholar]
- 47. Lange R, Hengge-Aronis R. 1994. The cellular concentration of the sigma S subunit of RNA polymerase in Escherichia coli is controlled at the levels of transcription, translation, and protein stability. Genes Dev. 8:1600–1612 [DOI] [PubMed] [Google Scholar]
- 48. Eisenstark A, Calcutt MJ, Becker-Hapak M, Ivanova A. 1996. Role of Escherichia coli rpoS and associated genes in defense against oxidative damage. Free Radic. Biol. Med. 21:975–993 [DOI] [PubMed] [Google Scholar]
- 49. Schellhorn HE. 1995. Regulation of hydroperoxidase (catalase) expression in Escherichia coli. FEMS Microbiol. Lett. 131:113–119 [DOI] [PubMed] [Google Scholar]
- 50. Strom AR, Kaasen I. 1993. Trehalose metabolism in Escherichia coli: stress protection and stress regulation of gene expression. Mol. Microbiol. 8:205–210 [DOI] [PubMed] [Google Scholar]
- 51. Alvarez-Peral FJ, Zaragoza O, Pedreno Y, Arguelles JC. 2002. Protective role of trehalose during severe oxidative stress caused by hydrogen peroxide and the adaptive oxidative stress response in Candida albicans. Microbiology 148:2599–2606 [DOI] [PubMed] [Google Scholar]
- 52. Benaroudj N, Lee DH, Goldberg AL. 2001. Trehalose accumulation during cellular stress protects cells and cellular proteins from damage by oxygen radicals. J. Biol. Chem. 276:24261–24267 [DOI] [PubMed] [Google Scholar]
- 53. Martinez A, Kolter R. 1997. Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J. Bacteriol. 179:5188–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Almiron M, Link AJ, Furlong D, Kolter R. 1992. A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli. Genes Dev. 6:2646–2654 [DOI] [PubMed] [Google Scholar]
- 55. Park S, You X, Imlay JA. 2005. Substantial DNA damage from submicromolar intracellular hydrogen peroxide detected in Hpx- mutants of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 102:9317–9322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eto DS, Sundsbak JL, Mulvey MA. 2006. Actin-gated intracellular growth and resurgence of uropathogenic Escherichia coli. Cell. Microbiol. 8:704–717 [DOI] [PubMed] [Google Scholar]
- 57. Vergne I, Constant P, Laneelle G. 1998. Phagosomal pH determination by dual fluorescence flow cytometry. Anal. Biochem. 255:127–132 [DOI] [PubMed] [Google Scholar]
- 58. Foster JW. 2004. Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2:898–907 [DOI] [PubMed] [Google Scholar]
- 59. Castanie-Cornet MP, Penfound TA, Smith D, Elliott JF, Foster JW. 1999. Control of acid resistance in Escherichia coli. J. Bacteriol. 181:3525–3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Calhoun LN, Kwon YM. 2011. Structure, function and regulation of the DNA-binding protein Dps and its role in acid and oxidative stress resistance in Escherichia coli: a review. J. Appl. Microbiol. 110:375–386 [DOI] [PubMed] [Google Scholar]
- 61. Small P, Blankenhorn D, Welty D, Zinser E, Slonczewski JL. 1994. Acid and base resistance in Escherichia coli and Shigella flexneri: role of rpoS and growth pH. J. Bacteriol. 176:1729–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cheville AM, Arnold KW, Buchrieser C, Cheng CM, Kaspar CW. 1996. rpoS regulation of acid, heat, and salt tolerance in Escherichia coli O157:H7. Appl. Environ. Microbiol. 62:1822–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Waterman SR, Small PL. 1996. Characterization of the acid resistance phenotype and rpoS alleles of Shiga-like toxin-producing Escherichia coli. Infect. Immun. 64:2808–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Le Bouguenec C, Schouler C. 2011. Sugar metabolism, an additional virulence factor in enterobacteria. Int. J. Med. Microbiol. 301:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Loughman JA, Hunstad DA. 2012. Induction of indoleamine 2,3-dioxygenase by uropathogenic bacteria attenuates innate responses to epithelial infection. J. Infect. Dis. 205:1830–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rouquet G, Porcheron G, Barra C, Reperant M, Chanteloup NK, Schouler C, Gilot P. 2009. A metabolic operon in extraintestinal pathogenic Escherichia coli promotes fitness under stressful conditions and invasion of eukaryotic cells. J. Bacteriol. 191:4427–4440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Himpsl SD, Lockatell CV, Hebel JR, Johnson DE, Mobley HL. 2008. Identification of virulence determinants in uropathogenic Proteus mirabilis using signature-tagged mutagenesis. J. Med. Microbiol. 57:1068–1078 [DOI] [PubMed] [Google Scholar]
- 68. Gonzalez-Flecha B, Demple B. 1997. Transcriptional regulation of the Escherichia coli oxyR gene as a function of cell growth. J. Bacteriol. 179:6181–6186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kostakioti M, Hadjifrangiskou M, Cusumano CK, Hannan TJ, Janetka JW, Hultgren SJ. 2012. Distinguishing the contribution of type 1 pili from that of other QseB-misregulated factors when QseC is absent during urinary tract infection. Infect. Immun. 80:2826–2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Poljakovic M, Karpman D, Svanborg C, Persson K. 2002. Human renal epithelial cells express iNOS in response to cytokines but not bacteria. Kidney Int. 61:444–455 [DOI] [PubMed] [Google Scholar]
- 71. Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088 [DOI] [PubMed] [Google Scholar]
- 72. McAdam KP, Ryan JL. 1978. C57BL/10/CR mice: nonresponders to activation by the lipid a moiety of bacterial lipopolysaccharide. J. Immun. 120:249–253 [PubMed] [Google Scholar]
- 73. Justice SS, Hung C, Theriot JA, Fletcher DA, Anderson GG, Footer MJ, Hultgren SJ. 2004. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 101:1333–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Svanborg C, Frendeus B, Godaly G, Hang L, Hedlund M, Wachtler C. 2001. Toll-like receptor signaling and chemokine receptor expression influence the severity of urinary tract infection. J. Infect. Dis. 183(Suppl 1):S61–S65 [DOI] [PubMed] [Google Scholar]
- 75. Ma Z, Gong S, Richard H, Tucker DL, Conway T, Foster JW. 2003. GadE (YhiE) activates glutamate decarboxylase-dependent acid resistance in Escherichia coli K-12. Mol. Microbiol. 49:1309–1320 [DOI] [PubMed] [Google Scholar]
- 76. Ma Z, Richard H, Foster JW. 2003. pH-dependent modulation of cyclic AMP levels and GadW-dependent repression of RpoS affect synthesis of the GadX regulator and Escherichia coli acid resistance. J. Bacteriol. 185:6852–6859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Raghavan R, Sage A, Ochman H. 2011. Genome-wide identification of transcription start sites yields a novel thermosensing RNA and new cyclic AMP receptor protein-regulated genes in Escherichia coli. J. Bacteriol. 193:2871–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zheng D, Constantinidou C, Hobman JL, Minchin SD. 2004. Identification of the CRP regulon using in vitro and in vivo transcriptional profiling. Nucleic Acids Res. 32:5874–5893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Farr SB, Kogoma T. 1991. Oxidative stress responses in Escherichia coli and Salmonella typhimurium. Microbiol. Rev. 55:561–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bus JS, Gibson JE. 1984. Paraquat: model for oxidant-initiated toxicity. Environ. Health Perspect. 55:37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lacour S, Landini P. 2004. SigmaS-dependent gene expression at the onset of stationary phase in Escherichia coli: function of sigmaS-dependent genes and identification of their promoter sequences. J. Bacteriol. 186:7186–7195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang Z, Gosset G, Barabote R, Gonzalez CS, Cuevas WA, Saier MH., Jr 2005. Functional interactions between the carbon and iron utilization regulators, Crp and Fur, in Escherichia coli. J. Bacteriol. 187:980–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wiles TJ, Bower JM, Redd MJ, Mulvey MA. 2009. Use of zebrafish to probe the divergent virulence potentials and toxin requirements of extraintestinal pathogenic Escherichia coli. PLoS Pathog. 5:e1000697 doi:10.1371/journal.ppat.1000697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kulesus RR, Diaz-Perez K, Slechta ES, Eto DS, Mulvey MA. 2008. Impact of the RNA chaperone Hfq on the fitness and virulence potential of uropathogenic Escherichia coli. Infect. Immun. 76:3019–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]