

Abstract
Renal cell carcinoma (RCC) is the most common type of kidney cancer and follows an unpredictable disease course. To improve prognostication, a better understanding of critical genes associated with disease progression is required. The objective of this review was to focus attention on 2 such genes, p53 and murine double minute 2 (MDM2), and to provide a comprehensive summary and critical analysis of the literature regarding these genes in RCC. Information was compiled by searching the PubMed database for articles that were published or e-published up to April 1, 2009. Search terms included renal cancer, renal cell carcinoma, p53, and MDM2. Full articles and any supplementary data were examined; and, when appropriate, references were checked for additional material. All studies that described assessment of p53 and/or MDM2 in renal cancer were included. The authors concluded that increased p53 expression, but not p53 mutation, is associated with reduced overall survival/more rapid disease progression in RCC. There also was evidence that MDM2 up-regulation is associated with decreased disease-specific survival. Two features of RCC stood out as unusual and will require further investigation. First, increased p53 expression is tightly linked with increased MDM2 expression; and, second, patients who have tumors that display increased p53 and MDM2 expression may have the poorest overall survival. Because there was no evidence to support the conclusion that p53 mutation is associated with poorer survival, it seemed clear that increased p53 expression in RCC occurs independent of mutation. Further investigation of the mechanisms leading to increased p53/MDM2 expression in RCC may lead to improved prognostication and to the identification of novel therapeutic interventions.
Keywords: renal cancer, renal cell carcinoma, p53, murine double minute 2
The latest available figures from the National Cancer Institute’s Surveillance, Epidemiology, and End Results Program predict that there will be 49,096 new cases of kidney cancer (renal cancer and cancer of the renal pelvis) and 11,033 deaths from the disease in the United States in 2009.1 In the United Kingdom, the latest figures for 2007 indicate that 7380 individuals were diagnosed with the disease and that 3752 died from it.2 Advances in our understanding of the molecular biology of renal cell carcinoma (RCC) have helped classify this heterogeneous disease and led to the development of several new “von Hippel-Lindau (VHL) pathway”-targeted molecular drug treatments.3 Nevertheless, patients with metastatic disease still have an extremely short life expectancy.3 Certainly other molecular pathways must contribute to the poor prognosis observed for those who have advanced RCC. The objective of the current review was to turn the molecular focus back to 1 of the most important genes in cancer biology, p53, and its counterpart, murine double minute 2 (MDM2), and to describe and critically review what is known of their role in RCC.
For the purposes of this review, the 3 most common histologic subtypes of RCC (as set out in the 2004 World Health Organization classification; see Lopez-Beltran et al4) are discussed. Clear cell RCC (CCRCC) is the commonest histologic subtype and is believed to account for 75% of all RCCs. Papillary RCC (PRCC) is the second most common and accounts for 10% of RCC: PRCC is subclassified into type I and type II. Chromophobe RCC is the third most common subtype and accounts for 5% of RCCs. Sarcomatoid RCC is a high-grade histologic variation that can arise from all types of RCC, although it is not a separate entity.
The p53 and MDM2 Pathway
In 50% of human cancers, p53 is mutated, and it has become 1 of the most studied molecules in science.5 The p53 protein accumulates at times of cellular or genotoxic stress, when it functions primarily as a transcription factor to promote cell cycle arrest and DNA repair, to initiate and maintain a senescent phenotype, or to promote apoptosis if the normal cellular conditions are not restored (Fig. 1). This latter function helps prevent conditions arising within the cell that can lead to the establishment of mutations with the consequent increased risk of malignant disease. The importance of the tumor suppressor function of p53 also is highlighted by the high frequency of tumors that occur in individuals with a monoallelic germline mutation of p53, as observed in patients with Li-Fraumeni syndrome.6
Figure 1.
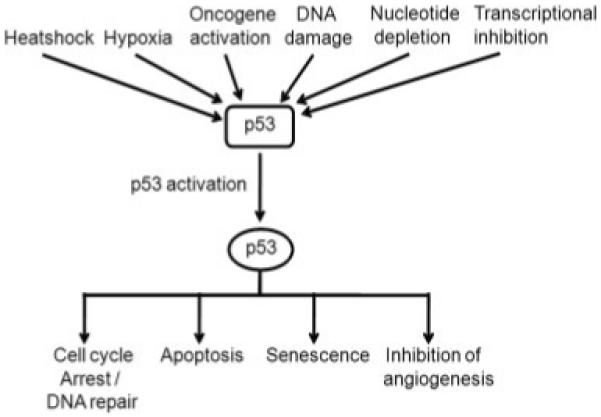
Pathways leading to and consequences of p53 activation are shown.
The potentially lethal activities of p53 are regulated by the proto-oncogene Mdm2 (murine double minute 2) or MDM2 in humans. Transgenic mice that are Mdm2 null display early embryonic lethality and die at around Day 5 or 6 of embryogenesis. This lethality can be rescued by concomitant deletion of the p53 gene,7 thus demonstrating that loss of Mdm2 is lethal because of the lethal effects of unregulated p53. Then, as levels of p53 rise, transcription of MDM2 is induced; thus p53 and MDM2 exist in an autoregulatory feedback loop.8 Binding of Mdm2 to p53 and can block p53 transcriptional activity by preventing it from interacting with the transcriptional machinery.9 MDM2 also causes p53 degradation by targeting it for destruction by the 26S proteasome. The latter effect is caused by the ability of MDM2 to act as an E3 ubiquitin ligase with specificity for p53 (among other targets).10 MDM2 also can target itself for ubiquitylation and, thus, can regulate its own stability, although how these competing activities (ubiquitylation with concomitant degradation of p53 and/or MDM2) are regulated remains unclear.11 Thus, increased levels of MDM2 can lead to a reduction in p53 levels as a result of this ubiquitin ligase activity, which, in turn, results in decreased p53-dependent MDM2 transcription, restoring the normal cellular status quo.
With respect to cancer, MDM2 over expression has been associated with increased metastasis and advanced disease in several cancers, including breast carcinoma.12 It is noteworthy that the oncogenic effects of MDM2 are not caused simply by the inhibition of p53 function, because they often are detectable in tumors that harbor p53 mutations,13 as indicated by further evidence provided from in vivo studies.14 How MDM2 elicits p53-independent oncogenic effects is unclear, although Yang et al12 demonstrated that MDM2 expression led to a decrease in E-cadherin levels and a subsequent increase in cell motility in breast carcinoma. Those authors also demonstrated that high expression of MDM2 with low E-cadherin expression was more frequent in metastatic tumor samples.
The balance between MDM2 and p53 is modulated in several ways, depending on the nature of cellular stress. For example, in response to ionizing radiation, p53 is phosphorylated by the ataxia telangiectasia mutated (ATM) kinase, which inhibits MDM2 binding14; whereas, after exposure to ultraviolet radiation, p53 is modified by the related ATM and Rad3-related (ATR) kinase.15 The complex interplay between p53 and MDM2 presumably has evolved to ensure that cells are able to respond rapidly and appropriately to a wide range of genotoxic stresses. One consequence of this sophistication in the regulation of p53 is that it can lead to unreliable conclusions when attempting to determine p53 status from simple assays, such as immunohistochemical (IHC) analyses.16,17
In addition to the variable outcomes induced by p53 in response to different types or amounts of stress/damage, there is also a high degree of spatial (ie, tissue) variability in response to genotoxic stress, for example, within an organism (for review, see Slee et al18). Studies of transgenic mice exposed to ionizing radiation have revealed broadly 3 classes of p53 response19: In the first class of tissues, p53 is up-regulated and elicits a dramatic apoptotic response, as typified by tissues like the small intestine, spleen, and thymus. In the second class of tissues, which includes the kidney, p53 is up-regulated, but little or no apoptotic response is detected. In the third class of tissues, there is little or no apparent induction of p53. There are also clear differences within tissues. Even when p53 is activated, cellular responses are determined by both tissue-specific and cell-intrinsic factors, which may vary according to the status of the individual cell.18,20
Therefore, in the kidney, it appears that there already may be factors at work that compromise the ability of p53 to activate an apoptotic response to genotoxic stress.19,21 This suggests that p53 tumor suppression may be less effective in the kidney than in some other tissues, and the reason for this remains a major, unanswered question. Moreover, the identification of such a tissue-specific mechanism might provide an opportunity for therapeutic “reactivation” of p53 in these cells. Nevertheless, studies in vitro have questioned this interpretation, demonstrating that p53 is normally functional in renal cancer cells and is regulated by MDM2 in a manner that is typical of cells from other tissues, findings that require further investigation.22,23
The majority of studies of p53 and MDM2 in clinical material from renal cancers have been based on IHC of these proteins. This approach is technically simple but is not quantitative and relies on the observation that mutant p53 often is present at higher levels in cells than the wild-type protein.16,17 The standard explanation for this genotype/phenotype correlation is that mutant p53 lacks the capacity to up-regulate MDM2; thus, an imbalance in p53/MDM2 homeostasis develops and leads to excess p53. In a study of the use of IHC to interrogate p53 status, Nenutil et al16 observed that combining highly sensitive IHC for p53 (with the ability to detect low levels of wild-type expression and, thus, distinguish this from an absence of expression) with IHC for downstream markers (MDM2 and p21[CDKN1A]) increased the reliability of predicting p53 status. In that study, high levels of p53 almost always were indicative of p53 mutation when MDM2 expression was low, thus according with the standard model for mutant p53 up-regulation. This is not the case in renal cancer, as detailed below.
p53 and MDM2 in Renal Cell Carcinoma
Relatively little is known with certainty about the status and role of p53 or MDM2 in RCC, in striking contrast to some other, albeit more common cancers. To date, at least 31 studies have investigated the expression of p53 in RCC (Table 1).
Table 1.
Studies Investigating the Expression of p53 in Renal Cell Carcinoma
| Study | No. of Specimens (% CCRCC) |
Antibody | Positive Criteria, % |
No. Positive (%) |
p53 Prognostic Value |
Comments |
|---|---|---|---|---|---|---|
| Klatte 200924 | 170 (100); All stages; M=0 | ?a | Any | ?a | Yes | Decreased DFS |
| Perret 200825 | 50 (0) All PRCC; 25 type I/25 type II; M=8 |
DO-7 | >20 | 24 (48); Type I=12/type 2=36 |
Yes | Decreased OS in type II PRCC |
| Phuoc 200726 | 119 (100); All stages; M=23 | DO-7 | >10 | 64 (54) | Yes | Decreased DSS in all patients |
| Kankuri 200627 | 117 (86); All stages; M=29 | DO-7 | >10 | 15 (12.8) | Yes | Decreased OS in patients with metastases |
| Kramer 200528 | 117 (89); All stages; M=21 | ?a | >5 | 16 (13.6) | No | |
| Langner 200529 | 95 (75); stage pT1 only; M=0 | DO-7 | ?a | 23 (24) | Not evaluated | |
| Cho 200530 | 92 (100); All stages; M=7 | ?a | >10 | 11 (12) | Yes | Decreased DSS |
| Shvarts 200531 | 193 (85); All stages; M=0 | DO-7 | Any, >20 | 111 (57.5), 14 (7.3) | Yes | 20% Cutoff predicted recurrence |
| Uzunlar 200532 | 57 (77.1); All stages and grades; M=? | ?a | >1 | 20 (35) | Yes | Decreased DSS |
| Zigeuner 200433 | 184 (70.7)/Metastases, 56 (94.8)b | DO-7 | >1 | 42 (22.8)/29 (51.8) | Yes | Decreased metastasis-free survival in CCRCC only |
| Kim 200434 | 318; All stages; M=155 | DO-7 | >15 | ?a | Yes | Decreased DSS |
| Uchida 200235 | 112 (78); All stages; M=? | DO-7 | >1 | 15 (13.4) | Yes | Decreased OS |
| Olumi 200136 | 43 (100); All stages; M=14 | DO-7/PAB240/both | >10, Either | 22 (51)/13 (30)/26 (60) | No | Combined antibody positivity was 60% |
| Ljungberg 200137 | 99 (74); All stages; M=? | DO-7 | >5 | 17 (19) | Yes | Decreased survival in non-CCRCC |
| Girgin 200138 | 50 (62); All stages; M=0 | DO-1 | >20 | 16 (20) | Yes | Decreased disease-specific death |
| Haitel 200039 | 97 (100); All stages; M=15 | DO-1 | >5 | 35 (36) | Yes | Decreased DSS |
| Rioux-Leclercq 200040 | 66 (?); All stages; M=10 | DO-7 | >20 | 11 (17) | Yes | Decreased DSS |
| Sejima & Miyagawa 199941 | 53 (?); All stages; M=25 | RSP53 | ?a | 1 (2) | No | |
| Vasavada 199842 | 39 (71); T1 and T2 only; M=0 | DO-7 | >1 | 0 | No | |
| Sinik 199743 | 39 (100); All stages; M=? | DO-7 | >10 | 7 (17.9) | No | |
| Papadopoulos 199744 | 90 (?); T1 and T2; M=14 | DO-1 | Any positive nuclei | 30 (33) | No | |
| Zhang 199745 | 70 (?); All stages; M=? | Ab-6 | >10 | 16 (23) | Not evaluated | |
| Gelb 199746 | 52 (100); T1 and T2 only; M=0 | DO-7 | >5 | 2 (2) | No | |
| Shiina 199747 | 72 (?); All stages; M=6 | DO-7 | >10 | 29 (40.3) | Yes | Decreased OS |
| Moch 199748 | 50 (100); T3 only; M=1 | DO-7 | ?a | 8 (16) | Yes | Decreased OS |
| Hofmockel 199649 | 31 (?); T1-T3; M=0 | DO-7 | >1% | 5 (16) | No | |
| Chemeris 199550 | 82 (40)c; M=? | DO-1 | ?a | 43 (52) | Not evaluated | |
| Lipponen 199451 | 123 (?); M=29 | CM1 | Any positive | 41 (33) | Yes | Increased recurrence-free survival |
| Kamel 199452 | 56 (?); All stages; M=13 | CM1 | >1 | 6 (11) | No | |
| Bot 199453 | 100 (74); T1-T3 only; M=0 | DO-7 | >50 | 32 (32) | No | |
| Uhlman 199454 | 175 (?); All stages; M=45 | ?a | >1 | 49 (28) | Yes | Decreased DSS |
CCRCC indicates clear cell renal cell carcinoma; PRCC, papillary renal cell carcinoma; M, metastatic disease; DFS, disease-free survival; OS, overall survival; DSS, disease-specific survival; pT1, pathologic T1 tumor classification.
Information not provided.
In addition to 184 renal cancers, 56 surgically removed metastatic tissues were analyzed.
Thirty-three CCRCCs and 47 clear/chromophilic renal cell carcinomas.
All of these studies used IHC staining of formalin-fixed, paraffin-embedded tumor samples. When they were available, the percentages of samples from CCRCC, the range of disease stages, and the number of metastatic samples are indicated. All samples contained a mixture of high-grade and low-grade tumors. Several different antibodies were used, although the majority of studies used the p53 DO-7 monoclonal antibody. When available, the criterion for dichotomizing p53 staining has been indicated. According to published data (excluding articles by Klatte et al24 and Kim et al,34 in which no details of p53 status were provided), 2519 tumors were stained for p53, and 618 tumors were deemed positive for p53 for p53-positive frequency of 24.5%; however the heterogeneity of the samples must be taken into consideration (see below). Variability between studies may be attributed in part to the lack of a consensus on p53 dichotomization (see Munro et al17) compounded by differences in antibody choice and also by processing techniques. One study36 reported the use of 2 different p53 antibodies, which led to a higher overall p53 detection level/expression rate of 60% (DO-7, 51% positive; p53 antibody 240, 30% positive). This highlights the finding that using different antibodies can result in apparent differences in p53 expression. Interpretation of these results also is hindered by variations in the numbers of different histologic subtypes of RCC tumors, tumor stages, and grades and the variable presence of metastases in the sample populations.
Analysis of p53 expression in primary and metastatic samples has demonstrated an increased frequency of staining of 51.8% in metastatic samples versus 22% in primary samples.33 This suggests that p53 expression may be a relatively late event in the evolution of RCC and may be associated with metastatic capabilities. If this is correct, then it seems reasonable to expect that p53 expression will be associated with a poorer prognosis regardless of a functional or causal connection. In the rarer histologic subtypes of RCC, considerably more heterogeneity is apparent; and, inevitably, the smaller number of samples analyzed makes interpretation more difficult.
p53 and MDM2 Expression and Prognostic Implications in Renal Call Carcinoma
The prognostic implications of p53 expression were evaluated, and the results are summarized in Table 1. Of the 27 studies that evaluated p53 expression as a clinical outcome predictor, 18 studies indicated that it predicted a poor outcome, and 10 studies did not. The 10 articles that did not had a smaller study size (mean, 62 patients per study compared with 119 patients per study in the positive articles). The 4 largest studies to date, all of which used tumor microarrays, indicated that p53 is a prognostic predictor. Kim et al evaluated 318 patients with CCRCC who had local and metastatic disease (49%; ie, 155 of 318 patients had metastatic disease). Those authors observed that p53 was an independent predictor of decreased disease-specific survival on univariate (P<.001) and on multivariate Cox regression analysis (P = .014) in which the presence of metastasis was included as a covariate.34 Klatte et al,24 in a similar study of 170 patients with CCRCC, also reported that p53 was retained in a multivariate Cox regression analysis for predicting disease-free survival. In the study by Klatte et al, the percentage of tumors that stained for p53 was evaluated, obviating the need to describe tumors as either positive or negative for p53 staining. Zigeuner and colleagues33 demonstrated that p53 was a predictor of disease progression (metastasis-free survival) on multivariate Cox regression analysis in their study of 130 patients with CCRCC. Those patients were followed for a median of 26 months, and 9 of 16 patients with p53-positive tumors progressed versus 20 of 114 patients with p53-negative tumors (P = .0005). In another substantial study, Shvarts et al31 evaluated p53 staining as a predictor of 5-year recurrence in 193 patients who underwent surgery for localized disease. Those authors also reported that a p53-positive cutoff of 20% expression detected by IHC was a predictor of recurrence (hazard ratio, 3.28; P = .0108) on univariate and multivariate Cox regression analysis. When considering such studies of biomarkers of disease outcome, the possibility of publication bias leading to the publication of fewer articles that demonstrate no disease outcome correlation should not be neglected. Nevertheless, the trend appears to be that more recent studies with higher numbers of patients indicate that p53 protein levels are prognostically significant in RCC.
p53 Mutational Analysis in Renal Cell Carcinoma
When examining the significance of p53 involvement in cancer in general and in RCC in particular, it is clear that IHC detection of p53 alone cannot reliably inform us whether the protein is functional or mutated.16,55 However, p53 mutational status often has been inferred in such studies, because high-level expression of p53 is used as a surrogate indicator of mutation. This interpretation certainly is not correct for renal cancer cells in culture, in which relatively high levels of p53 protein frequently are detected in the absence of p53 mutation.22 In addition, in some 10% to 20% of cases that harbor p53 mutations, tumors may harbor nonsense (truncating) mutations, which can lead to less stable mutant proteins that are unlikely to be detected by IHC but that would be inferred to possess wild-type p53 according to typical IHC analyses (not all truncated forms of p53 are unstable, although their expression still may be suppressed by nonsense-mediated decay56). In any event, these proteins will be expressed at low levels, and such cases would be grouped together with samples that have low-level wild-type p53 expression. Because recent studies in other cancers, such as squamous cell cancer of the head and neck,57 have indicated that deletion of p53 defines a group of patients with the worst outcome, such a grouping together of wild-type with deletions/nonsense mutations will obscure the significance of p53 in prognostication. To date, at least 14 articles have been published in which the p53 mutational rate was evaluated in RCC (Table 2).
Table 2.
Studies Evaluating the p53 Mutational Rate in Renal Cell Carcinoma
| Study | Sample Type | Technique | p53 Mutation Frequency, %a |
|---|---|---|---|
| Gad 200658 | 46 Chromophobe/19 clear cell/9 papillary |
Direct sequencing, exons 2-11 | 23.9/5.3/11.1 |
| Kawasaki 199959 | 5 | SSCP and direct sequencing, exons 4-8 | 20 |
| Zhang 199745 | 16; All p53 positive by IHCa | SSCP only, exons 5-8 | 44a |
| Contractor 199760 | 30 Clear cell/20 chromophobe | SSCP and direct sequencing or subclone sequencing, exons 5-8 |
3/30 |
| Dahiya 199861 | 40 | SSCP and direct sequencing, exons 5-9 | 35 |
| Dijkhuizen 199662 | 14 Papillary | SSCP only, exons 2-11 | 0 |
| Oda 199563 | 14 Sarcomatoid | Subclone sequencing, exons 1-8 | 79 |
| Chemeris 199550 | 29; All p53 positive by IHCa | SSCP and direct sequencing, exons 4-8 | 0a |
| Kuczyk 199564 | 33 | SSCP and direct sequencing, exons 5-8 | 6 |
| Uchida 199465 | 36 | SSCP and direct sequencing, exons 5-8 | 5 |
| Kikuchi 199466 | 118 | SSCP and subclone sequencing, exons 4-9 | 2 |
| Imai 199467 | 53 | SSCP and direct sequencing. exons 4-8 | 9 |
| Reiter 199368 | 31 RCC cell lines | SSCP and subclone sequencing, exons 5-9 | 33 |
| Suzuki 199269 | 23 | SSCP and direct sequencing, exons 5-8 | 4.8 |
| Torigoe 199270 | 21 | SSCP, exons 2-11 | 10 |
SSCP indicates single-strand conformation polymorphism; IHC, immunohistochemistry; RCC, renal cell carcinoma.
Positive IHC results were used to preselect samples for genetic analysis. Note that p53 mutation frequency refers only to the percentage of mutations identified in the samples that were analyzed in this manner.
In most of these studies, single-strand conformation polymorphism (SSCP) was used as an initial screen to detect mutations. The majority of studies analyzed the central core domain of the gene (exons 4-8 or 5-8), because this is the most common site of p53 mutation.71 Approximately 15% of p53 mutations occur outside exons 5 through 8, in exons 4, 9, and 1072; therefore, it is likely that there will be some underreporting of p53 mutations in these studies. The frequency of p53 mutations reported is between 0% and 44% (excluding the study by Oda et al,63 in which sarcomatoid tumors were evaluated). For comparison, in other tumors, the reported incidence of p53 mutations typically has been between 60% and 65% for lung and colon cancers; between 40% and 45% for stomach, esophageal, and bladder cancers; between 25% and 30% for breast, liver, and prostate cancers and lymphomas; and between 10% and 15% for leukemias (further information available at: http://www-p53.iarc.fr/ accessed June 15, 2009).71 One intriguing observation derives from the study by Chemeris and colleagues, who observed that 0 of 29 RCC samples, all positive for p53 by IHC, had a p53 point mutation.50 However, in another study, Zhang and colleagues observed that 44% of tumors with p53 staining (n = 16) had a p53 point mutation.45 It is possible that the contamination of samples with normal tissue might lead to reduced detection of p53 mutations by SSCP; thus, variations in the extent of this may explain the differences between these 2 studies. This conclusion is supported indirectly by the observation that 33% of RCC-derived tumor cell lines harbored p53 mutations.68 However, this assertion should be tempered by the possibility that the selection of cells to adapt to growth in vitro may have resulted in an increased frequency of p53 mutation. It is noteworthy that data from CCRCC cell lines contrast with data from the study by Dijkhuizen et al62 in which no p53 mutations were identified in 29 PRCC-derived tumor cell lines; thus, it seems possible that different mechanisms other than direct p53 mutation may inactivate p53 more frequently in PRCC than in RCC and that these also may result in higher expression.37,33 One other finding of note is the high p53 mutation rate reported in sarcomatoid tumors.62 Sarcomatoid change is a histologic finding associated with a poor prognosis in renal carcinoma in which a high p53 mutation rate (79%; n = 14)63 was detected. Notwithstanding these differences between individual studies and RCC subtypes, it seems clear that, in contrast to both other cancers and p53 protein detection in RCC, p53 mutational analysis has yet to demonstrate prognostic utility.
MDM2 Expression and Prognosis in Renal Cell Carcinoma
Four studies have evaluated MDM2 and its prognostic value in RCC. Imai et al67 screened 53 RCC tumor samples for MDM2 amplifications and identified none. In another study, IHC was used, and MDM2 expression was detected in 2 of 112 tumor samples (2%).35 When 50 consecutive T3 and T4 tumors were screened by IHC, MDM2 expression was identified in 30%, but it reportedly had no prognostic significance in terms of overall survival.48 However, it was observed that 7 of 8 patients who expressed p53 also expressed MDM2, raising the possibility that MDM2 expression may be linked to the up-regulation or activation of p53. This idea, that p53 and MDM2 expression may be linked in RCC, was supported by statistical analysis in that study (P = .0006). Another study that analyzed this question from Haitel and colleagues39 examined 97 CCRCCs of all stages. MDM2 expression was detected in 19% of tumors and was significantly more frequent in high-grade tumors (P = .0149). In addition, MDM2 staining was strongly associated with tumor progression (P = .00113), and p53 expression was detected in 36% of the samples and was correlated with decreased progression-free survival (P = .00291). When different p53 and MDM2 phenotypes were compared (Fig. 2), it was observed that patients who had tumors with both MDM2 expression and p53 expression had the shortest progression-free survival (P = .00179). Perhaps most interesting from a mechanistic perspective and in accordance with the study by Moch and colleagues, the authors observed a highly significant correlation between MDM2 expression and p53 expression (P < .00004). These studies of MDM2 in RCC examined relatively small numbers of patients using different cutoff values for MDM2 expression. Nevertheless, the association of p53 and MDM2 detected by Moch et al and Haitel et al suggests that tumor progression in RCC may present a tissue-specific pattern that has not been observed in many other cancers. For example, this link has not been observed in soft tissue sarcomas73 or in bladder cancer,13 although, in the latter, patients who had tumors that expressed mutant p53 and increased MDM2 had a poorer prognosis similar to that reported for patients with RCC.
Figure 2.
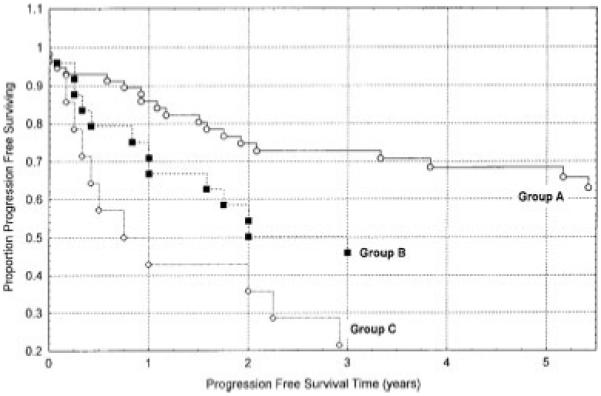
Kaplan-Meier disease-free survival curves are shown for patients with murine double minute 2 (mdm2)-negative/p53-negative tumors (Group A) versus patients with mdm2-negative/p53-positive or mdm2-positive/p53-negative tumors (Group B) versus patients with mdm2-positive/p53-positive tumors (Group C; P = .00179). Reproduced with permission from Haitel A, Wiener HG, Baethge U, Marberger M, Susani M. mdm2 Expression as a prognostic indicator in clear cell renal cell carcinoma: comparison with p53 overexpression and clinicopathological parameters. Clin Cancer Res. 2000; 6:1840-1844.39
Regarding MDM2 in RCC, an additional noteworthy point is the recently described single nucleotide polymorphism at codon 309 (SNP309 thymine/guanine [T/G]). This polymorphism lies in the intronic promoter region of the MDM2 gene and reportedly alters binding of the Sp1 transcription factor, with the G/G variant displaying increased binding and increased transcription of MDM2. It also has been reported that, in RCC, differences in the SNP309 genotype lead to different levels of MDM2 expression, as detected by IHC,74 and that the G/G SNP309 genotype is an independent predictor of poor prognosis. Clearly, further analysis of this polymorphism is warranted.
In Vitro Studies of p53 and MDM2 Function in Renal Cell Carcinoma
Several groups, including our own, have used in vitro analyses to investigate the functional relation between p53 and MDM2 in RCC cells. We previously investigated a panel of RCC cell lines and concluded that p53 is regulated by MDM2, several of which retain relatively high levels of both wild-type p53 and MDM2.22 p53 mutations have been detected in approximately 30% of RCC-derived cell lines, as discussed previously.68 This, together with other studies demonstrating that p53 expression is an independent prognostic indicator, suggests that p53 function (or, rather, its loss or aberration) contributes to tumor evolution in the kidney. It has been suggested, however, that an alternative, novel, dominant mechanism leads to the inactivation of p53 in RCC. Although our own studies and those of others have provided evidence that does not appear to support this conclusion,22,23 good reasons remain to continue investigating the possibility. Two arguments for this are immediately apparent. First, there is evidence from several studies that p53 is not mutated as frequently in RCC as it is in many other cancers. Second, the strong association between p53 expression and MDM2 expression may suggest a functional link between them, with 1 obvious possibility that MDM2 expression may be driven by wild-type p53. It is intriguing to note that in vitro studies have connected MDM2 with 2 critical phenotypes: the ability to promote both motility and invasiveness12 and the regulation of angiogenic factors, such as hypoxia-inducible factor 1 (HIF-1).22,23 Either or both of these phenotypic connections (MDM2 expression with metastasis and/or angiogenesis) may have important consequences.
Manipulating the p53 and MDM2 Pathway
Given the link between p53 and MDM2 expression with poor outcome, a key question becomes whether this is an association or a causal relation. In the event of the former, screening for p53 and/or MDM2 becomes justified for prognostic purposes and, in the future, might be used to stratify therapy according to an individual patient’s risk. In the event of the latter, therapies that target the p53/MDM2 axis also become desirable. The best characterized drug that targets this pathway at the present time is nutlin-3,75 which acts by competing with p53 for binding to the hydrophobic cleft in MDM2 and, thus, up-regulates wild-type p53. Other conceptually similar compounds that prevent MDM2-p53 interactions have been described recently that up-regulate wild-type p53.76 Alternatively, drugs like the HLI98 family of compounds, which inhibit the E3 ligase activity of MDM2, may play a role in RCC, because high levels of MDM2 are linked to tumor progression. However, the associated co-up-regulation of MDM2 with p53 raises questions regarding the activity of MDM2 in these cells77 with implications for the utility of MDM2 enzymatic inhibitors. Therefore, it will be important to determine whether the role of MDM2 in disease progression in RCC depends on its enzymatic activity as an E3 ligase or whether this relates to some other function of MDM2. An alternative strategy would be to focus on the wild-type p53 present in these cells, which, clearly, is not fully active; otherwise, the cells would undergo a classic antiproliferative response. Moreover, we have demonstrated that the introduction of wildtype p53 into RCC cells does elicit a response, at least with respect to a limited set of outputs.22 Thus, a potentially potent therapeutic option would be to reactivate the already high levels of p53 in these cells. RCC is notoriously insensitive to traditional chemotherapeutic and radiotherapeutic regimens that normally might activate p53,78 possibly indicating that signaling to p53 is defective in these cells. However, p53 coexists with high levels of MDM2, suggesting that there is a breakdown in the interaction of these proteins. For future identification of effective therapy, it would be advantageous to identify the actual mechanism that prevents p53 activity in these cells. Notwithstanding this, several compounds aimed at p53 have demonstrated effectiveness in vitro in activating p53 in renal cancer cells. Examples include RITA (reactivation of p53 and induction of tumor cell apoptosis) and derivatives of 9-aminoacridine (including quinacrine).79 It has been reported that RITA induces predominantly growth arrest rather than an apoptotic response in A498 RCC cells in vitro and that 9-aminoacridine and its derivatives, including quinacrine, indirectly inhibit MDM2 in RCC cells in vitro.79,80 It remains unclear whether these compounds specifically target the pathway(s) that regulates p53/MDM2 co-up-regulation in RCC cells, and, to date, none of these compounds or their derivatives have been evaluated in clinical trials for RCC.
In conclusion, for many years, the role of the p53 pathway in renal cancer has been the subject of seemingly conflicting results. However, recent studies appear to be generating a consensus, which suggests a clear link between p53 expression and disease progression, particularly in CCRCC. It is noteworthy that this p53 positivity and link with prognosis is not corroborated by studies of p53 mutation; indeed, there are data suggesting that mutation of p53 may not be linked to outcome or progression in renal cancer and, thus, that the defect in p53 leading to its up-regulation lies elsewhere in the pathway. To our knowledge, only 2 studies of p53 in RCC have examined both p53 protein expression and p53 gene mutations,45,50 and those results were conflicting. Given the disparity between the results from studies of p53 mutation and the more frequent observations of p53 up-regulation, it appears likely that p53 up-regulation is not caused by mutation in most cases of RCC. If this finding is correct, then it has important implications for understanding the nature of the defects in this pathway in RCC. For example, the up-regulation of wild-type p53 is linked to the up-regulation of MDM2, and it appears probable that this is because of p53 transcriptional activation of MDM2. Although it has been studied less than p53, MDM2 expression appears to be linked with disease progression. It remains unclear why this should be so when, clearly, it occurs in the context of high levels of p53 (ie, MDM2 is not performing its normal function to degrade p53).
The expression of p53 identifies a population of patients with RCC who are more likely to perform poorly. However, pathologists do not routinely monitor p53 in patients with RCC despite an increasing body of evidence suggesting that positive p53 status can predict recurrence and decreased survival. The ability to predict which patients will follow a poor disease course clearly would benefit clinicians, because more rigorous follow-up maybe indicated. Because small tumors (<4 cm; T1a) can present with metastases, more prognostic information clearly is required.81 The limitations of current follow-up practice and the potential for molecular markers to improve this situation are reviewed excellently by Rouviere et al.82 Moreover, 2 studies that included p53 status in their predictive nomograms demonstrated that it contributed to better prediction of survival.24,34 Although the tumor microenvironment and other host factors will have an impact on outcome, intrinsic tumor mutations predominantly determine tumor growth, invasion, and metastasis. Following the existing paradigm (which led to the discovery of the role of VHL mutations; the consequences of these for HIF-1α and HIF-2α expression and for oncogenic mediators, such as vascular endothelial growth factor; and recent developments in therapeutic targeting of such tumor determinants) should lead to the identification of these personalized indicators. Loss of p53 function is a key event in carcinogenesis; and the evidence suggests that p53 and MDM2 not only may provide 2 components of such improved prognostication but, at the same time, also may provide for novel, potentially tissue-specific therapeutic target(s) to improve treatment.
Acknowledgments
We apologize to colleagues whose articles we have not cited through lack of space.
Footnotes
CONFLICT OF INTEREST DISCLOSURES
Supported by Mersey Kidney Research, Cancer Research UK, and the North West Cancer Research Fund.
REFERENCES
- 1.Horner MJ, Ries LAG, Krapcho M, et al., editors. SEER Cancer Statistics Review, 1975-2006. National Cancer Institute; Bethesda, Md: [Accessed on June 15, 2009]. 2009. Based on the November 2008 SEER data submission; posted to the SEER website. Available at: http://info.cancerresearch.uk.org/cancerstats/types/kidney/incidence/ [Google Scholar]
- 2.Cancer Research UK . UK Kidney Cancer Incidence Statistics. Cancer Research UK; London, United Kingdom: 2009. 2009. [Google Scholar]
- 3.Lane BR, Rini BI, Novick AC, Campbell SC. Targeted molecular therapy for renal cell carcinoma. Urology. 2007;69:3–10. doi: 10.1016/j.urology.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Beltran A, Scarpelli M, Montironi R, Kirkali Z. 2004 WHO classification of the renal tumors of the adults. Eur Urol. 2006;49:798–805. doi: 10.1016/j.eururo.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 5.Hollstein M, Rice K, Greenblatt MS, et al. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994;22:3551–3555. [PMC free article] [PubMed] [Google Scholar]
- 6.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 7.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 8.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 9.Thut CJ, Goodrich JA, Tjian R. Repression of p53-mediated transcription by MDM2: a dual mechanism. Genes Dev. 1997;11:1974–1986. doi: 10.1101/gad.11.15.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 11.Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275:8945–8951. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- 12.Yang JY, Zong CS, Xia W, et al. MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Mol Cell Biol. 2006;26:7269–7282. doi: 10.1128/MCB.00172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu ML, Wikman F, Orntoft TF, et al. Impact of alterations affecting the p53 pathway in bladder cancer on clinical outcome, assessed by conventional and array-based methods. Clin Cancer Res. 2002;8:171–179. [PubMed] [Google Scholar]
- 14.Canman CE, Lim DS, Cimprich KA, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 15.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 16.Nenutil R, Smardova J, Pavlova S, et al. Discriminating functional and non-functional p53 in human tumours by p53 and MDM2 immunohistochemistry. J Pathol. 2005;207:251–259. doi: 10.1002/path.1838. [DOI] [PubMed] [Google Scholar]
- 17.Munro AJ, Lain S, Lane DP. P53 abnormalities and outcomes in colorectal cancer: a systematic review. Br J Cancer. 2005;92:434–444. doi: 10.1038/sj.bjc.6602358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slee EA, O’Connor DJ, Lu X. To die or not to die: how does p53 decide? Oncogene. 2004;23:2809–2818. doi: 10.1038/sj.onc.1207516. [DOI] [PubMed] [Google Scholar]
- 19.MacCallum DE, Hupp TR, Midgley CA, et al. The p53 response to ionising radiation in adult and developing murine tissues. Oncogene. 1996;13:2575–2587. [PubMed] [Google Scholar]
- 20.Lahav G, Rosenfeld N, Sigal A, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147–150. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- 21.Gurova KV, Hill JE, Razorenova OV, Chumakov PM, Gudkov AV. p53 pathway in renal cell carcinoma is repressed by a dominant mechanism. Cancer Res. 2004;64:1951–1958. doi: 10.1158/0008-5472.can-03-1541. [DOI] [PubMed] [Google Scholar]
- 22.Warburton HE, Brady M, Vlatkovic N, Linehan WM, Parsons K, Boyd MT. p53 regulation and function in renal cell carcinoma. Cancer Res. 2005;65:6498–6503. doi: 10.1158/0008-5472.CAN-05-0017. [DOI] [PubMed] [Google Scholar]
- 23.Carroll VA, Ashcroft M. Regulation of angiogenic factors by HDM2 in renal cell carcinoma. Cancer Res. 2008;68:545–552. doi: 10.1158/0008-5472.CAN-06-4738. [DOI] [PubMed] [Google Scholar]
- 24.Klatte T, Seligson DB, LaRochelle J, et al. Molecular signatures of localized clear cell renal cell carcinoma to predict disease-free survival after nephrectomy. Cancer Epidemiol Biomarkers Prev. 2009;18:894–900. doi: 10.1158/1055-9965.EPI-08-0786. [DOI] [PubMed] [Google Scholar]
- 25.Perret AG, Clemencon A, Li G, Tostain J, Peoc’h M. Differential expression of prognostic markers in histological subtypes of papillary renal cell carcinoma. BJU Int. 2008;102:183–187. doi: 10.1111/j.1464-410X.2008.07605.x. [DOI] [PubMed] [Google Scholar]
- 26.Phuoc NB, Ehara H, Gotoh T, et al. Immunohistochemical analysis with multiple antibodies in search of prognostic markers for clear cell renal cell carcinoma. Urology. 2007;69:843–848. doi: 10.1016/j.urology.2007.01.069. [DOI] [PubMed] [Google Scholar]
- 27.Kankuri M, Soderstrom KO, Pelliniemi TT, Vahlberg T, Pyrhonen S, Salminen E. The association of immunoreactive p53 and Ki-67 with T-stage, grade, occurrence of metastases and survival in renal cell carcinoma. Anticancer Res. 2006;26(5B):3825–3833. [PubMed] [Google Scholar]
- 28.Kramer BA, Gao X, Davis M, Hall M, Holzbeierlein J, Tawfik O. Prognostic significance of ploidy, MIB-1 proliferation marker, and p53 in renal cell carcinoma. J Am Coll Surg. 2005;201:565–570. doi: 10.1016/j.jamcollsurg.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 29.Langner C, Ratschek M, Rehak P, Tsybrovskyy O, Zigeuner R. The pT1a and pT1b category subdivision in renal cell carcinoma: is it reflected by differences in tumour biology? BJU Int. 2005;95:310–314. doi: 10.1111/j.1464-410X.2005.05289.x. [DOI] [PubMed] [Google Scholar]
- 30.Cho DS, Joo HJ, Oh DK, et al. Cyclooxygenase-2 and p53 expression as prognostic indicators in conventional renal cell carcinoma. Yonsei Med J. 2005;46:133–140. doi: 10.3349/ymj.2005.46.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shvarts O, Seligson D, Lam J, et al. p53 is an independent predictor of tumor recurrence and progression after nephrectomy in patients with localized renal cell carcinoma. J Urol. 2005;173:725–728. doi: 10.1097/01.ju.0000152354.08057.2a. [DOI] [PubMed] [Google Scholar]
- 32.Uzunlar AK, Sahin H, Yilmaz F, Ozekinci S. Expression of p53 oncoprotein and bcl-2 in renal cell carcinoma. Saudi Med J. 2005;26:37–41. [PubMed] [Google Scholar]
- 33.Zigeuner R, Ratschek M, Rehak P, Schips L, Langner C. Value of p53 as a prognostic marker in histologic subtypes of renal cell carcinoma: a systematic analysis of primary and metastatic tumor tissue. Urology. 2004;63:651–655. doi: 10.1016/j.urology.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 34.Kim HL, Seligson D, Liu X, et al. Using protein expressions to predict survival in clear cell renal carcinoma. Clin Cancer Res. 2004;10:5464–5471. doi: 10.1158/1078-0432.CCR-04-0488. [DOI] [PubMed] [Google Scholar]
- 35.Uchida T, Gao JP, Wang C, et al. Clinical significance of p53, mdm2, and bcl-2 proteins in renal cell carcinoma. Urology. 2002;59:615–620. doi: 10.1016/s0090-4295(01)01601-6. [DOI] [PubMed] [Google Scholar]
- 36.Olumi AF, Weidner N, Presti JC. p53 immunoreactivity correlates with Ki-67 and bcl-2 expression in renal cell carcinoma. Urol Oncol. 2001;6:63–67. doi: 10.1016/s1078-1439(00)00109-5. [DOI] [PubMed] [Google Scholar]
- 37.Ljungberg B, Bozoky B, Kovacs G, et al. p53 expression in correlation to clinical outcome in patients with renal cell carcinoma. Scand J Urol Nephrol. 2001;35:15–20. doi: 10.1080/00365590151030705. [DOI] [PubMed] [Google Scholar]
- 38.Girgin C, Tarhan H, Hekimgil M, Sezer A, Gurel G. P53 mutations and other prognostic factors of renal cell carcinoma. Urol Int. 2001;66:78–83. doi: 10.1159/000056575. [DOI] [PubMed] [Google Scholar]
- 39.Haitel A, Wiener HG, Baethge U, Marberger M, Susani M. mdm2 expression as a prognostic indicator in clear cell renal cell carcinoma: comparison with p53 overexpression and clinicopathological parameters. Clin Cancer Res. 2000;6:1840–1844. [PubMed] [Google Scholar]
- 40.Rioux-Leclercq N, Turlin B, Bansard J, et al. Value of immunohistochemical Ki-67 and p53 determinations as predictive factors of outcome in renal cell carcinoma. Urology. 2000;55:501–505. doi: 10.1016/s0090-4295(99)00550-6. [DOI] [PubMed] [Google Scholar]
- 41.Sejima T, Miyagawa I. Expression of bcl-2, p53 oncoprotein, and proliferating cell nuclear antigen in renal cell carcinoma. Eur Urol. 1999;35:242–248. doi: 10.1159/000019855. [DOI] [PubMed] [Google Scholar]
- 42.Vasavada SP, Novick AC, Williams BR. P53, bcl-2, and Bax expression in renal cell carcinoma. Urology. 1998;51:1057–1061. doi: 10.1016/s0090-4295(98)00132-0. [DOI] [PubMed] [Google Scholar]
- 43.Sinik Z, Alkibay T, Ataoglu O, et al. Nuclear p53 overexpression in bladder, prostate, and renal carcinomas. Int J Urol. 1997;4:546–551. doi: 10.1111/j.1442-2042.1997.tb00306.x. [DOI] [PubMed] [Google Scholar]
- 44.Papadopoulos I, Rudolph P, Weichert-Jacobsen K. Value of p53 expression, cellular proliferation, and DNA content as prognostic indicators in renal cell carcinoma. Eur Urol. 1997;32:110–117. [PubMed] [Google Scholar]
- 45.Zhang XH, Takenaka I, Sato C, Sakamoto H. p53 and HER-2 alterations in renal cell carcinoma. Urology. 1997;50:636–642. doi: 10.1016/S0090-4295(97)00258-6. [DOI] [PubMed] [Google Scholar]
- 46.Gelb AB, Sudilovsky D, Wu CD, Weiss LM, Medeiros LJ. Appraisal of intratumoral microvessel density, MIB-1 score, DNA content, and p53 protein expression as prognostic indicators in patients with locally confined renal cell carcinoma. Cancer. 1997;80:1768–1775. doi: 10.1002/(sici)1097-0142(19971101)80:9<1768::aid-cncr11>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 47.Shiina H, Igawa M, Urakami S, Shirakawa H, Ishibe T, Kawanishi M. Clinical significance of immunohistochemically detectable p53 protein in renal cell carcinoma. Eur Urol. 1997;31:73–80. doi: 10.1159/000474422. [DOI] [PubMed] [Google Scholar]
- 48.Moch H, Sauter G, Gasser TC, et al. p53 protein expression but not mdm-2 protein expression is associated with rapid tumor cell proliferation and prognosis in renal cell carcinoma. Urol Res. 1997;25(suppl 1):S25–S30. doi: 10.1007/BF00942044. [DOI] [PubMed] [Google Scholar]
- 49.Hofmockel G, Wittmann A, Dammrich J, Bassukas ID. Expression of p53 and bcl-2 in primary locally confined renal cell carcinomas: no evidence for prognostic significance. Anticancer Res. 1996;16(6B):3807–3811. [PubMed] [Google Scholar]
- 50.Chemeris G, Loktinov A, Rempel A, Schwarz M, Bannasch P. Elevated content of p53 protein in the absence of p53 gene mutations as a possible prognostic marker for human renal cell tumors. Virchows Arch. 1995;426:563–569. doi: 10.1007/BF00192110. [DOI] [PubMed] [Google Scholar]
- 51.Lipponen P, Eskelinen M, Hietala K, Syrjanen K, Gambetta RA. Expression of proliferating cell nuclear antigen (PC10), p53 protein and c-erbB-2 in renal adenocarcinoma. Int J Cancer. 1994;57:275–280. doi: 10.1002/ijc.2910570224. [DOI] [PubMed] [Google Scholar]
- 52.Kamel D, Turpeenniemi-Hujanen T, Vahakangas K, Paakko P, Soini Y. Proliferating cell nuclear antigen but not p53 or human papillomavirus DNA correlates with advanced clinical stage in renal cell carcinoma. Histopathology. 1994;25:339–347. doi: 10.1111/j.1365-2559.1994.tb01352.x. [DOI] [PubMed] [Google Scholar]
- 53.Bot FJ, Godschalk JC, Krishnadath KK, van der Kwast TH, Bosman FT. Prognostic factors in renal-cell carcinoma: immunohistochemical detection of p53 protein versus clinicopathological parameters. Int J Cancer. 1994;57:634–637. doi: 10.1002/ijc.2910570504. [DOI] [PubMed] [Google Scholar]
- 54.Uhlman DL, Nguyen PL, Manivel JC, et al. Association of immunohistochemical staining for p53 with metastatic progression and poor survival in patients with renal cell carcinoma. J Natl Cancer Inst. 1994;86:1470–1475. doi: 10.1093/jnci/86.19.1470. [DOI] [PubMed] [Google Scholar]
- 55.Boyd MT, Vlatkovic N. p53: a molecular marker for the detection of cancer. Expert Opin Med Diagn. 2008;2:1013–1024. doi: 10.1517/17530059.2.9.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anczukow O, Ware MD, Buisson M, et al. Does the nonsense-mediated mRNA decay mechanism prevent the synthesis of truncated BRCA1, CHK2, and p53 proteins? Hum Mutat. 2008;29:65–73. doi: 10.1002/humu.20590. [DOI] [PubMed] [Google Scholar]
- 57.Poeta ML, Manola J, Goldwasser MA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357:2552–2561. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gad S, Lefevre SH, Khoo SK, et al. Mutations in BHD and TP53 genes, but not in HNF1beta gene, in a large series of sporadic chromophobe renal cell carcinoma. Br J Cancer. 2007;96:336–340. doi: 10.1038/sj.bjc.6603492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawasaki T, Bilim V, Takahashi K, Tomita Y. Infrequent alteration of p53 pathway in metastatic renal cell carcinoma. Oncol Rep. 1999;6:329–333. doi: 10.3892/or.6.2.329. [DOI] [PubMed] [Google Scholar]
- 60.Contractor H, Zariwala M, Bugert P, Zeisler J, Kovacs G. Mutation of the p53 tumour suppressor gene occurs preferentially in the chromophobe type of renal cell tumour. J Pathol. 1997;181:136–139. doi: 10.1002/(SICI)1096-9896(199702)181:2<136::AID-PATH766>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 61.Dahiya R, Deng G, Selph C, Carroll P, Presti J., Jr. A novel p53 mutation hotspot at codon 132 (AAG→AGG) in human renal cancer. Biochem Mol Biol Int. 1998;44:407–415. doi: 10.1080/15216549800201422. [DOI] [PubMed] [Google Scholar]
- 62.Dijkhuizen T, Van den Berg E, Van den Berg A, et al. Chromosomal findings and p53-mutation analysis in chromophilic renal-cell carcinomas. Int J Cancer. 1996;68:47–50. doi: 10.1002/(SICI)1097-0215(19960927)68:1<47::AID-IJC9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 63.Oda H, Nakatsuru Y, Ishikawa T. Mutations of the p53 gene and p53 protein overexpression are associated with sarcomatoid transformation in renal cell carcinomas. Cancer Res. 1995;55:658–662. [PubMed] [Google Scholar]
- 64.Kuczyk MA, Serth J, Bokemeyer C, et al. Detection of p53 gene alteration in renal-cell cancer by micropreparation techniques of tumor specimens. Int J Cancer. 1995;64:399–406. doi: 10.1002/ijc.2910640609. [DOI] [PubMed] [Google Scholar]
- 65.Uchida T, Wada C, Wang C, Egawa S, Ohtani H, Koshiba K. Genomic instability of microsatellite repeats and mutations of H-, K-, and N-ras, and p53 genes in renal cell carcinoma. Cancer Res. 1994;54:3682–3685. [PubMed] [Google Scholar]
- 66.Kikuchi Y, Kishi T, Suzuki M, Furusato M, Aizawa S. Polymerase chain reaction-single strand conformation polymorphism analysis of the p53 gene in paraffin-embedded surgical material from human renal cell carcinomas. Virchows Arch. 1994;424:229–233. doi: 10.1007/BF00194605. [DOI] [PubMed] [Google Scholar]
- 67.Imai Y, Strohmeyer TG, Fleischhacker M, Slamon DJ, Koeffler HP. p53 mutations and MDM-2 amplification in renal cell cancers. Mod Pathol. 1994;7:766–770. [PubMed] [Google Scholar]
- 68.Reiter RE, Anglard P, Liu S, Gnarra JR, Linehan WM. Chromosome 17p deletions and p53 mutations in renal cell carcinoma. Cancer Res. 1993;53:3092–3097. [PubMed] [Google Scholar]
- 69.Suzuki Y, Tamura G, Satodate R, Fujioka T. Infrequent mutation of p53 gene in human renal cell carcinoma detected by polymerase chain reaction single-strand conformation polymorphism analysis. Jpn J Cancer Res. 1992;83:233–235. doi: 10.1111/j.1349-7006.1992.tb00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Torigoe S, Shuin T, Kubota Y, Horikoshi T, Danenberg K, Danenberg PV. p53 gene mutation in primary human renal cell carcinoma. Oncol Res. 1992;4:467–472. [PubMed] [Google Scholar]
- 71.Petitjean A, Mathe E, Kato S, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 72.Soussi T, Beroud C. Assessing TP53 status in human tumours to evaluate clinical outcome. Nat Rev Cancer. 2001;1:233–240. doi: 10.1038/35106009. [DOI] [PubMed] [Google Scholar]
- 73.Cordon-Cardo C, Latres E, Drobnjak M, et al. Molecular abnormalities of mdm2 and p53 genes in adult soft tissue sarcomas. Cancer Res. 1994;54:794–799. [PubMed] [Google Scholar]
- 74.Hirata H, Hinoda Y, Kikuno N, et al. MDM2 SNP309 polymorphism as risk factor for susceptibility and poor prognosis in renal cell carcinoma. Clin Cancer Res. 2007;13:4123–4129. doi: 10.1158/1078-0432.CCR-07-0609. [DOI] [PubMed] [Google Scholar]
- 75.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 76.Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang Y, Ludwig RL, Jensen JP, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell. 2005;7:547–559. doi: 10.1016/j.ccr.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 78.Motzer RJ, Bander NH, Nanus DM. Renal-cell carcinoma. N Engl J Med. 1996;335:865–875. doi: 10.1056/NEJM199609193351207. [DOI] [PubMed] [Google Scholar]
- 79.Guo C, Gasparian AV, Zhuang Z, et al. 9-Aminoacridine-based anticancer drugs target the PI3K/AKT/mTOR, NF-kappaB and p53 pathways. Oncogene. 2009;28:1151–1161. doi: 10.1038/onc.2008.460. [DOI] [PubMed] [Google Scholar]
- 80.Enge M, Bao W, Hedstrom E, Jackson SP, Moumen A, Selivanova G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell. 2009;15:171–183. doi: 10.1016/j.ccr.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 81.Nguyen MM, Gill IS. Effect of renal cancer size on the prevalence of metastasis at diagnosis and mortality. J Urol. 2009;181:1020–1027. doi: 10.1016/j.juro.2008.11.023. discussion 1027. [DOI] [PubMed] [Google Scholar]
- 82.Rouviere O, Bouvier R, Negrier S, Badet L, Lyonnet D. Nonmetastatic renal-cell carcinoma: is it really possible to define rational guidelines for post-treatment follow-up? Nat Clin Pract Oncol. 2006;3:200–213. doi: 10.1038/ncponc0479. [DOI] [PubMed] [Google Scholar]