

Abstract
Rationale
Accelerated lung function decline is a key COPD phenotype; however its genetic control remains largely unknown.
Methods
We performed a genome-wide association study using the Illumina Human660W-Quad v.1_A BeadChip. Generalized estimation equations were used to assess genetic contributions to lung function decline over a 5-year period in 4,048 European-American Lung Health Study participants with largely mild COPD. Genotype imputation was performed using reference HapMap II data. To validate regions meeting genome-wide significance, replication of top SNPs was attempted in independent cohorts. Three genes (TMEM26, ANK3 and FOXA1) within the regions of interest were selected for tissue expression studies using immunohistochemistry.
Measurements and Main Results
Two intergenic SNPs (rs10761570, rs7911302) on chromosome 10 and one SNP on chromosome 14 (rs177852) met genome-wide significance after Bonferroni. Further support for the chromosome 10 region was obtained by imputation, the most significantly associated imputed SNPs (rs10761571, rs7896712) being flanked by observed markers rs10761570 and rs7911302. Results were not replicated in four general population cohorts or a smaller cohort of subjects with moderate to severe COPD; however, we show novel expression of genes near regions of significantly associated SNPS, including TMEM26 and FOXA1 in airway epithelium and lung parenchyma, and ANK3 in alveolar macrophages. Levels of expression were associated with lung function and COPD status.
Conclusions
We identified two novel regions associated with lung function decline in mild COPD. Genes within these regions were expressed in relevant lung cells and their expression related to airflow limitation suggesting they may represent novel candidate genes for COPD susceptibility.
Keywords: COPD, lung function decline, GWAS, genome wide association, genes, polymorphisms
INTRODUCTION
COPD is the third leading cause of death in the United States and the fifth leading cause of death worldwide, and its prevalence is expected to increase in upcoming decades.1–3 The majority of COPD is caused by environmental exposures. In developed countries, this exposure is primarily cigarette smoke; however only a minority of all smokers develops COPD.4 Furthermore, strong familial aggregation of COPD suggests a large role for genetic susceptibility to the detrimental effects of smoking.5
Genome-wide association studies (GWAS) have identified several novel COPD candidate genes, including FAM13A, HHIP, and CHRNA 3/5.6;7 However, two pathogenetic pathways likely contribute to the development of COPD; attainment of lower than maximal lung size and function by young adulthood, and an accelerated decline in lung function over time, and it is likely that different genes impart susceptibility to these processes. Although heritability of lung function decline is well-established8 and several candidate gene association studies have supported a genetic basis for lung function decline9–19, the genes controlling loss of lung function in smokers remain largely unknown.20
The NHLBI-supported Lung Health Study (LHS) was a multi-center randomized clinical trial in the U.S. and Canada to determine whether or not a program of smoking intervention and use of an inhaled bronchodilator could slow the rate of decline in pulmonary function in smokers with mild airflow limitation. The LHS has been a landmark study in understanding the longitudinal effects of smoking on short and long-term outcomes in mild COPD. It is uniquely suited to investigate causes of lung function decline since subjects were comprehensively assessed for smoking habits and lung function at each annual visit over a 5-year period with exceptionally high follow-up rates.21 Here we report results from the first GWAS of lung function decline in individuals with mild COPD and report replication of our results and lung tissue expression of relevant genes.
METHODS
Population
The LHS was a multicenter clinical study of current smokers with mild COPD. Lung function was measured annually over a 5-year period21;22 and data from Annual Visits 1 to 5 were used for the current analyses. Additional details of LHS 23;24 provided in Online Resource. The GWAS included 4,251 European American LHS participants with adequate DNA.
Genotyping and Quality Control
Samples were genotyped using the Illumina Human660W-Quad v.1_A BeadChip (see Online Resource). Sample and SNP quality control (QC) was done using IlluminaBeadStudio (see Online Resource). Overall, 98.4% of samples (n=4,181) passed initial quality-control standards and genotypes were released for 559,766 SNPs. 133 additional samples were removed for quality control criteria for a final sample of n=4,048.
Tests for association
Generalized estimation equations were used to test for the genetic contribution to lung function decline.25 Baseline predictors included gender, age, and study site. BMI and smoking status were included as time-varying covariates. Smoking was encoded in three different time-varying covariates to model effects of starting/quitting smoking: 1) average number of cigarettes smoked per day over previous year, 2) smoking status, defined as smoker or non-smoker, for the current year and 3) for the previous year. Interactions of smoking and gender with time and the first five principal components computed via Eigenstrat, in order to address the potential effects of population stratification, were included. The genetic contribution to lung function decline was assessed by testing the parameter for SNP by time interaction (allowing for genotype main effect in the model, but not part of the hypothesis test), using dominant coding for genotype. This coding allows for two separate trajectories of FEV1 thus comparing lung function decline of subjects with and without variant alleles. Because of its known strong link with absolute FEV1, height was included as a covariate in sensitivity analysis. To account for multiple testing, a Bonferroni threshold of p<9.45 × 10−8 was considered statistically significant, based on the 528,817 SNPs passing QC.
Imputation
We imputed genotypes for all polymorphic HapMap Phase 2 SNPs using hidden Markov model with MACH version 1.0 (http://www.sph.umich.edu/csg/abecasis/MACH/). (see Online Resource).
Replication
Replication of the top 10 genotyped SNPs associated with lung function decline was attempted in independent cohorts sampled from distinct general populations (Cardiovascular Health Study (CHS), Framingham Heart Study (FHS) Offspring cohort, Baltimore Longitudinal Study of Aging (BLSA) and Dutch-Belgian Lung Cancer Screening Trial (NELSON) cohort) and one cohort of smokers with moderate-severe COPD (Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE)). (see Online Resource).
Immunohistochemistry
Immunohistochemistry was used to test for the pulmonary expression of genes within the two loci meeting genome-wide significance, (chr 10: ANK3, CDK1, RHOBTB1, TMEM26; chr14: FOXA1) in lung tissue obtained from both the Lung Tissue Research Consortium (LTRC) and a tissue bank from St. Paul’s Hospital, Vancouver, Canada.26 ANK3, TMEM26 and FOXA1 were selected based on their proximity to risk loci and evidence of significant expression in the lung in published data and real-time PCR (data not shown). Carriers of the risk alleles with and without airflow obstruction were compared using quantitative morphology (see Online Resource).
RESULTS
Clinical characteristics of subjects are presented in Table 1. The GWAS sample represents 69.7% of the 5,638 European American volunteers who participated in the LHS study. Importantly, LHS subjects included in the GWAS had demographics (including age, gender and BMI) and rates of lung function decline (mean annual change in FEV1% predicted) similar to those not included in the GWAS: −0.94 %/yr vs. −1.01 %/yr, p=0.23, reflecting little selection bias for our primary outcome. They were, however, more likely to have quit smoking after 5 years.
Table 1.
Lung Health Study Subject Characteristics
| n=4048 | |
|---|---|
| Baseline Characteristics | |
| Mean age (SD) | 48.6 (6.7) |
| Male, N (%) | 2554 (63.1) |
| BMI, kg/m2 | 25.5 (3.9) |
| Smoking, pack-years (SD) | 40.6 (18.7) |
| Post-BD Lung function | |
| FEV1, L (SD) | 2.8 (0.6) |
| FVC | 4.3 (1.0) |
| FEV1/FVC | 0.65 (0.06) |
| FEV1 % predicted, % | 78.6 (9.0) |
| Longitudinal Characteristics at 5 years | |
| Post-BD ΔFEV1, mL/year | −56.8 (79.3) |
| Post-BD ΔFEV1 % predicted, %/yr | −1.02 (2.26) |
A summary of the tests for association between individual SNPs across the entire genome and lung function decline are presented in Figure 1. A Q-Q plot of these results is presented in Figure 2, and the 10 most strongly associated SNPs (representing seven different regions) are listed in Table 2. Two SNPs (rs10761570 and rs7911302) on chromosome 10 and one SNP on chromosome 14 (rs177852) met a Bonferroni threshold for genome-wide significance (p<9.45×10−8). To refine these two regions, we imputed SNPs in these regions of chromosomes 10 and 14 (Figure 3). Observed and imputed genotypes for those two regions overall were in high (chr10) to moderate (chr14) linkage disequilibrium (LD). The most significantly associated imputed SNPs (rs10761571 and rs7896712, both p=3.7 × 10−8) on chromosome 10 are flanked by the two genotyped SNPs that reached genome-wide significance. Several additional SNPs in this region reached genome-wide or near genome-wide significance. One imputed SNP (rs12147245, p=2.1 × 10−8) in chromosome 14 was more significantly associated with lung function decline than the genotyped SNP, and two additional, imputed SNPs were equally associated (rs177858 and rs177859, p=9.9×10−7) (Figure 4).
Figure 1. Manhattan Plot.
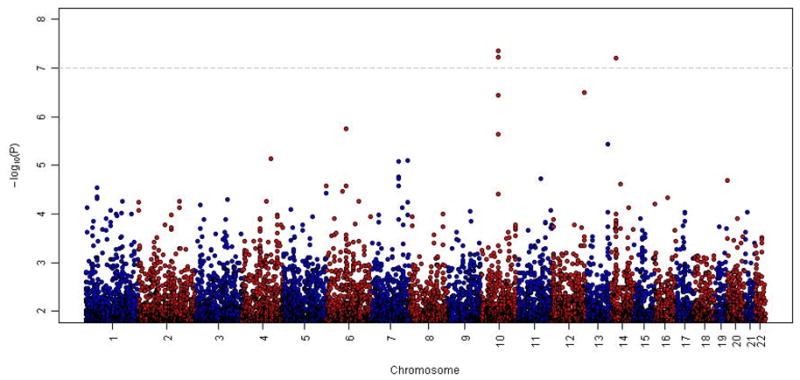
Genome-wide association for FEV1 decline ordered by chromosome position. The X-axis shows chromosome position and the Y-axis shows the −log10(P-value).
Figure 2. QQ plot.
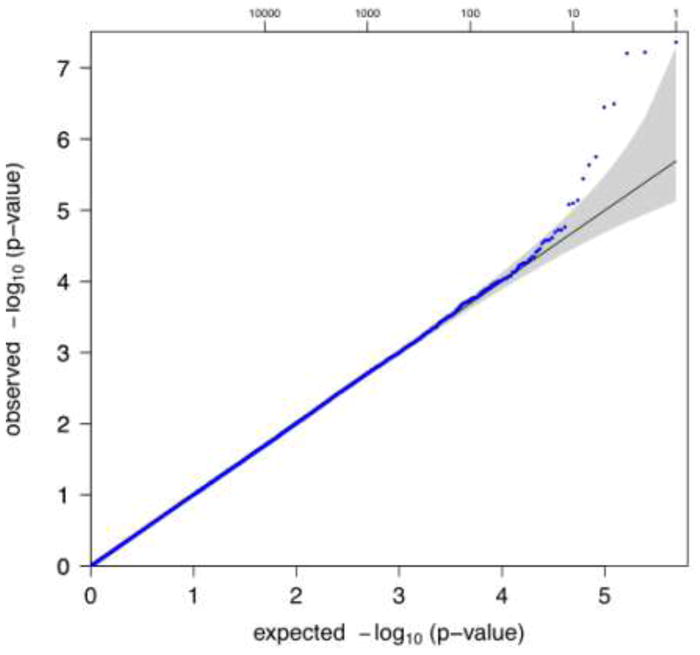
Quantile-quantile plot of GWA results for FEV1 decline
Table 2.
Top ten SNPs representing seven regions in Lung Health Study GWAS
| rs# | Chrom | Position | Minor allele | MAF | Beta (cc/yr) | P | gene | Left gene | Right gene |
|---|---|---|---|---|---|---|---|---|---|
| rs7922793 | 10 | 62496958 | C | 0.3614 | −8.2 | 2.3E-06 | NA | RHOBTB1 | TMEM26 |
| rs7911302 | 10 | 62577232 | A | 0.4372 | −9.9 | 4.4E-08 | NA | RHOBTB1 | TMEM26 |
| rs10761570 | 10 | 62576961 | T | 0.4367 | −9.8 | 6.0E-08 | NA | RHOBTB1 | TMEM26 |
| rs4948444 | 10 | 62600003 | G | 0.4474 | −9.3 | 3.6E-07 | NA | RHOBTB1 | TMEM26 |
| rs177852 | 14 | 37181621 | T | 0.3307 | −9.3 | 6.3E-08 | NA | LOC100128066 | TTC6 |
| rs2030436 | 12 | 128460517 | G | 0.1368 | −10.2 | 3.2E-07 | TMEM132D | NLRP9P | FLJ31485 |
| rs12194741 | 6 | 73433589 | C | 0.2224 | 8.3 | 1.8E-06 | KCNQ5 | LOC643067 | LOC100128189 |
| rs2275456 | 1 | 53308613 | C | 0.04329 | 12.9 | 3.1E-06 | PODN | SCP2 | LOC100129490 |
| rs7331728 | 13 | 104219264 | G | 0.1653 | −8.8 | 3.6E-06 | NA | LOC728183 | DAOA |
| rs4330400 | 4 | 136256860 | G | 0.3799 | −7.9 | 7.3E-06 | NA | LOC729551 | LOC100132574 |
Figure 3. Results of lung function decline analysis on chromosome 10 (Panel A) and chromosome 14 (Panel B) regions of association.
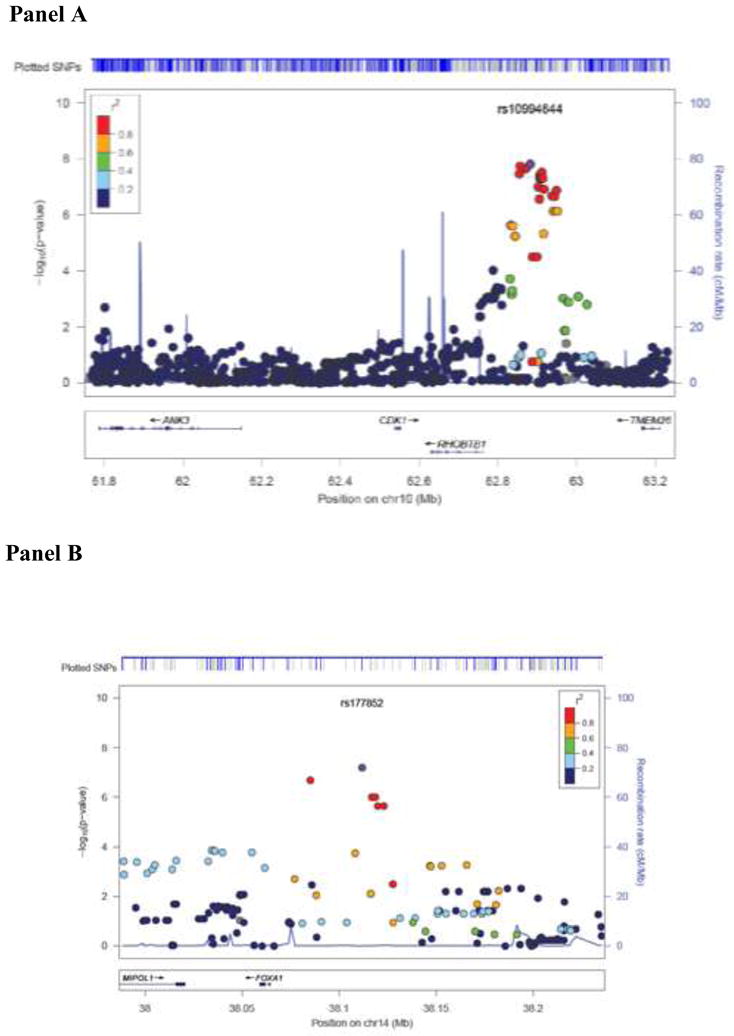
The genotyped SNPs in blue and the imputed SNPs in gray are shown in the upper track of the figure labeled as ‘plotted SNPs’. The relative location of the genes in each region and the direction of transcription are shown in the bottom track of the figure, and the chromosomal position is shown on the x axis. The light blue line shows the recombination rate across the region (right y axis), and the left y axis shows the significance of the associations. The purple circle shows the P value for the top signal in each region (rs10994644 and rs177852, respectively). The remaining circles show the P values for all other SNPs and are color coded according to the level of LD with the top SNP in the 1000 genome Nov 2010 CEU population (red, r2 > 0.8; orange, r2 = 0.6–0.8; green, r2 = 0.4–0.6; light blue, r2 = 0.2–0.4; dark blue, r2 = 0.0–0.2).
Figure 4. Linkage Disequilibrium plots.
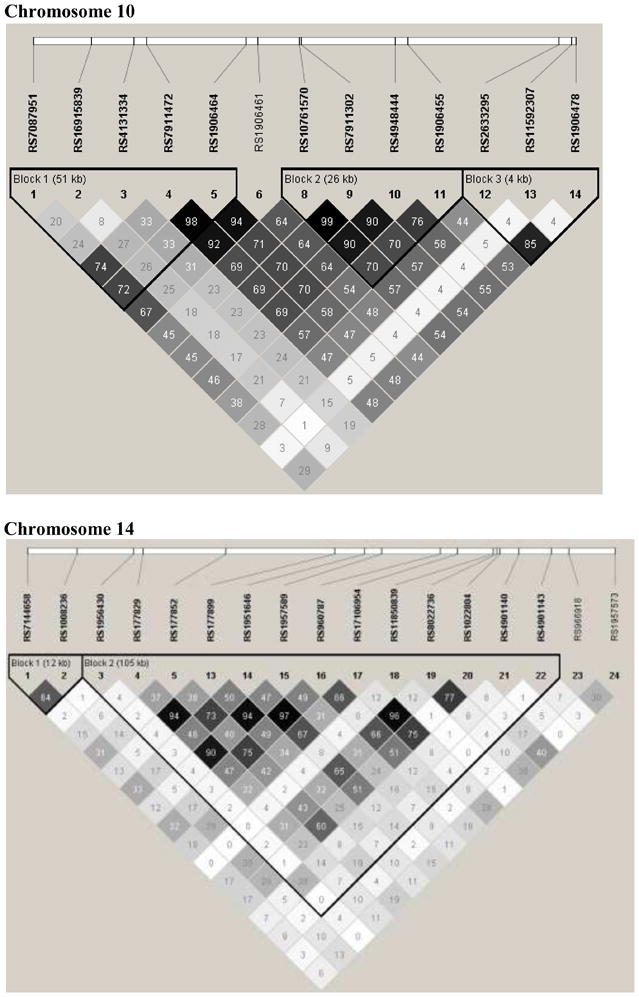
Plots of linkage disequilibrium (R2) patterns for chromosome 10 and 14 regions of interest: black squares for strong LD, gray squares nonsignificant LD, and white squares for little or no LD.
Height significantly explained some variability in the absolute measurement of FEV1, but was not associated with lung function decline in our preliminary analyses, and the results of association of genetic markers with lung function decline were not meaningfully different when height was included as a covariate in the model in sensitivity analyses (data not shown).
Replication
To test for generalizability of associations with lung function decline in general population cohorts (smokers and non-smokers without regard to pulmonary function), we compared our results with GWAS data from four independent general population samples with longitudinal data on lung function (Table 3). In addition, our results were compared to those of the ECLIPSE cohort which included subjects with GOLD stage II-IV COPD. In the NELSON cohort we observed nominal significance for rs2030436 on chromosome 12 (p=0.05) which did not hold its significance after Bonferroni correction. No significant evidence for association was observed in the CHS, FHS, BLSA or ECLIPSE cohorts.
Table 3.
Replication Results
| FHS N=850 |
CHS N=1299 |
BLSA N=473 |
NELSON N=1932 |
ECLIPSE N=1951 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| SNP | Chr | Position | Minor Allele | Beta (cc/yr) | p | Beta (cc/yr) | p | Beta (cc/yr) | p | Beta (cc/yr) | p | Beta (cc/yr) | p |
| rs7922793 | 10 | 62826952 | C | −2.2 | 0.51 | 3.5 | 0.29 | −0. 3 | 0.87 | 2.8 | 0.42 | 4.5 | 0.38 |
| rs10761570 | 10 | 62906955 | T | −1.9 | 0.60 | 3.2 | 0.37 | −0. 5 | 0.80 | 0.1 | 0.98 | 1.7 | 0.74 |
| rs7911302 | 10 | 62907226 | A | −1.9 | 0.60 | 3.0 | 0.39 | −0. 5 | 0.79 | −0.1 | 0.98 | 1.7 | 0.74 |
| rs4948444 | 10 | 62929997 | G | −1.0 | 0.79 | 3.6 | 0.30 | −0. 1 | 0.95 | 0. 2 | 0.96 | 1.6 | 0.77 |
| rs177852 | 14 | 38111870 | T | 0.9 | 0.74 | −1.1 | 0.73 | −1.0 | 0.67 | −2.6 | 0.46 | 4.6 | 0.36 |
| rs12194741 | 6 | 73376868 | C | 2.8 | 0.40 | −2.7 | 0.41 | −3.3 | 0.21 | −6.8 | 0.09 | −2.8 | 0.59 |
| rs2030436 | 12 | 129894564 | G | −1.4 | 0.72 | 3.1 | 0.42 | −2.8 | 0.33 | −9.8 | 0.05 | −1.9 | 0.76 |
| rs4330400 | 4 | 136037410 | G | 4.0 | 0.49 | −0.5 | 0.88 | 1.2 | 0.57 | 3.3 | 0.34 | 0.2 | 0.97 |
| rs7331728 | 13 | 105421263 | G | −0.6 | 0.85 | 0.3 | 0.93 | 2.3 | 0.32 | 1.9 | 0.70 | 3.8 | 0.50 |
| rs2275456* | 1 | 53536025 | C | 2.0 | 0.70 | − | − | 2.7 | 0.48 | − | − | 1.3 | 0.89 |
Not genotyped in CHS or NELSON
Tissue Validation for chromosome 14 locus
Staining for FOXA1 in the airway epithelium (a) and parenchyma (b) using tissue samples from the St. Paul’s’ Hospital repository is shown in supplementary Figure E1. In the airway epithelium, nuclei in all layers of the pseudostratified epithelium were stained positively for FOXA1. In the parenchyma, staining was confined to the alveolar epithelium and appeared to be associated with type II pneumocytes. There was a significant relationship between the extent of staining of the airway and parenchymal epithelium (r2=0.56, p<0.0001). FOXA1 expression was higher in the epithelium and parenchyma of smokers with airways obstruction compared with smokers without airways obstruction (p<0.001, both comparisons). There was no significant effect of FOXA1 genotype on FOXA1 protein expression (Figure E3). These results were not confounded by smoking status, as there was no differential expression by whether subjects were current or former smokers. There was no staining of alveolar macrophages. Similar findings were obtained in the LTRC cohort (data not shown).
Tissue validation for chromosome 10 locus
Using specimens from St. Paul’s Hospital cytoplasmic staining for TMEM26 occurred in the airway epithelium (a) and parenchyma (b), as shown in supplementary Figure E4 and there was a significant but weak to modest correlation between the extent of staining of the airway and parenchymal epithelium (r2=0.22, p=0.002). Subjects with airways obstruction had significantly decreased epithelial TMEM26 staining, and increased parenchymal TMEM26 staining, compared to those without airways obstruction (Figure E5). There was no effect of genotype and no evidence of interaction between genotype and lung function. Current smokers had significantly lower expression in the airway epithelium (31% vs. 45%, p=0.03) but there was no effect of current smoking on alveolar staining. Similarly, using the LTRC samples, immunohistochemical staining of lung sections for TMEM26 showed protein expression in the cytoplasm of the airway epithelium. Quantitative immunohistochemistry revealed no significant difference in staining between epithelium of subjects with COPD compared with epithelium of smokers without COPD (39.8% vs. 48.5%, p=0.17; Figure 5A). Subjects without COPD who were homozygous for the chromosome 10 risk allele (n=3) as compared to those without the risk allele (n=3) had increased TMEM26 staining (47% vs. 20%, p=0.02), but there was no statistical difference between genotypes in those with COPD.
Figure 5. Localization of TMEM26 and ANK3 in lungs of smokers with and without COPD.
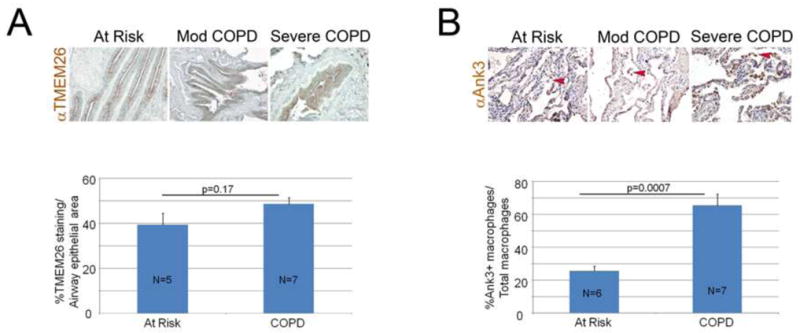
A. Representative TMEM26 immunohistochemical staining of lung sections from at risk and COPD patients shows staining in the cytoplasm of airway epithelium. Bottom panel shows quantitative immunohistochemistry demonstrating no significant difference in staining in epithelium of COPD patients and smokers without COPD. B. Representative ANK3 immunohistochemical staining of lung sections from at risk and COPD patients shows predominant staining in the alveolar macrophage compartment. Arrowheads denote macrophages. Bottom panel shows quantitative immunohistochemistry demonstrating increased staining in macrophages of COPD patients compared with smokers without COPD. All figures are 20X magnification. HZ-homozygosity for chromosome 10 at risk haplotype. WT-homozygosity for chromosome 10 wild-type haplotype. Numbers in columns indicate number of samples examined.
Representative ANK3 immunohistochemical staining of lung sections from at-risk and COPD patients from the LTRC shows predominant staining in the alveolar macrophage compartment. Quantitative immunohistochemistry demonstrated increased staining in macrophages of COPD specimens compared with at-risk smokers without COPD (25.6% vs. 65.4%, p=0.0007; Figure 5B). There was no significant effect of genotype at the chromosome 10 region with ANK3 macrophage expression.
DISCUSSION
In this first GWAS of lung function decline, we have identified two novel regions, one on chromosome 10 and one on chromosome 14, which were associated with decline in FEV1 in a cohort of European Americans with mild COPD. The Lung Health Study is uniquely suited to investigate genetic risk to smoking-induced lung function decline as it includes over 4,000 smokers with airflow limitation who have exquisitely characterized smoking status and annual lung function over 5 years and designed with rate of lung function decline as the primary endpoint of the study. Two intergenic SNPs (rs10761570 and rs7911302) on chromosome 10 and one SNP on chromosome 14 (rs177852) met a Bonferroni threshold of genome-wide significance for the outcome, rate of decline of lung function. Further support for the chromosome 10 locus was obtained by imputing additional markers, where the most significantly associated imputed SNPs (rs10761571 and rs7896712) were flanked by the two observed markers (rs10761570 and rs7911302). Immunohistochemistry performed on lung tissue from smokers demonstrated, for the first time, that ANK3, TMEM26 and FOXA1 proteins, which are encoded by genes neighboring the most significant SNPs in the GWAS, were expressed in the airway epithelium, lung parenchyma or alveolar macrophages, with differential expression in smokers with airways obstruction compared to smokers without airways obstruction. These results implicate these genes as novel biologically plausible candidate genes for lung function decline and COPD pathogenesis.
Using the ENCODE database (http://www.genome.gov/10005107), the chromosome 10q21.2 region has been determined to be a regulatory region with very specific binding sites for several regulatory proteins, including NFkB and STAT1, both of which have been implicated in COPD.27;28 Furthermore, the most significantly associated SNPs are flanked by RHOBTB1, CDK1 and ANK3 (upstream) and TMEM26 (downstream). Using both real-time PCR and immunohistochemistry, we saw protein expression of ANK3, TMEM26 and FOXA1, but negligible expression of RHOBTB1 and CDK1, in human lung tissue specimens irrespective of COPD status (data not shown). Ankyrins play a key role in activities such as cell motility, activation, proliferation, contact and the maintenance of specialized membrane domains. In humans, ANK3 proteins have been identified in a variety of epithelial cells, bone marrow macrophages and neurons.29;30 We demonstrate the novel finding that ANK3 protein is expressed in alveolar macrophages. Furthermore, its expression was higher in alveolar macrophages from subjects with COPD compared to at risk smokers without evidence of airways obstruction. Little is known about TMEM26 (transmembrane protein 26). It has previously been identified in a GWAS as being associated with blood pressure.31 This is the first demonstration that TMEM26 is expressed in airway epithelium and lung parenchyma, and smokers with airways obstruction had increased parenchymal, but decreased TMEM26 expression compared to smokers without airways obstruction. Except for subjects without COPD who were homozygous for the chromosome 10 risk allele (rs177852 (CC)) having increased TMEM26 staining compared to those without the risk allele in the LTRC samples, no associations were observed between genotype and protein expression. This may be because the susceptibility alleles do not influence risk by changing the level of protein expression, or because immunohistochemistry is insensitive to detect changes in the level of expression. Immunohistochemistry has utility for the detection of changes in the area fraction stained positive and but not the intensity of staining as would be the case if the same number of cells expressed more protein. In addition, we were likely underpowered to detect an association given our small sample size in the immunohistochemistry studies. Although the genetic associations observed in the current study do not implicate any of these genes directly, the location of the association signal and the novel finding of protein expression of ANK3 and TMEM26 in lung tissue and association with airways obstruction suggest these genes may be involved in COPD pathogenesis.
The chromosome 14 region also met genome-wide significance for association with lung function decline. The gene encoding forkhead box protein A1 (FOXA1), previously termed hepatocyte nuclear factor (HNF-3), is in the vicinity of the most significant SNPs and encodes a transcription factor previously known to be expressed in epithelial cells of the conducting airways and in type II alveolar epithelial cells.32 FOXA1 may regulate respiratory epithelial differentiation and structural maturation of the lung during development 32 and has recently been reported to play a role in regulation of apoptotic cell death in various diseases. Specifically, it has been shown to play a pro-apoptotic role during oxidative-stress induced apoptosis in type II pneumocytes.33 Over-expression of FOXA1 has been shown to promote apoptosis,34 which has been implicated as a mechanism contributing to COPD and emphysema progression.35 We demonstrate expression of FOXA1 protein in airway epithelium and lung parenchyma. Furthermore, FOXA1 expression was inversely associated with lung function, increased expression in alveolar and airway epithelium in subjects with severe airway obstruction compared with smokers without airways obstruction. An RNA seq-based survey of a limited number of small airway brush specimens confirmed expression of FOXA1 and ANK3 genes in the lungs of COPD patients (R.C., data not shown). Together these data support FOXA1 as a biologically plausible candidate gene for the pathogenesis of COPD.
There are several potential reasons for the lack of replication in general population cohorts. First, genetic factors influencing susceptibility to cigarette smoke might have little or no influence on the rate of lung function decline due to aging in general population cohorts. Accelerated lung function decline is likely to be strongly affected by environmental exposures, particularly cigarette smoking, and therefore requires both a sufficient environmental exposure and a genetically susceptible individual. Second, in the LHS accurately accounting for the effects of cessation and re-initiation of smoking greatly affected an individual’s lung function trajectory. For this reason, we included subjects’ smoking habits in three different time-varying covariates, including average number of cigarettes smoked per day over the previous year at each annual visit and a subject’s smoking status for each of the two previous years to model the effects of starting / quitting smoking. It is therefore not surprising that, in general populations not specifically designed to assess longitudinal changes in lung function, self-report of cigarette smoking ascertained at intervals spanning several years may not accurately reflect the intermittency of smoking exposure. Failure to incorporate effects of intermittent smoking may lead to a considerable residual confounding by smoking exposure, and consequently, an underestimation of the importance of genetic factors. Indeed, previous results from the FHS showed estimates of heritability in lung function decline among relatives increased when the analysis was restricted to relative pairs concordant for smoking status.8 Given the comprehensive evaluation of smoking status at each annual visit in LHS, this cohort of smokers is uniquely suited to identify genetic factors underlying smoking-related lung function decline. Third, despite high correlation between pre- and post-BD spirometry, pre-BD FEV1 may be reduced due to bronchoconstriction as well as by airway remodeling. Thus, genetic factors contributing to post-BD FEV1 may not be identified in a study where the outcome is pre-BD spirometry. The ECLIPSE cohort is most similar to the LHS cohort because it involves subjects who were recruited because they had COPD and because it incorporated regular assessment of post-BD spirometry and smoking status. However, ECLIPSE includes only subjects who have moderate, severe, or very severe COPD (mean baseline FEV1 of 48% predicted).36 The factors associated with lung function decline in the later stages of disease may be different from those associated with rates of decline in early-stage COPD. For instance, the rate of lung function decline may decelerate in those with more severe disease;37 or may be modified by medications used to treat those with more severe disease (i.e., combination therapy with inhaled corticosteroids and long-acting beta agonists).37 One notable difference between the studies was that bronchodilator reversibility was found to be associated with lung function decline in subjects with moderate-severe COPD in ECLIPSE but not in those with early stage COPD in LHS.36;38 Lack of available cohorts with mild-moderate COPD with longitudinal assessment of post-BD lung function and smoking status ascertained at annual visits was likely a key limiting factor in appropriate replication; however, despite correction for multiple testing, we cannot exclude the possibility that the signals were false-positive results of the GWAS.
In summary, we have identified two novel regions, one on chromosome 10 and one on chromosome 14, associated with decline in FEV1 in a longitudinal cohort of smokers with COPD. These results appear to be unique to smoking individuals who have mild to moderate COPD. In addition, immunohistochemistry results confirmed localization of proteins coded by genes in the vicinity of the most significant SNPs, including TMEM26, ANK3 and FOXA1, and suggest differential expression in the lungs of subjects with COPD compared to non-obstructed smokers. These results implicate two novel loci likely involved in lung function decline in smokers with early COPD.
Supplementary Material
Acknowledgments
This project was part of the Gene, Environment Association Studies (GENEVA) Consortium funded by the National Human Genome Research Institute (NHGRI) to enhance communication and collaboration among researchers conducting genome-wide studies of complex diseases. Our group benefited greatly from the work and efforts of the entire consortium, especially the Coordinating Center (directed by B. Weir and C. Laurie of the University of Washington) in data cleaning and preparation for submission to the Database for Genotypes and Phenotypes (dbGaP). Special thanks also to David Levine for additional, technical support. We also acknowledge the leadership of T. Manolio of NHGRI. We would also like to thank Helen Voelker and Kathy Farnell of the LHS Data Coordinating Center, University of Minnesota for assistance with the LHS database. We would also like to thank Corinne Boehm and Jane Romm of the Center for Inherited Disease Research, Johns Hopkins University, for technical support. The principal investigators and senior staff of the clinical and coordinating centers, the NHLBI, and members of the Safety and Data Monitoring Board of the Lung Health Study are as follows:
Case Western Reserve University, Cleveland, OH: M.D. Altose, M.D. (Principal Investigator), C.D. Deitz, Ph.D. (Project Coordinator); Henry Ford Hospital, Detroit, MI: M.S. Eichenhorn, M.D. (Principal Investigator), K.J. Braden, A.A.S. (Project Coordinator), R.L. Jentons, M.A.L.L.P. (Project Coordinator); Johns Hopkins University School of Medicine, Baltimore, MD: R.A. Wise, M.D. (Principal Investigator), C.S. Rand, Ph.D. (Co-Principal Investigator), K.A. Schiller (Project Coordinator); Mayo Clinic, Rochester, MN: P.D. Scanlon, M.D. (Principal Investigator), G.M. Caron (Project Coordinator), K.S. Mieras, L.C. Walters; Oregon Health Sciences University, Portland: A.S. Buist, M.D. (Principal Investigator), L.R. Johnson, Ph.D. (LHS Pulmonary Function Coordinator), V.J. Bortz (Project Coordinator); University of Alabama at Birmingham: W.C. Bailey, M.D. (Principal Investigator), L.B. Gerald, Ph.D., M.S.P.H. (Project Coordinator); University of California, Los Angeles: D.P. Tashkin, M.D. (Principal Investigator), I.P. Zuniga (Project Coordinator); University of Manitoba, Winnipeg: N.R. Anthonisen, M.D. (Principal Investigator, Steering Committee Chair), J. Manfreda, M.D. (Co-Principal Investigator), R.P. Murray, Ph.D. (Co-Principal Investigator), S.C. Rempel-Rossum (Project Coordinator); University of Minnesota Coordinating Center, Minneapolis: J.E. Connett, Ph.D. (Principal Investigator), P.L. Enright, M.D., P.G. Lindgren, M.S., P. O’Hara, Ph.D., (LHS Intervention Coordinator), M.A. Skeans, M.S., H.T. Voelker; University of Pittsburgh, Pittsburgh, PA: R.M. Rogers, M.D. (Principal Investigator), M.E. Pusateri (Project Coordinator); University of Utah, Salt Lake City: R.E. Kanner, M.D. (Principal Investigator), G.M. Villegas (Project Coordinator); Safety and Data Monitoring Board: M. Becklake, M.D., B. Burrows, M.D. (deceased), P. Cleary, Ph.D., P. Kimbel, M.D. (Chairperson; deceased), L. Nett, R.N., R.R.T. (former member), J.K. Ockene, Ph.D., R.M. Senior, M.D. (Chairperson), G.L. Snider, M.D., W. Spitzer, M.D. (former member), O.D. Williams, Ph.D.; Morbidity and Mortality Review Board: T.E. Cuddy, M.D., R.S. Fontana, M.D., R.E. Hyatt, M.D., C.T. Lambrew, M.D., B.A. Mason, M.D., D.M. Mintzer, M.D., R.B. Wray, M.D.; National Heart, Lung, and Blood Institute staff, Bethesda, MD: S.S. Hurd, Ph.D. (Former Director, Division of Lung Diseases), J.P. Kiley, Ph.D. (Former Project Officer and Director, Division of Lung Diseases), G. Weinmann, M.D. (Former Project Officer and Director, Airway Biology and Disease Program, DLD), M.C. Wu, Ph.D. (Division of Epidemiology and Clinical Applications).
Principal investigators and centers participating in ECLIPSE include: Bulgaria: Y Ivanov, Pleven; K Kostov, Sofia. Canada: J Bourbeau, Montreal; M Fitzgerald, Vancouver; P Hernández, Halifax; K Killian, Hamilton; R Levy, Vancouver; F Maltais, Montreal; D O’Donnell, Kingston. Czech Republic: J Krepelka, Praha. Denmark: J Vestbo, Hvidovre. The Netherlands: E Wouters, Horn. New Zealand: D Quinn, Wellington. Norway: P Bakke, Bergen, Slovenia: M Kosnik, Golnik. Spain: A Agusti, Jaume Sauleda, Palma de Mallorca. Ukraine: Y Feschenko, Kiev; V Gavrisyuk, Kiev; L Yashina, Kiev. UK: L Yashina, W MacNee, Edinburgh; D Singh, Manchester; J Wedzicha, London. USA: A Anzueto, San Antonio, TX; S Braman, Providence. RI; R Casaburi, Torrance CA; B Celli, Boston, MA; G Giessel, Richmond, VA; M Gotfried, Phoenix, AZ; G Greenwald, Rancho Mirage, CA; N Hanania, Houston, TX; D Mahler, Lebanon, NH; B Make, Denver, CO; S Rennard, Omaha, NE; C Rochester, New Haven, CT; P Scanlon, Rochester, MN; D Schuller, Omaha, NE; F Sciurba, Pittsburg, PA; A Sharafkhaneh, Houston, TX; T Siler, St Charles, MO; E Silverman, Boston, MA; A Wanner, Miami, FL; R Wise, Baltimore, MD; R ZuWallack, Hartford, CT. Steering Committee: H Coxson (Canada), C Crim (GlaxoSmithKline, USA), L Edwards (GlaxoSmithKline, USA), D Lomas (UK), W MacNee (UK), E Silverman (USA), R Tal Singer (Co-chair, GlaxoSmithKline, USA), J Vestbo (Co-chair, Denmark), J Yates (GlaxoSmithKline, USA). Scientific Committee: A Agusti (Spain), P Calverley (UK), B Celli (USA), C Crim (GlaxoSmithKline, USA), B Miller(GlaxoSmithKline, US), W MacNee (Chair, UK), S Rennard (USA), R Tal-Singer (GlaxoSmithKline, USA), E Wouters (The Netherlands), J Yates (GlaxoSmithKline, USA). Data deposition: Data can be obtained from dbGaP at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000335.v1.p1 through dbGaP accession number phs000335.v1.pl.
This research was supported by GENEVA (U01HG004738). The Lung Health Study I was supported by contract NIH/N01-HR-46002. Lung tissue validation studies were supported by the NHLBI HL095406-01. KCB was supported in part by the Mary Beryl Patch Turnbull Scholar Program. This CHS research was supported by NHLBI contracts HHSN268201200036C, N01-HC-85239, N01-HC-85079 through N01-HC-85086; N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133 and NHLBI grants HL080295, HL087652, HL105756 with additional contribution from NINDS. Additional support was provided through AG-023629, AG-15928, AG-20098, and AG-027058 from the NIA. See also http://www.chs-nhlbi.org/pi.htm. DNA handling and genotyping was supported in part by National Center of Advancing Translational Technologies CTSI grant UL 1TR000124 and National Institute of Diabetes and Digestive and Kidney Diseases grant DK063491 to the Southern California Diabetes Endocrinology Research Center. FHS was funded by N01 HC 25195 from NHLBI. ECLIPSE was supported by GlaxoSmithKline. The BLSA was supported in part by the Intramural Research Program of the NIH, National Institute on Aging. A portion of that support was through a R&D contract with MedStar Research Institute. COPACETIC (acronym of COPD Pathology: Addressing Critical gaps, Early Treatment & diagnosis and Innovative Concepts) is funded by the European Union FP7 program, grant agreement number: 201379. GWAS genotyping was performed at the Center for Inhreited Disease Research under the support of NIH GEI grant U01 HG004438.
Footnotes
Ethical standards
All experiments described here comply with the current laws of the country in which they were performed.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Minino AM. Death in the United States, 2009. NCHS. Data Brief. 2011:1–8. [PubMed] [Google Scholar]
- 2.Mannino DM, Homa DM, Akinbami LJ, Ford ES, Redd SC. Chronic obstructive pulmonary disease surveillance--United States, 1971–2000. Respir Care. 2002;47:1184–1199. [PubMed] [Google Scholar]
- 3.Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet. 1997;349:1498–1504. doi: 10.1016/S0140-6736(96)07492-2. [DOI] [PubMed] [Google Scholar]
- 4.Rennard SI, Vestbo J. COPD: the dangerous underestimate of 15% Lancet. 2006;367:1216–1219. doi: 10.1016/S0140-6736(06)68516-4. [DOI] [PubMed] [Google Scholar]
- 5.Khoury MJ, Beaty TH, Tockman MS, Self SG, Cohen BH. Familial aggregation in chronic obstructive pulmonary disease: use of the loglinear model to analyze intermediate environmental and genetic risk factors. Genet Epidemiol. 1985;2:155–166. doi: 10.1002/gepi.1370020206. [DOI] [PubMed] [Google Scholar]
- 6.Cho MH, Boutaoui N, Klanderman BJ, Sylvia JS, Ziniti JP, Hersh CP, Demeo DL, Hunninghake GM, Litonjua AA, Sparrow D, Lange C, Won S, Murphy JR, Beaty TH, Regan EA, Make BJ, Hokanson JE, Crapo JD, Kong X, Anderson WH, Tal-Singer R, Lomas DA, Bakke P, Gulsvik A, Pillai SG, Silverman EK. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42:200–202. doi: 10.1038/ng.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pillai SG, Ge D, Zhu G, Kong X, Shianna KV, Need AC, Feng S, Hersh CP, Bakke P, Gulsvik A, Ruppert A, Lodrup Carlsen KC, Roses A, Anderson W, Rennard SI, Lomas DA, Silverman EK, Goldstein DB. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gottlieb DJ, Wilk JB, Harmon M, Evans JC, Joost O, Levy D, O’Connor GT, Myers RH. Heritability of longitudinal change in lung function. The Framingham study. Am J Respir Crit Care Med. 2001;164:1655–1659. doi: 10.1164/ajrccm.164.9.2010122. [DOI] [PubMed] [Google Scholar]
- 9.Hansel NN, Gao L, Rafaels NM, Mathias RA, Neptune ER, Tankersley C, Grant AV, Connett J, Beaty TH, Wise RA, Barnes KC. Leptin receptor polymorphisms and lung function decline in COPD. Eur Respir J. 2009;34:103–110. doi: 10.1183/09031936.00120408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansel NN, Sidhaye V, Rafaels NM, Gao L, Gao P, Williams R, Connett JE, Beaty TH, Mathias RA, Wise RA, King LS, Barnes KC. Aquaporin 5 polymorphisms and rate of lung function decline in chronic obstructive pulmonary disease. PLoS One. 2010;5:e14226. doi: 10.1371/journal.pone.0014226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He JQ, Ruan J, Connett JE, Anthonisen NR, Pare PD, Sandford AJ. Antioxidant gene polymorphisms and susceptibility to a rapid decline in lung function in smokers. Am J Respir Crit Care Med. 2002;166:323–328. doi: 10.1164/rccm.2111059. [DOI] [PubMed] [Google Scholar]
- 12.He JQ, Connett JE, Anthonisen NR, Sandford AJ. Polymorphisms in the IL13, IL13RA1, and IL4RA genes and rate of decline in lung function in smokers. Am J Respir Cell Mol Biol. 2003;28:379–385. doi: 10.1165/rcmb.4885. [DOI] [PubMed] [Google Scholar]
- 13.He JQ, Connett JE, Anthonisen NR, Pare PD, Sandford AJ. Glutathione S-transferase variants and their interaction with smoking on lung function. Am J Respir Crit Care Med. 2004;170:388–394. doi: 10.1164/rccm.200312-1763OC. [DOI] [PubMed] [Google Scholar]
- 14.He JQ, Burkett K, Connett JE, Anthonisen NR, Pare PD, Sandford AJ. Interferon gamma polymorphisms and their interaction with smoking are associated with lung function. Hum Genet. 2006;119:365–375. doi: 10.1007/s00439-006-0143-z. [DOI] [PubMed] [Google Scholar]
- 15.Sandford AJ, Chagani T, Weir TD, Connett JE, Anthonisen NR, Pare PD. Susceptibility genes for rapid decline of lung function in the lung health study. Am J Respir Crit Care Med. 2001;163:469–473. doi: 10.1164/ajrccm.163.2.2006158. [DOI] [PubMed] [Google Scholar]
- 16.Siedlinski M, Boezen HM, Boer JM, Smit HA, Postma DS. ABCC1 polymorphisms contribute to level and decline of lung function in two population-based cohorts. Pharmacogenet Genomics. 2009;19:675–684. doi: 10.1097/FPC.0b013e32832f5eff. [DOI] [PubMed] [Google Scholar]
- 17.Siedlinski M, Postma DS, Boer JM, van der SG, Schouten JP, Smit HA, Boezen HM. Level and course of FEV1 in relation to polymorphisms in NFE2L2 and KEAP1 in the general population. Respir Res. 2009;10:73. doi: 10.1186/1465-9921-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Diemen CC, Postma DS, Vonk JM, Bruinenberg M, Schouten JP, Boezen HM. A disintegrin and metalloprotease 33 polymorphisms and lung function decline in the general population. Am J Respir Crit Care Med. 2005;172:329–333. doi: 10.1164/rccm.200411-1486OC. [DOI] [PubMed] [Google Scholar]
- 19.van Diemen CC, Postma DS, Siedlinski M, Blokstra A, Smit HA, Boezen HM. Genetic variation in TIMP1 but not MMPs predict excess FEV1 decline in two general population-based cohorts. Respir Res. 2011;12:57. doi: 10.1186/1465-9921-12-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molfino NA. Genetic predisposition to accelerated decline of lung function in COPD. Int J Chron Obstruct Pulmon Dis. 2007;2:117–119. [PMC free article] [PubMed] [Google Scholar]
- 21.Kanner RE, Connett JE, Williams DE, Buist AS. Effects of randomized assignment to a smoking cessation intervention and changes in smoking habits on respiratory symptoms in smokers with early chronic obstructive pulmonary disease: the Lung Health Study. Am J Med. 1999;106:410–416. doi: 10.1016/s0002-9343(99)00056-x. [DOI] [PubMed] [Google Scholar]
- 22.Anthonisen NR, Connett JE, Kiley JP, Altose MD, Bailey WC, Buist AS, Conway WA, Jr, Enright PL, Kanner RE, O’Hara P. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. JAMA. 1994;272:1497–1505. [PubMed] [Google Scholar]
- 23.Connett JE, Kusek JW, Bailey WC, O’Hara P, Wu M. Design of the Lung Health Study: a randomized clinical trial of early intervention for chronic obstructive pulmonary disease. Control Clin Trials. 1993;14:3S–19S. doi: 10.1016/0197-2456(93)90021-5. [DOI] [PubMed] [Google Scholar]
- 24.Kanner RE. Early intervention in chronic obstructive pulmonary disease. A review of the Lung Health Study results. Med Clin North Am. 1996;80:523–547. doi: 10.1016/s0025-7125(05)70452-1. [DOI] [PubMed] [Google Scholar]
- 25.Zeger SL, Liang KY, Albert PS. Models for longitudinal data: a generalized estimating equation approach. Biometrics. 1988;44:1049–1060. [PubMed] [Google Scholar]
- 26.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Pare PD. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 27.Bakke PS, Zhu G, Gulsvik A, Kong X, Agusti AG, Calverley PM, Donner CF, Levy RD, Make BJ, Pare PD, Rennard SI, Vestbo J, Wouters EF, Anderson W, Lomas DA, Silverman EK, Pillai SG. Candidate genes for COPD in two large data sets. Eur Respir J. 2011;37:255–263. doi: 10.1183/09031936.00091709. [DOI] [PubMed] [Google Scholar]
- 28.Clarke DL, Clifford RL, Jindarat S, Proud D, Pang L, Belvisi M, Knox AJ. TNFalpha and IFNgamma synergistically enhance transcriptional activation of CXCL10 in human airway smooth muscle cells via STAT-1, NF-kappaB, and the transcriptional coactivator CREB-binding protein. J Biol Chem. 2010;285:29101–29110. doi: 10.1074/jbc.M109.0999952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoock TC, Peters LL, Lux SE. Isoforms of ankyrin-3 that lack the NH2- terminal repeats associate with mouse macrophage lysosomes. J Cell Biol. 1997;136:1059–1070. doi: 10.1083/jcb.136.5.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kizhatil K, Yoon W, Mohler PJ, Davis LH, Hoffman JA, Bennett V. Ankyrin-G and beta2-spectrin collaborate in biogenesis of lateral membrane of human bronchial epithelial cells. J Biol Chem. 2007;282:2029–2037. doi: 10.1074/jbc.M608921200. [DOI] [PubMed] [Google Scholar]
- 31.Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, Najjar SS, Zhao JH, Heath SC, Eyheramendy S, Papadakis K, Voight BF, Scott LJ, Zhang F, Farrall M, Tanaka T, Wallace C, Chambers JC, Khaw KT, Nilsson P, van der HP, Polidoro S, Grobbee DE, Onland-Moret NC, Bots ML, Wain LV, Elliott KS, Teumer A, Luan J, Lucas G, Kuusisto J, Burton PR, Hadley D, McArdle WL, Brown M, Dominiczak A, Newhouse SJ, Samani NJ, Webster J, Zeggini E, Beckmann JS, Bergmann S, Lim N, Song K, Vollenweider P, Waeber G, Waterworth DM, Yuan X, Groop L, Orho-Melander M, Allione A, Di GA, Guarrera S, Panico S, Ricceri F, Romanazzi V, Sacerdote C, Vineis P, Barroso I, Sandhu MS, Luben RN, Crawford GJ, Jousilahti P, Perola M, Boehnke M, Bonnycastle LL, Collins FS, Jackson AU, Mohlke KL, Stringham HM, Valle TT, Willer CJ, Bergman RN, Morken MA, Doring A, Gieger C, Illig T, Meitinger T, Org E, Pfeufer A, Wichmann HE, Kathiresan S, Marrugat J, O’Donnell CJ, Schwartz SM, Siscovick DS, Subirana I, Freimer NB, Hartikainen AL, McCarthy MI, O’Reilly PF, Peltonen L, Pouta A, de Jong PE, Snieder H, van Gilst WH, Clarke R, Goel A, Hamsten A, Peden JF, Seedorf U, Syvanen AC, Tognoni G, Lakatta EG, Sanna S, Scheet P, Schlessinger D, Scuteri A, Dorr M, Ernst F, Felix SB, Homuth G, Lorbeer R, Reffelmann T, Rettig R, Volker U, Galan P, Gut IG, Hercberg S, Lathrop GM, Zelenika D, Deloukas P, Soranzo N, Williams FM, Zhai G, Salomaa V, Laakso M, Elosua R, Forouhi NG, Volzke H, Uiterwaal CS, van der Schouw YT, Numans ME, Matullo G, Navis G, Berglund G, Bingham SA, Kooner JS, Connell JM, Bandinelli S, Ferrucci L, Watkins H, Spector TD, Tuomilehto J, Altshuler D, Strachan DP, Laan M, Meneton P, Wareham NJ, Uda M, Jarvelin MR, Mooser V, Melander O, Loos RJ, Elliott P, Abecasis GR, Caulfield M, Munroe PB. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Besnard V, Wert SE, Kaestner KH, Whitsett JA. Stage-specific regulation of respiratory epithelial cell differentiation by Foxa1. Am J Physiol Lung Cell Mol Physiol. 2005;289:L750–L759. doi: 10.1152/ajplung.00151.2005. [DOI] [PubMed] [Google Scholar]
- 33.Song L, Wei X, Zhang B, Luo X, Liu J, Feng Y, Xiao X. Role of Foxa1 in regulation of bcl2 expression during oxidative-stress-induced apoptosis in A549 type II pneumocytes. Cell Stress Chaperones. 2009;14:417–425. doi: 10.1007/s12192-008-0095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song L, Zhang B, Feng Y, Luo X, Wei X, Xiao X. A role for forkhead box A1 in acute lung injury. Inflammation. 2009;32:322–332. doi: 10.1007/s10753-009-9139-x. [DOI] [PubMed] [Google Scholar]
- 35.Tuder RM, Yoshida T, Arap W, Pasqualini R, Petrache I. State of the art. Cellular and molecular mechanisms of alveolar destruction in emphysema: an evolutionary perspective. Proc Am Thorac Soc. 2006;3:503–510. doi: 10.1513/pats.200603-054MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vestbo J, Edwards LD, Scanlon PD, Yates JC, Agusti A, Bakke P, Calverley PM, Celli B, Coxson HO, Crim C, Lomas DA, Macnee W, Miller BE, Silverman EK, Tal-Singer R, Wouters E, Rennard SI. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011;365:1184–1192. doi: 10.1056/NEJMoa1105482. [DOI] [PubMed] [Google Scholar]
- 37.Celli BR, Thomas NE, Anderson JA, Ferguson GT, Jenkins CR, Jones PW, Vestbo J, Knobil K, Yates JC, Calverley PM. Effect of pharmacotherapy on rate of decline of lung function in chronic obstructive pulmonary disease: results from the TORCH study. Am J Respir Crit Care Med. 2008;178:332–338. doi: 10.1164/rccm.200712-1869OC. [DOI] [PubMed] [Google Scholar]
- 38.Anthonisen NR, Lindgren PG, Tashkin DP, Kanner RE, Scanlon PD, Connett JE. Bronchodilator response in the lung health study over 11 yrs. Eur Respir J. 2005;26:45–51. doi: 10.1183/09031936.05.00102604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.