

Background: Both tetrahydrobiopterin (BH4) and S-glutathionylation are important regulators of eNOS activity and coupling.
Results: S-Glutathionylation and BH4 deficiency induce eNOS uncoupling through distinct mechanisms but are mutually regulated by changes in BH4 oxidation and cellular GSH:GSSG ratio.
Conclusion: BH4-dependent and S-glutathionylation-induced eNOS uncoupling are mechanistically independent but functionally linked.
Significance: BH4 and S-glutathionylation exemplify eNOS as an integrated redox signaling “hub.”
Keywords: Endothelial Dysfunction, Glutathionylation, Nitric-oxide Synthase, Superoxide Ion, Tetrahydrobiopterin
Abstract
Endothelial nitric-oxide synthase (eNOS) is a critical regulator of vascular homeostasis by generation of NO that is dependent on the cofactor tetrahydrobiopterin (BH4). When BH4 availability is limiting, eNOS becomes “uncoupled,” resulting in superoxide production in place of NO. Recent evidence suggests that eNOS uncoupling can also be induced by S-glutathionylation, although the functional relationships between BH4 and S-glutathionylation remain unknown. To address a possible role for BH4 in S-glutathionylation-induced eNOS uncoupling, we expressed either WT or mutant eNOS rendered resistant to S-glutathionylation in cells with Tet-regulated expression of human GTP cyclohydrolase I to regulate intracellular BH4 availability. We reveal that S-glutathionylation of eNOS, by exposure to either 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) or glutathione reductase-specific siRNA, results in diminished NO production and elevated eNOS-derived superoxide production, along with a concomitant reduction in BH4 levels and BH4:7,8-dihydrobiopterin ratio. In eNOS uncoupling induced by BH4 deficiency, BCNU exposure further exacerbates superoxide production, BH4 oxidation, and eNOS activity. Following mutation of C908S, BCNU-induced eNOS uncoupling and BH4 oxidation are abolished, whereas uncoupling induced by BH4 deficiency was preserved. Furthermore, BH4 deficiency alone is alone sufficient to reduce intracellular GSH:GSSG ratio and cause eNOS S-glutathionylation. These data provide the first evidence that BH4 deficiency- and S-glutathionylation-induced mechanisms of eNOS uncoupling, although mechanistically distinct, are functionally related. We propose that uncoupling of eNOS by S-glutathionylation- or by BH4-dependent mechanisms exemplifies eNOS as an integrated redox “hub” linking upstream redox-sensitive effects of BH4 and glutathione with redox-dependent targets and pathways that lie downstream of eNOS.
Introduction
Nitric oxide, produced in the endothelium by endothelial nitric-oxide synthase (eNOS),2 is a critical regulator of vascular homeostasis (1, 2). NO has multiple antiatherogenic roles, inhibiting vascular smooth muscle cell proliferation, platelet aggregation, and leukocyte adhesion (1). Simultaneous loss of NO bioavailability and elevated production of superoxide is a hallmark of several vascular disease states including hypercholesterolemia, hypertension, diabetes, and atherosclerosis (3–5).
In the presence of Ca2+/calmodulin, eNOS produces NO from l-arginine by means of electron transfer from NADPH through a flavin-containing reductase domain to molecular oxygen, bound at the heme within the oxygenase domain (6). The essential NOS cofactor, tetrahydrobiopterin (BH4) is required to maintain enzymatic coupling of l-arginine oxidation, to produce NO. Loss or decreased levels of BH4 is associated with NOS “uncoupling,” resulting in production of superoxide rather than NO (7–9), by a mechanism generally attributed to the oxidation of BH4 to BH2 by peroxynitrite formed from the near diffusion rate reaction between superoxide and NO. This accumulation of BH2 is sufficient to displace BH4 from NOS, resulting in uncoupling of the enzyme at the heme group within the oxygenase domain (7). Until recently, the balance between NO and superoxide production by eNOS had been thought to be primarily determined by the availability of BH4. However, whereas NOS dysfunction occurs in diseases with redox stress, BH4 repletion is only sufficient to partially restore NOS activity and NOS-dependent vasodilation (10). This led to the important discovery that eNOS is also subject to post-translational regulation by S-glutathionylation (11), placing new emphasis on other redox-related mechanisms, independent of BH4 availability, and their role in regulating the function of eNOS.
S-Glutathionylation is a post-translational modification in which a glutathione tripeptide is reversibly bound to the protein via the formation of a disulfide bond with a protein thiol. The modification of target proteins by S-glutathionylation is directly linked to the redox status of the cell, because the modification of proteins can be promoted by physiological levels of reactive oxygen species and reactive nitrogen species (12). Indeed, GAPDH regulates the synthesis of the endothelial vasoconstrictor endothelin-1 by a novel, S-glutathionylation-based mechanism, and site specific S-glutathionylation in either the ATP binding domain or active site inhibits MAPK, ERK, and caspase-3 activity, respectively. Thiol oxidation and subsequent S-glutathionylation also occurs within mammalian thioredoxin, contributing to the regulation and functions of the protein (13).
Although recent evidence suggests that glutathionylation of eNOS at two highly conserved cysteine residues within the reductase domain is associated with impaired endothelium-dependent vasorelaxation (11), the exact mechanisms by which eNOS becomes uncoupled and the relationship between S-glutathionylation-induced and BH4-dependent superoxide production and their effect on eNOS function and tetrahydrobiopterin redox state remain unexplored.
Accordingly, we sought to address these questions using both pharmacologic and genetic manipulation of intracellular glutathione levels and by testing the effects of these interventions on eNOS uncoupling and BH4 oxidation. We used glutathione reductase (GR)-specific siRNA and 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) treatment to alter the intracellular GSH:GSSG ratio in endothelial cells and stably transfected cell lines with doxycycline-regulated expression of GTPCH (14). Moreover, cells also expressed either eNOS-GFP or mutant-eNOS-GFP fusion proteins rendered resistant to S-glutathionylation. Using these cells lines, we examined the interaction of enzymatic uncoupling of eNOS at the oxygenase and reductase domains and compared this with the availability and oxidation state of BH4. We report that BH4 dependent- and S-glutathionylation-induced mechanisms of eNOS uncoupling are mechanistically independent but functionally linked, acting in concert to regulate eNOS between the production of either NO or superoxide. These findings advance our understanding of eNOS coupling and describe new relationships and interplay between the regulatory pathways involved in modulating enzymatic coupling.
EXPERIMENTAL PROCEDURES
Generation of Tet-regulatable Cells
We used NIH 3T3 murine fibroblasts stably transfected with a Tet-Off transactivator construct as previously described (14). In the presence of doxycycline, binding of the transactivator is blocked, and gene expression is prevented. These 3T3-Tet-Off cells, previously shown not to express GTPCH (15) and also confirmed to be devoid of eNOS protein, were stably transfected with a plasmid encoding hemagglutinin antigen-tagged human GTPCH under the control of a tetracycline-responsive element. Individual colonies were isolated and analyzed for GTPCH expression, and a cell line termed “GCH cells” was established from expansion of a single colony. GCH/eNOS cells were produced by stable transfection of GCH cells with a plasmid encoding a human eNOS-eGFP fusion protein as described (16). In this model, the addition of doxycycline to GCH/eNOS cells results in diminished GCH mRNA, GTPCH protein, and BH4 levels leading to uncoupling of eNOS (14).
Cell Culture
The cells were cultured in DMEM (Invitrogen) supplemented with glutamine (2 mm), penicillin (100 units/ml), and streptomycin (0.1 mg/ml). Additionally GCH cells were maintained using media containing the antibiotics hygromycin (200 μg/ml) and genetecin (200 μg/ml), whereas eNOS-eGFP cell medium also included puromycin (2 μg/ml). Where appropriate, doxycycline (1 μg/ml) was added to cell culture media to abolish transcription of GCH1 mRNA.
Glutathione Reductase Knockdown by RNAi
GR-specific, ON-TARGETplus SMARTpool siRNA was purchased from Dharmacon Thermo Scientific. The siRNAs were used as a pool of four specific siRNA duplexes with the following sequences: Duplex 1, 5′-GGGUGGCACUUGCGUGAAU-3′; Duplex 2, 5′-GGUAGGAAGCCCACCACGA-3′; Duplex 3, 5′-CAGCAGUGCACUCGGAAUU-3′; and Duplex 4, 5′-CCACAUCCUAGUAGACGAA-3′.
sEnd.1 endothelial cells were seeded into 6-well plates 24 h prior to transfection. The cells were then transfected with GR-specific siRNA (100 nm), GAPDH-positive (100 nm), or nonspecific pooled duplex negative control siRNA (100 nm). The cells were cultured for 72 h, and gene silencing was detected by analysis of GR protein expression by Western blotting using GR-specific antibodies.
Generation of Glutathionylation-resistant Mutant eNOS
QuikChange site-directed mutagenesis (Stratagene) was used to create C689S and C908S human eNOS mutants. Mutant primers were designed and used as follows: C689S, 5′-GGCGACGAGCTGAGCGGCCAGGAGG-3′ (sense) and 5′-CCTCCTGGCCGCTCAGCTCGTCGCC-3′ (antisense); and C908S, 5′-GAAGTGGTTCCGCAGCCCCACGCTGC-3′ (sense) and 5′-GCAGCGTGGGGCTGCGGAACCACTTC-3′ (antisense). The sequence of each heNOS single and double mutant was confirmed by DNA sequencing (SourceBioscience). Mutant constructs were transiently expressed in GCH cells using FuGENE 6 (Roche Applied Science).
Analysis of NO Synthesis by eNOS
Cellular NO synthesis by eNOS was assessed by measuring the conversion of [14C]l-arginine to citrulline with HPLC detection, in the presence and absence of Nγ-monomethyl-l-arginine, as previously described (17).
Biopterin Quantification by HPLC with Electrochemical Detection
BH4, BH2, and biopterin levels in cell lysates were determined by HPLC followed by electrochemical and fluorescent detection, as previously described (14, 18). Briefly, the cells were grown to confluency and harvested by trypsinization. Sample pellets were resuspended in phosphate-buffered saline (50 mm), pH 7.4, containing dithioerythritol (1 mm) and EDTA (100 μm) and subjected to three freeze-thaw cycles. Following centrifugation (15 min at 13,000 rpm and 4 °C), the samples were transferred to new, cooled micro tubes and precipitated with cold phosphoric acid (1 m), TCA (2 m), and dithioerythritol (1 mm). The samples were vigorously mixed and then centrifuged for 15 min at 13,000 rpm and 4 °C. Samples were injected onto an isocratic HPLC system and quantified using sequential electrochemical (Dionex Coulochem III; Thermoscientific, Buckinghamshire, UK) and fluorescence (Jasco, Essex, UK) detection. HPLC separation was performed using a 250-mm, ACE C-18 column (Hichrom, Berkshire, UK) and a mobile phase comprising of sodium acetate (50 mm), citric acid (5 mm), EDTA (48 μm), and dithioerythritol (160 μm) (pH 5.2) (all ultrapure electrochemical HPLC grade) at a flow rate of 1.3 ml/min. Background currents of +500 and −50 μA were used for the detection of BH4 on electrochemical cells E1 and E2, respectively. 7,8-BH2 and biopterin were measured using a Jasco FP2020 fluorescence detector. Quantification of BH4, BH2, and biopterin was done by comparison with authentic external standards and normalized to sample protein content.
Quantification of Superoxide Production by HPLC
Measurement of 2-hydroxyethidium formation by HPLC was used to quantify superoxide production, as previously described (19, 20). The cells were washed three times in PBS and incubated in Krebs-Hepes buffer in the presence or absence of l-NAME (100 μm). After 30 min, dihydroethidium (25 μm) was added, and the cells were then incubated for an additional 20 min at 37 °C. The cells were then harvested by scraping, centrifuged, and lysed in ice-cold methanol. Hydrochloric acid (100 mm) was added (1:1 v/v) prior to loading into the autosampler for analysis. All of the samples were stored in darkened tubes and protected from light at all times. Separation of ethidium, oxyethidium, and dihydroethidium was performed using a gradient HPLC system (Jasco) with an ODS3 reverse phase column (250 mm, 4.5 mm; Hichrom), and quantified using a fluorescence detector set at 510 nm (excitation) and 595 nm (emission). A linear gradient was applied from Mobile phase A (0.1% TFA (v/v)) to Mobile phase B (0.1% TFA (v/v) in acetonitrile) over 23 min (30% acetonitrile to 50% acetonitrile).
Immunoprecipitation and Western Blotting
Following exposure of cells with the appropriate treatment, in the presence or absence of DTT (to remove the glutathionylation modification of eNOS), the cells were suspended in radioimmune precipitation assay lysis buffer (20 mm Tris-HCl, 150 mm NaCl, 20 mm N-ethylmaleimide, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton (v/v), 0.1% SDS (w/v), 0.1 sodium deoxycholate, pH 7.4), including a mixture of protease inhibitors (Roche Applied Science), and subjected to three freeze-thaw cycles in liquid nitrogen. For immunoprecipitation, eNOS was pulled down with agarose-conjugated anti-eNOS antibody (Santa Cruz). Western blotting was carried out using standard techniques with either anti-glutathione monoclonal antibody (ViroGen), anti-eNOS (BD Transduction Laboratories), anti-Glutathione reductase (Santa Cruz), and anti-GAPDH (Sigma) antibodies.
Quantification of GSSG and GSH
To assess the levels of GSH and the accumulation of GSSG following induction of oxidative stress, we used a glutathione assay kit from Cayman Chemicals as per the manufacturer's instructions.
Statistical Analysis
The data are presented as the means ± S.E. The data were subjected to the Kolmogorov-Smirnov test to determine distribution. Groups were compared using the Mann-Whitney U test for nonparametric data or the Student's t test for parametric data. When comparing multiple groups, the data were analyzed by analysis of variance with Newman-Keuls post test for parametric data or Kruskal-Wallis test with Dunns post-test for nonparametric data. A value of p < 0.05 was considered statistically significant.
RESULTS
Effects of GR Inhibition and Knockdown on eNOS in Endothelial Cells
We first investigated the protective role of GR on the S-glutathionylation of eNOS and set out to elucidate the effect of S-glutathionylation on enzymatic activity. Inhibition of GR activity by treatment of endothelial cells with BCNU was sufficient to induce a 3.5-fold decrease in the ratio of GSH:GSSG (Fig. 1A) and significant S-glutathionylation of proteins as detected by Western blotting. More specifically, glutathionylation of eNOS was increased by 5-fold following exposure of endothelial cells to 25 and 100 μm BCNU, an effect that was reversed following incubation with DTT (Fig. 1B). Phosphorylation of eNOS at Ser-1177 remained unchanged, and levels of eNOS-GSH, normalized to both total protein-GSH and total eNOS, were calculated. Correspondingly, eNOS activity was decreased in a dose-dependent manner, as evidenced by quantifying the conversion of radiolabeled arginine to citrulline (Fig. 1, C and D). Furthermore, eNOS produced superoxide in response to BCNU (56.3 ± 5.4 versus 26.3 ± 7.9 pmol/well; Fig. 1E), which was partially inhibitable with l-NAME, and greatly abrogated in the presence of DTT (Fig. 1F). These effects of BCNU treatment and subsequent uncoupling of eNOS resulted in marked oxidation of intracellular BH4 and striking BH2 accumulation, leading to a dose-dependent decrease in the ratio of BH4:BH2 (Fig. 2, A and B). Induction of BH4-dependent eNOS uncoupling by S-glutathionylation was evidenced by the significant decrease in intracellular BH4 oxidation following pretreatment of endothelial cells with l-NAME (1 mm) prior to GR inhibition by BCNU (Fig. 2, C and D).
FIGURE 1.

Inhibition of glutathione reductase S-glutathionylates and uncouples eNOS in endothelial cells. sEnd.1 endothelial cells were exposed to a dose range of the glutathione reductase inhibitor BCNU for 4 h with or without DTT for the final 30 min. Glutathionylation and activity of eNOS were determined by immunoprecipitation and HPLC as outlined under “Experimental Procedures.” A, following exposure of cells to BCNU, a significant decrease in the ratio of GSH:GSSG was observed (n = 3). B–D, eNOS was glutathionylated in a dose-dependent manner that was reversed by DTT (B, n = 3), which uncoupled eNOS as evidenced by a dose-dependent decrease in eNOS activity (C and D) and an increase in total superoxide production (E, *, p < 0.05; **, p < 0.01). F, this superoxide production was partially inhibited with l-NAME pretreatment and abolished in the presence of DTT (†, p < 0.05; n = 6).
FIGURE 2.

Inhibition of glutathione reductase leads to a dose-dependent oxidation of BH4. sEnd.1 endothelial cells were exposed to a range of doses of BCNU, and the effects on intracellular BH4 were determined by HPLC with electrochemical and fluorescence detection as under “Experimental Procedures.” BH4 is oxidized (A) to BH2 (B) in a dose-dependent manner following exposure of endothelial cells to BCNU. The changes in both BH4 and BH2 are partially reversed by l-NAME (C and D) (*, p < 0.05; n = 4).
To complement these experiments using pharmacological inhibition of GR by BCNU, we next sought to test the effects of knocking down GR mRNA and protein expression using GR-specific siRNAs. Following incubation of endothelial cells with GR-specific siRNAs for 72 h, GR protein levels were knocked down by greater than 95% (Fig. 3A). GR knockdown caused marked oxidation of BH4 to BH2, indicated by the change in BH4:BH2 ratio (28.0 ± 3.12 decreased to 11.9 ± 4). Furthermore, GR knockdown induced eNOS uncoupling, as evidenced by decreased eNOS activity and elevated superoxide production. Interestingly, as with BCNU treatment, eNOS-derived superoxide was partially inhibitable with l-NAME (Fig. 3), suggesting that S-glutathionylation-induced eNOS uncoupling also involves changes in BH4-dependent eNOS regulation. These changes in superoxide production mainly derive from the reductase domain but also trigger eNOS uncoupling at the oxygenase domain. Although this contribution is relatively small, this is an important finding to suggest cross-talk between glutathionylation-induced uncoupling and BH4-dependent uncoupling of eNOS.
FIGURE 3.
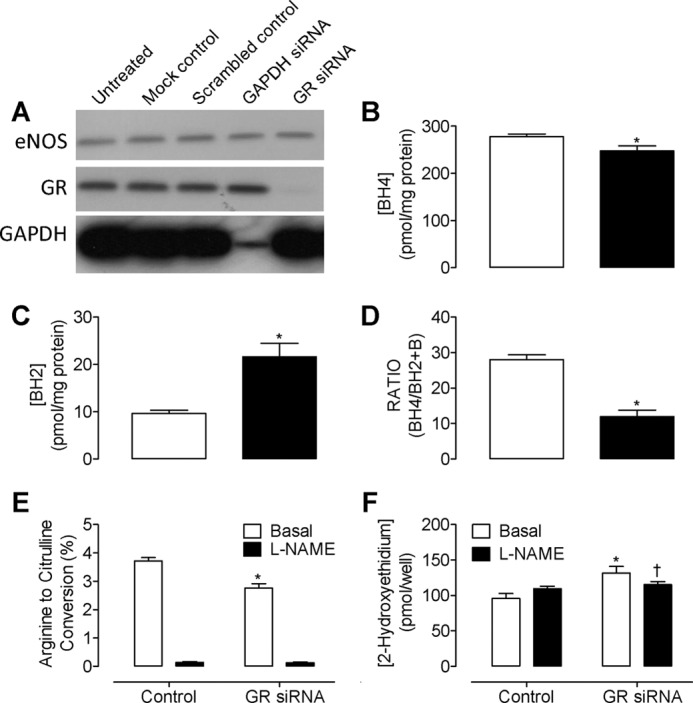
Genetic knockdown of glutathione reductase with siRNA results in uncoupling of eNOS. Murine endothelial cells were transfected with GR-specific, control GAPDH, or scrambled nonspecific siRNA, as described under “Experimental Procedures.” Protein expression and eNOS activity were then determined by Western blotting and HPLC. A, GR-specific siRNA induced an average of 95% knockdown of GR protein. Importantly, GAPDH knockdown and scrambled control siRNAs did not have any effect on eNOS protein levels. The blots shown are representative of three separate experiments. B, intracellular BH4 levels are significantly decreased in the presence of GR-specific siRNA but remain unchanged in the presence of GAPDH knockdown or scrambled control siRNAs (data not shown). C, knockdown of GR resulted in a 2.5-fold increase in BH2 and a corresponding decrease in BH4:BH2 ratio (D, *, p > 0.05). E, eNOS activity was diminished following exposure to GR-specific siRNAs, as demonstrated by measuring the conversion of radiolabeled arginine to citrulline by HPLC (*, p < 0.05). F, knockdown of GR resulted in a marked accumulation of 2-hydroxyethidium (*, p < 0.05), which was partially inhibited by l-NAME (200 μm) (†, p < 0.05; n = 6).
The Effects of BCNU on BH4 Are Dependent on eNOS
Having established that manipulation of intracellular GSH:GSSG ratio leads to eNOS S-glutathionylation and uncoupling in endothelial cells, we next sought to evaluate the contribution of eNOS to total BCNU-induced superoxide production, comparing the effects of GR inhibition in cells with or without eNOS expression. To this end, we used cell lines stably expressing human GTPCH under the control of the tetracycline response element, either with (GCH/eNOS) or without (GCH) eNOS-GFP coexpression, as described previously (14, 21). BCNU treatment decreased the GSH:GSSG ratio, which lead to the S-glutathionylation of eNOS and diminished eNOS activity (Fig. 4, A–C). Concomitantly, BCNU treatment induced a striking increase in the production of superoxide in eNOS expressing, but not in eNOS-deficient cells (85.2 ± 7.4 versus 34.6 ± 12.6 pmol/well; Fig. 4D). Furthermore, this elevation in superoxide in GCH/eNOS cells was partially inhibited with l-NAME and abolished in the presence of DTT, demonstrating that this production of superoxide is eNOS-dependent, BCNU-induced, and l-NAME-inhibitable (Fig. 4, E and F). BCNU decreased the ratio of BH4:BH2 even in cells lacking eNOS (Fig. 4G). However, in GCH/eNOS cells, but not in GCH cells without eNOS, BCNU further decreased the BH4:BH2 ratio that was reversed by DTT, indicating that eNOS-derived superoxide, induced by S-glutathionylation, contributes to the oxidation of BH4.
FIGURE 4.
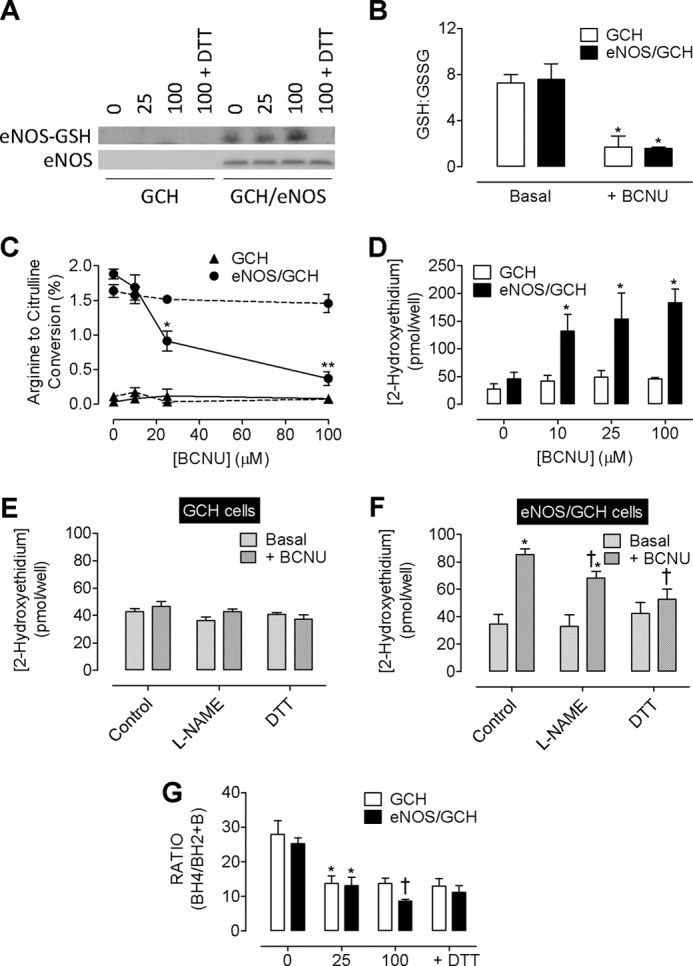
The effects of BCNU are dependent on eNOS. Cells expressing Tet-regulatable GTPCH (GCH cells) were subject to BCNU exposure, and the presence or absence or eNOS was assessed (eNOS/GCH cells). A, a dose-dependent S-glutathionylation of eNOS was observed that was removed following exposure to DTT. There was no change in overall eNOS levels following BCNU treatment. No eNOS was detectable in GCH cells lacking the eNOS transgene. B, in both GCH and eNOS/GCH cells, BCNU exposure attenuated the GSH:GSSG ratio by 4-fold (*, p < 0.05). C, eNOS activity was decreased in a dose-dependent manner following exposure to BCNU in eNOS/GCH cells (●, solid line). *, p < 0.05; **, p < 0.01). These changes in eNOS activity do not occur in the presence of DTT (●, dotted line). eNOS activity is undetectable in GCH cells lacking eNOS (▴). D, total superoxide production is elevated in eNOS/GCH, but not GCH cells following exposure to BCNU. †, p < 0.01 (n = 4). E, this elevation in superoxide production is not evident in GCH cells, and no effect of either l-NAME or DTT was observed. F, these elevated levels of superoxide (*, p < 0.05), induced by BCNU (100 μm) are partially inhibitable with l-NAME (200 μm) and almost totally abolished in the presence of DTT (†, p < 0.05). G, the subsequent effect of this superoxide is the oxidation of BH4. Following BCNU (100 μm) treatment, the diminished BH4:BH2 ratio (*, p < 0.05) is further exacerbated in eNOS/GCH but not GCH cells (†, p < 0.05; n = 6).
S-Glutathionylation Exacerbates BH4-dependent eNOS Uncoupling
Next we further probed the link between S-glutathionylation- and tetrahydrobiopterin-dependent production of superoxide by eNOS. As expected, DOX exposure decreased arginine to citrulline conversion. However, following BCNU (12.5 μm) exposure eNOS activity was attenuated 4-fold (2.06 ± 0.1 versus 0.41 ± 0.05% arginine-to-citrulline conversion) in DOX-treated cells, but not cells in the absence of DOX. Greater concentrations of BCNU were required to decrease eNOS activity in cells containing high BH4 levels (100 μm BCNU for no DOX versus 12.5 μm for DOX treated cells). Furthermore, at all BCNU concentrations, following DOX treatment to reduce BH4 levels, eNOS activity was significantly decreased (Fig. 5A). Comparing the activity of eNOS following exposure to DOX and/or DTT at a concentration of 12.5 μm, BH4 deficiency compounded the deleterious effects of BCNU (Fig. 5B). Interestingly, the GSH:GSSG ratio was augmented in the presence of DTT and diminished in BH4-deficient cells (Fig. 5C), suggesting that intracellular BH4 can modulate the effect of BCNU on GSH redox state. Accordingly, evidence of eNOS-derived superoxide production was observed, with markedly greater levels of superoxide produced when BH4-deficient cells were also exposed to BCNU (402.8 ± 70.9 versus 288.2 ± 82.33 pmol/well) (Fig. 5D). The effect of this increased superoxide production was to oxidize BH4, as evidenced by the decreased BH4:BH2 ratio observed in DOX-treated versus untreated cells, indicative of BH4 deficiency and eNOS uncoupling. Following inhibition of GR using BCNU, this oxidation of BH4 was exacerbated in both DOX-treated and untreated cells, with accumulation of BH2 reflected in the ratio of BH4:BH2 (Fig. 5, E and F). Prior to BCNU treatment, the ratio of BH4:BH2 in BH4-deficient, DOX-treated cells was 40% lower than that of the non-DOX-treated controls. Following exposure to BCNU (100 μm), the ratio of BH4:BH2 in DOX-treated cells was strikingly diminished compared with control cells (8.65 ± 0.9 versus 1.93 ± 0.17). This represents a 75% decrease in the ratio of BH4:BH2 in BH4-deficient cells following glutathionylation of eNOS, induced by BCNU (Fig. 5G).
FIGURE 5.
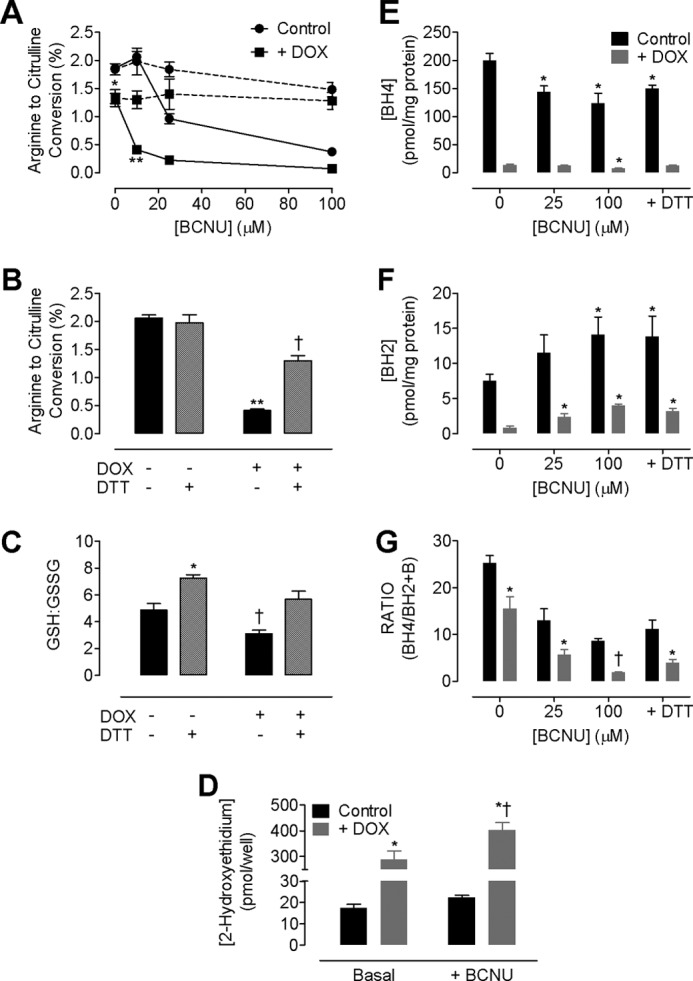
Glutathionylation of eNOS exacerbates BH4-dependent uncoupling. Attenuation of intracellular BH4 levels in Tet-regulatable eNOS/GCH with doxycycline (DOX) is a model of BH4 deficiency-induced eNOS uncoupling. A, basal eNOS activity is attenuated in eNOS/GCH cells following exposure to doxycycline for 7 days (*, p < 0.05). Upon exposure to BCNU, eNOS activity is further diminished in eNOS/GCH cells in both the presence (■) and the absence (●) of doxycycline (**, p < 0.01). This diminished eNOS activity is prevented by incubation of the cells with DTT (dotted line). Importantly, when cells are exposed to BCNU (12.5 μm), eNOS activity in the presence of doxycycline decreased 4-fold compared with in the absence of doxycycline (n = 6). B, 12.5 μm BCNU triggered a significant decrease in the conversion of arginine to citrulline in eNOS cells in the presence but not the absence of DOX that was attenuated in the presence of DTT. C, at 12.5 μm, changes in eNOS activity were mimicked by changes in the GSH:GSSG ratio; BH4-dependent uncoupling significantly diminished GSH:GSSG ratio, which was reversed following treatment with DTT. D, BH4 deficiency induced by doxycycline increases superoxide production by 15-fold in the absence and 20-fold in the presence of BCNU (*, p < 0.01). Significantly more superoxide is produced from BH4-deficient cells when exposed to BCNU (†, p < 0.01; n = 4). E and F, BH4 levels are diminished, and BH2 levels are increased, compared with control cells (E) and following BCNU exposure (F; *, p < 0.05). G, the effect of BCNU is greater in BH4-deficient cells such that BCNU (100 μm) exacerbates the already decreased ratio of BH4:BH2 (†, p < 0.01; n = 4).
BH4 Deficiency Induces Glutathionylation of eNOS
Having established that superoxide produced by eNOS occurs independently whether by glutathionylation or BH4 deficiency and having observed that glutathionylation of eNOS is sufficient to induce BH4 deficiency, we next sought to determine whether there is also a link between BH4 deficiency and glutathionylation of eNOS. DOX-induced eNOS uncoupling alone was sufficient to elevate the specific S-glutathionylation of eNOS as shown by immunoprecipitation. Moreover, the degree of S-glutathionylation of eNOS was greater in DOX-treated versus BCNU-treated cells and even greater when cells were treated with both DOX and BCNU (Fig. 6).
FIGURE 6.
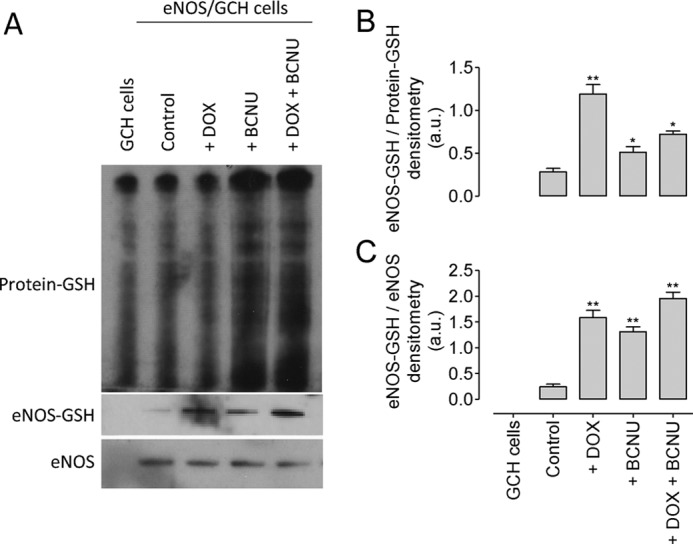
BH4 deficiency induces glutathionylation of eNOS. A, total protein glutathionylation was observed in GCH and eNOS/GCH control cells and was markedly elevated following exposure of BCNU, but not DOX. Treatment of GCH/eNOS cells with doxycycline and/or BCNU resulted in S-glutathionylation of eNOS. B, DOX treatment significantly increased the proportion of eNOS that was glutathionylated when compared with total protein-GSH. C, DOX and BCNU treatment elevated eNOS-GSH to equal levels when compared with the amount of unmodified eNOS. The blots shown are representative of three separate experiments (*, p < 0.05; **, p < 0.01).
eNOS Mutants Reveal Additive Effects of BH4-dependent and Glutathionylation-induced eNOS Uncoupling
We next examined the function of key cysteine residues within the eNOS reductase domain. S-glutathionylation of eNOS has been demonstrated to occur at residues cysteine 689 or cysteine 908, and mutation of these residues (Cys → Ser) has been shown to prevent the modification of eNOS in vitro. To investigate the role that these specific residues play in eNOS uncoupling, induced either by BH4 deficiency or glutathionylation, we transiently expressed either eNOS-WT or eNOS C908S in the tetracycline regulatable GCH cell line and then subjected them to BCNU (100 μm) and/or DOX, to induce BH4 deficiency as before (Fig. 7). In cells expressing eNOS-WT, DOX treatment diminished eNOS activity by 25%, indicative of BH4 deficiency and eNOS uncoupling as described previously (14, 21). BCNU exposure had a more striking effect and decreased eNOS activity by 65% and a further 15% (80% total) in the presence of both BCNU and DOX. Mutation of eNOS C908S prevented the effects of BCNU, but not DOX, on the conversion of arginine to citrulline (Fig. 7A). Similarly, DOX treatment uncoupled eNOS-WT as demonstrated by the quantification of 2-hydroxyethidium accumulation within cells by HPLC. BCNU treatment resulted in a 2-fold increase in superoxide production from eNOS-WT cells in either the presence or the absence of DOX. The effect of BCNU, in both the absence and the presence of DOX, was prevented by C908S mutation. Interestingly, DOX treatment alone, in the absence of eNOS, elevated superoxide production by 10-fold, suggesting an antioxidant effect of intracellular BH4 (Fig. 7B). The effects of eNOS uncoupling were reflected in BH4 oxidation; no effect of DOX was observed on the ratio of BH4:BH2 in GCH cells without eNOS. However, the ratio of BH4:BH2 was significantly reduced following BCNU exposure (10.26 ± 2.47 versus 19.7 ± 4.2). When GR was inhibited by BCNU in cells with low GTPCH activity and low BH4 levels, the BH4:BH2 ratio was further reduced, suggesting that the role of GR is more important to maintain BH4 redox state when absolute BH4 levels are low. Furthermore, in the presence of eNOS, DOX-induced BH4 deficiency decreased the BH4:BH2 ratio, and the effect of BCNU was markedly greater (than in the absence of eNOS). When eNOS was uncoupled in a BH4-dependent manner (in BH4-deficient, DOX-treated cells), the effect of BCNU was even more striking, with a further reduction in the ratio of BH4:BH2. In eNOS mutant cells (C908S), BCNU still decreased the BH4:BH2 ratio, but only to levels found in the control cells lacking eNOS. However, the additive effect of eNOS-derived superoxide was no longer detected, and the ratio of BH4:BH2 in BH4-deficient cells was restored (Fig. 7C). Similar findings with C689S and double (C689S and C908S) eNOS mutants were also seen on eNOS activity, superoxide production, and intracellular BH4 levels and therefore indicate that glutathionylation of both residues is required to stimulate superoxide production from the reductase domain (data not shown). Importantly, although DOX treatment-induced BH4 deficiency triggers glutathionylation of eNOS, mutation of eNOS preventing glutathionylation does not attenuate the detrimental effects of BH4 deficiency on eNOS activity.
FIGURE 7.
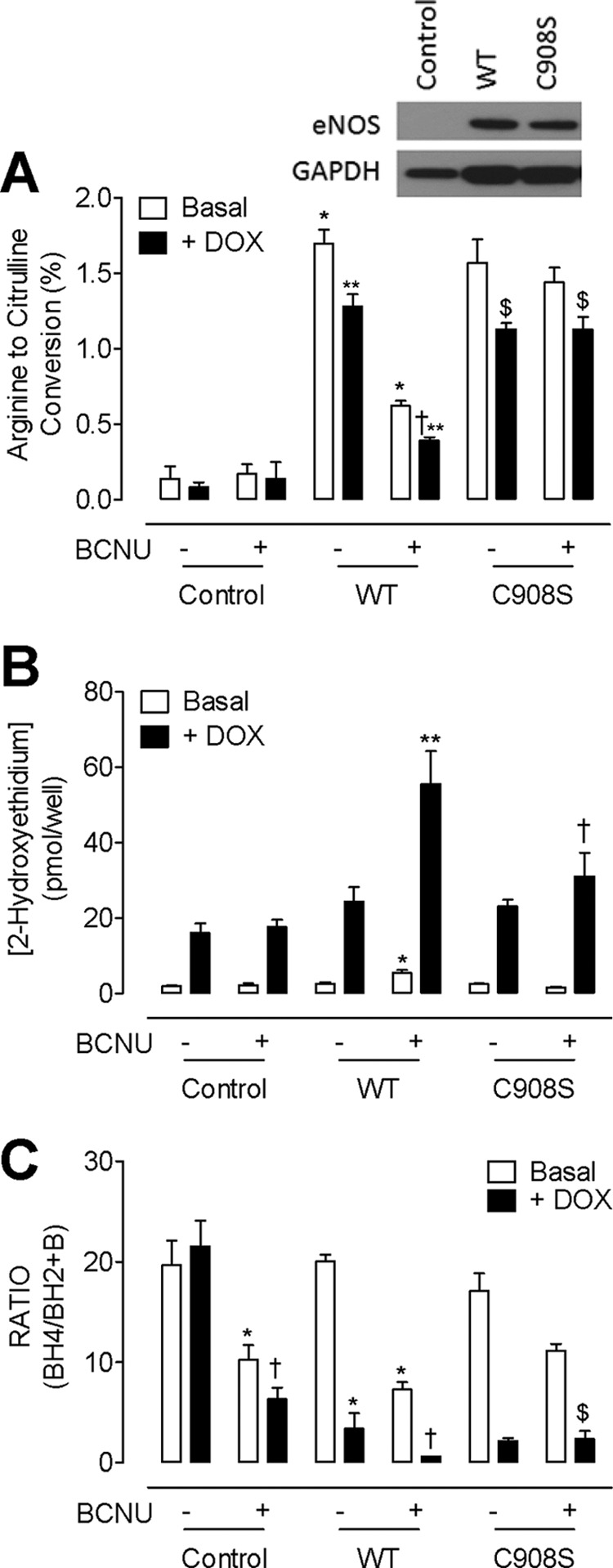
C908S mutation prevents S-glutathionylation-induced but not BH4-dependent uncoupling of eNOS. GCH cells were cultured in doxycycline (DOX) for 7 days and then transiently transfected with either eNOS WT or eNOS C908S mutant DNA. Transfected cells were then exposed to BCNU (100 μm) for 4 h prior to analysis of eNOS enzymatic activity by HPLC as under “Experimental Procedures.” A, eNOS activity, as measured by arginine to citrulline HPLC, was attenuated by doxycycline treatment (**, p < 0.05) but to an even greater extent by BCNU (*, p < 0.01). When BH4-deficient, doxycycline-treated cells are treated with BCNU, arginine to citrulline conversion is even further exacerbated (†, p < 0.01). Mutation of C908S attenuated the BCNU-induced but not the doxycycline-induced decrease in eNOS activity ($, p < 0.01; n = 6). B, eNOS mutants exhibit complementing changes in superoxide production. BCNU exposure triggers a 2-fold increase in superoxide production that is attenuated by mutation of C908S (*, p < 0.05). Accordingly, following 7 days of doxycycline treatment, superoxide is elevated 10-fold in untreated cells and 30-fold in the eNOS WT cells in the presence of BCNU (**, p < 0.01). C908S mutation prevented this accumulation of 2-hydroxyethidium, indicative of superoxide production (†, p < 0.05; n = 5). C, correspondingly, BH4:BH2 ratios were also altered by BCNU and doxycycline treatment. In untreated cells, both doxycycline (*, p < 0.05) and BCNU (†, p < 0.01) resulted in diminished BH4:BH2 ratios. BH4:BH2 ratio was further attenuated in eNOS WT expressing cells and even further exacerbated following exposure of BH4-deficient cells to BCNU (†, p < 0.01). The effects of BCNU but not doxycycline were totally abolished by C908S mutation ($, p < 0.05; n = 6).
DISCUSSION
In this study we compare two distinct mechanisms of eNOS uncoupling, S-glutathionylation and BH4 deficiency, and investigate how these two pathways may interact to regulate eNOS function. We demonstrate key, independent roles for both glutathionylation- and BH4-dependent uncoupling of eNOS and advance previous findings by demonstrating that reciprocal effects of intracellular BH4 levels and S-glutathionylaton link eNOS coupling versus uncoupling via these two mechanisms. The major findings of this study are as follows. First, manipulation of the GSH:GSSG ratio by either pharmacologic or genetic means, such that intracellular GSSG accumulates, induces S-glutathionylation and uncoupling of eNOS. Second, this uncoupling of eNOS is sufficient to promote BH4 oxidation. Third, eNOS is required for the induction of superoxide production by glutathione reductase inhibition. Fourth, S-glutathionylation of both of the critical cysteine residues is required to induce a feed forward cascade of eNOS uncoupling and BH4 oxidation. Finally, uncoupling of eNOS via glutathionylation or BH4 deficiency occur independently, but their effects are additive and mutually linked. Superoxide produced by eNOS from the heme within the oxygenase domain, induced by BH4 deficiency, causes S-glutathionylation of eNOS and superoxide production from the reductase domain. Conversely, S-glutathionylation within the reductase domain can trigger superoxide production, BH4 oxidation, and subsequently superoxide production from the oxygenase domain (Fig. 8). Taken together, our findings provide clear evidence to support a role for eNOS glutathionylation as part of a complex and a tightly controlled mechanism that regulates BH4 homeostasis and increases our understanding of the interaction between pathways that determine eNOS function. We propose eNOS to be a redox “hub,” being regulated by and contributing to the regulation of intracellular redox homeostasis, through mutually interacting BH4-dependent and GSH-dependent pathways.
FIGURE 8.

Schematic representation of the interaction between oxygenase and reductase domain derived superoxide from eNOS. Superoxide produced from the oxygenase domain (triggered by BH4 deficiency) has a marked effect to decrease the GSH:GSSG ratio, thus initiating glutathionylation of the enzyme (blue) and a feed forward cascade of eNOS uncoupling. The reverse is also true; superoxide produced from the reductase domain in response to S-glutathionylation oxidizes BH4, thus activating superoxide production from the oxygenase domain (red). DOX, doxycycline.
Other examples of redox-sensitive signaling activity and pathways must also be considered; a critical cysteine is responsible for DNA binding to the redox-sensitive transcription factor NF-κB, a molecular switch has been identified on p21 (Ras), and reactive oxygen species directly alter MnSOD expression and are involved in the induction of MnSOD by TNF-α (22). Thiol metabolism and antioxidant systems have complementary roles in antioxidant defense, cell signaling, and cytoprotection. Impairment of the thiol-metabolizing enzymes thioredoxin and GR have previously been shown to have detrimental effects on oxidative and nitrosative pathways (23), and previous studies have reported that the production of eNOS-derived superoxide can be regulated by S-glutathionylation (11). However, no studies have addressed the impact of this post-translational modification on BH4-dependent eNOS uncoupling and the availability of the NOS cofactor. In this study we provide the first evidence that BH4 deficiency- and S-glutathionylation-induced mechanisms of eNOS uncoupling occur independently but act together to regulate the switching of eNOS between the production of either NO or superoxide. We also speculate that BH4 plays an important role in regulating the cellular effects of BCNU; high BH4 levels appear to be protective against the detrimental effects of BCNU on eNOS activity and the GSH:GSSG ratio. Thus, high levels of BH4 within the cell may also protect eNOS from S-glutathionylation and BH4-independent uncoupling. It is important to consider that the levels of eNOS-GSH may reflect glutathionylation-dependent signaling across the whole cell. However, this is not the case; DOX treatment elevated eNOS-GSH levels more than BCNU treatment when compared with total protein-GSH, and thus inhibition of BH4 synthesis by treatment of cells with DOX occurs independently of elevated protein-GSH. These data provide key evidence that (i) GSH-eNOS is not solely a reflection of total protein-GSH and (ii) BH4 is protective against eNOS-GSH modification but not global cellular glutathionylation.
Pharmacological inhibition of thioredoxin and GR have previously been shown to decrease VEGF-stimulated NO production in endothelial cells that was not restored following addition of BH4 (23). Discordance between the effects of GR inhibition and eNOS activity was observed, suggesting a further thiol-regulated signaling mechanism, such as glutathionylation in regulating eNOS activity. We clearly show that attenuation of the protective mechanisms actioned by GR using pharmacological and genetic means (in both endothelial cells and a cell culture model of eNOS uncoupling) results in the production of superoxide from both the reductase and oxygenase domains of eNOS as a direct result of direct post-translational modification by S-glutathionylation rather than merely by broad changes in cellular redox state.
There are several proposed mechanisms for the modification of proteins by S-glutathionylation. Thiol-disulfide exchange with oxidized GSSG is one of the main mechanisms, but ROS and RNS may produce protein-thiyl radicals that can react with GSH to generate an S-glutathionylated protein. It is believed that thiol-disulfide exchange can only occur when the ratio of GSH:GSSG is diminished, and GSSG accumulates within the cell. Moreover, the formation of thiyl intermediates such as the thiyl radical, sulfenic acid, or S-nitrosyl is a more rapid and efficient mechanism for protein S-glutathionylation in vivo, and these mechanisms can play a role in signal transduction. Indeed, formation of an eNOS-thiyl radical has been shown to occur in response to exogenous ROS and self-propagated superoxide production from eNOS. Our data support this finding and reveal that induction of eNOS uncoupling with doxycycline in Tet-regulated cells promotes eNOS glutathionylation and that this superoxide, derived from eNOS, is capable of oxidizing BH4. Our data also reveal for the first time that the reverse is also true; glutathionylation of the enzyme even in the presence of saturating levels of BH4 promotes elevated ROS production such that BH4 oxidation is exacerbated. When eNOS becomes glutathionylated in conditions where the enzyme is already uncoupled, as in GCH-Tet cells following DOX exposure, oxygenase and reductase domain-derived superoxide is additive, creating a condition where eNOS is producing significantly more substantial amounts of ROS (Fig. 8).
Two cysteine residues critical to the modulation of eNOS by glutathione at the reductase domain also have significant effects on BH4-dependent uncoupling at the heme within the oxygenase domain. Indeed, cysteine 908 within the FAD/FMN-binding site of the reductase domain (responsible for regulating electron transfer between FAD and FMN) has been shown to be the site of thiyl radical formation and putative glutathionylation. C908S mutation rendered eNOS resistant to modification by glutathionylation and prevented in the induction of superoxide production, derived from both the heme and the reductase domain. We demonstrate that expression of C908S rather than WT eNOS was able to prevent glutathione-induced superoxide production and BH4 oxidation but not the uncoupling of eNOS triggered by BH4 deficiency. This suggests that although uncoupling of eNOS at both the reductase and oxygenase domains occurs independently of one another, once uncoupled, the effects of eNOS-derived superoxide are self-perpetuating, contributing to one pool of superoxide. Importantly, in this situation, restoration of both intracellular GSH and BH4 levels will be required to fully recouple the enzyme and restore normal NO synthesis. These findings are important in the clinical setting and may provide a possible explanation for the disappointing results obtained from clinical trials; a recent report on sapropterin treatment in patients with coronary artery disease demonstrated that oral BH4 treatment failed to improve brachial flow-mediated vasodilation and acetylcholine-induced vasodilation in ex vivo saphenous vein rings. The ability of BH4 to recouple NOS in patients with cardiovascular disease may therefore be limited by BH4 oxidation, BH2 accumulation, and failure to improve BH4:BH2 ratios (24). Although intracellular GSH was not measured in this study, elevation of GSH levels and/or other compounds to moderate thiol-redox signaling, as well as enhancing BH4 levels, may have proved beneficial.
Interrelations among thiol oxidation, S-glutathionylation, and S-nitrosylation might serve to translate oxidative or nitrosative stimuli into a functional response. In some proteins the competition between S-glutathionylation and S-nitrosylation has also been compared, showing that there are large differences among them regarding their tendencies to undergo each modification. The localization and relative selectivity of proteins for both modifications will be a determinant of the cellular proteins that will be more easily or more stably modified. Accordingly, S-nitrosylation of eNOS has been demonstrated at several cysteine residues, although these sites are distinct from those that are S-gluthathionylated (modification by S-nitrosylation does occur at cysteines −660, −801, and −113), all of which lie within the FAD/FMN-binding domain of the reducase domain of the protein. Modification of these residues may affect or disturb the modification of cysteine 908 by glutathione, and thus competition between nitrosylation and glutathionylation may play a pivotal role in determining protein function and activity.
We propose that rather than characterize uncoupled eNOS as “dysfunctional,” uncoupling by either glutathionylation of BH4 deficiency is in fact a tightly regulated mechanism that renders eNOS as a redox hub. This hypothesis links the upstream redox-sensitive effects of BH4 and glutathione that determine eNOS function, with of a plethora of targets and pathways that lie downstream of eNOS that have been demonstrated to be modulated by cellular redox state. Observations suggest that changes in redox status can exert a powerful influence on cellular homeostasis via eNOS, and further studies are required to elucidate the mechanisms for this redox-sensitive, downstream signaling.
This work was supported by British Heart Foundation Centre of Research Excellence Intermediate Fellowship Award RE/08/004 (to M. J. C.) and British Heart Foundation Programme Grant RG/07/003/23133.
- eNOS
- endothelial nitric-oxide synthase
- BH4
- tetrahydrobiopterin
- BH2
- 7,8-dihydrobiopterin
- BCNU
- 1,3-bis(2-chloroethyl)-1-nitrosourea
- GTPCH
- GTP cyclohydrolase I
- GR
- glutathione reductase
- l-NAME
- l-nitroarginine methyl ester.
REFERENCES
- 1. Ignarro L. J. (2002) Nitric oxide as a unique signaling molecule in the vascular system. A historical overview. J. Physiol. Pharmacol. 53, 503–514 [PubMed] [Google Scholar]
- 2. Furchgott R. F., Zawadzki J. V. (1980) The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288, 373–376 [DOI] [PubMed] [Google Scholar]
- 3. Panza J. A., García C. E., Kilcoyne C. M., Quyyumi A. A., Cannon R. O., 3rd (1995) Impaired endothelium-dependent vasodilation in patients with essential hypertension. Evidence that nitric oxide abnormality is not localized to a single signal transduction pathway. Circulation 91, 1732–1738 [DOI] [PubMed] [Google Scholar]
- 4. Ohara Y., Peterson T. E., Harrison D. G. (1993) Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Invest. 91, 2546–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. White C. R., Brock T. A., Chang L.-Y., Crapo J., Briscoe P., Ku D., Bradley W. A., Gianturco S. H., Gore J., Freeman B. A., Tarpey M. M. (1994) Superoxide and peroxynitrite in atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 91, 1044–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hurshman A. R., Krebs C., Edmondson D. E., Huynh B. H., Marletta M. A. (1999) Formation of a pterin radical in the reaction of the heme domain of inducible nitric oxide synthase with oxygen. Biochemistry 38, 15689–15696 [DOI] [PubMed] [Google Scholar]
- 7. Crabtree M. J., Smith C. L., Lam G., Goligorsky M. S., Gross S. S. (2008) Ratio of 5,6,7,8-tetrahydrobiopterin to 7,8-dihydrobiopterin in endothelial cells determines glucose-elicited changes in NO vs. superoxide production by eNOS. Am. J. Physiol. Heart Circ. Physiol. 294, H1530–H1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stroes E., Hijmering M., van Zandvoort M., Wever R., Rabelink T. J., van Faassen E. E. (1998) Origin of superoxide production by endothelial nitric oxide synthase. FEBS Lett. 438, 161–164 [DOI] [PubMed] [Google Scholar]
- 9. Vásquez-Vivar J., Martásek P., Whitsett J., Joseph J., Kalyanaraman B. (2002) The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase. An EPR spin trapping study. Biochem. J. 362, 733–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dumitrescu C., Biondi R., Xia Y., Cardounel A. J., Druhan L. J., Ambrosio G., Zweier J. L. (2007) Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc. Natl. Acad. Sci. U.S.A. 104, 15081–15086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen C. A., Wang T. Y., Varadharaj S., Reyes L. A., Hemann C., Talukder M. A., Chen Y. R., Druhan L. J., Zweier J. L. (2010) S-Glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468, 1115–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Townsend D. M. (2007) S-glutathionylation. Indicator of cell stress and regulator of the unfolded protein response. Mol. Interv. 7, 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dalle-Donne I., Rossi R., Colombo G., Giustarini D., Milzani A. (2009) Protein S-glutathionylation. A regulatory device from bacteria to humans. Trends Biochem. Sci. 34, 85–96 [DOI] [PubMed] [Google Scholar]
- 14. Crabtree M. J., Tatham A. L., Al-Wakeel Y., Warrick N., Hale A. B., Cai S., Channon K. M., Alp N. J. (2009) Quantitative regulation of intracellular endothelial nitric oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status. Insights from cells with Tet-regulated GTP cyclohydrolase I expression. J. Biol. Chem. 284, 1136–1144 [DOI] [PubMed] [Google Scholar]
- 15. Tzeng E., Billiar T. R., Robbins P. D., Loftus M., Stuehr D. J. (1995) Expression of human inducible nitric oxide synthase in a tetrahydrobiopterin (H4B)-deficient cell line. H4B promotes assembly of enzyme subunits into an active enzyme. Proc. Natl. Acad. Sci. U.S.A. 92, 11771–11775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McDonald D. M., Alp N. J., Channon K. M. (2004) Functional comparison of the endothelial nitric oxide synthase Glu298Asp polymorphic variants in human endothelial cells. Pharmacogenetics 14, 831–839 [DOI] [PubMed] [Google Scholar]
- 17. de Bono J. P., Warrick N., Bendall J. K., Channon K. M., Alp N. J. (2007) Radiochemical HPLC detection of arginine metabolism. Measurement of nitric oxide synthesis and arginase activity in vascular tissue. Nitric Oxide 16, 1–9 [DOI] [PubMed] [Google Scholar]
- 18. Heales S., Hyland K. (1989) Determination of quinonoid dihydrobiopterin by high-performance liquid chromatography and electrochemical detection. J. Chromatogr. 494, 77–85 [DOI] [PubMed] [Google Scholar]
- 19. Zhao H., Joseph J., Fales H. M., Sokoloski E. A., Levine R. L., Vasquez-Vivar J., Kalyanaraman B. (2005) Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc. Natl. Acad. Sci. U.S.A. 102, 5727–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fink B., Laude K., McCann L., Doughan A., Harrison D. G., Dikalov S. (2004) Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay. Am. J. Physiol. Cell. Physiol. 287, C895–C902 [DOI] [PubMed] [Google Scholar]
- 21. Crabtree M. J., Tatham A. L., Hale A. B., Alp N. J., Channon K. M. (2009) Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling. Relative importance of the de novo biopterin synthesis versus salvage pathways. J. Biol. Chem. 284, 28128–28136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sen C. K. (1998) Redox signaling and the emerging therapeutic potential of thiol antioxidants. Biochem. Pharmacol. 55, 1747–1758 [DOI] [PubMed] [Google Scholar]
- 23. Sugiyama T., Michel T. (2010) Thiol-metabolizing proteins and endothelial redox state. Differential modulation of eNOS and biopterin pathways. Am. J. Physiol. Heart Circ. Physiol. 298, H194–H201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cunnington C., Van Assche T., Shirodaria C., Kylintireas I., Lindsay A. C., Lee J. M., Antoniades C., Margaritis M., Lee R., Cerrato R., Crabtree M. J., Francis J. M., Sayeed R., Ratnatunga C., Pillai R., Choudhury R. P., Neubauer S., Channon K. M. (2012) Systemic and vascular oxidation limits the efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation 125, 1356–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]