

Abstract
The molecular mechanisms underlying preconditioning (PC), a powerful endogenous neuroprotective phenomenon, remain to be fully elucidated. Once identified, these endogenous mechanisms could be manipulated for therapeutic gain. We investigated whether lymphocyte cell kinase (Lck), a member of the Src kinases family, mediates PC. We used both in vitro primary cortical neurons and in vivo mouse cerebral focal ischemia models of preconditioning, cellular injury, and neuroprotection. Genetically engineered mice deficient in Lck, gene silencing using siRNA, and pharmacological approaches were used. Cortical neurons preconditioned with sublethal exposure to NMDA or oxygen glucose deprivation (OGD) exhibited enhanced Lck kinase activity, and were resistant to injury on subsequent exposure to lethal levels of NMDA or OGD. Lck gene silencing using siRNA abolished tolerance against both stimuli. Lck−/− mice or neurons isolated from Lck−/− mice did not exhibit PC-induced tolerance. An Lck antagonist administered to wild-type mice significantly attenuated the neuroprotective effect of PC in the mouse focal ischemia model. Using pharmacological and gene silencing strategies, we also showed that PKCε is an upstream regulator of Lck, and Fyn is a downstream target of Lck. We have discovered that Lck plays an essential role in PC in both cellular and animal models of stroke. Our data also show that the PKCε-Lck-Fyn axis is a key mediator of PC. These findings provide new opportunities for stroke therapy development.
Introduction
Ischemic tolerance induced by preconditioning (PC) is an endogenous protective mechanism whereby short exposure to sublethal levels of a noxious stimulus results in protection from subsequent more severe levels of exposure to the stimulus (Zemke et al., 2004; Obrenovitch, 2008). PC occurs in a variety of organs including the heart and brain (Lo et al., 2003). Numerous preclinical in vitro and in vivo models of PC are available, and clinical data supporting preconditioning in human diseases also exist. For example, transient ischemic attacks (TIAs), caused by brief periods of interruption in blood flow to the brain, are associated with decreased stroke severity and improved outcome (Moncayo et al., 2000; Dirnagl et al., 2009). To date, all acute neuroprotective stroke therapies have failed in clinical trials, and, consequently, no neuroprotective therapies for acute stroke exist. An alternative strategy to reduce stroke severity may involve enhancing endogenous neuroprotection such as PC (Fisher and Ratan, 2003; Dirnagl et al., 2009). Although several mediators of PC have been proposed, such as hypoxia-inducible factor, heat shock proteins, erythropoietin, and ion channels (Kennedy and Buchan, 2005; Malhotra et al., 2006; Obrenovitch, 2008; Dirnagl et al., 2009), the exact mechanisms that mediate preconditioning are not yet fully understood.
Lymphocyte cell kinase (Lck), a member of the Src family kinases, has been shown to mediate diverse cellular pathways including cell survival, proliferation, differentiation, and cell death in many different cell types (Salmond et al., 2009). Previous studies using Lck knock-out mice demonstrated that Lck gene deletion abolished the cardioprotective effect of PC in myocardium (Ping et al., 1999, 2002). Although, Lck is expressed in the brain (Omri et al., 1996; Salter and Kalia, 2004), its role in neuroprotection and PC has not been previously investigated.
We investigated the role of Lck in PC in the brain using in vitro and in vivo ischemia models and show that Lck is a critical mediator of PC in brain. Furthermore, we show that Lck-associated PC is mediated through its interaction with PKCε and activation of another Src kinase, Fyn. In this study, we provide new insights into the mechanisms underlying endogenous neuroprotection, and suggest a novel therapeutic candidate pathway for stroke prevention or treatment.
Materials and Methods
Materials.
Cell culture media and reagents (Neurobasal A, B27, glutamine, and penicillin/streptomycin), Lipofectamine 2000, Dynabeads Protein G Immunoprecipitation kit, mouse anti-p59Fyn Ab, and rabbit anti-Src [pY418] Ab were obtained from Invitrogen. Poly-d-lysine, NMDA, 4′,6-diamidino-2-phenylindole (DAPI), and propidium iodide (PI) were purchased from Sigma. Lactate dehydrogenase (LDH) assay kit (CytoTox 96 Non-Radioactive cytotoxicity assay) and ProFluor Src-family kinase assay kit were obtained from Promega. BCA Protein assay reagents, SuperSignal West Pico Chemiluminescent Substrate, RIPA cell lysis buffer, Halt protease and phosphatase inhibitor mixture, and On-Target plus SMARTpool siRNA against mouse Lck, Fyn, or Yes and its nontargeting negative control siRNA were purchased from Thermo Fisher Scientific. RNeasy Mini kit was obtained from Qiagen, and BioPORTER reagent was from Genlantis. Mouse anti-protein kinase C ε (PKCε) Ab and mouse anti-p56Lck Ab were from BD Biosciences. Mouse anti-β-actin Ab, rabbit anti-Yes Ab, and mouse anti-glyderaldehyde-3-phosphate dehydrogenase (GAPDH) Ab were purchased from Abcam, Cell Signaling Technology, and Millipore, respectively. All other reagents were used with highest purity available.
Primary cortical neuronal cultures.
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Michigan State University. Primary cortical neuronal cultures were established as previously described (Yim et al., 2010). Briefly, cerebral cortices were isolated from C57BL/6 newborn mice at postnatal day 0 and dissociated in dissection media (81.8 mm Na2SO4, 30 mm K2SO4, 5.8 mm MgCl2, 0.252 mm CaCl2, 1.5 mm HEPES, 20 mm glucose, and 0.001% phenol red, pH 7.6) supplemented with 4 mm l-cysteine, 10 U/ml papain (Worthington), and 1000 U/ml DNase (Roche) for 30 min at 37°C. After dissociation, cells were washed with Neurobasal A and then triturated with pipette. Cells (1 × 106) were plated onto poly-d-lysine-precoated 12 well plates. Three days after plating, 50% of the medium was changed, and subsequently replaced every 3 d. Neuronal cultures were maintained in CO2 incubator (5% CO2/ 95% air balance) at 37°C, and used between days in vitro (DIV) 7 and 11.
Determination of NMDA-induced cytotoxicity.
Delayed NMDA-induced cytotoxicity was measured at 24 h after NMDA exposure using PI-staining or LDH assay as previously described (Tauskela et al., 2003; Wetzel et al., 2008). On DIV 9, cultured neurons were exposed to NMDA for 20 min and maintained in CO2 incubator for 24 h. Cells were stained with 5 μg/ml PI at 37°C for 30 min, and examined by fluorescent microscopy (Nikon) or fluorescent microplate reader (Ascent, Thermo Lab Systems). The extent of LDH leakage was measured in conditioned media using a Cytotox 96 Non-Radioactive cytotoxicity assay. The cell viability in sister cells treated with 100 μm NMDA was used as the total cell death (100%).
In experiments with NMDA-induced PC, cells were exposed to sublethal dose of NMDA (10 μm) for 20 min on DIV 8, and subsequently exposed to lethal dose of NMDA (50 μm) at 24 h after PC on DIV 9. Cell viability was measured as described above on DIV 10.
PKCε activity modulating peptides.
The peptide inhibitor for PKCε translocation (EAVSLKPT; V1-2) (Chen et al., 2005) and PKCε selective agonist (HDAPIGYD; ψεRACK) (Inagaki et al., 2005) were synthesized by Macromolecular Structure Facility (Department of Biochemistry, Michigan State University). εV1-2 or ψεRACK was diluted and mixed with BioPORTER reagent (Alano et al., 2010). Cells were incubated with BioPORTER-peptide complex for 4 h at 37°C. After transfection, cell were washed with complete culture media and immediately exposed to NMDA. Cell transfection efficiency was confirmed using parallel transfections with fluorescein-labeled control IgG, as suggested by the manufacturer.
Gene silencing with siRNAs.
Primary cortical neurons were transfected with 10 nm siRNAs using Lipofectamine 2000 transfection reagent (Ueda et al., 2007). On DIV 5, neurons were incubated with Neurobasal A containing Lipofectamine 2000 (0.2%) and 10 nm siRNAs against mouse Lck, Yes, or Fyn, or nontargeting negative control for 4 h at 37°C. After transfection, the original medium collected before transfection was replaced. To ensure gene silencing efficiency, target mRNA or protein samples were collected after 48 and 96 h after siRNA transfection, and measured by quantitative real time PCR or Western blot, respectively.
Immunoprecipitation.
Neuronal lysates were applied to immunoprecipitation using magnetic beads (Dynabeads, Invitrogen) (Gudz et al., 2006). Cell lysates were harvested using RIPA buffer, and centrifuged at 14,000 × g for 15 min. Protein concentrations were determined by BCA assay, and then adjusted to 1 mg/ml. Five hundred microliters of lysates were applied to immunoprecipitation using specific anti-Lck or Fyn Ab-conjugated magnetic beads (Dynabeads, Invitrogen). Eluted immunoprecipitates were used for kinase assay or Western blot.
Lck/Fyn kinase activity.
After immunoprecipitation with anti-Lck or Fyn Ab, eluted immunoprecipitates were analyzed using a ProFluor Src-Family Kinase Assay (Watanabe et al., 2010). The total kinase amounts in immunoprecipitates were examined by Western blot, and the kinase activity from the corresponding control was defined as one-fold.
Western blot.
After the cell lysate was collected using RIPA buffer supplemented with Halt protease/phosphatase inhibitor mixture, the protein concentrations were determined using BCA assay. The protein samples (20 μg/lane) were separated in 12% Tris-HCl SDS-polyacrylamide Ready Gels (Bio-Rad) and were transferred to PVDF membrane (Millipore). After blocking with 5% BSA, membranes were incubated overnight at 4°C with primary antibodies of anti-Lck (1:1000), anti-Fyn (1:500), anti-p-Src/Fyn (1:1000), anti-PKCε (1:1000), anti-Yes (1:1000), anti-GAPDH (1:5000), or anti-β-actin (1:5000). Horseradish peroxidase-conjugated secondary antibodies (1:5000, Cell Signaling Technology) were used as secondary antibodies. The immune complexes were visualized by enhanced chemiluminescence using SuperSignal West Pico Chemiluminescent Substrate. Bands were quantified by NIH ImageJ program, and normalized by the corresponding loading controls of β-actin or GAPDH. All immunoblots were repeated for at least three independent experiments.
Quantitative real time PCR.
Total RNA was extracted using RNeasy mini kit (Qiagen), and converted into cDNA using TaqMan reverse transcription kit (Applied Biosystems) for quantitative real-time PCR (qRT-PCR). The specific primer sequences were as follows: 18S, forward: ACC GCA GCT AGG AAT AAT GGA; reverse: GCC TCA GTT CCG AAA ACC A; Lck, forward: TGG AGA ACA TTG ACG TGT GTG; reverse: ATC CCT CAT AGG TGA CCA GTG; and Fyn, forward: ACC TCC ATC CCG AAC TAC AAC; reverse: CGC CAC AAA CAG TGT CAC TC. Quantification of gene copies was performed on the 7500 Real-Time PCR system, using SYBR Green master mix (Applied Biosystems). PCR cycles consisted of three stages with an initial step at 95°C for 10 min followed by 40 cycles at 95°C for 15 s and 60°C for 1 min, and a final stage for dissociation curve. Relative mRNA expressions were calculated by the comparative CT method (2−ΔΔCt), and normalized to the endogenous 18S control.
Oxygen glucose deprivation.
Oxygen glucose deprivation (OGD) on primary cortical neurons was performed as previously described (Plesnila et al., 2001; Wetzel et al., 2008). On DIV 9, cells were washed, and the medium was replaced by glucose free Earl's balanced salt solution (5.4 mm KCl, 26.2 mm NaHCO3, 116 mm NaCl, 1 mm NaH2PO4, 0.8 mm MgSO4, 1.8 mm CaCl2, and 0.01 mm glycine, pH 7.4). Cell plates were placed in a closed chamber (Billups-Rothenberg) filled with 5% CO2/95% N2 for 2 h at 37°C. OGD was terminated by returning the cultures to normal condition of complete Neurobasal A media and 5%CO2/ 95% air. After 24 h, OGD-induced cell death was examined by PI staining and LDH assay as described above. To induce OGD-PC, cells were exposed to sublethal OGD for 30 min on DIV 8, and lysed for Western blot/kinase assay or further exposed to lethal OGD for 2 h on DIV 9 for cytotoxicity assay.
Permanent middle cerebral artery occlusion.
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the Michigan State University. Focal ischemia was induced by permanent middle cerebral artery occlusion (pMCAO) in male C57BL/6 mice (22–27 g; Charles River Laboratory) as previously described (Rajanikant et al., 2007). Mice were kept under isoflurane anesthesia during the entire procedure and their body temperature was maintained at 37°C. A skin-incision was made to create a small subtemporal craniotomy to expose the middle cerebral artery, and the artery was occluded using a bipolar coagulator. To ensure the completeness of the occlusion, the cerebral blood flow in the MCA territory was measured before and after MCAO by a laser doppler (Perimed PF-3). After incision closure, the animals were allowed to recover from the anesthesia. At 24 h after pMCAO, animals were killed, and then the brains were removed and sliced into 1 mm coronal sections, and stained with 2,3,5-triphenyl tetrazolium chloride (TTC) solution (Ruscher et al., 2011). The infarct area was measured using the NIH ImageJ program from the scanned images of stained brain slices. The infarct volume in each slice was calculated by taking the average of the infarct areas on both sides of the slice and multiplying it by the section thickness.
In vivo preconditioning.
PC was induced 48 h before pMCAO by exposing the animals for 2 h to a continuously flushed gas mixture of 8% oxygen/92% nitrogen at a rate of 1.5 L/min. Control animals were exposed to ambient air. After 2 h, the mice were returned to normal atmospheric conditions. At 48 h after PC, the mice were subjected to pMCAO and the differences in infarct volumes between the groups were determined at 24 h after pMCAO by TTC staining as described above. To determine the effect of A420983 on PC protection against brain infarction, mice were orally administered with vehicle or 18 mg/kg A420983 immediately after PC and every 12 h thereafter for 48 h before pMCAO.
Statistics.
We calculated the means and SEM for all treatment groups. The data were subjected to Student's t test to determine the significant differences between treatment groups. Statistical analysis was performed using SPSS software. In all cases, a p value of <0.05 was considered significant.
Results
Lck mediates preconditioning in NMDA-induced neuronal death
Only cultures which were >90% positive for neuronal specific markers were used. NMDA-induced delayed cytotoxicity in primary cortical neurons has been used as an in vitro model of ischemic neuronal cell death (Manzerra et al., 2001; Tauskela et al., 2003; Lin et al., 2008). NMDA-induced (10–100 μm) cytotoxicity was observed in a dose-dependent manner. Confirming previous reports, cells that were preconditioned by preexposure to sublethal NMDA (10 μm) exhibited decreased vulnerability to subsequent exposure to lethal concentrations of NMDA (50 μm) (Fig. 1A,B).
Figure 1.
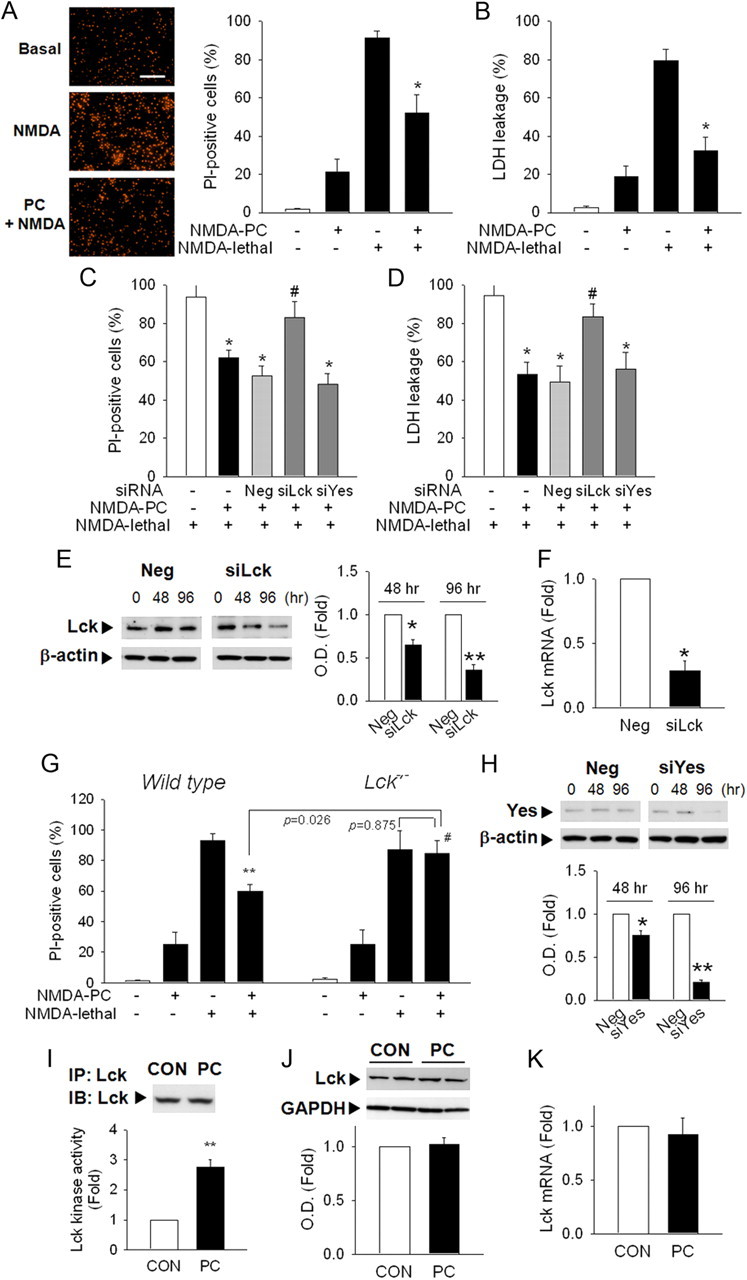
Lck mediates PC neuroprotection against NMDA-induced cytotoxicity in primary cortical neurons. A, PC with NMDA (10 μm) decreased vulnerability to lethal levels of NMDA (50 μm). Neurons were stained with PI at 24 h after lethal NMDA exposure. Scale bar, 200 μm. B, Lactate dehydrogenase (LDH) released into the medium was measured. C, D, Transfection of siRNA against Lck but not Yes reversed PC neuroprotection in PI-staining (C) or LDH leakage (D) at 24 h after lethal NMDA exposure. Neg, Nontargeting negative siRNA; siYes, siRNA against Yes. *p < 0.05 versus NMDA lethal; #p < 0.05 versus Neg+NMDA PC+NMDA lethal. E, Lck protein levels were significantly decreased after siLck transfection. β-actin, Loading control. F, Lck mRNA was reduced at 48 h after siLck transfection. *p < 0.05, **p < 0.01 versus Neg. G, NMDA PC neuroprotection was not observed in cortical neurons from Lck−/− mice. **p < 0.01 versus NMDA lethal; #p < 0.05 versus wild-type NMDA PC+NMDA lethal. H, siYes transfection decreased Yes protein level. I, Lck kinase activity was significantly increased after NMDA PC. Cell lysates were immunoprecipitated with Lck Ab and applied to kinase assay. Total level of Lck in each immunoprecipitates was determined by Western blot. *p < 0.05 versus control without PC; #p < 0.05 versus NMDA PC. J, NMDA PC did not change total Lck protein levels. GAPDH, A loading control. K, Gene transcription levels of Lck were not affected by NMDA PC. A, n = 6; B, I, n = 4; C–H, K, n = 3; J, n = 4–5. All values are means ± SEM and analyzed by Student's t test.
We then examined the effect of decreasing Lck protein on PC using siRNA against Lck and mutant mice deficient for the Lck gene. Lck gene silencing using siRNA abolished PC (Fig. 1C,D), suggesting that Lck plays a critical role in neuronal PC. Silencing efficiency was confirmed by demonstrating both decreased Lck protein and mRNA levels (Fig. 1E,F). Similarly, although cortical neurons isolated from Lck−/− mice exhibited a cytotoxic response to NMDA in a similar fashion to wild-type neurons, neurons from Lck−/− mice could not be preconditioned (Fig. 1G). Significantly, silencing another Src kinase, Yes, by using siRNA against Yes did not affect PC (Fig. 1C,D,H), suggesting that not all Src family members mediate PC.
To determine whether changes in Lck kinase activity mediate PC, neuronal lysates were immunoprecipitated with Lck antibody. The total Lck amount in each immunoprecipitate was not different (Fig. 1I, top), but Lck kinase activity was significantly increased by NMDA PC (Fig. 1I). Neither the total Lck protein levels nor the mRNA levels were affected by NMDA PC (Fig. 1J,K), suggesting that enhanced Lck activity rather than enhanced enzyme levels mediate PC.
Lck mediates PC in the OGD model of PC
To confirm our findings in another in vitro model of PC, we used OGD, a well established in vitro model, for ischemic neuronal damage and PC (Stenzel-Poore et al., 2004; Wetzel et al., 2008). Neuronal cells were preconditioned with exposure to 30 min OGD. Confirming previous reports, preconditioned neurons exhibited decreased vulnerability to subsequent lethal exposure to 2 h OGD (Fig. 2A). Silencing Lck using siRNA abolished OGD PC protection (Fig. 2B), and similarly, neurons from Lck−/− mice, mutant mice deficient for the Lck gene, could not be preconditioned by OGD (Fig. 2C). Lck kinase activation but not mRNA or protein levels (Fig. 2D) was increased after OGD PC in a similar pattern to NMDA PC. Together, these findings suggest that Lck mediates PC regardless of type of the preconditioning stimuli.
Figure 2.

Lck is important in OGD PC neuroprotection. A, Primary cortical neurons were incubated in glucose free Earl's balanced salt solution under 5% CO2/95% N2 at 37°C for OGD. OGD PC (30 min) reduced lethal OGD (2 h)-induced neuronal cytotoxicity in PI-staining. OGD PC was performed 24 h before lethal OGD stimulation. Cytotoxicity was examined at 24 h after lethal OGD stimulation. B, Silencing Lck significantly reversed OGD PC neuroprotection. siRNAs were transfected 48 h before OGD PC, and OGD PC/lethal OGD exposure was performed as described above. *p < 0.05, **p < 0.01 versus OGD lethal; #p < 0.05 versus Neg+OGD PC+OGD lethal. C, Protective effect of OGD PC was not found in primary neurons from Lck−/− mice. D, Lck kinase activity was increased during OGD PC. Neuronal lysates were collected after OGD PC and used for Lck immunoprecipitation and kinase assay. Total level of Lck in each immunoprecipitates was determined by Western blot. *p < 0.05 versus control without PC. A, n = 4; B, D, n = 3; C, n = 5. All values are means ± SEM and analyzed by Student's t test.
Lck mediates PC in focal cerebral ischemia
Next, we sought to determine the role of Lck in PC in vivo using the mouse pMCAO model. PC (exposure to 8% oxygen for 2 h) of mice followed by pMCAO at 48 h after PC significantly reduced the infarct volume compared with the control group, as measured by TTC staining at 24 h after MCAO (Fig. 3A). To determine whether Lck influences in vivo PC neuroprotection, we used Lck−/− mice and an orally active Lck antagonist, A420983 (Waegell et al., 2002). First, we examined whether the vulnerability to pMCAO is affected by Lck gene deletion (Fig. 3B) and found no significant difference in infarct volumes between wild-type, Lck+/− or Lck−/− mice (WT vs Lck+/−, p = 0.319; WT vs Lck−/−, p = 0.229; Lck+/− vs Lck−/−, p = 0.961). Unlike wild-type mice that demonstrated decreased infarct volumes following PC, Lck−/− mice did not exhibit decreased vulnerability to focal ischemia after PC (Fig. 3C). Physiological parameters during surgical procedure were not different between the groups as shown in Table 1. Similarly, mice treated with A420983 did not exhibit decreased infarct volumes after PC compared with vehicle treated mice (Fig. 3D).
Figure 3.
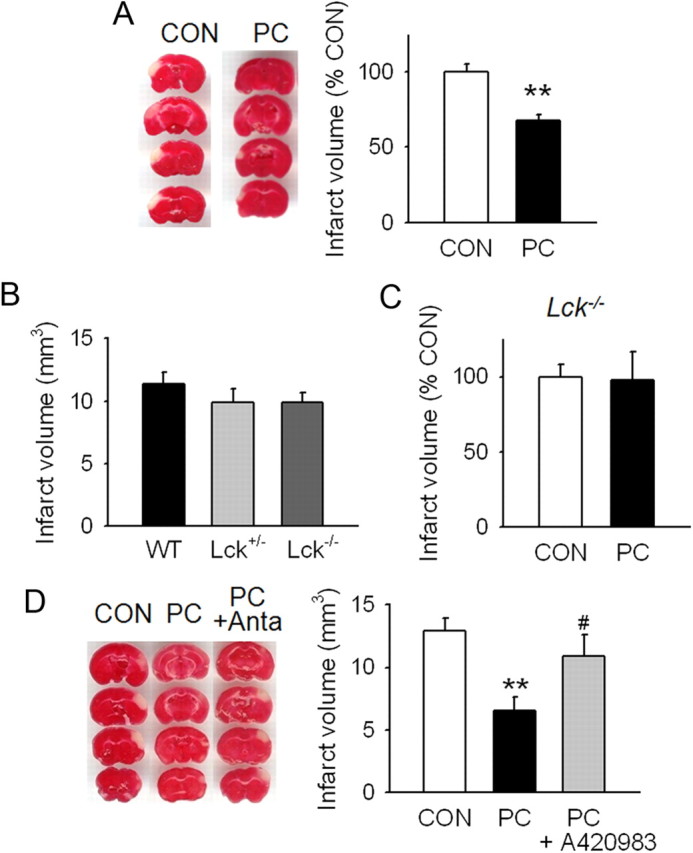
In vivo PC protection is abolished in Lck−/− mice or by Lck antagonist. A, In vivo PC was performed by exposing mice to 8% O2/92% N2 for 2 h. At 48 h after PC, the middle cerebral artery was permanently occluded for the induction of focal ischemia. Brain damage was determined by the infarct volumes at 24 h after ischemia using 2,3,5-triphenyl tetrazolium chloride staining. The representative brain slides are shown. B, Vulnerability to pMCAO was compared among wild-type (WT), Lck+/−, and Lck−/− mice and no significant difference was found. C, Protective effect of in vivo hypoxic PC against ischemic brain damage was not observed in Lck−/− mice. D, Lck antagonism by A420983, an orally active Lck antagonist, reversed in vivo PC neuroprotection. A420983 (18 mg/kg) was orally administered to mice immediately after PC and every 12 h thereafter for 48 h before focal ischemia. Anta, Lck antagonist A420983. **p < 0.01 versus control without PC; #p < 0.05 versus PC. A, n = 10–11; B, n = 11–15; C, n = 8–9; D, n = 14–16. All values are means ± SEM and analyzed by Student's t test.
Table 1.
Physiological parameters during pMCAO
| Wild type |
Lck−/− |
|||
|---|---|---|---|---|
| Control | Preconditioned | Control | Preconditioned | |
| Body weight (g) | 24.0 ± 0.60 | 25.5 ± 0.25 | 24.6 ± 0.99 | 24.8 ± 1.25 |
| Body temperature (°C) | 37.4 ± 0.04 | 37.3 ± 0.02 | 37.3 ± 0.07 | 37.3 ± 0.03 |
| CBF before pMCAO | 333.0 ± 18.3 | 306.4 ± 2.03 | 311.1 ± 12.0 | 315.0 ± 13.5 |
| CBF after pMCAO | 58.0 ± 4.90 | 49.5 ± 1.96 | 50.0 ± 5.00 | 52.5 ± 4.53 |
| Reduction of CBF (%) | 82.8 ± 0.66 | 83.8 ± 0.60 | 84.1 ± 1.10 | 83.4 ± 0.91 |
Values are means ± SEM. CBF, Cerebral blood flow.
Protein kinase C ε regulates Lck activation in PC neuroprotection
Previous data suggest that Lck mediates cardioprotection through the formation of PKCε-Lck signaling module (Ping et al., 1999, 2002). To investigate the upstream regulators of Lck in PC in the brain, we investigated the involvement of PKCε in the NMDA PC model. Pretreatment with a PKCε inhibitor (εV1-2;10 μm) abolished PC, suggesting that PC is mediated by PKCε activation (Fig. 4A,B). Moreover, pharmacological activation of PKCε by ψεRACK (10 μm) simulated NMDA PC, resulting in decreased vulnerability to lethal NMDA toxicity in cortical neuronal cells (Fig. 4A,B).
Figure 4.

PKCε mediates Lck activation in PC. A, B, Pretreatment with εV1-2 (PCKε inhibitor; 10 μm) using BioPORTER abolished NMDA PC neuroprotection in PI-staining (A) or LDH leakage (B). Pharmacological activation of PKCε by ψεRACK (10 μm) demonstrated a similar effect as NMDA PC. *p < 0.05, **p < 0.01 versus NMDA lethal; #p < 0.05 versus NMDA PC+NMDA lethal. C, Colocalization of PKCε with Lck was increased by NMDA PC. Lck or PKCε in each Lck-immunoprecipitates were examined by Western blot. D, Inhibition of PKCε by εV1-2 reversed NMDA PC-induced Lck kinase activation. εV1-2 (10 μm) was treated by BioPORTER before PC, and cell lysates were collected for Lck immunoprecipitation and kinase assay. *p < 0.05 versus control without PC; #p < 0.05 versus NMDA PC. A–D, n = 3. All values are means ± SEM and analyzed by Student's t test.
Next, we sought to determine whether interaction between PKCε and Lck occurs in preconditioned neurons using coimmunoprecipitation (Ping et al., 2002). PKCε was found to coreside in Lck-immunoprecipitates, and this interaction was significantly increased after NMDA PC (Fig. 4C). After inhibiting PKCε by εV1-2, NMDA-induced Lck kinase activation during PC was abolished (Fig. 4D), suggesting that Lck activity is regulated by PKCε activation during PC.
Fyn is the downstream target of Lck
Published data suggest that Lck can regulate Fyn, another member of the Src family kinases (Suzuki and Okumura-Noji, 1995; Filipp et al., 2008; Isosaka et al., 2008). To determine whether Fyn is the downstream target of Lck in PC, we first examined Fyn phosphorylation and activity in NMDA PC. Immunoblotting to Fyn phosphorylated at Tyr 417, a positive regulatory residue for its activation (Filipp et al., 2008), revealed that p-Fyn levels are significantly enhanced during NMDA PC (Fig. 5A). Similarly, Fyn kinase activity from Fyn-immunoprecipitates was increased by NMDA PC (Fig. 5B), suggesting that Fyn is activated during PC. While inhibition of Fyn by silencing Fyn (siFyn) transfection did not affect NMDA-induced cytotoxicity (Fig. 5C), siFyn significantly abolished PC (Fig. 5D), reflecting that Fyn activation is critical for PC. Silencing efficiency was determined (Fig. 5E). We also investigated the role of Fyn in PC using OGD ischemia model, and found similar effects to NMDA PC (Fig. 5F,G).
Figure 5.

Fyn is activated by Lck during PC neuroprotection. A, The active form of Fyn was increased after NMDA PC. Phosphorylation of Fyn at Tyr 417 was examined by Western blot after immunoprecipitation with Fyn Ab. Total levels of Fyn in each immunoprecipitate was also determined. B, Fyn kinase activity was enhanced by NMDA PC. *p < 0.05 versus control without PC. C, NMDA-induced cytotoxicity was examined after siFyn. D, Silencing Fyn reversed PC neuroprotection. siFyn was transfected 48 h before PC, and PC was applied at 24 h before the lethal stimuli. Cytotoxicity was determined at 24 h after lethal NMDA exposure. *p < 0.05 versus NMDA lethal; #p < 0.05 versus Neg+NMDA PC+NMDA lethal. E, Fyn levels were decreased by siFyn transfection. *p < 0.05, **p < 0.01 versus Neg. F, Silencing Fyn significantly reversed OGD PC neuroprotection. siRNAs were transfected 48 h before OGD PC, and OGD PC/lethal OGD exposure was performed as described above. **p < 0.01 versus OGD lethal; #p < 0.05 versus Neg+OGD PC+OGD lethal. G, Active form of p-Fyn was enhanced by OGD PC in cortical neurons. *p < 0.05 versus CON without OGD PC. H, Fyn activation during PC was reversed by silencing Lck. siLck was transfected 48 h before PC. p-Fyn or Fyn levels were examined by Western blot after Fyn-immunoprecipitation. N, Nontargeting negative siRNA; siL, siLck. *p < 0.05 versus Neg without PC; #p < 0.05 versus Neg with PC. I, Fyn activation by PC was not observed in neurons from Lck−/− mice. J, Silencing Fyn did not influence Lck kinase activation during PC. Lck kinase activity was determined in Lck-immunoprecipitates after siFyn transfection and PC. siF, Silencing Fyn. *p < 0.05 versus Neg without PC. K, Inhibition of PKCε decreased Fyn activation during PC. εV1-2 was treated by BioPORTER before PC, and cell lysates were collected after PC for Fyn immunoprecipitation and Western blot. εV, εV1-2; Veh, vehicle control. *p < 0.05 versus vehicle control without PC; #p < 0.05 versus Vehicle+NMDA PC. A–C, E, G–K, n = 3; D, F, n = 4. All values are means ± SEM and analyzed by Student's t test.
Next we examined changes in p-Fyn levels after silencing Lck using siRNA or in neurons from Lck−/− mice. NMDA PC-induced Fyn phosphorylation was not increased after Lck gene silencing with siRNA or in neurons from Lck−/− mice (Fig. 5H,I), implying that Lck is the upstream regulator of Fyn activation. While Fyn is regulated by Lck, siFyn did not affect PC-induced Lck kinase activation (Fig. 5J), confirming that Fyn is the downstream target of Lck. Notably, εV1-2 reversed PC-induced Fyn phosphorylation (Fig. 5K), suggesting that the PKCε-Lck-Fyn axis is critical in PC.
Discussion
Our data provide new insights into the role of Lck in PC in the brain. Using in vitro models of PC, we showed that enhanced Lck kinase activity mediates PC. Neurons derived from Lck−/− mice or neurons in which the Lck gene was silenced by siRNA could not be preconditioned. We also showed that PKCε, a kinase that has been shown to be an important mediator of PC, is an upstream activator of Lck. Significantly, we have identified Fyn as a downstream target for Lck. Lck activation during PC leads to the activation of Fyn and silencing Fyn abolished PC. The important role of Lck was verified in vivo using an animal model of stroke. Lck gene deletion or pharmacological Lck antagonism blocked PC in vivo. Together, our data show that the PKCε-Lck-Fyn signaling axis is an important mediator of PC. We cannot, however, exclude the possibility that Lck and/or Fyn could be activated by other yet-to be-identified upstream signals.
We used sublethal NMDA, OGD, and hypoxia to induce tolerance, but other models also exist (McLaughlin et al., 2003; Hoyte et al., 2006; Dave et al., 2008; Hu et al., 2010). Whether Lck plays a role in those preconditioning paradigms is not known. Although we used highly purified neuronal cultures, the potential influence of small amounts of other non-neuronal cells such as astrocytes and microglia cannot be entirely discounted and need further investigation. For the in vivo studies, we determined 24 h outcomes (infarct volume). Long-term outcomes (both histological and behavioral) remain to be determined in future studies.
Future studies will also need to explore the relative contribution of Lck and Fyn at various time intervals after PC, because rapid tolerance and delayed tolerance may be mechanistically distinct (Bright et al., 2008; Obrenovitch, 2008).
The role of PKCε in PC
Previous studies showed that PKCε is an important mediator of PC in both heart and brain (Raval et al., 2003; Chou and Messing, 2005; Jia et al., 2007; Bright et al., 2008; DeFazio et al., 2009). PKCε is activated by a number of preconditioning stimuli, including hypoxia and transient ischemia. Downstream targets of PKCε are many and include GABA signaling, mitochondrial function alteration, and extracellular signal-regulated kinases (ERKs) (Jia et al., 2007; Dave et al., 2008; DeFazio et al., 2009). However, the precise molecular targets of PKCε still remain unclear. It is unlikely that PC involves a single obligatory mediator, and it is likely that there is activation of a constellation of different pathways that ultimately leads to tolerance (Brooks and Hearse, 1996). It is possible that other mediators and pathways may play a greater role depending on the PC stimulus. Studying other PC models will be important to better understand this complicated endogenous neuroprotective mechanism.
Lck—a novel mediator of PC in brain
Lck has been primarily investigated in the immune system in T lymphocytes. Lck plays an essential role in T cell receptor (TCR) signaling, modulating T cell activation and differentiation (Palacios and Weiss, 2004; Salmond et al., 2009). Recent data also suggest that Lck plays a role in mitochondrial signaling in apoptosis (Samraj et al., 2006; Kim et al., 2008). Until now, most studies have focused on the selective inhibition of Lck for therapeutic immunosuppression and treatment of immunological diseases, such as rheumatoid arthritis, based on its role in the immune system (Benati and Baldari, 2008; Meyn and Smithgall, 2008). Our data show that Lck activation contributes to PC and that inhibition of Lck abolishes PC. Whether activation of Lck would induce neuroprotection is not known and selective Lck activators are not available at this time. Although recent data suggest that Unc 119 can activate Lck in T cells (Gorska et al., 2004), it can also activate other Src kinases that would make it difficult to tease out the precise role of Lck activation. Another strategy worthy of further investigation may involve using PKCε activators to activate Lck.
Lck can also modulate the activity of other Src family kinase members such as Fyn, by phosphorylation of its active site Tyr 417 (Filipp et al., 2003). Previous studies suggest that Lck and Fyn have overlapping cellular functions and are closely related to each other (Filipp et al., 2003, 2008). However, this has not been previously examined in PC or in the brain. Our data show that neurons from Lck−/− mice or wild-type neurons transfected with siRNA against Lck (siLck) exhibited significantly reduced Fyn activation and phosphorylation induced by PC, suggesting that Lck modulates Fyn activation in neurons. While it appears that Lck is responsible for Fyn activation during PC, silencing Fyn did not affect PC-induced Lck activation, suggesting that Fyn activation is downstream of Lck.
Downstream targets of Fyn
The precise function of Fyn in the brain is not known. Fyn regulates NMDA receptors by the phosphorylation of its subunits (Salter and Kalia, 2004; Wu et al., 2007), which modulate NMDA receptor channel activity (Takasu et al., 2002). Following transient ischemia in rats, phosphorylation of the NR2A and, to a lesser extent, NR2B subunits of the NMDA receptor is increased (Takagi et al., 1999). The association between Fyn and NR2A following ischemia is accompanied by increased NR2A phosphorylation (Hou et al., 2003; Jiang et al., 2008). Ischemia also enhances the association between Fyn and other proteins in the NMDA receptor complex, such as PSD-95 and the l-type voltage gated calcium channel (Hou et al., 2003). Administration of a selective NR2A antagonist increased neuronal death and abolished preconditioning, whereas administration of an NR2B antagonist decreased neuronal death and enhanced the preconditioning effect (Chen et al., 2008). Fyn has been implicated in the phosphorylation and activation of both of these subunits, therefore it is possible that Fyn activity modulates preconditioning by influencing NR2A and NR2B activity. Another potential downstream candidate target is carveolin, a scaffolding protein that may also influence the activation of NMDA receptors (Head et al., 2008). Although our laser Doppler studies did not show any differences in blood flow between experimental groups in the in vivo studies, we cannot, however, discount the possibility that changes in blood flow may have occurred in the penumbra of the infarct. The precise downstream pathways that are modulated by Fyn activation are not known and need further investigation.
Other Src family members
Although the role of Src family kinases in neurons has been investigated in previous studies, the individual importance of each family member is not fully understood due to the lack of selective inhibitors/activators. Conventional inhibitors such as PP1 and PP2 lack selectivity (Hanke et al., 1996). To overcome this, we used RNA silencing to inhibit specific Src family members, Lck, Fyn, and Yes. We demonstrated that Lck and Fyn, but not Yes, are involved in PC. It is possible that other Src kinases may also be involved in PC and their function may not be mutually exclusive. An important point for consideration when manipulating Src kinases is that they may enhance angiogenesis that may facilitate recovery after stroke (Schlessinger, 2000; Slevin et al., 2006).
Implications
A desperate need exists for new stroke therapies (Fagan, 2010). Although PC is a powerful endogenous neuroprotective mechanism, translation of preclinical findings into therapies still remains a challenge (Keep et al., 2010). Our data suggest that selective activation of Lck or Fyn may represent a novel therapeutic strategy for stroke therapy development.
Footnotes
This study was supported by an American Heart Association grant to A.M. and by a postdoctoral fellowship from the Korea Research Foundation (KRF-2007-357-E00036) to O.-N.B.
The authors report no competing financial interests.
References
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benati D, Baldari CT. SRC family kinases as potential therapeutic targets for malignancies and immunological disorders. Curr Med Chem. 2008;15:1154–1165. doi: 10.2174/092986708784310404. [DOI] [PubMed] [Google Scholar]
- Bright R, Sun GH, Yenari MA, Steinberg GK, Mochly-Rosen D. epsilonPKC confers acute tolerance to cerebral ischemic reperfusion injury. Neurosci Lett. 2008;441:120–124. doi: 10.1016/j.neulet.2008.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks G, Hearse DJ. Role of protein kinase C in ischemic preconditioning: player or spectator? Circ Res. 1996;79:627–630. doi: 10.1161/01.res.79.3.628. [DOI] [PubMed] [Google Scholar]
- Chen M, Lu TJ, Chen XJ, Zhou Y, Chen Q, Feng XY, Xu L, Duan WH, Xiong ZQ. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke. 2008;39:3042–3048. doi: 10.1161/STROKEAHA.108.521898. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cantrell AR, Messing RO, Scheuer T, Catterall WA. Specific modulation of Na+ channels in hippocampal neurons by protein kinase C epsilon. J Neurosci. 2005;25:507–513. doi: 10.1523/JNEUROSCI.4089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WH, Messing RO. Protein kinase C isozymes in stroke. [Review] Trends Cardiovasc Med. 2005;15:47–51. doi: 10.1016/j.tcm.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Dave KR, DeFazio RA, Raval AP, Torraco A, Saul I, Barrientos A, Perez-Pinzon MA. Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase C epsilon. J Neurosci. 2008;28:4172–4182. doi: 10.1523/JNEUROSCI.5471-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFazio RA, Raval AP, Lin HW, Dave KR, Della-Morte D, Perez-Pinzon MA. GABA synapses mediate neuroprotection after ischemic and epsilonPKC preconditioning in rat hippocampal slice cultures. J Cereb Blood Flow Metab. 2009;29:375–384. doi: 10.1038/jcbfm.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan SC. Stroke: measuring disease-free life after thrombolysis. Nat Rev Neurol. 2010;6:361–362. doi: 10.1038/nrneurol.2010.79. [DOI] [PubMed] [Google Scholar]
- Filipp D, Zhang J, Leung BL, Shaw A, Levin SD, Veillette A, Julius M. Regulation of Fyn through translocation of activated Lck into lipid rafts. J Exp Med. 2003;197:1221–1227. doi: 10.1084/jem.20022112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipp D, Moemeni B, Ferzoco A, Kathirkamathamby K, Zhang J, Ballek O, Davidson D, Veillette A, Julius M. Lck-dependent Fyn activation requires C terminus-dependent targeting of kinase-active Lck to lipid rafts. J Biol Chem. 2008;283:26409–26422. doi: 10.1074/jbc.M710372200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M, Ratan R. New perspectives on developing acute stroke therapy. Ann Neurol. 2003;53:10–20. doi: 10.1002/ana.10407. [DOI] [PubMed] [Google Scholar]
- Gorska MM, Stafford SJ, Cen O, Sur S, Alam R. Unc119, a novel activator of Lck/Fyn, is essential for T cell activation. J Exp Med. 2004;199:369–379. doi: 10.1084/jem.20030589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudz TI, Komuro H, Macklin WB. Glutamate stimulates oligodendrocyte progenitor migration mediated via an alphav integrin/myelin proteolipid protein complex. J Neurosci. 2006;26:2458–2466. doi: 10.1523/JNEUROSCI.4054-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Head BP, Patel HH, Tsutsumi YM, Hu Y, Mejia T, Mora RC, Insel PA, Roth DM, Drummond JC, Patel PM. Caveolin-1 expression is essential for N-methyl-d-aspartate receptor-mediated Src and extracellular signal-regulated kinase 1/2 activation and protection of primary neurons from ischemic cell death. FASEB J. 2008;22:828–840. doi: 10.1096/fj.07-9299com. [DOI] [PubMed] [Google Scholar]
- Hou XY, Zhang GY, Yan JZ, Liu Y. Increased tyrosine phosphorylation of alpha(1C) subunits of l-type voltage-gated calcium channels and interactions among Src/Fyn, PSD-95 and alpha(1C) in rat hippocampus after transient brain ischemia. Brain Res. 2003;979:43–50. doi: 10.1016/s0006-8993(03)02845-2. [DOI] [PubMed] [Google Scholar]
- Hoyte LC, Papadakis M, Barber PA, Buchan AM. Improved regional cerebral blood flow is important for the protection seen in a mouse model of late phase ischemic preconditioning. Brain Res. 2006;1121:231–237. doi: 10.1016/j.brainres.2006.08.107. [DOI] [PubMed] [Google Scholar]
- Hu H, Yamashita S, Hua Y, Keep RF, Liu W, Xi G. Thrombin-induced neuronal protection: role of the mitogen activated protein kinase/ribosomal protein S6 kinase pathway. Brain Res. 2010;1361:93–101. doi: 10.1016/j.brainres.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki K, Begley R, Ikeno F, Mochly-Rosen D. Cardioprotection by epsilon-protein kinase C activation from ischemia: continuous delivery and antiarrhythmic effect of an epsilon-protein kinase C-activating peptide. Circulation. 2005;111:44–50. doi: 10.1161/01.CIR.0000151614.22282.F1. [DOI] [PubMed] [Google Scholar]
- Isosaka T, Hattori K, Kida S, Kohno T, Nakazawa T, Yamamoto T, Yagi T, Yuasa S. Activation of Fyn tyrosine kinase in the mouse dorsal hippocampus is essential for contextual fear conditioning. Eur J Neurosci. 2008;28:973–981. doi: 10.1111/j.1460-9568.2008.06405.x. [DOI] [PubMed] [Google Scholar]
- Jia J, Wang X, Li H, Han S, Zu P, Li J. Activations of nPKCepsilon and ERK1/2 were involved in oxygen-glucose deprivation-induced neuroprotection via NMDA receptors in hippocampal slices of mice. J Neurosurg Anesthesiol. 2007;19:18–24. doi: 10.1097/01.ana.0000211020.88431.e2. [DOI] [PubMed] [Google Scholar]
- Jiang X, Mu D, Biran V, Faustino J, Chang S, Rincón CM, Sheldon RA, Ferriero DM. Activated Src kinases interact with the N-methyl-d-aspartate receptor after neonatal brain ischemia. Ann Neurol. 2008;63:632–641. doi: 10.1002/ana.21365. [DOI] [PubMed] [Google Scholar]
- Keep RF, Wang MM, Xiang J, Hua Y, Xi G. Is there a place for cerebral preconditioning in the clinic? Transl Stroke Res. 2010;1:4–18. doi: 10.1007/s12975-009-0007-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy J, Buchan AM. C-EPO: ready for prime-time preconditioning? [Review] Cerebrovasc Dis. 2005;19:272–273. doi: 10.1159/000084140. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Park MT, Yoon CH, Byun JY, Lee SJ. Activation of Lck is critically required for sphingosine-induced conformational activation of Bak and mitochondrial cell death. Biochem Biophys Res Commun. 2008;370:353–358. doi: 10.1016/j.bbrc.2008.03.084. [DOI] [PubMed] [Google Scholar]
- Lin CH, Chen PS, Gean PW. Glutamate preconditioning prevents neuronal death induced by combined oxygen-glucose deprivation in cultured cortical neurons. Eur J Pharmacol. 2008;589:85–93. doi: 10.1016/j.ejphar.2008.05.047. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Malhotra S, Savitz SI, Ocava L, Rosenbaum DM. Ischemic preconditioning is mediated by erythropoietin through PI-3 kinase signaling in an animal model of transient ischemic attack. J Neurosci Res. 2006;83:19–27. doi: 10.1002/jnr.20705. [DOI] [PubMed] [Google Scholar]
- Manzerra P, Behrens MM, Canzoniero LM, Wang XQ, Heidinger V, Ichinose T, Yu SP, Choi DW. Zinc induces a Src family kinase-mediated up-regulation of NMDA receptor activity and excitotoxicity. Proc Natl Acad Sci USA. 2001;98:11055–11061. doi: 10.1073/pnas.191353598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc Natl Acad Sci USA. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyn MA, 3rd, Smithgall TE. Small molecule inhibitors of Lck: the search for specificity within a kinase family. Mini Rev Med Chem. 2008;8:628–637. doi: 10.2174/138955708784534454. [DOI] [PubMed] [Google Scholar]
- Moncayo J, de Freitas GR, Bogousslavsky J, Altieri M, van Melle G. Do transient ischemic attacks have a neuroprotective effect? Neurology. 2000;54:2089–2094. doi: 10.1212/wnl.54.11.2089. [DOI] [PubMed] [Google Scholar]
- Obrenovitch TP. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev. 2008;88:211–247. doi: 10.1152/physrev.00039.2006. [DOI] [PubMed] [Google Scholar]
- Omri B, Crisanti P, Marty MC, Alliot F, Fagard R, Molina T, Pessac B. The Lck tyrosine kinase is expressed in brain neurons. J Neurochem. 1996;67:1360–1364. doi: 10.1046/j.1471-4159.1996.67041360.x. [DOI] [PubMed] [Google Scholar]
- Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23:7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- Ping P, Zhang J, Zheng YT, Li RC, Dawn B, Tang XL, Takano H, Balafanova Z, Bolli R. Demonstration of selective protein kinase C-dependent activation of Src and Lck tyrosine kinases during ischemic preconditioning in conscious rabbits. Circ Res. 1999;85:542–550. doi: 10.1161/01.res.85.6.542. [DOI] [PubMed] [Google Scholar]
- Ping P, Song C, Zhang J, Guo Y, Cao X, Li RC, Wu W, Vondriska TM, Pass JM, Tang XL, Pierce WM, Bolli R. Formation of protein kinase C (epsilon)-Lck signaling modules confers cardioprotection. J Clin Invest. 2002;109:499–507. doi: 10.1172/JCI13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, Chiarugi A, Thomas SS, Kohane DS, Korsmeyer SJ, Moskowitz MA. BID mediates neuronal cell death after oxygen/ glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci USA. 2001;98:15318–15323. doi: 10.1073/pnas.261323298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajanikant GK, Zemke D, Senut MC, Frenkel MB, Chen AF, Gupta R, Majid A. Carnosine is neuroprotective against permanent focal cerebral ischemia in mice. Stroke. 2007;38:3023–3031. doi: 10.1161/STROKEAHA.107.488502. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Pérez-Pinzón MA. Epsilon PKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. J Neurosci. 2003;23:384–391. doi: 10.1523/JNEUROSCI.23-02-00384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscher K, Shamloo M, Rickhag M, Ladunga I, Soriano L, Gisselsson L, Toresson H, Ruslim-Litrus L, Oksenberg D, Urfer R, Johansson BB, Nikolich K, Wieloch T. The sigma-1 receptor enhances brain plasticity and functional recovery after experimental stroke. Brain. 2011;134:732–746. doi: 10.1093/brain/awq367. [DOI] [PubMed] [Google Scholar]
- Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R. T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol Rev. 2009;228:9–22. doi: 10.1111/j.1600-065X.2008.00745.x. [DOI] [PubMed] [Google Scholar]
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Samraj AK, Stroh C, Fischer U, Schulze-Osthoff K. The tyrosine kinase Lck is a positive regulator of the mitochondrial apoptosis pathway by controlling Bak expression. Oncogene. 2006;25:186–197. doi: 10.1038/sj.onc.1209034. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. New roles for Src kinases in control of cell survival and angiogenesis. Cell. 2000;100:293–296. doi: 10.1016/s0092-8674(00)80664-9. [DOI] [PubMed] [Google Scholar]
- Slevin M, Kumar P, Gaffney J, Kumar S, Krupinski J. Can angiogenesis be exploited to improve stroke outcome? Mechanisms and therapeutic potential. Clin Sci. 2006;111:171–183. doi: 10.1042/CS20060049. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Simon RP. Genomics of preconditioning. Stroke. 2004;35:2683–2686. doi: 10.1161/01.STR.0000143735.89281.bb. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Okumura-Noji K. NMDA receptor subunits epsilon 1 (NR2A) and epsilon 2 (NR2B) are substrates for Fyn in the postsynaptic density fraction isolated from the rat brain. Biochem Biophys Res Commun. 1995;216:582–588. doi: 10.1006/bbrc.1995.2662. [DOI] [PubMed] [Google Scholar]
- Takagi N, Cheung HH, Bissoon N, Teves L, Wallace MC, Gurd JW. The effect of transient global ischemia on the interaction of Src and Fyn with the N-methyl-d-aspartate receptor and postsynaptic densities: possible involvement of Src homology 2 domains. J Cereb Blood Flow Metab. 1999;19:880–888. doi: 10.1097/00004647-199908000-00007. [DOI] [PubMed] [Google Scholar]
- Takasu MA, Dalva MB, Zigmond RE, Greenberg ME. Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science. 2002;295:491–495. doi: 10.1126/science.1065983. [DOI] [PubMed] [Google Scholar]
- Tauskela JS, Brunette E, Monette R, Comas T, Morley P. Preconditioning of cortical neurons by oxygen-glucose deprivation: tolerance induction through abbreviated neurotoxic signaling. Am J Physiol Cell Physiol. 2003;285:C899–C911. doi: 10.1152/ajpcell.00110.2003. [DOI] [PubMed] [Google Scholar]
- Ueda H, Fujita R, Yoshida A, Matsunaga H, Ueda M. Identification of prothymosin-alpha1, the necrosis-apoptosis switch molecule in cortical neuronal cultures. J Cell Biol. 2007;176:853–862. doi: 10.1083/jcb.200608022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waegell W, Babineau M, Hart M, Dixon K, McRae B, Wallace C, Leach M, Ratnofsky S, Belanger A, Hirst G, Rossini A, Appel M, Mordes J, Greiner D, Banerjee S. A420983, a novel, small molecule inhibitor of LCK prevents allograft rejection. Transplant Proc. 2002;34:1411–1417. doi: 10.1016/s0041-1345(02)02909-3. [DOI] [PubMed] [Google Scholar]
- Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K, Sato S, Tedder TF, Fujimoto M. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J Immunol. 2010;184:4801–4809. doi: 10.4049/jimmunol.0902385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel M, Li L, Harms KM, Roitbak T, Ventura PB, Rosenberg GA, Khokha R, Cunningham LA. Tissue inhibitor of metalloproteinases-3 facilitates Fas-mediated neuronal cell death following mild ischemia. Cell Death Differ. 2008;15:143–151. doi: 10.1038/sj.cdd.4402246. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hsu FC, Gleichman AJ, Baconguis I, Coulter DA, Lynch DR. Fyn-mediated phosphorylation of NR2B Tyr-1336 controls calpain-mediated NR2B cleavage in neurons and heterologous systems. J Biol Chem. 2007;282:20075–20087. doi: 10.1074/jbc.M700624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim YI, Sun T, Wu LG, Raimondi A, De Camilli P, Eisenberg E, Greene LE. Endocytosis and clathrin-uncoating defects at synapses of auxilin knockout mice. Proc Natl Acad Sci USA. 2010;107:4412–4417. doi: 10.1073/pnas.1000738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemke D, Smith JL, Reeves MJ, Majid A. Ischemia and ischemic tolerance in the brain: an overview. Neurotoxicology. 2004;25:895–904. doi: 10.1016/j.neuro.2004.03.009. [DOI] [PubMed] [Google Scholar]