

Abstract
c-IAP1 and c-IAP2 are ubiquitin protein ligases (E3s) that repress non-canonical NF-κB activation. We have created mice that bear a mutation in c-IAP2 that inactivates its E3 activity and interferes, in a dominant negative fashion, with c-IAP1 E3 activity (c-IAP2H570A). The immune response of these animals was explored by infecting them with the Th1-inducing parasite Toxoplasma gondii. Surprisingly, c-IAP2H570A mice succumbed due to T cell production of high levels of pro-inflammatory cytokines. Unlike naïve wild type (WT) cells, which require signals generated by the T cell receptor (TCR) and costimulatory receptors to become fully activated, naïve c-IAP2H570A T cells proliferated and produced high levels of IL-2 and IFN-γ to stimulation via TCR alone. c-IAP2H570A T cells had constitutive non-canonical NF-κB activation, and IKK inhibition reduced their proliferation to anti-TCR alone to WT levels but had no effect when costimulation via CD28 was provided. Notably, T cells from nfkb2−/− mice, which cannot generate the p52 component of non-canonical NF-κB, were also costimulation-independent, consistent with the negative role of this unprocessed protein in canonical NF-κB activation. Whereas T cells from nfkb2+/− mice behaved like WT, co-expression of a single copy of c-IAP2H570A resulted in cleavage of p100, upregulation of p52, and T-cell costimulation-independence. Thus, p100 represses and p52 promotes costimulation, the ratio regulating T cell dependence on costimulatory signals.
Introduction
The nuclear factor-κB (NF-κB) family of transcription factors plays critical roles in a variety of biological processes (1, 2). There are two mechanisms that activate NF-κB. The majority of stimuli that activate NF-κB, such as tumor necrosis factor α (TNFα), interleukin (IL)-1β, and lipopolysaccharide (LPS), do so via the so-called canonical pathway (1). In resting cells IκBα constitutively binds NF-κB dimers and retains them in the cytosol. Upon stimulation, the IκB kinase (IKK) β subunit is activated, leading to phosphorylation and subsequent ubiquitination and proteasome-mediated degradation of IκBα, freeing NF-κB to migrate to the nucleus and activate transcription. Activation of the canonical NF-κB pathway depends exclusively on IκBα degradation, does not require protein synthesis, and is rapid and transient (2, 3). Non-canonical NF-κB signaling is initiated by TRAF-2-mediated K63-linked ubiquitination of c-IAP1 and c-IAP2. This modification diverts their activity from NIK to K48-linked ubiquitination of TRAF3, inducing its proteasomal-mediated degradation and release of NIK from the complex. As a result of both protein stabilization and de novo synthesis, NIK accumulates in the cytosol where it phosphorylates and activates IKKα, which in turn phosphorylates the NF-κB family member p100, inducing its ubiquitination and partial proteolysis to p52. The resulting RelB/p52 heterodimers migrate to the nucleus where they activate transcription of target genes (4–8). Such a regulatory mechanism ensures the absence of receptor-unrelated signals by suppressing the level of NIK in unstimulated cells, and is characterized by relatively slow kinetics (on the order of hours (3)).
c-IAP1 and -2 are highly homologous members of the IAP family, characterized by the presence of one or more Baculovirus IAP Repeat (BIR) domains (a ~70 amino acid region that mediates protein-protein interactions) and a RING domain conferring E3 activity (9). B cells from knock-in mice containing a point mutation inactivating the E3 activity of c-IAP2 (c-IAP2H570A) have constitutive upregulation of NIK with activation of the non-canonical NF-κB, B cell hyperplasia, and enlarged gut-associated lymphoid tissue (10). We choose to explore the effect of c-IAP2 E3-inactivation in T cells by infecting c-IAP2H570A mice with Toxoplasma gondii, an obligate intracellular protozoan that in immuno-compromised hosts can cause encephalitis (11). We find that c-IAP2H570A mice succumb to an avirulent strain of T. gondii due to excessive proinflammatory cytokine production, the result of an exuberant response by T cells. c-IAP2H570A T cell no longer require costimulation to become fully active and initiate effector functions. Using pharmacological and genetic approaches, we find that this state of readiness results from a decrease in the inhibitory unprocessed form of p100 and a corresponding accumulation of its activating fragment, p52.
Methods
Mice
RAG1−/− mice were obtained from Taconic, C57BL/6 (B6) mice were obtained from The Jackson Laboratory, c-IAP2H570A mice were generated in our lab (10), cIAP2−/− mice were kindly provided by Robert Korneluk and Eric LaCasse (Children’s Hospital of Eastern Ontario, Ottawa, Canada), and nfkb2−/− mice by Uli Siebenlist (NIAID, NIH). All mice were bred and maintained in a National Cancer Institute pathogen-free animal facility. For adoptive transfer experiment, RAG1−/− mice were injected iv with 107 T cells from age-matched WT or c-IAP2H570A mice. Study protocols were approved by the Institutional Animal Care and Use Committee of the National Cancer Institute.
T. gondii infection and parasite burden evaluation
T. gondii cysts from the avirulent strain ME-49 were prepared from the brains of infected C57BL/6 mice. For experimental infections, mice were inoculated ip with an average of 20 cysts/animal and monitored daily for survival. To measure parasite burden, peritoneal cells were harvested on day 7 post-infection and the number of infected cells was assessed evaluating Diff-Quik-stained Cytospin smears of exudates.
Cell Preparation and Purification
T cells were purified from lymph nodes using Easy Sep T cell enrichment kit (StemCell Technologies) following the manufacturer’s protocol and the number of live cells was assessed by trypan blue exclusion. Purity was determined by flow cytometry, and for all experiments was >90%. T cells were cultured in RPMI 1640 supplemented with 10% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, and 50 μM-β-mercaptoethanol.
Reagents and antibodies
Hoechst 33342 and Pyronin Y were purchased from Molecular Probes and Polysciences, respectively. IKK Inhibitor XII (12). was from Calbiochem. Anti–mouse CD3 (145-2C11), anti-mouse CD28 (37.51), anti-mouse CD40 (HM40-3) and all antibodies for flow cytometry were purchased from BD Biosciences. PMA, ionomycin (Iono), and anti-β-actin were purchased from Sigma-Aldrich, carboxyfluorescein diacetate succinimidyl ester (CFSE) from Invitrogen, antibodies to p100/p52, phospho-Ser866/870 p100, NIK, phospho-Ser473 Akt, and Akt were from Cell Signaling, and antibodies to IκBα, cyclin D3, RelA, RelB and Lamin A from Santa Cruz.
Flow cytometry and sorting
All stainings were performed in PBS plus 1% FCS and 0.01% NaN3 in the presence of 1:500 Fc-blocking antibodies (24G.2). CFSE was used at a concentration of 500 nM and cells were stained following the manufacturer’s instructions. For DNA/RNA staining, cells were incubated with Hoechst 33342 and pyronin Y, respectively, as described (13). Sorting experiments were performed on a BD FACS Aria. All data were analyzed with FlowJo software (TreeStar).
Proliferation assay
Assays were performed in 96-well flat-bottom plates in a final volume of 200 μl. Wells were coated with anti-CD3 alone or in combination with anti-CD28 at the indicated concentrations for 1 hr at 37°C or overnight at 4°C in PBS. Cells were cultured for 48 hr, pulsed with 1 μCi [3H]-thymidine, and harvested 18 hr later. [3H]-thymidine uptake was determined with a Wallac 1450 MicroBeta Liquid Scintillation Counter. All experimental points were performed in triplicate and the error bars represent the standard error of the mean.
Immunoblotting
T cells were normalized to cell number and lysed in sample buffer (50 mM Tris pH 6.8, 10% glycerol, 2% SDS, 2% β-mercaptoethanol, and 0.04% bromophenol blue), resolved by SDS-PAGE, and immunoblotted with the indicated antibodies. To detect phospho-Ser473 Akt and Akt, samples were lysed in RIPA buffer (Sigma-Aldrich) and normalized to protein concentration. Nuclear extracts were prepared using NE-PER® Nuclear and Cytoplasmic Extraction Reagents (Pierce). Densitometry was performed using the ImageJ gel analysis tool.
Cytokine and AST quantification
Supernatants from T cells stimulated with plate-bound anti-CD3 or anti-CD3 plus anti-CD28 were collected at the indicated time points and the level of IL-2 and IFN-γ measured using the IL-2 or IFN-γ ELISA Ready-SET-Go! kit (e-Bioscience). For T. gondii infection experiments, mice were bled at day 7 or 8 after infection and levels of AST were determined using a commercial kit (Boehringer) in an automatic analyzer (model 917; Hitachi). Levels of IL-12, IFN-γ, and IL-10 were determined by ELISA.
Real-Time PCR
Total RNA was extracted from purified T cells using RNeasy Mini kits (Qiagen) and reverse transcribed using the Superscript II Reverse Transcriptase kit (Invitrogen) following the manufacturers’ protocol. IL-2 and hypoxanthine phosphoribosyltransferase (HPRT) mRNA was quantified using the respective primers, SYBR Green PCR Master Mix (Applied Biosystems), and the 7500 Real Time PCR System (Applied Biosystems). Values were normalized to HPRT and the percent increase relative to WT was calculated by dividing c-IAP2H570A values by WT values.
Results
c-IAP2H570A mice succumb to T. gondii infection
To explore the biological consequences of c-IAP2H570A mutation in T cells, we infected WT, c-IAP2H570A, and c-IAP2−/− mice with an avirulent T. gondii strain. This model was chosen because protective immunity is B cell-independent, relying on vigorous T helper 1 (Th1)-mediated immunity mediated primarily by dendritic cell-derived IL-12 and T cell-derived interferon-γ (IFN-γ) and IL-10 (14). Whereas the majority of WT mice survived, c-IAP2H570A mice succumbed in the acute phase of the infection with a median of day 9.5 (Fig. 1A). To distinguish between a requisite role for c-IAP2 and a role for c-IAP2 E3 activity, mice deficient in c-IAP2 were also examined and were found to survive infection. The death of c-IAP2H570A mice was not due to uncontrolled parasite replication, because the parasite burden was similar in WT and c-IAP2H570A mice (Figure 1B). In contrast, there was a high serum level of aspartate aminotransferase (AST), indicating acute liver damage (Fig. 1C). This excessive response to T. gondii was also reflected in high amounts of IFN-γ and IL-10 in the sera of c-IAP2H570A compared to WT and c-IAP2−/− mice (Fig. 1D). The amount of IL-12 and IL-6 was increased as well, although to a lesser extent. Each of these cytokines was found at low to undetectable levels in the sera of uninfected mice of all genotypes. To determine if the exaggerated response to T. gondii was T-cell intrinsic, RAG1−/− mice were reconstituted with WT or c-IAP2H570A T cells and infected. Whereas WT T cells protected four of five RAG1−/− mice for up to 25 days, mice reconstituted with c-IAP2H570A T cells died within 20 days post-infection (Fig. 1E), demonstrating that the death of c-IAP2H570A mice was due to the T cell response.
Figure 1. c-IAP2H570A mice succumb to T. gondii infection.

(A) WT (n=8), c-IAP2−/− (n=5), and c-IAP2H570A (n=8) mice were infected i.p. with 20 cysts of the ME49 strain of T. gondii and animal survival was monitored daily. (B–D) Peritoneal exudate cells (PEC) and sera were collected on day 7 post-infection. (B) Number of tachyzoite-infected PEC in three WT and three c-IAP2H570A mice. (C) Level of AST was determined in serum from one uninfected animal per group, two WT and four c-IAP2H570A mice. (D) Serum levels of IFN-γ, IL-10, IL-12-p40 and IL-6 in infected WT (n=4), c-IAP2−/− (n=5), and c-IAP2H570A (n=3) mice. Bars represent the mean ± or SD value in three to five mice per group from one representative of two experiments performed. (E) RAG1−/− animals reconstituted with c-IAP2H570A T cells fail to control acute T. gondii infection. Ten million purified T cells from either WT or c-IAP2H570A mice were adoptively transferred into recipients. 7 days later, reconstituted RAG1−/− animals (n = 5 for each group) were infected and monitored as described in (A).
c-IAP2H570A T cells are costimulation-independent
The functional responses of WT versus c-IAP2H570A T cells were compared in vitro. As expected, TCR-driven WT T cell proliferation was largely dependent on the presence of costimulation. Strikingly, c-IAP2H570A T cells proliferated after stimulation with anti-TCR alone, as shown by [3H]-thymidine incorporation and CFSE dilution (Fig. 2A and B). This was not due to differences in their intrinsic proliferative capacity, because WT and c-IAP2H570A T cells proliferated similarly in the presence of antibodies to the TCR and CD28, or phorbol 12-myristate 13-acetate (PMA) and ionomycin, a mitogenic stimulus that bypasses the TCR. Upregulation of the activation markers CD44 and CD25, but not CD69, was much greater in WT T cells stimulated with anti-CD3/CD28 compared to anti-CD3 alone (Fig. 2C). In contrast, CD44 and CD25 upregulation on c-IAP2H570A T cells was the same whether or not costimulation was present. The expression of these markers at rest was similar between WT and c-IAP2H570A T cells (Conze et al., 2010 and Fig. 4A). If c-IAP2H570A T cells have a costimulation advantage, WT T cells should proliferate to the same extent as the mutant T cells if they are exposed to a sufficient amount of costimulation. To test this, anti-CD28 was titrated in the presence of fixed amounts of anti-CD3 (Fig. 2D). Whereas c-IAP2H570A T cell proliferation did not change as a function of anti-CD28 at any anti-CD3 concentration, the dependence of WT T cell proliferation on anti-CD28 increased as the amount of anti-CD3 decreased. The question of whether this costimulation advantage was cell-intrinsic was addressed by activating co-cultured WT and c-IAP2H570A T cells with anti-CD3 alone (Fig. 2E). Proliferation of a 1:1 ratio of WT and c-IAP2H570A T cells in response to anti-CD3 alone was intermediate between the two unmixed populations, indicating that the c-IAP2H570A T cells did not produce a factor that made WT T cells less dependent by costimulation. The dominant-negative effect of E3-defective c-IAP2 on c-IAP1 was needed for this phenotype, because c-IAP2−/− and WT T cells proliferated equally poorly to anti-CD3 alone (Fig. 2F). Hence, it is not the loss of c-IAP2 E3 activity per se, but the resulting upregulation of non-canonical NF-κB that is responsible for costimulation-independence.
Figure 2. c-IAP2H570A are costimulation-independent.

(A, D, E, and F) Purified T cells were cultured with the indicated stimuli for 48 hr, pulsed with [3H]-thymidine, and harvested 18 hr later. Anti-CD28 was coated at 2 μg/ml, except for panel D, were a titration was done. PMA was used at the indicated concentrations and ionomycin was used at 500ng/ml. (B) CFSE-labeled T cells were stimulated with anti-CD3 (3 μg/ml) in the presence or absence of anti-CD28 (2μg/ml), and CFSE dilution was monitored after 3 days. (C) The expression of activation markers was monitored on WT (gray filled histogram) and c-IAP2H570A (open histogram) purified T cells after 3 days of stimulation as indicated. The dotted line represents the expression profile of unstimulated WT cells. The results showed in this figure are representative of, respectively: (A) more than five; (B), (C), and (E) three; (D) and (F) two independent experiments.
Figure 4. Costimulation of naïve versus memory c-IAP2H570A T cells.
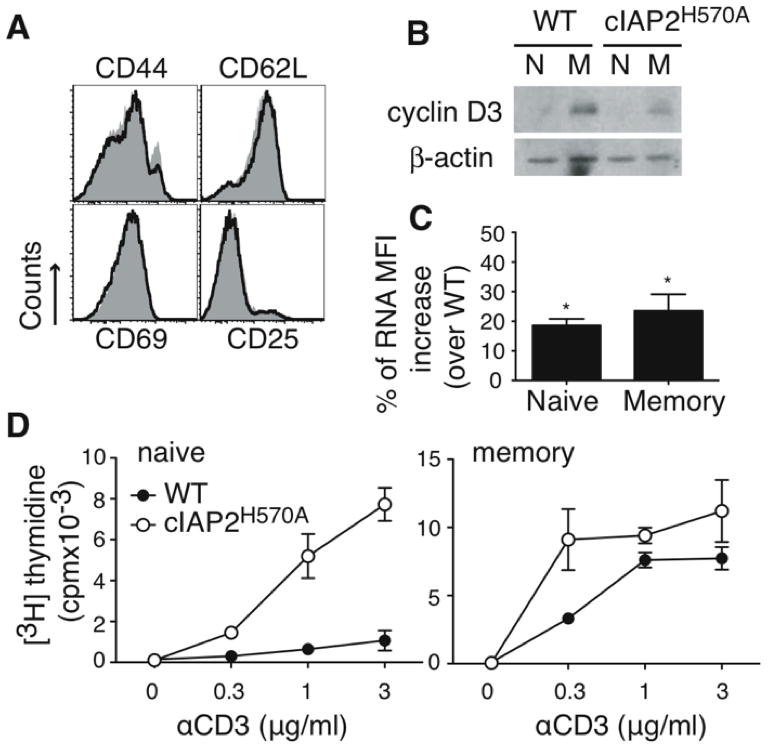
(A) Expression of the indicated markers on WT (gray filled histogram) and c-IAP2H570A (open histogram) T cells (gated on TCRβ+) from lymph nodes. The data shown is representative of more than five independent experiments. (B) Sorted naïve (CD44lo) or memory (CD44hi) cells were cultured with plate-bound anti-CD3 for 48 hr, pulsed with [3H]-thymidine, and harvested 18 hr later. This experiment is representative of three independent experiments. (C) T cells were stained with pyronin Y and gated naïve (CD44lo) or memory (CD44hi) cells were analyzed. The bars represent the percentage increase of pyronin Y MFI over that of WT cells. The results are the average of five independent experiments and the error bars represent the standard error of the mean. Statistical analysis was performed using an unpaired Student t test. * p < 0.05. (D) Immunoblot of sorted naïve (N; CD44lo) or memory (M; CD44hi) cells lysed immediately after purification. β-actin expression was used as loading control. The data shown are representative of three independent experiments.
Increased IL-2 and IFN-γ production by c-IAP2H570A T cells
Costimulatory signals are required for the production of effector cytokines such as IL-2 and IFN-γ(15). Purified T cells were stimulated with anti-CD3 in the presence or absence of anti-CD28 and cytokine production measured (Fig. 3A). Whereas WT T cells produced little IL-2 unless CD28 was engaged, anti-CD3 alone was sufficient to induce c-IAP2H570A T cells to produce as much IL-2 as WT cells in the presence of both anti-CD3 and anti-CD28, which was increased another twofold by anti-CD28. IL-2 mRNA was also increased in c-IAP2H570A T cells (Fig. 3B). As with IL-2, WT T cells secreted IFN-γ only in the presence of a costimulatory signal, whereas a comparable amount of IFN-γ was detected in supernatants of c-IAP2H570A T cells stimulated with anti-CD3 alone (Fig. 3A). Thus, lack of c-IAP2 E3 activity allows TCR signals alone to induce cytokine expression, the high levels of IL-2 likely being responsible for proliferation in the absence of costimulation.
Figure 3. Increased IL-2 and IFN-γ production by c-IAP2H570A T cells.
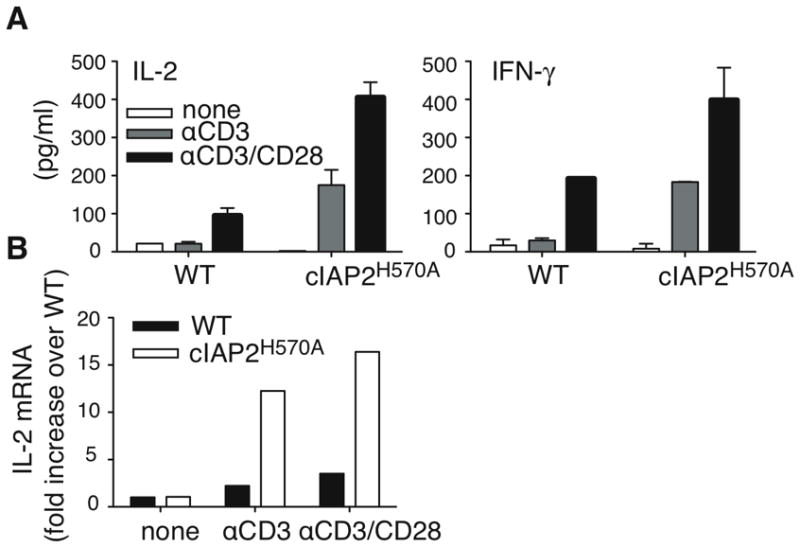
(A) WT and c-IAP2H570A T cells were stimulated as indicated and IL-2 and IFN-γ were analyzed by ELISA after 24 and 48 hr, respectively. Each point was performed in triplicate and the error bars represent the standard error of the mean. This experiment is representative of three independent experiments. (B) IL-2 mRNA expression was determined by real-time PCR in freshly purified T cells (none) or after stimulation for 6 hr as indicated. Bars represent the mean increase of IL-2 mRNA over unstimulated WT T cells. The data are representative of two independent experiments.
Costimulation of naïve versus memory c-IAP2H570A T cells
The requirement for costimulation in TCR-mediated proliferation is more pronounced in naïve than memory cells (reviewed in reference 15) To determine if the proportion of memory cells in c-IAP2H570A mice was altered, T cell subpopulations were analyzed. There was no difference in the CD44, CD62L, CD25, and CD69 expression between WT and c-IAP2H570A T cells (Fig. 4A). Two important characteristics of memory cells are elevated expression of cyclin D3 and a higher content of total RNA compared to naïve T cells (13, 16). Although cyclin D3 expression was not increased in naïve c-IAP2H570A T cells (Fig. 4B), they did have higher total RNA levels than their WT counterparts (Fig. 4C), consistent with their being in a state of readiness similar to memory cells. Accordingly, TCR stimulation alone was sufficient to induce proliferation of naive (CD44lo) c-IAP2H570A but not naive WT T cells (Fig 4D). In contrast, memory T cells (CD44hi) of both genotypes responded similarly, demonstrating that it was predominantly naïve T cells that were affected by activation of non-canonical NF-κB.
Constitutive activation of non-canonical NF-κB in c-IAP2H570A T cells
Lack of c-IAP2 E3 activity caused constitutive activation of non-canonical NF-κB in resting B cells, an effect that was difficult to see in T cells because of the much higher levels of p52 in WT B compared to WT T cells (10). Therefore, we addressed the state of NF-κB activation specifically in T cells. Freshly isolated c-IAP2H570A T cells expressed higher levels of NIK and p52, and lower levels of the unprocessed p52 precursor p100, than WT T cells (Fig. 5A). The p100/p52 ratio was 16-fold lower relative to WT, as determined by densitometry. As expected, c-IAP2−/− T cells did not display any changes in the state of non-canonical NF-κB activation (Fig. 5B) and had a p100/p52 ratio of 0.8 relative to WT T cells, confirming that c-IAP1 compensates for the lack of c-IAP2 in the T cell inhibitory complex. To ask if the c-IAP2H570A mutation affects other pathways that are known to regulate costimulation, we analyzed Akt activation. At rest, phospho-Akt was undetectable in both WT and c-IAP2H570A T cells, and it increased in both in a similar fashion after activation by TCR or TCR/CD28 (Fig. 5C). Other potential candidates, including mTOR, which prevents anergy, and p27, which is required for induction of T cell anergy (17), were analyzed and no differences were found between c-IAP2H570A and WT T cells (data not shown). Similarly, canonical NF-κB activation as measured by IκBα degradation was comparable between WT and c-IAP2H570A T cells as well (data not shown). These results suggest that the dysregulation of non-canonical NF-κB accounts for c-IAP2H570A T cells costimulation-independency.
Figure 5. Constitutive activation of non-canonical NF-κB in c-IAP2H570A T cells.
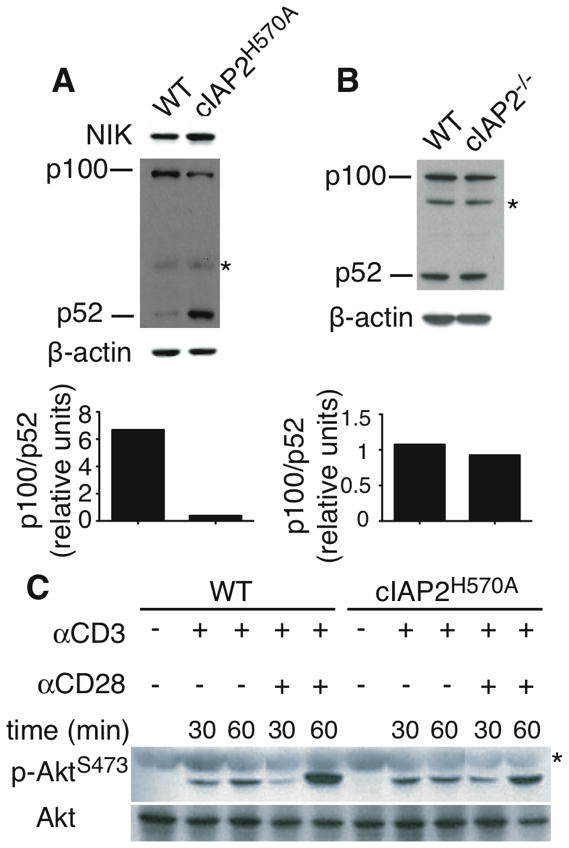
Immunoblot of whole cell lysate of freshly purified WT and c-IAP2H570A T cells (A) and c-IAP2−/− T cells (B). β-actin expression was used as loading control. The results in (A) and (B) are representative of more than three independent experiments. (C) T cells freshly purified or stimulated as indicated were lysed and Akt phosphorylation was detected by immunoblotting. Lanes were rearranged for clarity. This result is representative of two independent experiments. * Nonspecific band.
IKK inhibition prevents costimulation-independent proliferation
To determine if the c-IAP2H570A costimulation advantage was a consequence of NF-κB activation, we used a synthetic IKK inhibitor with activity toward both IKKα and β, IKK XII. Inhibition of IKK completely blocked the costimulation-independence of c-IAP2H570A T cells (Fig. 6A and B). This effect was highly specific, because at the concentrations used the IKK inhibitor had little effect when costimulation was mediated by CD28, which mediates costimulation by activation of Akt and is independent of non-canonical NF-κB. Higher concentrations of IKK XII blunted both CD28-dependent and independent proliferation, without affecting cell viability, consistent with the known role of canonical NF-κB activation via IKKβ (17) (data not shown). To characterize the effect of IKK XII on IKKα, we incubated WT splenocytes with anti-CD40 to activate non-canonical NF-κB. CD40-induced p100 phosphorylation, a hallmark of IKKα activity, was decreased by IKK XII, confirming its negative effect on IKKα, as was, to a lesser extent, constitutive p-100 phosphorylation in c-IAP2H570A T cells (Supplemental Fig. 1A and 1B). RelA (p65) and RelB nuclear translocation in anti-CD40-stimulated splenocytes was also inhibited (Supplemental Fig. 1C and 1D). These results indicate that costimulation provided by c-IAP2H570A, but not CD28, relies on NF-κB activation.
Figure 6. IKK inhibition prevents costimulation-independent proliferation.
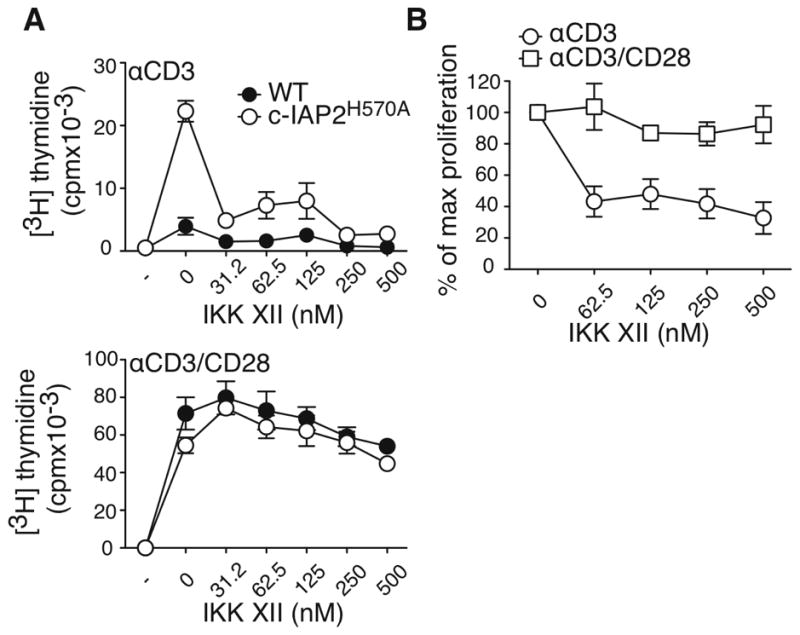
(A) Purified T cells were cultured with the indicated stimuli for 48 hr, pulsed with [3H]-thymidine, and harvested 18 hr later. Anti-CD3 was coated at 1μg/ml and anti-CD28 at 2μg/ml. (B) The average of the percent of maximum proliferation of c-IAP2H570A T cells in the presence of the indicated concentrations of IKK XII from three independent experiments. Bars represent the standard error of the mean.
The balance between p100 and p52 regulates costimulation-dependence
Non-canonical NF-κB activation is the result of processing p100 to p52, which was reflected in c-IAP2H570A T cells by a change in the ratio of these two proteins. p100 has been shown to have an inhibitory role on naïve T cell proliferation, presumably via its binding to, and inhibition of, RelA (18, 19). To evaluate the possible contribution of decreased p100 to costimulation-independence, we analyzed T cells from mice in which the gene encoding p100 (nfkb2) was disrupted. nfkb2−/− T cells proliferated in response to TCR stimulation in the absence of overt costimulation, similar to the proliferation observed with c-IAP2H570A T cells, and proliferated like WT T cells when costimulation was provided via CD28 (Fig. 7A). This is consistent with the notion that p100 restrains the T cell proliferative response to TCR engagement alone, and its lack results in T cell costimulation-independence.
Figure 7. The balance between p100 and p52 amount determines costimulation-dependence.
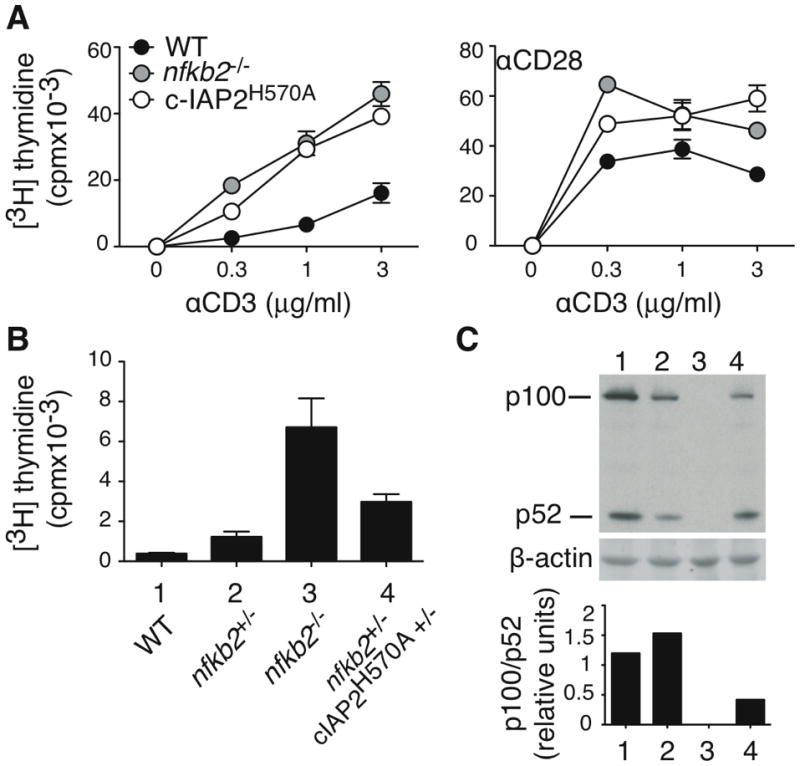
(A) Purified T cells were cultured with the indicated stimuli for 48 hr, pulsed with [3H]-thymidine, and harvested 18 hr later. Anti-CD28 was coated at 2μg/ml. This result is representative of three independent experiments. (B) Purified T cells were cultured with anti-CD3 (3μg/ml) for 48 hr, pulsed with [3H]-thymidine, and harvested 18 hr later. (C) p100/p52 expression was detected by immunoblot in freshly purified T cells. For each sample, densitometry of p100 and p52 was expressed as the ratio between it and β-actin. This result is representative of two independent experiments.
The analysis of nfkb2−/− T cells is complicated by the fact that both p100 and p52, which may have opposing effects, are absent. To address how these proteins might independently regulate T cell responses, we examined T cells expressing no, haploid, or normal levels of p100. nfkb2+/− T cells express about 50% less p100 (Figure 7C). If c-IAP2H570A T cells costimulation-independence relies only on reduced p100-mediated inhibition, nfkb2+/− T cells would be expected to have a similar phenotype. As shown in Fig. 7B, nfkb2+/− T cells were not costimulation-independent, whereas nfkb2−/− T cells were. Therefore, a decrease in p100 is not sufficient to deregulate the TCR response. Introduction of one c-IAP2H570A allele into nfkb2+/− T cells was sufficient to induce processing of p100 to p52, thereby decreasing the p100/p52 ratio by approximately half (Fig. 7B and C). This resulted in a more robust TCR-mediated response, although one that was still lower than that of nfkb2−/− T cells, demonstrating that is the imbalance of the p100/p52 ratio that affects costimulation-dependence. These results indicate that activation of non-canonical NF-κB causes costimulation-independence by two related but independent mechanisms: a decrease in the inhibitory protein p100, and an increase in its processed and activating form, p52.
Discussion
T cell activation is one of the fundamental processes determining the outcome of an immune response. Full T cell activation is achieved when the MHC-peptide complex recognized by the TCR is accompanied by a costimulatory signal generated by receptors such as CD28. Lack of costimulation not only prevents full activation but promotes a state of clonal anergy characterized by lack of cytokine secretion. T cell anergy is one means by which potentially self-reactive T cells are rendered unresponsive in the periphery, and also one of the mechanisms that tumor cells use to escape the immune response(20, 21). Occupancy of the most well-studied costimulatory molecule, CD28, activates phosphatidylinositol 3-kinase (PI3K), Akt, and canonical NF-κB leading to cytokine production and T cell survival (15, 22). Other costimulatory molecules belong to the TNF receptor family: OX40, 4-1BB, CD27, TNF-R2 (23, 24). Upon engagement with their respective ligand, these molecules activate canonical NF-κB, causing enhanced cell survival and effector cytokine production. In addition they activate non-canonical NF-κB(25–27), although the contribution of this pathway to biological outcomes is not well understood.
Although role of non-canonical NF-κB has been characterized mostly in B cells, there are a few reports that deal with non-canonical NF-κB activation and T cells. aly/aly mice bear a mutation in NIK that interferes with the activation of IKKα and the generation of p52, and T cells from these animals have a slight decrease in TCR-induced proliferation and produce lower amounts of IL-2 than WT cells (28). In line with this, T cells from RelB−/− mice have a profound defect in IL-2 and IFN-γ production upon activation (29). Conversely, p100ΔCT/ΔCT mice, in which the inhibitory ankyrin domain of p100 is deleted resulting in a constitutively active N-terminal fragment, have enlarged lymph nodes, increased T cell proliferation in response to anti-CD3 and anti-CD3/CD28, and enhanced cytokine production (30). More recently, it was suggested that p100 represses canonical NF-κB by inhibiting RelA nuclear translocation, and that a low level of p100 makes naïve T cell costimulation-independent (18). Thus, the role of the non-canonical NF-κB in T cell activation is not well understood. The data in the present report show that isolated upregulation of non-canonical NF-κB in T cells does not result in overt activation or alteration of the distribution of naive and memory T cells, but synergizes with TCR signals to cause naïve T cells to proliferate in a memory-like fashion. We refer to this phenomenon as “costimulation-independence” to mean that c-IAP2H570A T cells are substantially more responsive to TCR-mediated activation than wild-type cells (≥ 10-fold shift in the dose-response curve and a much higher level of proliferation at any concentration of anti-CD3 used). This depended on NF-κB activation, as demonstrated with the IKK inhibitor, and was very likely contributed to by the corresponding decrease in p100 levels, because T cells completely lacking p100 were costimulation-independent as well. In line with this result, the hyperactivation of p100ΔCT/ΔCT T cells may also depend on the lack of p100 inhibitory activity, which requires the ankyrin repeats. Consistent with our results with a genetic model, it has recently been reported that treatment of naïve T cells with SMAC-mimetics, a class of compounds that induces rapid degradation of both c-IAP1 and c-IAP2, activates non-canonical NF-κB and enables them to proliferate in response to TCR stimulation alone (31). Because the c-IAP2H570A T cells express c-IAP1 and (E3-dead) c-IAP2, we can conclude that it is the E3 activity of the c-IAPs rather than some other property (for example, BIR-dependent interactions with other proteins such as caspases) that is responsible for the costimulation-independent phenotype. This is supported by the finding that c-IAP2−/− T cells behave like the WT T cells. In this case, the lack of c-IAP2 is presumably compensated for by c-IAP1, preventing activation of the non-canonical NF-κB pathway. Our results indicate that T cell requirement for costimulation is regulated by the ratio between p100 and p52. Indeed, the approximately 50% decrease in p100 levels in nfkb2+/− T cells was not sufficient to deregulate the response to TCR signaling. This was not the case when p52 was increased (with only a small further decrease in p100) by co-expression of c-IAP2H570A. Because the decrease of p100 was not sufficient to induce a costimulation advantage in T cells without a concurrent imbalance with p52, these data argue strongly that it is the relative levels of p100 and p52 that determine whether T cells can proliferate to TCR signals alone.
Naïve T cells exist in a state of quiescence (G0) with low RNA content and slow activation kinetics. Conversely, memory cells are in G1 and have higher RNA content (13, 16, 32, 33), which includes mRNA of cytokines (e.g., IFN-γ, IL-2) and effector molecules (e.g. perforin, granzyme B) (34), and higher levels of ribosomal RNA, which allows for the rapid translation of effector molecule mRNAs (35). Although we found no evidence for constitutive expression of IL-2 mRNA in c-IAP2H570A T cells, they did express higher levels of RNA than WT T cells. The activation of c-IAP2H570A T cells by TCR stimulation alone, therefore, may be because non-canonical NF-κB is always active and the cells are poised to translate upregulated mRNAs rapidly upon stimulation.
There has been a great deal of interest in targeting c-IAP molecules with SMAC mimetics as an anti-cancer therapy (36). Although there may be initial benefit to activating non-canonical NF-κB, the long-term sequelae of such an intervention must be considered. There are several lines of evidence that chronic non-canonical NF-κB activation can contribute to lymphoproliferative disorders. Gene rearrangement of nfκb2 leading to transcription of constitutively active fragments lacking the ankyrin inhibitory domain have been described in some cases of chronic lymphocytic leukemia, multiple myeloma, and cutaneous T-cell lymphoma (37–40). In another example, mucosa-associated lymphoid tissue (MALT) lymphoma, c-IAP2-MALT1 fusion proteins lacking E3 activity induce constitutive p100 to p52 processing (41), and activation of non-canonical NF-κB in c-IAP2H570A B cells leads to a MALT lymphoma-like disease (10). Human T-cell leukemia virus type 1 (HTLV1) Tax protein triggers non-canonical NF-κB activation in T cells, causing their transformation (8, 42, 43). Although not tested in T cells, in fibroblasts stable expression of p100 blocked Tax-dependent transformation (44). In addition to promoting tumors, the observation that c-IAP2H570A mice died after infection with T. gondii due to an exaggerated pro-inflammatory response adds another potentially deleterious outcome to activating T cell non-canonical NF-κB. Therefore, therapies in which c-IAPs expression is targeted may have to contend with broader consequences of non-canonical NF-κB expression that include potentially harmful enhancement of T cell activation.
Supplementary Material
Acknowledgments
We are grateful to Eric C. La Casse and Robert Korneluk for providing the c-IAP2−/−mice, to Uli Siebenlist for providing the p100−/− mice, and to Ehydel Castro for expert technical assistance.
This work was supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute.
Footnotes
Contribution: M.L.G.T., D.B.C., and D.J. performed experiments and analyzed results; M.L.G.T. and J.D.A. designed the research and wrote the paper.
Conflict-of- interest disclosure: The authors declare no competing financial interests.
References
- 1.Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 2.Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 3.Shih VF, Tsui R, Caldwell A, Hoffmann A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011;21:86–102. doi: 10.1038/cr.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 5.Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, Brink R, Leverkus M, Tergaonkar V, Schneider P, Callus BA, Koentgen F, Vaux DL, Silke J. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–693. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 6.Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, Korneluk RG, Cheng G. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun SC. Non-canonical NF-κB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srinivasula SM, Ashwell JD. IAPs: what’s in a name? Mol Cell. 2008;30:123–135. doi: 10.1016/j.molcel.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conze DB, Zhao Y, Ashwell JD. Non-canonical NF-kappaB activation and abnormal B cell accumulation in mice expressing ubiquitin protein ligase-inactive c-IAP2. PLoS Biol. 2010;8:e1000518. doi: 10.1371/journal.pbio.1000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubey JP. Advances in the life cycle of Toxoplasma gondii. Int J Parasitol. 1998;28:1019–1024. doi: 10.1016/s0020-7519(98)00023-x. [DOI] [PubMed] [Google Scholar]
- 12.Christopher JA, Avitabile BG, Bamborough P, Champigny AC, Cutler GJ, Dyos SL, Grace KG, Kerns JK, Kitson JD, Mellor GW, Morey JV, Morse MA, O’Malley CF, Patel CB, Probst N, Rumsey W, Smith CA, Wilson MJ. The discovery of 2-amino-3,5-diarylbenzamide inhibitors of IKK-alpha and IKK-beta kinases. Bioorg Med Chem Lett. 2007;17:3972–3977. doi: 10.1016/j.bmcl.2007.04.088. [DOI] [PubMed] [Google Scholar]
- 13.Allam A, Conze DB, Giardino Torchia ML, Munitic I, Yagita H, Sowell RT, Marzo AL, Ashwell JD. The CD8+ memory T-cell state of readiness is actively maintained and reversible. Blood. 2009;114:2121–2130. doi: 10.1182/blood-2009-05-220087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jankovic D, Kullberg MC, Feng CG, Goldszmid RS, Collazo CM, Wilson M, Wynn TA, Kamanaka M, Flavell RA, Sher A. Conventional T-bet(+)Foxp3(−) Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. 2007;204:273–283. doi: 10.1084/jem.20062175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kane LP, Andres PG, Howland KC, Abbas AK, Weiss A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-gamma but not TH2 cytokines. Nat Immunol. 2001;2:37–44. doi: 10.1038/83144. [DOI] [PubMed] [Google Scholar]
- 16.Munitic I, Ryan PE, Ashwell JD. T cells in G1 provide a memory-like response to secondary stimulation. J Immunol. 2005;174:4010–4018. doi: 10.4049/jimmunol.174.7.4010. [DOI] [PubMed] [Google Scholar]
- 17.Wells AD. New insights into the molecular basis of T cell anergy: anergy factors, avoidance sensors, and epigenetic imprinting. J Immunol. 2009;182:7331–7341. doi: 10.4049/jimmunol.0803917. [DOI] [PubMed] [Google Scholar]
- 18.Ishimaru N, Kishimoto H, Hayashi Y, Sprent J. Regulation of naive T cell function by the NF-κB2 pathway. Nat Immunol. 2006;7:763–772. doi: 10.1038/ni1351. [DOI] [PubMed] [Google Scholar]
- 19.Basak S, Kim H, Kearns JD, Tergaonkar V, O’Dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. 2007;128:369–381. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 21.Appleman LJ, V, Boussiotis A. T cell anergy and costimulation. Immunol Rev. 2003;192:161–180. doi: 10.1034/j.1600-065x.2003.00009.x. [DOI] [PubMed] [Google Scholar]
- 22.Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1:220–228. doi: 10.1038/35105024. [DOI] [PubMed] [Google Scholar]
- 23.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 24.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 25.Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9:271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hauer J, Puschner S, Ramakrishnan P, Simon U, Bongers M, Federle C, Engelmann H. TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-kappaB pathway by TRAF-binding TNFRs. Proc Natl Acad Sci U S A. 2005;102:2874–2879. doi: 10.1073/pnas.0500187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rauert H, Wicovsky A, Muller N, Siegmund D, Spindler V, Waschke J, Kneitz C, Wajant H. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2) J Biol Chem. 2010;285:7394–7404. doi: 10.1074/jbc.M109.037341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamada T, Mitani T, Yorita K, Uchida D, Matsushima A, Iwamasa K, Fujita S, Matsumoto M. Abnormal immune function of hemopoietic cells from alymphoplasia (aly) mice, a natural strain with mutant NF-κB-inducing kinase. J Immunol. 2000;165:804–812. doi: 10.4049/jimmunol.165.2.804. [DOI] [PubMed] [Google Scholar]
- 29.Caamano J, Alexander J, Craig L, Bravo R, Hunter CA. The NF-kappa B family member RelB is required for innate and adaptive immunity to Toxoplasma gondii. J Immunol. 1999;163:4453–4461. [PubMed] [Google Scholar]
- 30.Ishikawa H, Carrasco D, Claudio E, Ryseck RP, Bravo R. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-κB2. J Exp Med. 1997;186:999–1014. doi: 10.1084/jem.186.7.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dougan M, Dougan S, Slisz J, Firestone B, Vanneman M, Draganov D, Goyal G, Li W, Neuberg D, Blumberg R. IAP inhibitors enhance co-stimulation to promote tumor immunity. The Journal of Experimental Medicine. 2010;207:2195. doi: 10.1084/jem.20101123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veiga-Fernandes H, Rocha B. High expression of active CDK6 in the cytoplasm of CD8 memory cells favors rapid division. Nat Immunol. 2004;5:31–37. doi: 10.1038/ni1015. [DOI] [PubMed] [Google Scholar]
- 33.Parretta E, Cassese G, Santoni A, Guardiola J, Vecchio A, Di Rosa F. Kinetics of in vivo proliferation and death of memory and naive CD8 T cells: parameter estimation based on 5-bromo-2′-deoxyuridine incorporation in spleen, lymph nodes, and bone marrow. J Immunol. 2008;180:7230–7239. doi: 10.4049/jimmunol.180.11.7230. [DOI] [PubMed] [Google Scholar]
- 34.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 35.Pardee AB. G1 events and regulation of cell proliferation. Science. 1989;246:603–608. doi: 10.1126/science.2683075. [DOI] [PubMed] [Google Scholar]
- 36.Chen DJ, Huerta S. Smac mimetics as new cancer therapeutics. Anticancer Drugs. 2009;20:646–658. doi: 10.1097/CAD.0b013e32832ced78. [DOI] [PubMed] [Google Scholar]
- 37.Nishikori M. Classical and alternative NF-κB activation pathways and their roles in lymphoid malignancies. Journal of Clinical and Experimental Hematopathology. 2005;45:15–24. [Google Scholar]
- 38.Chang CC, Zhang J, Lombardi L, Neri A, Dalla-Favera R. Rearranged NFKB-2 genes in lymphoid neoplasms code for constitutively active nuclear transactivators. Mol Cell Biol. 1995;15:5180–5187. doi: 10.1128/mcb.15.9.5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neri A, Fracchiolla NS, Migliazza A, Trecca D, Lombardi L. The involvement of the candidate proto-oncogene NFKB2/lyt-10 in lymphoid malignancies. Leuk Lymphoma. 1996;23:43–48. doi: 10.3109/10428199609054800. [DOI] [PubMed] [Google Scholar]
- 40.Heusch M, Lin L, Geleziunas R, Greene WC. The generation of nfkb2 p52: mechanism and efficiency. Oncogene. 1999;18:6201–6208. doi: 10.1038/sj.onc.1203022. [DOI] [PubMed] [Google Scholar]
- 41.Hu S, Du MQ, Park SM, Alcivar A, Qu L, Gupta S, Tang J, Baens M, Ye H, Lee TH, Marynen P, Riley JL, Yang X. cIAP2 is a ubiquitin protein ligase for BCL10 and is dysregulated in mucosa-associated lymphoid tissue lymphomas. J Clin Invest. 2006;116:174–181. doi: 10.1172/JCI25641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lanoix J, Lacoste J, Pepin N, Rice N, Hiscott J. Overproduction of NFKB2 (lyt-10) and c-Rel: a mechanism for HTLV-I Tax-mediated trans-activation via the NF-κB signalling pathway. Oncogene. 1994;9:841–852. [PubMed] [Google Scholar]
- 43.Qu Z, Qing G, Rabson A, Xiao G. Tax deregulation of NF-κB2 p100 processing involves both beta-TrCP-dependent and -independent mechanisms. J Biol Chem. 2004;279:44563–44572. doi: 10.1074/jbc.M403689200. [DOI] [PubMed] [Google Scholar]
- 44.Yamaoka S, Inoue H, Sakurai M, Sugiyama T, Hazama M, Yamada T, Hatanaka M. Constitutive activation of NF-κB is essential for transformation of rat fibroblasts by the human T-cell leukemia virus type I Tax protein. EMBO J. 1996;15:873–887. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.