

Abstract
CD86 engagement on a CD40L/IL-4-primed murine B cell activates signaling intermediates that promote NF-κB activation to increase Oct-2 and mature IgG1 mRNA and protein expression, as well as the rate of IgG1 transcription, without affecting class switch recombination. One of the most proximal signaling intermediates identified is phospholipase Cγ2 (PLCγ2), a protein reported to bind tyrosine residues, which are absent in the cytoplasmic domain of CD86. Using a proteomics-based identification approach, we show that the tyrosine-containing transmembrane adaptor proteins, prohibitin-1 (Phb1) and prohibitin-2 (Phb2), bind to CD86. The basal expression of Phb1/2 and association with CD86 was low in resting B cells, while the level of expression and association increased primarily after priming with CD40. The CD86-induced increase in Oct-2 and IgG1 was less when either Phb1/2 expression was reduced by shRNA or the cytoplasmic domain of CD86 was truncated or mutated at serine/threonine PKC-phosphorylation sites, which did not affect Phb1/2 binding to CD86. Using this approach, we also show that Phb1/2 and the CD86 cytoplasmic domain are required for the CD86-induced phosphorylation of IκBα, which we previously reported leads to NF-κB p50/p65 activation; whereas, only Phb1/2 was required for the CD86-induced phosphorylation of PLCγ2 and PKCα/βII, which we have previously reported leads to NF-κB (p65) phosphorylation and subsequent nuclear translocation. Together, these findings suggest that Phb1/2 and the CD86 cytoplasmic domain cooperate to mediate CD86 signaling in a B cell through differential phosphorylation of distal signaling intermediates required to increase IgG1.
Introduction
CD86, also referred to as B7-2, is a 70 kDa transmembrane glycoprotein expressed primarily on APCs including macrophages, dendritic cells, and B cells (1, 2). CD86 is a well-known costimulatory molecule that ligates CD28 and CTLA-4 expressed on a CD4+ T cell, to increase or decrease, respectively, T cell activation signals (3–6), and essential for germinal center formation (7, 8). CD86 expression is low on resting B cells (1), but increases in response to engagement of the BCR (1), CD40 (9), the IL-4R (10), LPS receptor (11, 12) or the beta-2 adrenergic receptor (13, 14). CD86 contains a short cytoplasmic domain that lacks tyrosine phosphorylation sites and was thought not to signal directly. However, the CD86 cytoplasmic domain contains three putative PKC serine/threonine phosphorylation sites. In addition, a proposal by Lenschow and colleagues reported that the CD86 cytoplasmic domain might become phosphorylated due to cellular activation stimuli (15) suggesting that CD86 may signal directly. Studies have reported that CD86 engagement induced a signal directly within the B cell that increased IgG4 production in anti-CD40/IL-4 primed human B cells (16), and the murine IgG4 homolog IgG1 production in CD40L/IL-4 (13, 17–20), or LPS (21) primed murine B cells in vitro, as well as in B cells from mice immunized with either Trinitrophenyl hapten (TNP)-keyhole limpet hemocyanin (KLH) (20), or influenza virus (22). It has also been reported that CD86 also signals to regulate other Ig-isotypes including IgE (13, 16), and IgG2a (21) an affect that may be controlled by the priming antigen or stimulus. Collectively, these findings suggested that CD86 on a B cell plays a role in regulating the level of IgG1 produced.
The initial functional results from these studies led to the search for signaling intermediates and transcription factors activated by CD86 engagement to mediate the increase in IgG1 production. CD86 engagement on the surface of a CD40L/IL-4-primed B cell was found to activate two cascades of signaling intermediates that ultimately allowed for NF-κB p50/p65 activation via phosphorylation of IκBα and p65 phosphorylation, respectively (18). Inhibition or loss of these signaling intermediates in a B cell eliminated the CD86-induced increase in Oct-2 expression (18, 19), Oct-2 binding to the 3′-IgH enhancer (18, 19), the rate of mature IgG1 transcription (17), and the increase in IgG1 protein per cell (13), confirming their roles in mediating CD86 signals to affect the level of IgG1 produced. Importantly, CD86 engagement on primed B cells failed to affect class switch recombination (13, 17–20), indicating that the increase in IgG1 was due to an effect on the amount of IgG1 produced per cell and not the number of cells that switched to IgG1. The increased level of signaling intermediate activation and/or Oct-2 that was induced by CD86 engagement on primed B cells resulted in a 2–3 fold increase in IgG1 as compared to primed B cells in the absence of CD86 engagement. Notably, clinical findings have shown that a 2–3 fold increase in serum IgG correlates to a 3–9 fold increase in protection against Streptococcus pneumoniae and pertussis (23, 24), suggesting that increases of this magnitude in the level of Ab produced are potentially relevant clinically.
This study sought to identify a potential signaling intermediate(s) associated directly with CD86, and to verify its role in mediating CD86 signaling. Such a directly associated protein(s) must exist because one of the most proximal CD86-dependent signaling intermediates, phospholipase Cγ2 (PLCγ2)2, functions classically by recruitment to proteins containing tyrosine-residues (25, 26), which are lacking in the CD86 cytoplasmic domain. Using a proteomics approach, we demonstrate a novel protein/protein interaction that occurred in a CD40L/IL-4-primed B cell between CD86 and prohibitin (Phb)1 and Phb2, which are transmembrane adaptor proteins that contain tyrosine residues. Furthermore, we report that both Phb1/2 and an intact CD86 cytoplasmic domain are required to mediate CD86 signaling that regulates the level of IgG1 produced by a B cell via differential activation of distal signaling intermediates.
Materials and Methods
Animals
Female pathogen-free BALB/c mice were obtained from Taconic (Germantown, NY), and were housed in the American Association Accreditation of Laboratory Animal Care (AAALAC)-accredited Animal Research Facility at The Ohio State University (Columbus, OH). All mice were provided autoclaved food and deionized water ad libitum and used at 8–10 weeks of age.
Cell Lines/Transgenic B cells
CH12.LX is a murine B cell lymphoma line previously described (27) kindly provided by Dr. G. Bishop (University of Iowa, Iowa City, Iowa). WT, transgenic B cells (Line 7 Tg) that overexpress CD86 on a C57BL/6 background were previously described (28). Transgenic B cells that express a CD86 cytoplasmic truncation (Line I2) within a CD80/CD86-double deficient mouse on a C57BL/6 background were previously described (29).
B cell Isolation/Priming
Naive B cells were isolated using magnetic cell sorting (MACS) anti-mouse CD43 beads following manufacturer’s directions (Miltenyi Biotec, Auburn, CA). Cells were primed with CD40L-expressing Sf9 cells or control Sf9 cells lacking CD40L expression at a B cell to Sf9 cell ratio of 10:1, IL-4 [1 ng/ml (eBioscience)], anti-IgM (0.1 μg/ml), terbutaline (Terb) (10−6 M; Sigma-Aldrich), or LPS (50 μg/ml, Sigma-Aldrich) for 16 hours. Total mRNA and total protein were collected and analyzed via quantitative real time PCR (qRT-PCR) and immunoblot, respectively. All reagents used were negative for the presence of endotoxin, as determined by Etoxate (Sigma), a Limulus lysate assay with a level of detection < 0.1U/ml.
Immunoprecipitation
Immunoprecipitations were performed using the Profound Mammalian Co-Immunoprecipitation Kit (Pierce). Briefly, 25 × 106 CH12.LX B cells were cultured and primed as described above, lysed with 0.5% Triton-X100 buffer, immunoprecipitated with an anti-CD86 GL1 (eBioscience), or species- and isotype-matched control Ab (Rat IgG2a, ebioscience) following manufacturer’s directions. The precipitates were analyzed via immunoblot for CD86, Phb2, and Phb1. Alternatively, 20 × 106 CH12.LX B cells, and 50×106 WT B cells were activated and lysed as described above, and immuoprecipitated with 50 μg of either anti-FLAG Ab (M2; Sigma) or anti-CD86 Ab (GL1; eBioscience) overnight at 4°C, followed by the addition of 50 μl of protein G agarose beads (Invitrogen).
Mass Spectrometry
Mass Spectrometry and protein identification were performed by the Mass Spectrometry and Proteomics Core Facility at The Ohio State University (Columbus, OH). The unique band present at ~35 kDa in an anti-CD86 immunoprecipitate of CH12LX cells from a gel stained with Sypro Ruby (Invitrogen) was excised and digested with sequencing grade trypsin from Promega (Madison, WI) using the Multiscreen Solvinert Filter Plates from Millipore (Bedford, MA). Capillary-liquid chromatography-nanospray tandem mass spectrometry (Nano-LC/MS/MS) was performed on a Thermo Finnigan LTQ mass spectrometer equipped with a nanospray source operated in positive ion mode. The LC system was an UltiMate™ 3000 system from Dionex (Sunnyvale, CA). Sequence information from the MS/MS data was processed by converting the .raw files into a merged file (.mgf) using an in-house program, RAW2MZXML_n_MGF_batch (merge.pl, a Perl script). The resulting mgf files were searched using Mascot Daemon by Matrix Science version 2.3.2 (Boston, MA).
Immunoblot Analysis
Resting B cells (5–7×106) were primed as described above. CH12.LX B cells were transfected with scrambled negative control shRNA or Phb1/2-specific shRNA plasmids (1–5 μg/106 cells) Phb1 (clones 1, 3, and 4), Phb2 (clones 1 and 3) (SA Biosciences) for 24 hours followed by priming with CD40L/IL-4 for 16 hours or with a FLAG-CD86 expression plasmid (1 μg/106 cells) via nucleofection (Amaxa, program O-003), followed by priming with CD40L/IL-4 for 16 hours. In some experiments, cells were cultured under serum-free conditions for at least 30 minutes, and engagement of CD86 with anti-CD86 (P03, ebioscience), species- and isotype-matched control Ab (Rat IgG2b, ebioscience), anti-FLAG (M2, Sigma) or species- and isotype- matched control Ab (mouse IgG1, Southern Biotec). Cell lysates were prepared as described previously (18). Nuclear-enriched protein lysates were prepared by washing 5×106 CH12.LX B cells with 1X PBS following by the addition of 0.5% NP-40 lysis buffer containing 10 mM NaCl, 10 mM Tris-HCl pH (7.4), and 3 mM MgCl2. Nuclei were pelleted and resuspended in 1X lysis buffer (20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton-X100, 2.5 mM sodium pyrophosphate, 1 mM Na2VO4, 1 μg/ml leupeptin, 10 nM okadaic acid, and 10 nM tautomycin). Protein samples, (5–12 μg) were run on a denaturing 10% polyacrylamide gel and transferred to Immobilon-PVDF membranes (Millipore). Membranes were blocked with tris buffered saline-tween 20 (TBST) as described previously (18) and incubated overnight with primary antibodies at 4°C. Membranes were probed with HRP-labeled secondary antibodies, developed using LumiGlo Detection Kit (Cell Signaling) and specific bands were visualized on Kodak Biomax MS film. Antibodies used were anti-CD86 (M-20), anti-CD86 (H-200), anti-GAPDH (FL-335), anti-PKCβII (C-18) (Santa Cruz Biotechnology), anti-Bap37 (Phb2) (Poly6118) (BioLegends), anti-Phb1, anti-phospho-PLCγ2 (Y1217), anti-PLCγ2, anti-phospho PKCα/βII (T638/641), anti-PKCα, anti-phospho-IκBα (S32) (14D4), anti-IκBα (44D4), anti-phospho-p65 (S536) (93H1), anti-p65 (C22B4), anti-Lamin A/C (4C11), anti-GAPDH (14C10), anti-β-Actin (13E5) (Cell Signaling), anti-α-tubulin (DM1A) (Sigma).
Immunoblot Densitometric Analysis
Specific protein bands detected via Immunoblot Analysis were quantified via Densitometry (ImageJ Software, NIH). The scanned image was inverted in order to measure the optical density (OD) of a specific protein band. A measurement box was created around the broadest band on a given gel and used to measure each band on the gel. The numerical value was recorded as an optical density (OD). Background measurements of each band were recorded from the corresponding gel lanes and subtracted from the original ODs. The ODs of Phb1/2 total protein bands were normalized to GAPDH protein loading control band ODs. The ODs of phospho-PLCγ2 (Y1217) protein bands were normalized to ODs of corresponding total PLCγ2 protein bands. ODs from phospho-PKCα/βII (T638/641) protein bands were normalized to OD values from both PKCα and PKCβII total protein bands. ODs obtained from phospho-IκBα (S32) and total IκBα protein bands were normalized to GAPDH total protein band ODs. The ODs recorded from phospho-p65 (S536) protein bands were normalized to both total p65 protein band ODs and ODs obtained from GAPDH protein bands. The ODs measured from nuclear phospho-p65 (S536) and nuclear p65 were normalized to both Lamin A and Lamin C protein band ODs. The data are expressed as mean Fold Change + standard deviation (SD)/standard error of the mean (SEM) relative to resting, CD40L/IL-4-priming alone, or species- and isotype-matched control Ab conditions.
Flow Cytometry
The level of CD86 on GFP+ Phb1/2-shRNA+ CH12.LX B cells was determined as previously described (13) using allophycocyanin (APC)-conjugated rat-anti-mouse CD86 (GL1). Samples were analyzed using a FACS CantoII (BD Biosciences). Expression of FLAG-CD86 was measured via FITC-conjugated anti-mouse FLAG (M2), (Sigma). The data were analyzed using FlowJo Software (Tree Star).
RNA-interfering Phb1 and Phb2 Knock-down and qRT-PCR analysis
CH12.LX B cells were transfected with scrambled negative control shRNA, or Phb1 (clones 1, 3, and 4) or Phb2 (clones 1 and 3)-specific shRNA (SABiosciences) (1–5 μg/106 cells), or a FLAG-CD86 expression vector (1 μg/106 cells) via nucleofection (Amaxa, program O-003) according to manufacturer’s instructions, cultured for 24 hours followed by CD40L/IL-4 priming for an additional 16 hours. In some experiments, GFP+ cells were sorted (ICy-Reflection) and re-cultured in the presence of either anti-CD86 (PO3 eBioscience) or species- and isotype-matched control Ab (RatIgG2b eBioscience), or CD28/Fc chimera (CD28/Ig) (R & D Biosciences) for 24 hours. Total RNA was isolated via Trizol (Invitrogen), cDNA was created via reverse transcription with random hexamer primers (Qiagen) and qRT-PCR (SYBRGreen, Roche) was conducted as previously described (17) for Phb1 mRNA levels, forward primer: 5′-ATGGCTGCCAAAGTGTTTGAGTC-5′ reverse primer: 5′-TCACTGGGGAAGCTGGAGAAGC-3′; Phb2 mRNA levels, forward primer: 5′-GTAGAAGCCAAGCAAGTGGC-3′ reverse primer: 5′-TTCAGCACAAGGTTGTCAGC-3′; CD86 mRNA levels, forward primer: 5′-CGAGCACTATTTGGGCACAGAG-3′ reverse primer: 5′-TTTCCAGAACACACACAACGGTC-3′; cytoplasmic CD86 mRNA levels, forward primer: 5′-GTCAATGAAGATTTCCTCCAAA-5′ reverse primer: 5′-CAGGTTGATAGTCTCTCTGTCAG-3′; O c t-2 mRNA levels, forward primer: 5′-ATCAAGGCTGAAGACCCCAGTG-5′ reverse primer: 5′-TGGAGGAGTTGCTGTATGTCCC-3′; germline IgG1 mRNA levels, forward primer: 5′-CATCCTATCACGGGAGATTGGG-3′ reverse primer: 5′-ATCCTCGGGGCTCAGGTTTG-3′; and mature IgG1 mRNA levels, forward primer: 5′-TATGGACTACTGGGGTCAAG-3′ reverse primer: 5′-CCTGGGCACAATTTTCTTGT-3′ relative to actin mRNA levels, forward primer: 5′-TACAGCTTCACCACCACAGC-3′ reverse primer: 5′-AAGGAAGGCTGGAAAAGAGC-3′or GAPDH mRNA levels, forward primer: 5′-ACCACAGTCCATGCCATCAC-3′ reverse primer: 5′-TCCACCACCCTGTTGCTGTA-3′.
cDNA constructs and Site-Directed Mutagenesis
Total RNA was isolated by Trizol (Invitrogen) from CD40L (10 B cells:1 Sf9) and recombinant IL-4 (1 ng/ml) (eBioscience)-primed murine splenocytes for 24 h and cDNA was generated using Accuscript RT (Stratagene). The murine CD86 cDNA was amplified by PCR using the Easy-A High Fidelity PCR Cloning (Stratagene) and the following primers. The forward primer containing EcoRI site: 5′-GAATTCCGAGACGCAAGCTTATTTCAATGGGACTGCATAT-3′ and the reverse primer containing BamHI site 5′-TTTTGGATCCTCACTCTGCATTTGGTTTTGCTGAAGC-3′. The CD86 cDNA was then subcloned into the pCR2.1 TOPO TA vector. To generate a FLAG-CD86 expression construct, murine CD86 cDNA was subsequently cloned into the pFLAG-CMV3 vector (Sigma-Aldrich), introducing an N-terminal FLAG epitope tag (DYKDDDK) before the start of the mature polypeptide receptor. Mutant plasmids containing either FLAG-CD86 cytoplasmic truncations at KKPΔ or 282Δ and or single alanine point mutations at positions S285, T291, and S303 were created by using the Quick Change Site-Directed Mutagenesis Kit (Stratagene) according to manufacturer’s instructions. The following primers were used to generate specific mutant plasmids: KKPΔ forward primer: 5′-ATTGTATGTCACAAGAAGTAGAATCAGCCTAGCAGGCCC-3′ reverse primer: 5′-GGGCCTGCTAGGCTGATTCTACTTCTTGTGACATACAAT-3′; 282Δ forward primer: 5′-AACACAGCCTCTAAGTTATAGCGGGATAGTAACGCTGACAG-3′ reverse primer: 5′-CTGTCAGCGTTACTATCCCGCTATAACTTAGAGGCTGTGTT-3′; S285 A forward primer: 5′-GCCTCTAAGTTAGAGCGGGATGCTAACGCTGACAGAGAGACTATC-3′ reverse primer: 5′-GGTTGATAGTCTCTCTGTCAGCGTTAGCATCCCGCTCTAACTTAG-5′; T 291 A forward primer: 5′-ATAGTAACGCTGACAGAGAGGCTATCAACCTGAAGGAACT-3′ reverse primer: 5′-AGTTCCTTCAGGTTGATAGCCTCTCTGTCAGCGTTACTAT-3′; S303A forward primer: 5′-AACTTGAACCCCAAATTGCTGCAGCAAAACCAAATGCAGA-3′ reverse primer: 5′-TCTGCATTTGGTTTTGCTGCAGCAATTTGGGGTTCAAGTT-3′. Integrity of all constructs was confirmed by sequencing at the Nucleic Acid Shared Resource facility at The Ohio State University (Columbus, OH). Surface expression of all constructs was confirmed by Flow Cytometry.
In-Cell ELISA
In-Cell ELISAs were performed to detect in-cell levels of phospho-PLCγ2 (Y1217) and total PLCγ2 protein using the In-Cell Colorimetric ELISA kit (Pierce). Briefly, 1×106 CH12.LX B cells were transfected under Mock conditions or with either scrambled negative control shRNA or Phb1/2-specific shRNA plasmids for 24 hours followed by priming with CD40L/IL-4, or with a WT or cytoplasmic-deficient (KKPΔ) FLAG-CD86 expression plasmid followed by priming with CD40L/IL-4 for 16 hours. Cells were cultured under serum-free conditions for at least 30 minutes, followed by engagement of either CD86 with anti-CD86, or FLAG-CD86 with anti-FLAG and appropriate controls as described above for 15–30 minutes. The cells were then washed in 1X PBS. 2×104 cells were plated on a 96-well culture plate and fixed with 8% formaldehyde. The remainder of the assay was performed according to manufacturer’s instructions. Antibodies used to detect phospho-PLCγ2 (Y1217) and total PLCγ2 in-cell protein levels were anti-phospho-PLCγ2 (Y1217), and anti-PLCγ2 (Cell Signaling). The HRP-conjugate antibody was used at a (1:2000) dilution.
Statistical Analysis
Data from multiple treatment groups were analyzed using a one-way analysis of variance to determine if an overall statistical change existed. Certain p values were calculated using a Bonferroni post hoc analysis or a two-sided Student t test for comparison of two treatment groups. A p value of ≤ 0.05 indicated statistically significant results.
RESULTS
CD86 associates with Phb1 and Phb2 in B Lymphocytes
CD86 engagement on a CD40L/IL-4-primed B cell is known to activate PLCγ2 (19), a signaling intermediate that requires binding to proteins containing phospho-tyrosine residues for recruitment/activation and its ability to implement receptor function. We reasoned that CD86, which does not contain tyrosine residues in the cytoplasmic domain, must associate with a tyrosine-containing adaptor/scaffolding protein to mediate the activation of PLCγ2. Therefore, a proteomics-based approach was used to identify any protein that associated with CD86 in primed B cells. The murine B cell line CH12.LX was cultured overnight in the presence of CD40L/IL-4 and then exposed to either an anti-CD86 or species- and isoytpe-matched control Ab. CD86 was immunoprecipitated from cell lysates, and proteins were separated via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and stained with Sypro Ruby (Fig. 1A). Three unique bands were present at molecular weights of ~50 kDa, ~35 kDa, and ~28 kDa in the anti-CD86 antibody immunoprecipitate, but were absent when the control Ab was used. The unknown ~35 kDa protein was excised since it was clearly isolated from other bands and identified by Mass Spectrometry as the adaptor protein prohibitin-2 (Phb2) (Fig. 1B). In all, 14 non-redundant tryptic peptide fragments of murine Phb2 were identified.
FIGURE 1.

CD86 associates with Phb1 and Phb2 in B Lymphocytes. A, CH.12LX B cells were primed with CD40L/IL-4 for 16 hours followed by the addition of a species- and isotype-matched control Ab (anti-rat IgG2a ctrl Ab), or rat anti-mouse CD86 (GL1) (anti-CD86) for 15 minutes. Lysates were prepared, followed by immunoprecipitation (Pierce). Immunoprecipitates were separated via SDS-PAGE followed by total protein staining with Sypro Ruby. Unique bands in the CD86 immunoprecipitate are indicated by arrows at ~50, ~35, and ~28 kDa. B, The band marked with an arrow at ~35 kDa was sequenced via LC/MS Mass Spectrometry. Tryptic peptide fragments were sequenced and matched with known tryptic peptides via a MASCOT search. 14 non-redundant tryptic peptides were identical to murine prohibitin-2 (Phb2). C, CH12.LX B cells alone, CH12.LX B cells expressing a FLAG-CD86 recombinant protein, or primary splenic B cells were primed as described above. Immunoprecipitates were immunoblotted for CD86, Phb2, and Phb1. Gels are representative of three independent experiments. D, FLAG-CD86 transfected CH12.LX B cells with either Full Length FLAG-CD86 or a Truncated FLAG-CD86 (KKPΔ) or non-transfected B cells from WT or Trunc CD86-expressing mice were primed as described above, immunoprecipitated with either anti-FLAG or anti-CD86, and were analyzed for Phb1/2 and CD86 protein levels via immunoblot. The gels shown are representative of three independent experiments (left panel) or one experiment (right panel).
Although sequence analysis was not performed on the other two bands, we predict that the CD86-associated ~28 kDa protein band is Phb1 since the literature suggests that Phb2 associates closely with the highly-related Phb1 in a variety of intracellular locales (30). The identity of the CD86-associated protein band at ~50 kDa remains unclear, but may be another adaptor/scaffolding protein similar to that reported for other B cell surface receptors, including CD40 (31–34) and the BCR (35–39) that are also devoid of tyrosine residues within the cytoplasmic domain, but use adaptor/scaffolding complexes to function.
We tested whether Phb1 and/or Phb2 could be specifically co-immunoprecipitated from B cells under primed vs. resting conditions. Parent CH12.LX cells, CH12.LX cells transfected with FLAG-tagged CD86, or primary B cells were cultured overnight in the presence or absence of CD40L/IL-4-priming. Lysates were prepared and immunoprecipitated with control and anti-CD86 Abs, and then analyzed for level of Phb1/2 and CD86 by immunoblot. As shown in (Fig. 1C), levels of Phb1 and Phb2 were low or undetectable in CD86 immunoprecipitates from resting CH12.LX B cells, while co-immunoprecipitation of both proteins increased substantially in primed B cells. This increase in Phb1/2 binding to CD86 may be due to a specific CD40L/IL-4-dependent recruitment event, or an increased level of CD86 present in the CD86 immunoprecipitates. Similar results were observed when CH12.LX B cells were transfected with a FLAG epitope-tagged CD86 construct, and primary B cells were used (Fig. 1C). To determine if Phb1/2 associated with the cytoplasmic domain of CD86, we tested CH12.LX B cells transfected with a cytoplasmic truncated form of FLAG-CD86 (KKPΔ), as well as primary B cells from transgenic mice that express a truncated form of the CD86 cytoplasmic domain. Phb1/2 were found to associate with CD86 in the absence or presence of the cytoplasmic domain (Fig. 1D), suggesting that a transmembrane-specific association occurred between CD86 and Phb1/2. Taken together, these findings indicated that low levels of Phb1 and Phb2 associated with CD86 in resting B cells, but that the levels increased after CD40L/IL-4-priming, and was independent of most of the CD86 cytoplasmic domain.
The expression of Phb1 and Phb2 is regulated primarily by CD40 engagement
A number of studies have reported that Phb1 and Phb2 associate with each other to form a large complex within the inner mitochondrial membrane, and function by stabilizing proteins of the electron transport chain (40). If a similar stabilizer/molecular chaperone function existed for Phb1/2 to facilitate CD86 stability, transport, and insertion into the plasma membrane, then Phb1/2 protein expression would be expected to increase concomitantly with CD86 protein expression when the B cell was primed with CD40L/IL-4, as well as with other B cell stimuli that are known to increase CD86 protein expression. Primary resting B cells were cultured in the absence or presence of IL-4 overnight with either CD40L, anti-IgM to engage the BCR, the beta-2 adrenergic receptor agonist terbutaline (Terb), or LPS, all of which have been reported to increase CD86 expression on a B cell (1, 9–14), and as shown in (Supplemental Fig. 1A). Total mRNA and protein were collected and analyzed for the level of Phb1/2 and CD86 expression by qRT-PCR and immunoblot, respectively. Engagement of CD40 alone promoted a robust increase in the level of expression for both Phb1 and Phb2 mRNA (Fig. 2A) and protein (Fig. 2B) as compared to resting B cells. IL-4R engagement on a resting B cell had no effect on the level of Phb1/2 expression while concomitant engagement of the IL-4R with CD40 further increased the level of Phb2, but not Phb1, mRNA, and increased the level of Phb1/2 protein as compared to CD40 engagement alone. Treatment with LPS alone also increased levels of Phb1/2 mRNA relative to resting B cells, although the increase was less than that caused by CD40 engagement, and this LPS-induced increase in mRNA was further increased by the addition of IL-4. In contrast, only a weak increase in Phb1/2 protein was detected in LPS exposed B cells in the absence or presence of IL-4. Other stimuli that were able to increase CD86, i.e., IgM and beta-2 adrenergic receptor engagement on a B cell, were unable to upregulate Phb1/2 expression to any significant level in comparison to resting levels. Importantly, control Sf9 cells lacking CD40L expression failed to induce Phb1/2 mRNA or protein levels (Fig. 2C). Together, these findings suggested that Phb1/2 protein expression may be regulated primarily by a CD40-dependent mechanism that is independent from the mechanism involved in upregulating expression of CD86.
FIGURE 2.

The expression of Phb1 and Phb2 is regulated primarily by CD40 engagement. Naive splenic B cells were isolated and cultured for 16 hours in the absence or presence of CD40L, anti-IgM, terbutaline (Terb), or LPS in the absence or presence of IL-4. A, Total mRNA was collected and analyzed via qRT-PCR analysis Phb2, Phb1, and GAPDH. Phb1/2 values were normalized to GAPDH and the data are represented as a mean Fold Change in Phb1/2 mRNA from primed B cells relative to resting B cells and are expressed as the mean ± SEM of quadruplicate samples/condition from three independent experiments. B, Total protein was collected and immunoblot analysis was used to measure total Phb2, Phb1, and GAPDH protein levels. Gels are representative of three independent experiments. Densitometry was performed and measured Phb1/2 band intensity/GAPDH band intensity and data are presented as the mean Fold Change in Phb1/2 protein from primed B cells relative to resting and expressed as the mean Fold Change ± SEM from three independent experiments. C, Naive splenic B cells were isolated and cultured for 16 hours in the presence or absence of control Sf9 cells lacking expression of CD40L (ctrl Sf9 CD40L neg). mRNA and total protein was collected and analyzed for Phb1/2 expression levels relative to actin or GAPDH via qRT-PCR or immunoblot, respectively. The data are expressed as the mean Fold Change ± SEM from either three (mRNA) or Fold Change ± SD from 2–3 (protein) independent experiments. Statistical differences are shown relative to resting. *, p < 0.05.
Phb1 and Phb2 play a role in mediating the effect of CD86 engagement on B cell function
Although the data thus far showed that Phb1/2 associated with CD86, the functional significance of this association was unclear. Recent studies have reported that Phb1 interacts with various signaling intermediates to modulate cellular processes, and thus, the possibility was supported that by associating with CD86 in primed B cells, Phb1 and Phb2 could be providing CD86 with a cytoplasmic signaling platform to stimulate B cell responses. To test this possibility directly, CH12.LX B cells were transfected with GFP-containing shRNA expression vectors designed to suppress Phb1/2 expression. The effect of transfection on B cell CD86 expression was measured before functional studies were performed. Flow Cytometric analysis showed that CD86 surface expression following CD40L/IL-4 priming was comparable in the absence or presence of Phb1/2 knockdown (Supplemental Fig. 1B). qRT-PCR analysis of FACS-sorted GFP+ and GFP− cells showed that both Phb1 and Phb2 expression was reduced by at least 50% in shRNA-expressing GFP+ cells (Fig. 3A), where reduction of Phb1 resulted in the loss of Phb2 and vice versa (Supplemental Fig. 2).
FIGURE 3.

Phb1 and Phb2 play a role in mediating the effect of CD86 engagement on B cell function. A and B, CH12.LX B cells were transfected with either a GFP-containing Phb1, Phb2, or scrambled, negative control shRNA (neg ctrl shRNA) vector for 36 hours to silence Phb1 and Phb2 gene expression. At 24 hours post transfection, cells were primed with CD40L/IL-4 for 16 hours. GFP cells containing the Phb1 or Phb2 shRNA vector were separated by FACS then re-cultured with either an anti-CD86 Ab, or a species- and isotype-matched control Ab (iso ctrl). On day 2 or day 6-post cell priming, Phb1/Phb2mRNA (A), or Oct-2/germline IgG1 and mature IgG1 mRNA, (B) respectively, were measured via qRT-PCR analysis and normalized to actin. GFP negative cells and scrambled negative control shRNA are depicted as (gray bars) and GFP+ cells containing Phb1/2 shRNA are depicted as (black bars). Data are expressed as mean Fold Change ± SEM relative to either No GFP (A) or iso ctrl of each group (B) and represent at least quadruplicate samples/condition from three independent experiments. Statistical differences are shown relative to No GFP (A) or iso ctrl of each group (B). *, p < 0.05.
We next determined if both Phb1/2 play roles in mediating the CD86-induced increases in Oct-2 and IgG1 without a change in class switch recombination, as reported previously (13,17–20). CD86 engagement on primed GFP+ B cells resulted in comparable levels of germline IgG1, Oct-2, and mature IgG1 mRNA relative to isotype control-treated B cells, while significant increases in Oct-2 and mature IgG1 mRNA were observed due to CD86 engagement in GFP− B cells (Fig. 3B). To ensure the comparable levels of Oct-2 and mature IgG1 mRNA were not due to an off target effect of the Phb1/2 shRNA plasmids, scrambled negative control shRNA plasmids were transfected into CH12.LX B cells as described. CD86 engagement promoted significant increases in the level of Oct-2 and mature IgG1 mRNA produced (Fig. 3B), whereas germline IgG1 mRNA remained unchanged. Together, these findings indicated that Phb1 and Phb2 are both necessary for CD86 engagement on a primed B cell to enhance the level of B cell function.
The cytoplasmic domain of CD86 also plays a role in mediating the effect of CD86 engagement on B cell function
The evidence thus far suggested that CD86 associated with Phb1/2 through transmembrane-specific interactions and that Phb1/2 were essential to couple CD86 engagement to a downstream functional increase in IgG1. However, it remained unclear whether or not the cytoplasmic domain of CD86 played any role in mediating the CD86-dependent increase in the B cell functional response. It was proposed previously that the cytoplasmic domain of CD86 had the potential to be phosphorylated at one or more of several putative PKC phosphorylation sites (15), which might provide a mechanism for CD86 to recruit and/or activate downstream signaling intermediates that are known to participate in the CD86-induced increases in Oct-2 and IgG1.
To test this possibility, a series of mutations/truncations were introduced into the FLAG-CD86 cytoplasmic domain that included single-alanine point mutations of the three putative PKC serine/threonine phosphorylation sites, as well as either partial or full truncation of the cytoplasmic domain as diagramed in Fig. 4A. CH12.LX B cells were transfected with the FLAG-CD86 plasmids, and cells were primed with CD40L/IL-4 in culture for 16 hours, at which time either a species- and isotype-matched control Ab or anti-FLAG Ab (M2) was added. Cells were assessed, via qRT-PCR analysis, for levels of Oct-2/germline IgG1 mRNA on day 2 or mature IgG1 mRNA on day 5 (Fig. 4B). The introduction of alanine-point mutations at all PKC phosphorylation sites had no effect on germline IgG1, but significantly reduced Oct-2 and mature IgG1 mRNA levels, except for Oct-2 induction by the T291A and S285A mutants, as compared to WT controls. Partial and full truncation of the CD86 cytoplasmic domain resulted in significant reductions of Oct-2 and mature IgG1 mRNA levels compared to WT controls. Importantly, levels of either WT or mutant FLAG-CD86 expressed on the B cell surface remained comparable (data not shown). These results emphasize that PKC phosphorylation of the cytoplasmic domain of CD86 is important for signaling function.
FIGURE 4.

The cytoplasmic domain of CD86 also plays a role in mediating the effect of CD86engagement on B cell function. A, A series a single-alanine point mutations (S285A, T291A, S303A) or cytoplasmic truncations (KKPΔ and 282Δ) were introduced into a FLAG-CD86 expression plasmid. B, Either WT FLAG-CD86 or mutant/truncated plasmids were transfected into CH12.LX B cells before the cells were primed with CD40L/IL-4. Cells were cultured for 16 hours before an anti-FLAG Ab (M2) or species- and isotype-matched control Ab (mouse IgG1 iso ctrl) was added to the culture. 24 hours and 4 days later, respectively, the level of germline IgG1/Oct-2 mRNA and mature IgG1 mRNA was determined by qRT-PCR. Values were normalized to actin and data are expressed as mean Fold Change ± SEM relative to WT mouse IgG1 iso ctrl and represent data of quadruplicate replicates/condition from three independent experiments. Statistical analysis was used to determine statistical significance relative to WT anti-FLAG. *, p < 0.05.
In addition to the use of CD86-transfected B cells, B cells were also used that had been isolated from mice transgenic for either WT CD86 (Tg WT) that overexpressed CD86 or a truncated form of CD86 (Trunc CD86) that lacked the cytoplasmic domain. In B cells from the Trunc CD86 mice that were primed, Phb1/2 proteins remained associated with CD86 (Fig. 1D), but the level of mRNA expressed for the cytoplasmic domain of CD86, Oct-2, and mature IgG1 was decreased in comparison to WT-expressing B cells, while germline IgG1 was comparable (Supplemental Fig. 3). Thus, to our knowledge, these data collectively provide evidence that, in addition to Phb1/2, the cytoplasmic domain of CD86 is also required to mediate CD86 signaling to regulate B cell function, likely via phosphorylation of the cytoplasmic domain at serine and/or threonine residues, as the present site-directed mutagenesis data show.
Phb1 and Phb2 are necessary for the CD86-induced activation of PLCγ2
Although findings thus far suggested that both Phb1/2 and the CD86 cytoplasmic domain are required to mediate CD86 signaling to regulate a B cell functional response, the proximal molecular mechanism was unclear. One of the most proximal CD86 signaling intermediates to date, PLCγ2 (19), is classically recruited to phosphorylated tyrosine-containing protein/protein complexes (25, 26). In addition, it was reported that Phb1 binds the protein tyrosine kinase (PTK) spleen tyrosine kinase (Syk) upon BCR engagement in murine B cells (41), indicating that Phb1 may be involved with the activation of PTKs specifically in B cells, and perhaps other membrane-associated signaling intermediates after receptor engagement. To address this, CH12.LX B cells were transfected with scrambled negative control or Phb1/2-specific shRNA plasmids, and either a WT, or cytoplasmic-deficient FLAG-CD86 expression plasmid (KKPΔ) followed by priming with CD40L/IL-4 and CD86 or FLAG-CD86 engagement. Phb1/2 protein knockdown efficiency and the level of PLCγ2 phosphorylation was measured by immunoblot. The presence of Phb1/2-specific shRNA resulted in modest reductions of Phb1/2 protein where reduction of Phb1 resulted in the loss of Phb2 (Fig. 5A), and vice versa. Importantly, levels of Phb1/2 among Mock transfected cells vs. cells transfected with scrambled negative control shRNA were comparable (Fig. 5A). While Mock and scrambled negative control shRNA transfected CH12.LX B cells promoted a time-dependent CD86-induced phosphorylation of PLCγ2 relative to primed cells alone (Fig. 5B), PLCγ2 phosphorylation upon CD86 engagement in the presence of Phb1/2 shRNA remained comparable to priming alone (Figs. 5C, 5D). Engagement of FLAG-CD86 on B cells transfected with both WT and KKP FLAG-CD86 expression plasmids caused an increase in PLCγ2 phosphorylation relative to a species- and isotype-matched control Ab (Fig. 5E). To extend our immunoblot findings, the level of PLCγ2 phosphorylation was measured via In-Cell ELISA. While Mock and scrambled negative control shRNA transfected CH12.LX B cells allowed for a CD86-induced increase in PLCγ2 phosphorylation relative to priming alone, PLCγ2 phosphorylation in the presence of Phb1/2 shRNA remained comparable to priming alone (Fig. 5F). Similar to our immunoblot findings, engagement of FLAG-CD86 on B cells transfected with both WT and KKP FLAG-CD86 expression plasmids caused an increase in PLCγ2 phosphorylation relative to a species- and isotype-matched control Ab (Fig. 5G). Together, these findings suggested that while Phb1/2 are critical for the CD86-induced activation of PLCγ2, the CD86 cytoplasmic domain is not involved, indicating that Phb1/2 and the CD86 cytoplasmic domain must differentially activate signaling intermediates upon CD86 engagement.
FIGURE 5.

Phb1 and Phb2 are necessary for the CD86-induced activation of PLCγ2. A, CH12.LX B cells were either Mock transfected, with scrambled negative control, or Phb1/2-specific shRNA plasmids via nucleofection for 24 hours followed by priming with CD40L/IL-4 for 16 hours. Phb1/2 protein levels were measured via immunoblot relative to GAPDH. Densitometry was performed and the data are expressed as the mean % Change + SEM relative to Mock and are representative of three independent experiments. B, CH12.LX B cells were either Mock transfected or with scrambled negative control shRNA (neg ctrl shRNA) via nucleofection for 24 hours followed by priming with CD40L/IL-4 for 16 hours. A CD86 Ab (anti-CD86) was administered for 5, 15, 30, and 60 minutes. Levels of phospho-PLCγ2 (pPLCγ2) protein relative to total PLCγ2 were measured via immunoblot. C and D, Phb1 or Phb2 shRNA plasmids were transfected into CH12.LX B cells followed by CD40L/IL-4 priming, and a CD86 Ab (anti-CD86) was added as described above. Levels of pPLCγ2 protein were measured relative to total PLCγ2 via immunoblot. E, WT, and CD86 cytoplasmic-deficient (KKPΔ) FLAG-CD86 plasmids were transfected into CH12.LX B cells via nucleofection and primed with CD40L/IL-4 for 16 hours. Either an anti-FLAG Ab, or species- and isotype-matched control Ab (iso ctrl Ab) was added for 15 minutes. Levels of pPLCγ2 protein were measured relative to total PLCγ2 via immunoblot. Representative gels (B to E) are shown from three independent experiments. Densitometry was performed and measured pPLCγ2 relative to PLCγ2 band intensity and the data are presented as the mean Fold Change in pPLCγ2 from primed B cells where CD86 was engaged relative to priming alone (B to D) or iso ctrl Ab (E) and expressed as the mean Fold Change ± SEM from three independent experiments. Statistical differences are shown relative to Mock (A), priming alone (B to D) or iso ctrl Ab (E). *, p < 0.05. F, CH12.LX B cells were either Mock transfected or with scrambled negative control shRNA (neg ctrl shRNA) via nucleofection for 24 hours followed by priming with CD40L/IL-4 for 16 hours. A CD86 Ab (anti-CD86) was administered for 30 minutes. Levels of phospho-PLCγ2 (pPLCγ2) protein relative to total PLCγ2 were measured via In-Cell ELISA. G, WT, and CD86 cytoplasmic-deficient (KKPΔ) FLAG-CD86 plasmids were transfected into CH12.LX B cells via nucleofection and primed with CD40L/IL-4 for 16 hours. Either an anti-FLAG Ab, or species-and isotype-matched control Ab (iso ctrl Ab) was added for 15 minutes. Levels of pPLCγ2 protein were measured relative to total PLCγ2 via In-Cell ELISA. (F & G) pPLCγ2 protein OD values were normalized to total pPLCγ2 protein OD values and the data are represented as a mean Fold Change in pPLCγ2 protein from CD86-engaged B cells relative to primed alone B cells of each group (F) or iso ctrl (G) and are expressed as the mean ± SD of quintuplicate samples/condition from at least two independent experiments. Statistical differences are shown relative to priming alone of each group (F) or iso ctrl (G). *, p < 0.05.
Phb1 and Phb2 are necessary for the CD86-induced activation of PKCα/βII
Although our evidence suggested that Phb1/2 are necessary for the CD86-induced activation of PLCγ2 independent of the CD86 cytoplasmic domain, whether or not Phb1/2 were essential for the CD86-dependent downstream activity of PLCγ2 remained unknown. Since PLC classically allows for the activation of PKC via cleavage of phosphoinositides into inositol-3 phosphate and diacylglycerol, leading to an increase in Ca2+ (42), and our previous findings showed that CD86 engagement on a CD40L/IL-4-primed B cell promoted the activation of PLCγ2 and the downstream activation of PKCα/βII (19), we considered PKCα/βII phosphorylation a viable measurement of PLCγ2 activity. We predicted that Phb1/2 alone would be necessary to induce CD86-dependent PKCα/βII phosphorylation independent of the CD86 cytoplasmic domain since Phb1/2 alone were sufficient to allow for CD86-induced PLCγ2 activation. To address this, CH12.LX B cells were either Mock transfected, or with scrambled negative control shRNA, or with Phb1/2-specific shRNA plasmids followed by priming with CD40L/IL-4 and the addition of a CD86 Ab over a 60 minute time course. The levels of PKCα/βII phosphorylation (pPKCα/βII)relative to total PKCα and total PKCβII protein were measured via immunoblot. While CH12.LX B cells that were Mock transfected and scrambled negative control shRNA produced a time-dependent CD86-induced PKCα/βII phosphorylation relative to priming alone, the effect was prevented in the presence of Phb1/2 specific shRNA relative to priming alone (Fig. 6A). Engagement of FLAG-CD86 on B cells transfected with both WT and KKP FLAG-CD86 expression plasmids caused an increase in PKCα/βII phosphorylation relative to a species- and isotype-matched control Ab (Fig. 6B). Collectively, these findings indicate that while Phb1/2 are necessary for CD86-dependent activation of PKCα/βII, a downstream signaling intermediate promoted through PLCγ2 activity, the CD86 cytoplasmic domain is not involved, providing further support that CD86 engagement allows for differential activation of distal signaling intermediates.
FIGURE 6.

Phb1 and Phb2 are necessary for the CD86-induced activation of PKCα/βII. A, CH12.LX B cells were either Mock transfected, with a scrambled negative control shRNA (neg ctrl shRNA), or with Phb1 or Phb2 shRNA plasmids into CH12.LX B cells via nucleofection for 24 hours followed by priming with CD40L/IL-4 for 16 hours. A CD86 Ab (anti-CD86) was administered for 5, 15, 30, and 60 minutes. Levels of phospho-PKCα/βII (pPKCα/βII) protein relative to total PKCα and PKCβII were measured via immunoblot. B, WT, and CD86 cytoplasmic-deficient (KKPΔ) FLAG-CD86 plasmids were transfected into CH12.LX B cells via nucleofection and primed with CD40L/IL-4 for 16 hours. Either an anti-FLAG Ab, or species-and isotype-matched control Ab (iso ctrl Ab) was added for 30 minutes. Levels of pPKCα/βII protein relative to total PKCα and PKCβII were measured via immunoblot. Representative gels are shown from three independent experiments. Densitometry was performed and measured pPKCα/βII relative to PKCα and PKCβII band intensity and the data are presented as the mean Fold Change in pPKCα/βII from primed B cells where CD86 was engaged relative to priming alone (A) or iso ctrl Ab (B) and expressed as the mean Fold Change ± SEM from three independent experiments. Statistical differences are shown relative to Mock priming alone (A) or iso ctrl Ab (B). *, p < 0.05.
Phb1/2 and the CD86 cytoplasmic domain are each required for the CD86-dependent activationof NF-κB
Although our findings suggested that Phb1/2 alone is sufficient to promote CD86-dependent PLCγ2 and PKCα/βII activation, the molecular role of the CD86 cytoplasmic domain remained unknown. Previous reports demonstrated that engagement of CD86 on the surface of a CD40L/IL-4-primed B cell initiates two signaling cascades that converge to activate the p50/p65 subunits of NF-κB (18, 19). While one cascade is essential for CD86-dependent IκBα phosphorylation (pIκBα), the other promotes the phosphorylation of p65 (p-p65) in a PKCα/βII- and PLCγ2-dependent manner (18, 19). To determine which CD86-induced signaling intermediate controlled NF-κB activation, i.e., IκBα and p65 phosphorylation, CH12.LX B cells were either Mock transfected, or transfected with scrambled negative control, or Phb1/2-specific shRNA plasmids, and either WT-FLAG CD86 or CD86 cytoplasmic domain-deficient, (KKPΔ) expression plasmids followed by CD40L/IL-4-priming and engagement of either CD86 or FLAG-CD86 over a 60–90 minute time course. The level of IκBα and p65 phosphorylation, and total levels of IκBα protein was determined via immunoblot. We observed a time dependent increase in the level of phosphorylated IκBα and time-dependent decrease in the level of total IκBα protein upon engagement of CD86 when both Phb1/2 and the CD86 cytoplasmic domain were intact; whereas, Phb1/2 depletion and the lack of the CD86 cytoplasmic domain prevented a CD86-dependent increase in the level of IκBα phosphorylation and decrease in the level of total IκBα protein (Figs. 7A, 7B). Furthermore, the CD86-induced increase in p65 phosphorylation was prevented when Phb1/2 was depleted (Fig. 7A), and when CD86 lacked the cytoplasmic domain (Fig. 7B), relative to either priming alone or isotype control Ab, respectively. However, we speculated that the signaling intermediate Phb1/2, which was essential for PLCγ2 and PKCα/βII activation, may also prevent CD86-induced p65 phosphorylation since previous findings showed that PKCα/βII-dependent p65 phosphorylation occurred distal to IκBα phosphorylation (18). Together, these findings indicated that both Phb1/2 and the CD86 cytoplasmic domain are necessary for the CD86-dependent activation of NF-κB via IκBα phosphorylation/degradation, followed by p65 phosphorylation.
FIGURE 7.

Phb1/2 and the CD86 cytoplasmic domain are each required for the CD86- dependent activation of NF-κB. A, CH12.LX B cells were Mock transfected, transfected with either scrambled negative control shRNA, or with Phb1, or Phb2-specific shRNA plasmids via nucleofection for 24 hours followed by priming with CD40L/IL-4 for 16 hours. The cells were then resuspended in serum-free conditions for at least 30 minutes. A CD86 Ab (anti-CD86) was added to cell cultures for 5, 15, 30, 60, and 90 minutes. Levels of phospho-IκBα (pIκBα), total IκBα, and phospho-p65 (p-p65) protein were measured relative to GAPDH or total p65 via immunoblot. B, WT, or CD86-cytoplasmic deficient (KKPΔ) FLAG-CD86 plasmids were transfected into CH12.LX B cells via nucleofection and primed with CD40L/IL-4 for 16 hours. An anti-FLAG Ab was added for 5, 15, 30, and 60 minutes relative to a species- and isotype- matched control Ab (iso ctrl Ab). Levels of pIκBα, total IκBα, and p-p65 protein were measured relative to GAPDH or total p65 via immunoblot. Representative gels are shown from three independent experiments. Densitometry was performed and measured pIκBα, total IκBα, and p-p65 band intensity relative to GAPDH, and p-p65 band density relative to total p65 and the data are presented as the mean Fold Change in pIκBα, total IκBα, or p-p65 from primed B cells where CD86 was engaged relative to priming alone (A) or iso ctrl Ab (B) and expressed as the mean Fold Change ± SEM from at least three independent experiments. Statistical differences are shown relative to priming alone (A) or iso ctrl Ab (B). *, p < 0.05.
Phb1/2 and the CD86 cytoplasmic domain are each required for the CD86-dependent nuclear localization of NF-κB (p65)
Although our findings suggested that both Phb1/2 and the CD86 cytoplasmic domain were required for the CD86-induced activation of NF-κB, what remained unclear was whether or not the intermediates were necessary for NF-κB (p65) nuclear entry promoted via CD86 engagement. Since previous findings from our laboratory showed that CD86 engagement promoted an increase in the level of p65 phosphorylation present in the nucleus (18), and findings in the present study indicated that both Phb1/2 and the CD86 cytoplasmic domain were required to promote CD86-dependent NF-κB activation, it was likely that both Phb1/2 and the CD86 cytoplasmic domain were required to promote CD86-dependent p65 nuclear localization. To address this, CH12.LX B cells were either Mock transfected or with scrambled negative control shRNA and Phb1/2-specific shRNA plasmids to deplete Phb1/2, and either WT-FLAG CD86 or CD86 cytoplasmic domain-deficient, (KKPΔ) plasmids followed by CD40L/IL-4-priming and either CD86 or FLAG-CD86 engagement for 90 minutes. Nuclear enriched protein lysates were prepared and levels of p65 phosphorylation and total p65 protein levels relative to nuclear membrane proteins Lamin A/C was measured via immunoblot. While phospho- and total p65 nuclear localization increased upon CD86 engagement when both Phb1/2 and the CD86 cytoplasmic were intact, the effect failed to occur upon CD86 engagement in the absence of the intermediates (Figs. 8A, 8B). Furthermore, successful Cytoplasmic and Nuclear-enriched protein lysate fractionation was demonstrated via the presence of α-tubulin, β-Actin, and GAPDH in Cytoplasmic-enriched protein lysates, and the presence of Lamin A/C in Nuclear-enriched protein lysates (Supplemental Fig. 4). Thus, our findings indicated that Phb1/2 and the CD86 cytoplasmic domain were necessary to promote the CD86-dependent nuclear localization of p65, and, therefore, extended the finding that both Phb1/2 and the CD86 cytoplasmic domain are each required for the CD86-dependent NF-κB activation.
FIGURE 8.

Phb1/2 and the CD86 cytoplasmic domain are each required for the CD86-dependent nuclear localization of NF-κB (p65). A, CH.12LX B cells were transfected via either under Mock conditions, or with scrambled negative control shRNA, or Phb1/2-specific shRNA plasmids via nucleofection for 24 hours followed by CD40L/IL-4-priming for an additional 16 hours. The cells were then resuspended in serum-free conditions for at least 30 minutes. A CD86 Ab (anti-CD86) was added to cell cultures for 90 minutes. Nuclear-enriched protein lysates were prepared and the level of p65 phosphorylation (p-p65) and total levels of p65 protein present in the nucleus were measured via immunoblot relative to Lamin A/C. B, WT, or CD86-cytoplasmic deficient (KKPΔ) FLAG-CD86 plasmids were transfected into CH12.LX B cells via nucleofection and primed with CD40L/IL-4 for 16 hours. An anti-FLAG Ab was added for 90 minutes relative to a species- and isotype-matched control Ab (iso ctrl Ab). The level of p65 phosphorylation and total levels of p65 present in the nucleus were determined via immunoblot. Densitometry was performed and measured p-p65 and total p65 band intensity present in the nucleus relative to Lamin A/C and the data are presented as the mean Fold Change in p-p65 or total p65 from primed B cells where CD86 was engaged relative to priming alone (A) or iso ctrl Ab (B) and expressed as the mean Fold Change ± SEM from three independent experiments. Statistical differences are shown relative to priming alone (A) or iso ctrl Ab (B). *, p < 0.05.
Discussion
Here, we show for the first time that expression of the tyrosine-containing adaptor/scaffolding proteins Phb1 and Phb2 increases in a CD40L/IL-4-primed B cell to associate with CD86, and that depletion of these proteins renders CD86 unable to mediate an enhancement in the level of Oct-2 and IgG1 produced. We also show for the first time that Phb1/2 and the CD86 cytoplasmic domain are required for the CD86-induced phosphorylation of IκBα, which we previously reported leads to NF-κB p50/p65 activation (18); whereas, only Phb1/2 was required for the CD86-induced phosphorylation of PLCγ2, which we previously reported leads to PKCα/βII activation and NF-κB (p65) phosphorylation (19). Thus, Phb1/2 association with CD86 and expression of an intact CD86 cytoplasmic domain are each necessary to mediate CD86 signaling function to enhance the level of the Oct-2/IgG1 response in a primed B cell through the differential activation of distal signaling intermediates.
Phb2, and its closely related homolog prohibitin-1 (Phb1), are evolutionarily-conserved, multi-functional proteins that are expressed in most cell types and typically reside in a variety of cellular compartments, including the inner mitochondrial and plasma membranes (30). Both Phbs have previously been found to be associated with the BCR (43), and were recently found to co-localize with CD3 of the TCR complex (44). Phb2 is also known as either Bap37 (B cell receptor-associated protein 37-kDa) or REA (nuclear repressor of estrogen receptor activity), while Phb1 is also referred to as Bap32 (B cell receptor-associated protein 32-kDa). Phbs are a family of proteins that possess various cellular and molecular functions (30, 45, 46) that have been found to function independently of each other (30, 45), except within the context of the mitochondrial membrane, where they seem to function together (40, 47–49), and when they associate with the BCR and CD3 of the TCR complex, where their function remains less well-understood. Taken together with findings that Phb1/2 residues can be phosphorylated, including tyrosines (50–53), we considered Phb1/2 as viable candidates to mediate CD86 signaling function.
Previous reports demonstrated that NF-κB (p65) phosphorylation is regulated via PKCα/βII, and that PKCα/βII activation is PLCγ2-dependent upon CD86 engagement on a B cell (18, 19). Data in the present study showed that the CD86-induced PLCγ2 phosphorylation failed to occur when Phb1/2 was reduced, but not when the CD86 cytoplasmic tail was deleted. On the other hand, CD86-induced IκBα phosphorylation failed to occur when either Phb1/2 was reduced or the CD86 cytoplasmic domain was deleted, indicating that both were required for NF-κB activation. The present evidence indicated that both Phb1/2 and the cytoplasmic domain of CD86 on a B cell, although not directly associated, appear to each be necessary to elicit CD86-induced back signaling to enhance the level of Oct-2 and IgG1 responses. Previous reports showed in B cells that engagement of cell surface receptors that lack cytoplasmic tyrosine residues, including the BCR (54, 55) and CD40 (56, 57) used adaptor protein complexes to signal intracellularly to activate NF-κB. Since the CD86 cytoplasmic domain is devoid of tyrosine residues, and since our evidence suggested that a novel association exists between CD86 and Phb1/2, which does expresses tyrosine residues, we initially thought is was possible that Phb1/2 alone might mediate CD86-dependent IκBα phosphorylation, independently of the CD86 cytoplasmic domain. However, data in the present study showed that both an intact CD86 cytoplasmic domain and Phb1/2 are required to activate IκBα upon CD86 engagement, suggesting that cross talk may exist between CD86-associated Phb1/2 and the cytoplasmic domain for IκBα activation. Data also showed that all alanine substitutions of the PKC phosphorylation sites within the CD86 cytoplasmic domain impaired CD86 function, with S303A causing the most significant reduction of CD86-induced Oct-2 and mature IgG1 mRNA, suggesting that phosphorylation of the CD86 cytoplasmic domain may serve as a potential docking site(s) for an unknown common signaling intermediate which may also interact with Phb1/2 to regulate phosphorylation of IκBα. Although the mechanistic link between Phb1/2 and the CD86 cytoplasmic domain is unclear at present, each appears to be required to promote CD86-dependent phosphorylation of IκBα and the activation of NF-κB p50/p65.
Previous reports showed that both Phb1 and Phb2 become tyrosine phosphorylated within their C-terminal domains upon engagement of cell surface receptors such as the TCR and CD28 (50), while Phb1 alone was shown to undergo tyrosine phosphorylation upon insulin receptor engagement (51–53, 58). Recently, it was found that Phb1 and Phb2 are linked to the MAP kinase signaling cascade upon CD3 engagement in T cells (44). Therefore, it was possible that Phb1/2 may serve as critical intermediates for the CD86-induced activation of PLCγ2. We also proposed that the CD86 cytoplasmic domain may not be involved with the CD86-induction of PLCγ2 due to the lack of tyrosine residues within this domain that would be necessary for PLCγ2 binding. Since prior reports revealed that PLCγ2 becomes recruited to tyrosine-containing proteins upon cell surface receptor engagement (25, 26), our findings indicate a potential role for Phb1/2 alone in mediating CD86-induced PLCγ2 phosphorylation, independent of the CD86 cytoplasmic domain, since Phb1/2 contain tyrosine residues, while the cytoplasmic domain does not. Also, it was reported that Phb1 associates with Syk upon BCR engagement (41). Since Syk has been reported to regulate the activation of PLCγ2 via the adaptor protein, B cell linker protein (BLNK), upon BCR engagement (59), it is possible that Phb1/2 serve as a scaffolding complex to link CD86 engagement to PLCγ2 activation. Nonetheless, the modest interdependent reduction in Phb1/2 protein levels in the current study was sufficient to prevent CD86 induction of IgG1 and proximal signaling intermediates, suggesting that Phb1/2 are each required to mediate CD86-induced signaling in B cells.
In the present study, we used RNA-interference to silence Phb1/2 because both Phb1- and Phb2-deficient mice are embryonic lethal. Previous reports revealed that Phb1 and Phb2 protein expression levels were interdependent, with depletion of one causing the loss of the other (60, 61), which is consistent with findings in the current study. Phb1/2 mRNA levels, however, were reported to be comparable in the presence of non gene-specific Phb1/2 siRNA (61), which is in contrast with findings of the current study. The discrepancy could be due to a mixed population of Phb1/2 shRNA plasmids, as were used in the present study that targeted multiple regions of Phb1/2 transcripts, as Phb1/2 are highly homologous (40), thus promoting a higher likelihood of suppressing mRNA translation.
To our knowledge, the present findings using RNA-interference are the first to identify a role for Phb1 and Phb2 in mediating the CD86-induced enhancement of the levels of Oct-2 and IgG1 produced by a CD40L/IL-4-primed B cell. Phb1/2 shRNA-mediated depletion in the current study led to modest reductions in Phb1/2 protein levels, suggesting that only a modest amount of Phb1/2 protein in the cell is required to mediate signaling events induced by CD86; whereas, a greater proportion is necessary to regulate mitochondrial function. This is supported by previous reports that showed Phb1/2 depletion resulted in the loss of mitochondrial membrane potential (50, 62), even though other reports have shown Phb1/2 depletion did not affect mitochondrial membrane potential (61, 63). Also, using gene silencing, Phb1 was shown to have functional roles in cell migration (64), regulation of angiogenic activity and cellular senescence (62), and cellular metabolic activity (65) upon engagement of various cell surface receptors. Likewise, gene-silencing of Phb2 indicated a functional role for Phb2 in muscle cell differentiation (66). Also, Phb1/2 have been reported to associate with the BCR (43) and, recently, CD3 of the TCR complex (44), although the exact functional roles for the associations remain unclear. Furthermore, a direct protein/protein interaction has been shown to occur between Phb1 and Syk upon BCR engagement (41), while another report showed a decrease in MAP kinase signaling upon CD3 engagement when Phb1/2 function was blocked, indicating that Phb1/2 may serve functional roles in BCR/TCR signaling. Thus, the present findings using RNA-interference provide further support that Phb1/2 participate in mediating cell surface receptor-induced regulation of cellular functions.
We originally hypothesized that the cytoplasmic domain of CD86 associated directly with Phb1/2 to mediate CD86 signaling function. However, our data showed that Phb1/2 associated with CD86 even in the absence of the CD86 cytoplasmic domain, suggesting a transmembrane-specific interaction. The ability of CD86 to bind to transmembrane proteins is supported by a recent study in dendritic cells where it was shown that CD86 associated with an E3 ubiquitin ligase in the presence or absence of the CD86 cytoplasmic domain (67). It is also supported by previous reports showing that Phb1/2 exist as transmembrane proteins that function to stabilize proteins in the electron transport chain and to mediate functional changes induced by cell surface receptors (40, 47–49, 62, 64–66). Taken together, these findings support the proposal that CD86 associates with Phb1/2 via transmembrane-specific interactions in B cells.
Based on the findings of the current study and our previous reports, we propose the following CD86 signaling model (Fig. 9): Upon CD86 engagement, Phb1/2 and the CD86 cytoplasmic domain promote the phosphorylation of IκBα, allowing for the activation of NF-κB p50/p65. Concurrently, Phb1/2 alone promote CD86-induced PLCγ2 phosphorylation, allowing for subsequent PKCα/βII activation and phosphorylation of p65. NF-κB p50/p65 then initiates Oct-2 transcription, which ultimately regulates 3′IgH-enhancer activity to elevate the rate of IgG1 transcription and the level of IgG1 produced by a B cell. Findings from the current study may help us to understand the mechanism by which CD86 signals directly to a B cell to increase not only IgG1, but also IgE production, as shown previously (13, 16). In addition, understanding this novel signaling mechanism may help direct the rational design of CD86-targeted therapeutic strategies to increase the level of protective antibody in immuno-compromised patients. Conversely, CD86-targeted therapeutic strategies might also be developed to decrease the level of harmful antibodies produced in certain cases of autoimmunity or allergic asthma.
FIGURE 9.
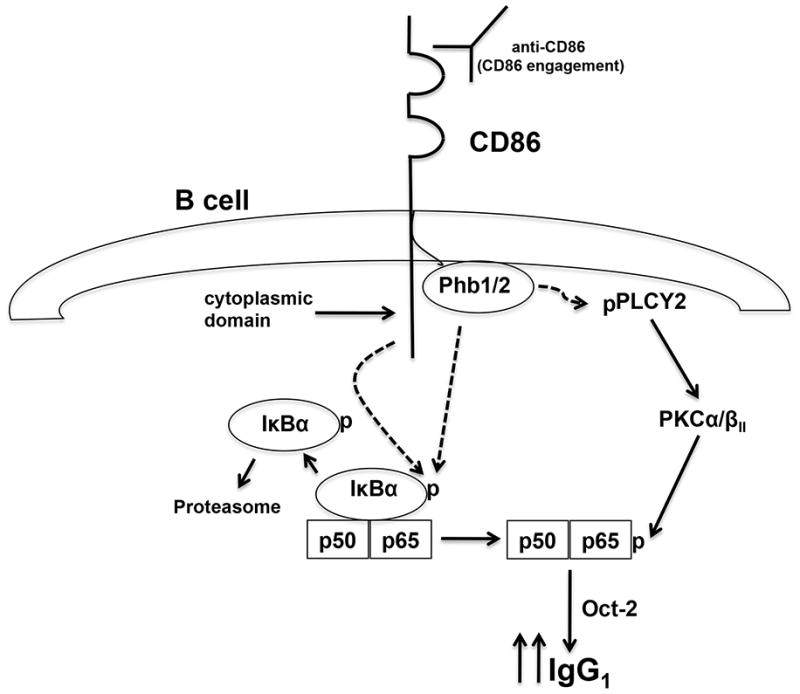
CD86 Signaling Model. (Dotted arrows indicate proposed novel signaling events identified in the current study). CD86 engagement promotes the phosphorylation of IκBα via mechanisms regulated by Phb1/2 and the CD86 cytoplasmic domain allowing for the activation of NF-κB p50/p65. Concurrently, Phb1/2 alone promotes the CD86-induced PLCγ2 phosphorylation, allowing for subsequent PKCα/βII activation and phosphorylation of p65. NF- κB p50/p65 is now poised to initiate Oct-2 transcription, which ultimately regulates 3′IgH- enhancer activity to increase the rate of IgG1 transcription and the level of IgG1 produced by a B cell.
Supplementary Material
Acknowledgments
We thank Dr. Alan Smith and Matthew Gormley for critical experimental support, and Todd Shawler for assistance with Flow Cytometry. We would also like to thank Caroline Padro and Sara McMaken for critical evaluation of our manuscript.
Footnotes
This work was supported by National Institutes of Health R01-AI037326 and R56-AI073663.
Abbreviations used in this paper were: PLCγ2, phospholipase Cγ2; Phb1, prohibitin-1; Phb2, prohibitin-2; Terb, terbutaline; Tg, transgenic; qRT-PCR, quantitative real time PCR; Syk, spleen tyrosine kinase
References
- 1.Freeman GJ, Gribben JG, Boussiotis VA, Ng JW, Restivo VAJ, Lombard LA, Gray GS, Nadler LM. Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science. 1993;262:909–911. doi: 10.1126/science.7694363. [DOI] [PubMed] [Google Scholar]
- 2.Azuma M, Ito D, Okumara K, Phillips JH, Lanier LL, Somoza C. B70 antigen is a second ligand for CTLA-4 and CD28. Nature. 1993;366:76–79. doi: 10.1038/366076a0. [DOI] [PubMed] [Google Scholar]
- 3.Freeman GJ, Borriello F, Hodes RJ, Reiser H, Gribben JG, Ng JW, Kim J, Goldberg JM, Hathcock K, Laszlo G, Lombard LA, Wang S, Gray GS, Nadler LM, Sharpe AH. Murine B7-2, an alternative CTLA4 counter-receptor that costimulates T cell proliferation and interleukin 2 production. J Exp Med. 1993;178:2185–2192. doi: 10.1084/jem.178.6.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- 5.Freeman GJ, Borriello F, Hodes RJ, Reiser H, Hathcock KS, Laszlo G, McKnight AJ, Kim J, Du L, Lombard DB, Gray GS, Nadler LM, Sharpe AH. Uncovering of functional alternative CTLA-4 counter-receptor in B7-deficient mice. Science. 1993;262:907–909. doi: 10.1126/science.7694362. [DOI] [PubMed] [Google Scholar]
- 6.Freeman GJ, Boussiotis VA, Anumanthan A, Bernstein GM, Ke XY, Rennert PD, Gray GS, Gribben JG, Nadler LM. B7-1 and B7-2 do not deliver identical costimulatory signals, since B7-2 but not B7-1 preferentially costimulates the initial production of IL-4. Immunity. 1995;2:523–532. doi: 10.1016/1074-7613(95)90032-2. [DOI] [PubMed] [Google Scholar]
- 7.Borriello F, Sethna MP, Boyd SD, Schweitzer AN, Tivol EA, Jacoby D, Strom TB, Simpson EM, Freeman GJ, Sharpe AH. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity. 1997;6:303–313. doi: 10.1016/s1074-7613(00)80333-7. [DOI] [PubMed] [Google Scholar]
- 8.Salek-Ardakani S, Choi YS, Rafii-El-Idrissi Benhnia M, Flynn R, Arens R, Shoenberger S, Crotty S, Croft M, Salek-Ardakani S. B cell-specific expression of B7-2 is required for follicular Th cell function in response to vaccinia virus. J Immunol. 2011;186:5294–5303. doi: 10.4049/jimmunol.1100406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy M, Aruffo A, Ledbetter J, Linsley P, Kehry M, Noelle R. Studies on the interdependence of gp39 and B7 expression and function during antigen-specific immune responses. Eur J Immunol. 1995;25:596–603. doi: 10.1002/eji.1830250243. [DOI] [PubMed] [Google Scholar]
- 10.Stack RM, Lenschow DJ, Gray GS, Bluestone JA, Fitch FW. IL-4 treatment of small splenic B cells induces costimulatory molecules B7-1 and B7-2. J Immunol. 1994;152:5723–5733. [PubMed] [Google Scholar]
- 11.Hathcock KS, Laszlo G, Dickler HB, Bradshaw J, Linsley P, Hodes RJ. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science. 1993;262:905–907. doi: 10.1126/science.7694361. [DOI] [PubMed] [Google Scholar]
- 12.Lenschow DJ, Su GHT, Zuckerman LA, Nabavi N, Jellis CL, Gray GS, Miller J, Bluestone JA. Expression and functional significance of an additional ligand for CTLA-4. Proc Natl Acad Sci USA. 1993;90:11054–11058. doi: 10.1073/pnas.90.23.11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kasprowicz DJ, Kohm AP, Berton MT, Chruscinski AJ, Sharpe AH, Sanders VM. Stimulation of the B cell receptor, CD86 (B7-2), and the beta 2-adrenergic receptor intrinsically modulates the level of IgG1 and IgE produced per B cell. J Immunol. 2000;165:680–690. doi: 10.4049/jimmunol.165.2.680. [DOI] [PubMed] [Google Scholar]
- 14.Kohm AP, Mozaffarian A, Sanders VM. B cell receptor- and beta 2-adrenergic receptor-induced regulation of B7-2 (CD86) expression in B cells. J Immunol. 2002;168:6314–6322. doi: 10.4049/jimmunol.168.12.6314. [DOI] [PubMed] [Google Scholar]
- 15.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Ann Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 16.Jeannin P, Delneste Y, Lecoanet-Henchoz S, Gauchat J-F, Ellis J, Bonnefoy J-Y. CD86 (B7-2) on human B cells. A functional role in proliferation and selective differentiation into IgE- and IgG4-producing cells. J Biol Chem. 1997;272:15613–15619. doi: 10.1074/jbc.272.25.15613. [DOI] [PubMed] [Google Scholar]
- 17.Podojil JR, Sanders VM. Selective regulation of mature IgG1 transcription by CD86 and beta 2-adrenergic receptor stimulation. J Immunol. 2003;170:5143–5151. doi: 10.4049/jimmunol.170.10.5143. [DOI] [PubMed] [Google Scholar]
- 18.Podojil JR, Kin NW, Sanders VM. CD86 and beta2-adrenergic receptor signaling pathways, respectively, increase Oct-2 and OCA-B Expression and binding to the 3′-IgH enhancer in B cells. J Biol Chem. 2004;279:23394–23404. doi: 10.1074/jbc.M313096200. [DOI] [PubMed] [Google Scholar]
- 19.Kin NW, Sanders VM. CD86 stimulation on a B cell activates the phosphatidylinositol 3-kinase/Akt and phospholipase C gamma 2/protein kinase C alpha beta signaling pathways. J Immunol. 2006;176:6727–6735. doi: 10.4049/jimmunol.176.11.6727. [DOI] [PubMed] [Google Scholar]
- 20.Kin N, Sanders VM. CD86 regulates IgG1 production via a CD19-dependent mechanism. J Immunol. 2007;179:1516–1523. doi: 10.4049/jimmunol.179.3.1516. [DOI] [PubMed] [Google Scholar]
- 21.Suvas S, Singh V, Sahdev S, Vohra H, Agrewala JA. Distinct role of CD80 and CD86 in the regulation of the activation of B cell and B cell lymphoma. J Biol Chem. 2002;277:7766–7775. doi: 10.1074/jbc.M105902200. [DOI] [PubMed] [Google Scholar]
- 22.Rau FC, Dieter J, Luo Z, Priest SO, Baumgarth N. B7-1/2 (CD80/CD86) direct signaling to B cells enhances IgG secretion. J Immunol. 2009;183:7661–7671. doi: 10.4049/jimmunol.0803783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fine DP, Kirk JL, Schiffman G, Schweinle JE, Guckian JC. Analysis of humoral and phagocytic defenses against Streptococcus pneumoniae serotypes 1 and 3. J Lab Clin Med. 1988;112:487–497. [PubMed] [Google Scholar]
- 24.Taranger J, Trollfors B, Lagergard T, Sundh V, Bryla DA, Schneerson R, Robbins JB. Correlation between pertussis toxin IgG antibodies in postvaccination sera and subsequent protection against pertussis. J Infect Dis. 2000;181:1010–1013. doi: 10.1086/315318. [DOI] [PubMed] [Google Scholar]
- 25.Gauld SB, Cambier JC. Src-family kinases in B-cell development and signaling. Oncogene. 2004;23:8001–8006. doi: 10.1038/sj.onc.1208075. [DOI] [PubMed] [Google Scholar]
- 26.Kurosaki T, Hikida M. Tyrosine kinases and their substrates in B lymphocytes. Immunol Rev. 2009;228:132–148. doi: 10.1111/j.1600-065X.2008.00748.x. [DOI] [PubMed] [Google Scholar]
- 27.Haughton G, Arnold LW, Bishop GA, Mercolino TJ. The CH series of murine B cell lymphomas: neoplastic analogues of Ly-1+ normal B cells. Immunol Rev. 1986;93:35–51. doi: 10.1111/j.1600-065x.1986.tb01501.x. [DOI] [PubMed] [Google Scholar]
- 28.Fournier S, Rathmell JC, Goodnow CC, Allison JP. T cell-mediated elimination of B7.2 transgenic B cells. Immunity. 1997;6:327–339. doi: 10.1016/s1074-7613(00)80335-0. [DOI] [PubMed] [Google Scholar]
- 29.Williams JA, Lumsden JM, Yu X, Feigenbaum L, Zhang J, Steinberg SM, Hodes RJ. Regulation of thymic NKT cell development by the B7-CD28 costimulatory pathway. J Immunol. 2008;181:907–917. doi: 10.4049/jimmunol.181.2.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mishra S, Murphy LC, Murphy LJ. The Prohibitins: emerging roles in diverse functions. J Cell Mol Med. 2006;10:353–363. doi: 10.1111/j.1582-4934.2006.tb00404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- 32.Cheng G, Cleary AM, Ye ZS, Hong DI, Lederman S, Baltimore D. Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science. 1995;267:1494–1498. doi: 10.1126/science.7533327. [DOI] [PubMed] [Google Scholar]
- 33.Ishida T, Tojo T, Aoki T, Kobayashi N, Ohishi T, Watanabe T, Yamamoto T, Inoue J. TRAF5, a novel tumor necrosis factor receptor-associated factor family protein, mediates CD40 signaling. Proc Natl Acad Sci USA. 1996;93:9437–9442. doi: 10.1073/pnas.93.18.9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishida T, Mizushima S, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, Yamamoto T, Inoue J. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem. 1996;271:28745–28748. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]
- 35.Campbell KS, Cambier JC. B lymphocyte antigen receptors (mIg) are non-covalently associated with a disulfide linked, inducibly phosphorylated glycoprotein complex. EMBO J. 1990;9:441–448. doi: 10.1002/j.1460-2075.1990.tb08129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campbell KS, Hager EJ, Friedrich RJ, Cambier JC. IgM antigen receptor complex contains phosphoprotein products of B29 and mb-1 genes. Proc Natl Acad Sci USA. 1991;88:3982–3986. doi: 10.1073/pnas.88.9.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell KS, Hager EJ, Cambier JC. Alpha-chains of IgM and IgD antigen receptor complexes are differentially N-glycosylated MB-1-related molecules. J Immunol. 1991;147:1575–1580. [PubMed] [Google Scholar]
- 38.Clark MR, Campbell KS, Kazlauskas A, Johnson SA, Hertz M, Potter TA, Pleiman C, Cambier JC. The B cell antigen receptor complex: association of Ig-alpha and Ig-beta with distinct cytoplasmic effectors. Science. 1992;258:123–126. doi: 10.1126/science.1439759. [DOI] [PubMed] [Google Scholar]
- 39.Campbell KS. Signal transduction from the B cell antigen-receptor. Current Opinion in Immunology. 1999;11:256–264. doi: 10.1016/s0952-7915(99)80042-9. [DOI] [PubMed] [Google Scholar]
- 40.Artal-Sanz M, Tavernarakis N. Prohibitin and mitochondrial biology. Trends Endocrinol Metab. 2009;20:394–401. doi: 10.1016/j.tem.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 41.Paris LL, Hu J, Galan J, Ong SS, Martin VA, Ma H, Tao WA, Harrison ML, Geahlen RL. Regulation of Syk by phosphorylation on serine in the linker insert. J Biol Chem. 2010;285:39844–39854. doi: 10.1074/jbc.M110.164509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 43.Terashima M, Kim KM, Adachi T, Nielsen PJ, Reth M, Kohler G, Lamers MC. The IgM antigen receptor of B lymphocytes is associated with prohibitin and a prohibitin-related protein. EMBO J. 1994;13:3782–3792. doi: 10.1002/j.1460-2075.1994.tb06689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yurugi H, Tanida S, Ishida A, Akita K, Toda M, Inoue M, Nakada H. Expression of prohibitins on the surface of activated T cells. Biochem Biophys Res Commun. 2012;420:275–280. doi: 10.1016/j.bbrc.2012.02.149. [DOI] [PubMed] [Google Scholar]
- 45.Mishra S, Murphy LC, Nyomba BL, Murphy LJ. Prohibitin: a potential target for new therapeutics. Trends Mol Med. 2005;11:192–197. doi: 10.1016/j.molmed.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 46.Rajalingam K, Rudel T. Ras-Raf signaling needs prohibitin. Cell Cycle. 2005;4:1503–1505. doi: 10.4161/cc.4.11.2142. [DOI] [PubMed] [Google Scholar]
- 47.Nijtmans LG, Artal SM, Grivell LA, Coates PJ. The mitochondrial PHB complex: roles in mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell Mol Life Sci. 2002;59:143–155. doi: 10.1007/s00018-002-8411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osman C, Merkwirth C, Langer T. Prohibitins and the functional compartmentalization of mitochondrial membranes. J Cell Sci. 2009;122:3823–3830. doi: 10.1242/jcs.037655. [DOI] [PubMed] [Google Scholar]
- 49.Merkwirth C, Langer T. Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 50.Ross JA, Nagy ZS, Kirken RA. The PHB1/2 phosphocomplex is required for mitochondrial homeostasis and survival of human T cells. J Biol Chem. 2008;283:4699–4713. doi: 10.1074/jbc.M708232200. [DOI] [PubMed] [Google Scholar]
- 51.Ande SR, Moulik S, Mishra S. Interaction between O-GlcNAc modification and tyrosine phosphorylation of prohibitin: implication for a novel binary switch. PLoS One. 2009;4:e4586. doi: 10.1371/journal.pone.0004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ande SR, Gu Y, Nyomba BL, Mishra S. Insulin induced phosphorylation of prohibitin at tyrosine 114 recruits Shp1. Biochim Biophys Acta. 2009;1793:1372–1378. doi: 10.1016/j.bbamcr.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 53.Ande SR, Mishra S. Prohibitin interacts with phosphatidylinositol 3,4,5-triphosphate (PIP3) and modulates insulin signaling. Biochem Biophys Res Commun. 2009;390:1023–1028. doi: 10.1016/j.bbrc.2009.10.101. [DOI] [PubMed] [Google Scholar]
- 54.Flaswinkel H, Reth M. Dual role of the tyrosine activation motif of the Ig-alpha protein during signal transduction via the B cell antigen receptor. EMBO J. 1994;13:83–89. doi: 10.1002/j.1460-2075.1994.tb06237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moreno-Garcia ME, Sommer KM, Bandaranayake AD, Rawlings DJ. Proximal signals controlling B-cell antigen receptor (BCR) mediated NF-kappaB activation. Adv Exp Med Biol. 2006;584:89–106. doi: 10.1007/0-387-34132-3_7. [DOI] [PubMed] [Google Scholar]
- 56.Tsukamoto N, Kobayashi N, Azuma S, Yamamoto T, Inoue J. Two differently regulated nuclear factor kappaB activation pathways triggered by the cytoplasmic tail of CD40. Proc Natl Acad Sci USA. 1999;96:1234–1239. doi: 10.1073/pnas.96.4.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Propst SM, Estell K, Schwiebert LM. CD40-mediated activation of NF-kappa B in airway epithelial cells. J Biol Chem. 2002;277:37054–37063. doi: 10.1074/jbc.M205778200. [DOI] [PubMed] [Google Scholar]
- 58.Mishra S, Ande SR, Nyomba BL. The role of prohibitin in cell signaling. FEBS J. 2010;277:3937–3946. doi: 10.1111/j.1742-4658.2010.07809.x. [DOI] [PubMed] [Google Scholar]
- 59.Ishiai M, Kurosaki M, Pappu R, Okawa K, Ronko I, Fu C, Shibata M, Iwamatsu A, Chan AC, Kurosaki T. BLNK required for coupling Syk to PLC gamma 2 and Rac1-JNK in B cells. Immunity. 1999;10:117–125. doi: 10.1016/s1074-7613(00)80012-6. [DOI] [PubMed] [Google Scholar]
- 60.Kasashima K, Ohta E, Kagawa Y, Endo H. Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J Biol Chem. 2006;281:36401–36410. doi: 10.1074/jbc.M605260200. [DOI] [PubMed] [Google Scholar]
- 61.Sievers C, Billig G, Gottschalk K, Rudel T. Prohibitins are required for cancer cell proliferation and adhesion. PLoS One. 2010;5:e12735. doi: 10.1371/journal.pone.0012735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schleicher M, Shepherd BR, Suarez Y, Fernandez-Hernando C, Yu J, Pan Y, Acevedo LM, Shadel GS, Sessa WC. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J Cell Biol. 2008;180:101–112. doi: 10.1083/jcb.200706072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, von Kleist-Retzow JC, Waisman A, Westermann B, Langer T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rajalingam K, Wunder C, Brinkmann V, Churin Y, Hekman M, Sievers C, Rapp UR, Rudel T. Prohibitin is required for Ras-induced Raf-MEK-ERK activation and epithelial cell migration. Nat Cell Biol. 2005;7:837–843. doi: 10.1038/ncb1283. [DOI] [PubMed] [Google Scholar]
- 65.Vessal M, Mishra S, Moulik S, Murphy LJ. Prohibitin attenuates insulin-stimulated glucose and fatty acid oxidation in adipose tissue by inhibition of pyruvate carboxylase. FEBS J. 2006;273:568–576. doi: 10.1111/j.1742-4658.2005.05090.x. [DOI] [PubMed] [Google Scholar]
- 66.Sun L, Liu L, Yang XJ, Wu Z. Akt binds prohibitin 2 and relieves its repression of MyoD and muscle differentiation. J Cell Sci. 2004;117:3021–3029. doi: 10.1242/jcs.01142. [DOI] [PubMed] [Google Scholar]
- 67.Corcoran K, Jabbour M, Bhagwandin C, Deymier MJ, Theisen DL, Lybarger L. Ubiquitin-mediated regulation of CD86 protein expression by the ubiquitin ligase membrane-associated RING-CH-1 (MARCH1) J Biol Chem. 2011;286:37168–37180. doi: 10.1074/jbc.M110.204040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.