

Abstract
Objectives
Depot formulation as a carrier for cytotoxic chemotherapeutic drugs is not well studied. The objective of this study is to test the feasibility of using a subcutaneous depot formulation to administer a cytotoxic anti-cancer drug for systemic therapy.
Methods
A fatty-acid amide prodrug of the nucleoside analogue gemcitabine (4-(N)-stearoyl gemcitabine (GemC18)) was incorporated into poly(lactic-co-glycolic acid) (PLGA) nanoparticles or microspheres. A GemC18 solution was used as a control. The anti-tumour activity was evaluated after subcutaneous injection of the different formulations in C57BL/6 mice with pre-established model tumours. The clearance of GemC18 from the injection site was determined by measuring the percentage of GemC18 remaining at the injection site at different times after the injection.
Key findings
The depot formulation based on the GemC18-loaded PLGA nanoparticles showed the strongest anti-tumour effect, likely due to the proper ‘release’ of GemC18 from the injection site.
Conclusions
It is feasible to dose cytotoxic anti-cancer drugs as a nanoparticle-based depot formulation, especially when combined with an advanced prodrug strategy.
Keywords: depot formulation, microspheres, nanoparticles, prodrugs, tumour growth inhibition
Introduction
Depot formulations can be in the form of nanoparticles,[1] microspheres,[2] implants,[3] semisolid hydrogels[4] or solid boluses and are often injected subcutaneously or intramuscularly to achieve extended release of the incorporated drug. The advantages of using a depot formulation include improved patient compliance due to reduction in dosing frequency, as well as a more consistent serum concentration of the drug of interest.[5,6] Depot formulations are often used to treat chronic diseases, such as psychotic disorders[1,3] and diabetes,[4] and to incorporate antigens to enable their release in a controlled manner and induce sustained immune responses.[7] Depot formulations have been used successfully in cancer hormone therapy. For example, Lupron Depot (7.5 mg/vial for monthly depot injection), a microsphere formulation of leuprorelin (leuprolide) acetate, was approved by the US Food and Drug Administration (FDA) in 1989 to treat advanced prostate cancer. In cancer chemotherapy, depot formulation is often designed to bypass barriers that prevent chemotherapeutic drugs from reaching or entering tumour cells. For instance, several polymeric devices have been developed for local delivery of chemotherapeutic drugs to bypass the blood–brain barrier when treating malignant glioma.[8–10] As an example, PLGA microparticles have been used to locally deliver imatinib mesilate, a small molecule tyrosine kinase inhibitor, to inhibit the growth of intracranial xenograft glioma.[10] However, the application of depot formulation is less well studied when a systemic effect is desired in cancer chemotherapy.
Gemcitabine (2′,2′-difluoro-2′-deoxycytidine) is a deoxycytidine nucleoside analogue that has been approved for the treatment of pancreatic, non-small cell lung, breast and ovarian cancers.[11] However, gemcitabine suffers from several drawbacks, such as rapid deamination to the inactive 2′,2′-difluorodeoxyuridine by cytidine deaminase after intravenous injection,[12] which prompted efforts to amidify or esterify gemcitabine (with fatty acids) to improve its stability by preventing the deamination.[13–15] In this study, we tested the feasibility of using a subcutaneous depot formulation of the stearic acid amide derivative of gemcitabine, 4-(N)-stearoyl gemcitabine (GemC18), for cancer chemotherapy. GemC18 is not as sensitive to deamination as gemcitabine. In addition, GemC18, an amide derivative of gemcitabine, will likely induce less local toxicity than gemcitabine. Poly(lactic-co-glycolic acid) (PLGA) particulates were used as the GemC18 carrier because PLGA is an FDA-approved biodegradable and biocompatible polymer, and the drug release rate from particulates prepared with PLGA can be easily modified by altering the size of the formed particulates.[16] To this end, GemC18-loaded PLGA nanoparticles and microspheres with different drug release profiles were prepared, and their anti-tumour activity was evaluated when injected as a subcutaneous depot into tumour-bearing mice.
Materials and Methods
Materials
PLGA (752H), lauric acid, trehalose, polysorbate 80 (Tween 80), Pluronic F68, polyvinylalcohol 4–88 (PVA, molecular weight 31 K), sodium carboxymethyl cellulose (CMC-Na), ethanol, dichloromethane, HPLC-grade tetrahydrofuran (THF) and methanol were from Sigma-Aldrich (St Louis, MO, USA). GemC18 was synthesized according to previously published methods.[1] Mouse TC-1 lung cancer cell line was from American Type Culture Collection (Manassas, VA, USA). TC-1 cells were grown in RPMI1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, 100 U/ml of penicillin and 100 μg/ml of streptomycin, all from Invitrogen.
Preparation and characterization of GemC18-loaded PLGA nanoparticles and microspheres
A solvent displacement method was used to prepare GemC18-loaded PLGA nanoparticles. Briefly, 0.75 ml of THF containing 7.55 mg of PLGA 752H, 0.45 mg of GemC18 and 1.21 mg of lauric acid was added drop-wise into 3.75 ml of Pluronic F68 aqueous solution (1% w/v). The mixture was stirred overnight and centrifuged (18 000g, 15 min, 4°C) to collect nanoparticles (in the pellet), which were re-suspended in 5% trehalose (w/v) and lyophilized. GemC18-loaded PLGA microspheres were prepared using a homogenization method. Briefly, 0.5 ml of dichloromethane containing 20 mg of PLGA 752H, 0.3 mg of GemC18 and 3 mg of lauric acid was added into 5 ml 1% PVA (w/v), followed by homogenization at 15,000 rpm for 3 min. The resultant emulsions were stirred at 250 rpm for 3–4 h to form microspheres, which were centrifuged (1500g, 10 min, 4°C) and then lyophilized. The third GemC18 formulation was GemC18 in Tween 80/ethanol solution, which was prepared following a previously reported method.[14] Briefly, GemC18 was dissolved in Tween 80 and then diluted 1 : 4 with ethanol (v/v) to give a final GemC18 concentration of 8.33 mg/ml, which was further diluted before injection.
Characterization of GemC18-loaded PLGA particles
The GemC18-loaded PLGA nanoparticles and microspheres were first characterized using a Zeiss Supra 40 VP Scanning Electron Microscope (SEM; Carl Zeiss, Munich, Germany) to examine their morphology. Their particle size, polydispersity (PDI) and zeta potential were then measured using a Malvern Zetasizer Nano ZS (Malvern Instruments Ltd, Westborough, MA, USA). To determine the concentration of GemC18, the lyophilized GemC18-loaded nanoparticles or microspheres were dissolved in THF and subjected to HPLC analysis using an Agilent 1260 Infinity Quaternary Liquid Chromatographic System with an UV detector operated at 248 nm and an Agilent ZORBAX Eclipse Plus C18 column (5 μm, 4.6 mm × 150 mm; Agilent Technologies, Inc., Santa Clara, CA, USA). The mobile phase was methanol. The flow rate was 1 ml/min. The drug loading and the entrapment efficiency were calculated using the following equation:
| (1) |
| (2) |
In-vitro release
The in-vitro release of GemC18 from PLGA particles was determined using a centrifugation method.[17] Briefly, GemC18-loaded PLGA nanoparticles or microspheres (0.1 mg GemC18) were dispersed into 7 ml of phosphate-buffered saline (PBS, 10 mm, pH 7.4, 0.1% Tween 80 (v/v)) and incubated in a shaker-incubator (37°C, 50 rpm). At predetermined time points, 0.5 ml of the suspension was withdrawn and centrifuged to collect the supernatant fraction, which was then subjected to HPLC to quantify GemC18.
In-vivo anti-tumour activity
Female C57BL/6 mice (6–8 weeks old; Charles River Laboratories, Wilmington, MA, USA) with subcutaneously implanted mouse lung cancer cells (TC-1) (5 × 105 cells/mouse, in the right flank) were used to test the in-vivo anti-tumour activity of the GemC18 formulations. National Institutes of Health guidelines for animal care and use were followed, and the animal protocol was approved by the Institutional Animal Care and Use Committee at the University of Texas at Austin (AUP-2009-00123, 11 February 2010). When tumours reached 4–5 mm (6 days after injection), mice were subcutaneously injected in the upper dorsal side (back neck area) with GemC18 formulations dispersed in 0.5% of CMC-Na (w/v) and 0.1% of Tween 80 (w/v) (0.5 mg of GemC18 in 0.5 ml), and the injection was repeated once 6 days later. Tumour sizes were measured with a caliper in two perpendicular diameters every day and reported as tumour volume (V = ½ ([a × b2); a = longest diameter, b = shortest diameter). At the end of treatment, the hair at and around the injection site was carefully trimmed, and the skin condition in the injection site was examined and imaged using a digital camera.
In-vivo ‘release’ of GemC18 from the injection site
The clearance of GemC18 from the injection site was estimated by determining the percentage of GemC18 remaining in the injection site at different time points after the injection. GemC18 solution, GemC18-PLGA nanoparticles or GemC18-PLGA microspheres (50 μg of GemC18 in 50 μl) were subcutaneously injected into the footpad of one of the hind legs of the mice. Footpad injection was used because of the ease with which the injection site can be identified. Mice were sacrificed 24 or 48 h after the injection to harvest the footpads. The amount of GemC18 remaining in the injection site was determined by HPLC after the extraction of the GemC18.[18] Briefly, the footpad was homogenized in dichloromethane using a bead beater (Biospec Products, Inc., Bartlesville, OK, USA) at 4800 rpm for 80 s. The organic layer was collected after centrifugation (15 500g, 10 min) and dried under vacuum. The residue was re-dissolved in 100 μl of methanol and centrifuged (15 500g, 10 min) to collect the supernatant fraction. The concentration of GemC18 in the supernatant was determined using the same method as described above, while the detection wavelength was 308 nm.
Statistical analyses
The in-vivo release data were analysed using a non-parametric Kruskal–Wallis test, followed by Dunn's post-hoc test. Otherwise, statistical analyses were completed by performing analysis of variance followed by Fisher's protected least significant difference procedure. P ≤ 0.05 was considered significant.
Results
Characterization of GemC18 formulations
Both nanoparticles and microspheres were spherical, with a narrow size distribution (Figure 1). The size of nanoparticles was around 200 nm (Figure 1a), which is in agreement with the size determined using the particle sizer (206.1 ± 0.6 nm) (Table 1). The microspheres displayed a smooth surface, with an average diameter of around 3 μm (Figure 1b), which is also in agreement with results from the particle sizer (3587 ± 839 nm) (Table 1). Both nanoparticle and microsphere formulations showed negative zeta potentials when dispersed in PBS (10 mm, pH 7.4) (Table 1). The percentage of GemC18 loaded in the PLGA nanoparticles and microspheres was determined to be 2.4 ± 0.6% and 1.5 ± 0.01%, respectively, with a respective entrapment efficiency of 72.6 ± 12.0% and 73.2 ± 12.0% (Table 1).
Figure 1.

Scanning electron microscopic pictures of GemC18-loaded PLGA nanoparticles (a) and microspheres (b).
Table 1.
Characterization of GemC18-loaded PLGA nanoparticles and microspheres
| Formulation | Particle size (nm) | Polydispersity index | Zeta potential (mV) | Entrapment efficiency (%) | Drug loading percentage (%) |
|---|---|---|---|---|---|
| Nanoparticles | 206.1 ± 0.6 | 0.180 ± 0.017 | −7.91 ± 0.74 | 72.6 ± 12.0 | 2.4 ± 0.6 |
| Microspheres | 3587 ± 839 | 0.316 ± 0.026 | −2.76 ± 0.63 | 73.2 ± 12.0 | 1.5 ± 0.01 |
Data shown are mean ± SD, n = 3.
In-vitro release of GemC18 from PLGA nanoparticles and microspheres
The release of the GemC18 from the nanoparticles was significantly faster than from the microspheres. Within 8 h, 85.4 ± 2.1% of GemC18 was released from the nanoparticles, and the release reached maximum (96.0 ± 2.1%) after 48 h (Figure 2); only 33.2 ± 1.1% of GemC18 was released from the microspheres within 8 h, increasing to approximately 65% after 96 h.
Figure 2.
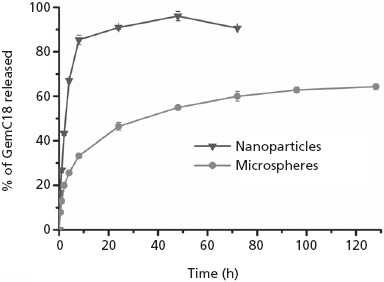
In-vitro release profiles of GemC18 from GemC18-loaded PLGA nanoparticles and microspheres. Data shown are mean ± SD, n = 3.
In-vivo anti-tumour activity
Subcutaneous injection to a site distal to the tumour site, instead of intratumoral injection, was chosen so that the GemC18 or its parent drug, gemcitabine, has to travel to the tumour site to show an anti-tumour activity. As shown in Figure 3a, tumours grew in an aggressive and uncontrolled manner when mice were injected with normal saline, while all three GemC18 formulations showed significant anti-tumour activity. The GemC18-PLGA nanoparticles were more effective than the GemC18-PLGA microspheres and the GemC18 solution, with the microspheres and solution showing similar anti-tumour activity.
Figure 3.
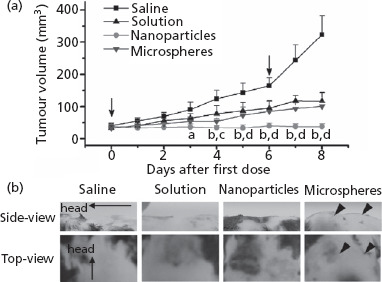
(a) The effect of normal saline, GemC18 solution and GemC18-loaded nanoparticles or microspheres on the growth of TC-1 tumours in C57BL/6 mice. The arrows indicate the time of injection. Data shown are mean ± SD, n = 5 mice. aP < 0.05, nanoparticles vs solution; bP < 0.01, nanoparticles vs solution; cP < 0.05, nanoparticles vs microspheres; dP < 0.01, nanoparticles vs microspheres. (b) Images of mouse skin in sites that were injected with normal saline, GemC18 solution, GemC18-loaded nanoparticles or GemC18-loaded microspheres. Arrow heads indicate the bumps formed in the injection site.
As shown in Figure 3b, injection of normal saline did not cause any local skin damage, while all three GemC18 formulations resulted in visible local skin damage. The depot formed after the injection of the GemC18-PLGA nanoparticles was flat and spread, with no obvious ‘bump’ in the injection site (Figure 3b). However, the depots formed after the injection of the GemC18-PLGA microspheres seemed to be more compact, with two obvious ‘bumps’ formed at the injection sites (Figure 3b).
‘Release’ of GemC18 from the injection site
As shown in Figure 4, 24 h after the injection of the GemC18 solution, only 8.9 ± 0.6% of the GemC18 was recovered from the injection site, while 50.7 ± 9.1% and 76.6 ± 11.7% of the GemC18 was recovered for the GemC18-loaded PLGA nanoparticles and microspheres, respectively. After 48 h, the recovered percentage of GemC18 decreased to 30.4 ± 4.2% and 58.9 ± 9.5% for nanoparticles and microspheres, respectively, while significant further decrease was not observed for the GemC18 solution.
Figure 4.
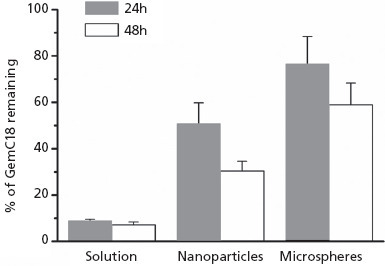
The percentage of GemC18 remaining in mouse footpads 24 and 48 h after the injection of GemC18 solution or GemC18-loaded PLGA nanoparticles or microspheres. Data shown are mean ± SD, n = 3–4. Kruskal–Wallis test revealed that there was a significant difference among the three treatments at both time points (P ≤ 0.05). Post-hoc Dunn's test showed that the values between the ‘solution’ and ‘microspheres’ are different at both time points (P ≤ 0.05). t-test showed that the values of the ‘nanoparticles’ and ‘microspheres’ are different at both time points.
Discussion
In this study, two depot formulations of a fatty-acid amide prodrug of gemcitabine, GemC18, were prepared and evaluated. The GemC18 nanoparticle depot formulation showed a stronger anti-tumour activity than the microsphere formulation, likely due to the proper ‘release’ of GemC18 from the injection site.
Different in-vitro release profiles of GemC18 were observed when comparing the PLGA nanoparticles and microspheres, with the nanoparticle formulation releasing GemC18 at a much faster rate. The higher release rate of GemC18 from the PLGA nanoparticles may be attributed to several factors. First, the higher loading percentage of GemC18 in the PLGA nanoparticles may have resulted in a larger osmotic pressure, which can potentially be the driving force for the diffusion of GemC18 from the solid inner hydrophobic core of the particles to the release medium.[19] Second, the larger surface-area-to-mass ratio of the nanoparticles likely also contributed to the faster release of GemC18.[16,20,21] Third, the residual Pluronic F68 in the nanoparticle formulation may have a synergistic effect with the Tween 80 in the release medium on solubilizing GemC18, and thus increasing its release from the nanoparticles, because it was reported that combined surfactant systems have a synergistic effect on the solubility of poorly water-soluble drugs.[22] This synergistic effect did not exist in the microsphere system because Pluronic F68 was not used in the preparation of the microspheres.
Importantly, in a mouse model with pre-established tumours, the GemC18-PLGA nanoparticle depot formulation showed a stronger anti-tumour activity than the GemC18-PLGA microsphere formulation. This is likely because the GemC18 was ‘released’ at a faster rate from the depot site formed by the injection of the GemC18-PLGA nanoparticles and reached the tumour cells. In fact, as seen in Figure 4, only 30.4 ± 4.2% of the GemC18 was recovered in the injection site 48 h after the injection of the GemC18-PLGA nanoparticles, whereas 58.9 ± 9.5% of GemC18 still remained in the injection site 48 h after the injection of the GemC18-PLGA microspheres. For the gemcitabine to effectively control tumour growth, it is likely that the GemC18 needs to move away from the depot site and reach the tumour site at a proper rate. The slow and limited release of GemC18 from the depot site formed by the GemC18-PLGA microspheres may have allowed the tumour cells to grow too extensively (Figures 2 and 3). The relatively faster ‘release’ of the GemC18 from the depot site formed by the GemC18-PLGA nanoparticles may be attributed to multiple mechanisms. For example, data in Figure 2 clearly show that the GemC18 release from the GemC18-PLGA nanoparticles was faster and more extensive than from the GemC18-PLGA microspheres. Moreover, it is known that nanoparticles are more prone than microspheres (< 10 μm) to penetrate the interstitial space around the injection site, move through the lymphatic capillaries into the lymphatic system[7,16,17] and then enter the circulatory system, eventually accumulating in tumour sites.[18] Finally, we also observed that the GemC18 in Tween 80/ethanol solution was not as effective as the GemC18-PLGA nanoparticles in controlling the tumour growth. As shown in Figure 4, GemC18 in solution diffused away from the injection site very rapidly; over 90% of the GemC18 that was injected into the footpad diffused away from the footpad within 24 h. It is possible that the GemC18 that diffused away from the injection site was quickly eliminated or distributed to tissues other than the tumours and thus became unavailable to control tumour growth. In fact, data from our previous studies showed that GemC18 alone was not effective, or only showed very limited anti-tumour activity, when intravenously injected into tumour-bearing mice.[15,18]
In this study, we showed that the GemC18-loaded PLGA nanoparticle depot formulation successfully inhibited the growth of subcutaneously implanted TC-1 tumours. As previously mentioned, this depot formulation for cancer chemotherapy is different from previously reported formulations, because we aimed at achieving the systemic delivery of GemC18 after depot injection, whereas others were designed for local delivery.[8–10] Therefore, our GemC18-PLGA nanoparticles represent an early attempt to systemically deliver a chemotherapeutic drug using a depot formulation. In fact, we have also compared the effectiveness of this GemC18-PLGA nanoparticle depot formulation with that of the GemC18 solid-lipid nanoparticle (SLN) intravenous formulation previously developed in our laboratory.[15] In C57BL/6 mice with subcutaneous TC-1 tumours, the GemC18-PLGA nanoparticle depot formulation and the GemC18-SLN intravenous formulation showed similar anti-tumour activity, when given at the same dose and dose frequency (Zhu and Cui, unpublished data).
GemC18, instead of gemcitabine, was used in this study partially to avoid the rapid deamination of gemcitabine upon injection. Moreover, GemC18 is a fatty-acid amide prodrug of gemcitabine, which needs to be hydrolysed by amidase to release the parent drug gemcitabine. Therefore, we assumed the GemC18 depot formulation would cause minimal local tissue damage at the injection site. However, it appears that the GemC18 in PLGA nanoparticles and microspheres still induced local skin tissue damage, since a local adverse effect was not observed in the site injected with normal saline (Figure 3b), and the biocompatibility of PLGA is well established.[1] Amidases are abundant in many tissues.[12,19]Thus, GemC18 may have been readily hydrolysed to regenerate gemcitabine once released at the injection site. To further improve the feasibility of using our nanoparticle depot formulation to deliver cytotoxic chemotherapeutic drugs, a more sophisticated prodrug strategy is likely needed. For example, a prodrug that can only be activated by enzymes which are exclusively expressed or overexpressed by tumour cells will likely induce a minimum local toxicity at the injection site.
Conclusions
In this study, we have demonstrated that it is feasible to use a polymeric nanoparticle depot formulation to systemically deliver the prodrug of a traditional cytotoxic chemotherapeutic drug and achieve strong anti-tumour activity. However, a more advanced prodrug strategy has to be integrated into future depot formulations to minimize potential local toxicity.
Declarations
Conflict of interest
The Author(s) declare(s) that they have no conflicts of interest to disclose.
Funding
This work was supported in part by a National Cancer Institute grant CA135274 (to ZC).
References
- Muthu MS et al. PLGA nanoparticle formulations of risperidone: preparation and neuropharmacological evaluation. Nanomedicine 2009; 5: 323–333. [DOI] [PubMed] [Google Scholar]
- Hazekawa M et al. Single injection of ONO-1301-loaded PLGA microspheres directly after ischaemia reduces ischaemic damage in rats subjected to middle cerebral artery occlusion. J Pharm Pharmacol 2012; 64: 353–359. [DOI] [PubMed] [Google Scholar]
- Rabin C et al. In vitro and in vivo demonstration of risperidone implants in mice. Schizophr Res 2008; 98: 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barichello JM et al. Absorption of insulin from pluronic F-127 gels following subcutaneous administration in rats. Int J Pharm 1999; 184: 189–198. [DOI] [PubMed] [Google Scholar]
- Crawford ED et al. A 12-month clinical study of LA-2585 (45.0 mg): a new 6-month subcutaneous delivery system for leuprolide acetate for the treatment of prostate cancer. J Urol 2006; 175: 533–536. [DOI] [PubMed] [Google Scholar]
- Sharifi R et al. Leuprolide acetate 22.5 mg 12-week depot formulation in the treatment of patients with advanced prostate cancer. Clin Ther 1996; 18: 647–657. [DOI] [PubMed] [Google Scholar]
- Lima KM, Rodrigues JM Jr. Poly-DL-lactide-co-glycolide microspheres as a controlled release antigen delivery system. Braz J Med Biol Res 1999; 32: 171–180. [DOI] [PubMed] [Google Scholar]
- Manome Y et al. Local delivery of doxorubicin for malignant glioma by a biodegradable PLGA polymer sheet. Anticancer Res 2006; 26: 3317–3326. [PubMed] [Google Scholar]
- Ranganath SH et al. Hydrogel matrix entrapping PLGA-paclitaxel microspheres: drug delivery with near zero-order release and implantability advantages for malignant brain tumour chemotherapy. Pharm Res 2009; 26: 2101–2114. [DOI] [PubMed] [Google Scholar]
- Benny O et al. Local delivery of poly lactic-co-glycolic acid microspheres containing imatinib mesylate inhibits intracranial xenograft glioma growth. Clin Cancer Res 2009; 15: 1222–1231. [DOI] [PubMed] [Google Scholar]
- Barton-Burke M. Gemcitabine: a pharmacologic and clinical overview. Cancer Nurs 1999; 22: 176–183. [DOI] [PubMed] [Google Scholar]
- Brusa P et al. Antitumor activity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. Anticancer Res 2007; 27: 195–199. [PubMed] [Google Scholar]
- Arias JL et al. Magnetoresponsive squalenoyl gemcitabine composite nanoparticles for cancer active targeting. Langmuir 2008; 24: 7512–7519. [DOI] [PubMed] [Google Scholar]
- Immordino ML et al. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Control Release 2004; 100: 331–346. [DOI] [PubMed] [Google Scholar]
- Sloat BR et al. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int J Pharm 2011; 409: 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel) 2011; 3: 1377–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandor M et al. A novel polyethylene depot device for the study of PLGA and P(FASA) microspheres in vitro and in vivo. Biomaterials 2002; 23: 4413–4423. [DOI] [PubMed] [Google Scholar]
- Zhu S et al. Lysosomal delivery of a lipophilic gemcitabine prodrug using novel acid-sensitive micelles improved its antitumor activity. Bioconjug Chem 2012; 23: 966–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais PC et al. Tailoring magnetic nanoparticle for transformers application. J Nanosci Nanotechnol 2010; 10: 1251–1254. [DOI] [PubMed] [Google Scholar]
- Almond BA et al. Efficacy of mitoxantrone-loaded albumin microspheres for intratumoral chemotherapy of breast cancer. J Control Release 2003; 91: 147–155. [DOI] [PubMed] [Google Scholar]
- Chakravarthi SS et al. Comparison of anti-tumor efficacy of paclitaxel delivered in nano- and microparticles. Int J Pharm 2010; 383: 37–44. [DOI] [PubMed] [Google Scholar]
- Nazzal S et al. Preparation and characterization of coenzyme Q10-Eudragit solid dispersion. Drug Dev Ind Pharm 2002; 28: 49–57. [DOI] [PubMed] [Google Scholar]